

Abstract
P2X4 receptors are calcium-permeable cation channels gated by extracellular ATP. They are found close to subsynaptic sites on hippocampal CA1 neurons. We compared features of synaptic strengthening between wild-type and P2X4 knockout mice (21–26 days old). Potentiation evoked by a tetanic presynaptic stimulus (100 Hz, 1 s) paired with postsynaptic depolarization was less in P2X4−/− mice than in wild-type mice (230 vs. 50% potentiation). Paired-pulse ratios and the amplitude and frequency of spontaneous excitatory postsynaptic currents (EPSCs) were not different between wild-type and knockout mice. Prior hyperpolarization (ten 3 s pulses to −120 mV at 0.17 Hz) potentiated the amplitude of spontaneous EPSCs in wild-type mice, but not in P2X4−/− mice; this potentiation was not affected by nifedipine, but was abolished by 10 mm 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA) in the recording pipette. The amplitude of N-methyl-d-aspartate EPSCs (in 6-cyano-7-nitroquinoxaline-2,3-dione, 10 or 30 μm, at −100 mV) facilitated during 20 min recording in magnesium-free solution. In wild-type mice, this facilitation of the N-methyl-d-aspartate EPSC was reduced by about 50% by intracellular BAPTA (10 mm), ifenprodil (3 μm) or 4-(4-fluorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)1H-imidazole (5 μm). In P2X4−/− mice, the facilitation was much less, and was unaffected by intracellular BAPTA, ifenprodil (3 μm) or mitogen-activated protein (MAP) kinase inhibitor 4-(4-fluorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)1H-imidazole (5 μm). This suggests that the absence of P2X4 receptors limits the incorporation of NR2B subunits into synaptic N-methyl-d-aspartate receptors.
Keywords: brain slice, hippocampus, NR2B receptors, P2X4 receptors, plasticity, whole-cell recording
Introduction
N-methyl-d-aspartate (NMDA) receptor-dependent long-term potentiation (LTP) is a widely studied cellular model of synaptic plasticity in the CA1 region of the hippocampus, and it may be related to spatial memory (Bliss & Lomo, 1973; Whitlock et al., 2006). In CA1 neurons, NMDA receptor-dependent LTP is typically monitored by recording changes in the amplitude of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor-mediated synaptic currents (or potentials) following a tetanic stimulus to the Schaffer collateral commissure fiber tract. A consistent observation for the induction of this form of LTP is a rise in postsynaptic intracellular calcium ([Ca2+]i), and it is well established that the main source of the [Ca2+]i is entry through the calcium-permeable NMDA receptors (Bliss & Collinridge, 1993; Malenka & Nicoll, 1993). This rise in [Ca2+]i results in the increased delivery of AMPA receptor subunits to the synapse, by a process requiring several interacting proteins and phosphorylation of glutamate receptor type 1 subunits by Ca2+/calmodulin-dependent protein kinase II (reviewed by Kessels & Malinow, 2009).
The P2X4 receptors have been shown to be located in the perisynaptic region of the postsynaptic membrane of CA1 neurones (Rubio & Soto, 2001). P2X4 receptors have a high permeability to calcium (PCa/PNa = 4) (Soto et al., 1996; North, 2002; Egan & Khakh, 2004). There is evidence in various brain regions that ATP is co-released at glutamate synapses (Pankratov et al., 2006, 2007). These three findings prompted the hypothesis that calcium entry through postsynaptic P2X4 receptors might contribute in some way to synaptic strengthening during repetitive stimulation. The features of calcium entry through P2X4 receptors differ from that through NMDA receptors, because P2X receptors do not show the voltage dependence (resulting from magnesium block) that characterizes most NMDA receptor currents (Mayer et al., 1984; Nowak et al., 1984).
The first studies of LTP in mice genetically modified to lack P2X4 receptors (P2X4−/−) used extracellular recording of field potentials from the CA1 region of hippocampal slices (Sim et al., 2006). These showed that synaptic facilitation was less than that observed in wild-type mice. It was also noticed that ivermectin, which potentiates currents at P2X4 receptors, increased LTP in wild-type but not in knockout mice. This finding was broadly consistent with the hypothesis that calcium entry through P2X4 receptors contributed to synaptic strengthening. However, it provided no mechanistic insight, and left open several possible alternative explanations. In the present work we have used whole-cell recording from CA1 pyramidal neurons to compare more systematically the synaptic currents evoked by Schaffer collateral stimulation between wild-type and P2X4−/− mice.
Materials and methods
Slice preparation and recording
We used male C57/BL6 mice at postnatal day 21–26, either wild-type or P2X4−/− (Sim et al., 2006). Mice were killed as Schedule 1 procedures under a United Kingdom Home Office License and in accordance with the regulations of the United Kingdom Animal (Scientific Procedures) Act (1986). Transverse hippocampal slices (300–400 μm thick) were prepared (Vibraslicer, VT1000S; Leica, Solms, Germany) in an artificial cerebrospinal fluid containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 4 MgCl2, 0.5 CaCl2, 26 NaHCO3, 25 glucose, 1 mm kyurenic acid, bubbled with 95% O2/5% CO2 (pH 7.4 and 300–315 mOsm/l). Experimenters were unaware of the genetic background of the mouse from which brain slices were prepared. Slices were incubated at 20–23 °C in artificial cerebrospinal fluid containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1.3 MgCl2, 2 CaCl2, 26 NaHCO3, 25 glucose, saturated with 95% O2/5% CO2 (pH 7.4 and 300–315 mOsm/l) for at least 1 h before recording. For recording, slices were fully submerged in a flowing solution (4 mL/min) of the same artificial cerebrospinal fluid with added picrotoxin (50 μm) and 8-cyclopentyl-1,2-dipropylxanthine (100 nm), to block GABAA and adenosine A1 receptors, respectively. CA1 pyramidal neurons were visualized with infrared differential interference contrast optics (BX50WI; Olympus, Southend, UK), and whole-cell currents recorded with the integrated HEKA EPC9/2 amplifier system (HEKA, Lambretch, Germany). Patch pipettes were made from thin-walled borosilicate glass (Harvard Apparatus, Kent, UK) on a puller (PP-830; Narishige, Tokyo, Japan) and polished on a microforge (Narishige). The pipettes had resistances of 5–10 MΩ when filled with intracellular solution containing (in mM): 130 CsCH3SO4, 8 NaCl, 10 HEPES, 0.2 ethylene glycol bis(2-aminoethylether)-N,N,N′,N′-tetra-acetic acid (EGTA), 5 N-(2,6-dimethyl-phenylcarbamoylmethyl)triethylammonium chloride, pH adjusted to 7.3 with CsOH and an osmolarity of 280–295 mOsm/l. N-(2,6-dimethyl-phenylcarbamoylmethyl)triethylammonium chloride was included to prevent action potential generation in the neuron from which the recording was made. In some experiments, 0.2 mm EGTA was replaced by 10 mm 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA). Recordings were performed at room temperature (20–23 °C). Data were acquired using Pulse software (HEKA) for subsequent analysis.
The NMDA receptor-mediated excitatory postsynaptic currents (EPSCs) were isolated pharmacologically. EPSCs were first elicited at −70 mV at a rate of 0.05 Hz, and the stimulus strength was adjusted to evoke AMPA receptor EPSCs of 250–300 pA. The artificial cerebrospinal fluid was then changed to one containing reduced MgCl2 (0.1 mm) with added 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (30 μm). After establishing stable NMDA EPSCs, a 30 min period commenced in which EPSCs were evoked every 20 s, each during a 1 s step hyperpolarization to −100 mV. After 10 min baseline, the solution was changed from one containing 0.1 mm MgCl2 to one with no added MgCl2 (also with CNQX), and recording continued for the remaining 20 min.
The drugs used were: N-(2,6-dimethyl-phenylcarbamoylmethyl)triethylammonium chloride, picrotoxin, nifedipine (Sigma-Aldrich, Poole, UK), d-2-amino-5-phosphonopentanoic acid, (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine maleate, 8-cyclopentyl-1,3-dipropylxanthine, CNQX, ifenprodil, 4-(4-fluorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)1H-imidazole (all from Tocris Biosciences, Bristol, UK) and tetrodotoxin (Calbiochem, Nottingham, UK).
Spontaneous excitatory postsynaptic currents
Spontaneous EPSCs (sEPSCs) were acquired at −60 mV with intracellular N-(2,6-dimethyl-phenylcarbamoylmethyl)triethylammonium chloride (5 mm). In some experiments, trains of depolarizing potential steps were applied to increase the EPSC frequency (Wyllie et al., 1994); these were trains of 10 steps from −60 to −20 mV, each lasting 3 s with an interval of 6 s. In other experiments, we applied trains of hyperpolarizing pulses from −60 to −120 mV (each of 3 s duration, at intervals of 6 s), reasoning that this would increase calcium entry through P2X4 receptors but reduce calcium entry through NMDA receptors.
Data analysis
The EPSC amplitudes were calculated using Pulsefit software (HEKA) and kinetic parameters were measured using Axograph 4.8 (Molecular Devices, USA), plotted either in Kaleidagraph version 4 (Synergy Software, Reading, PA, USA) or Prism version 4 (Graphpad Software Inc., San Diego, CA, USA) and illustrated using Canvas X (ACD Systems, Victoria, BC, Canada). For sEPSCs, individual current traces were selected using Pulsefit software (HEKA) and concatenated using Axograph software. The amplitudes, frequencies and kinetics of sEPSCs were measured using MiniAnalysis 6.01 (Synaptosoft, Fort Lee, NJ, USA). The threshold for the detection of EPSCs was typically 4–6 pA. To be included for analysis, EPSCs had to have a monotonic rising phase with a 10–90% rise time of < 6 ms and a single exponential decay with time constant < 25 ms. The great majority of EPSCs had rise times and decay time constants that were considerably less than these upper limits. It is likely that these criteria allow the inclusion of events not in close proximity to the neuron soma (Smith et al., 2003).
The sEPSC amplitudes were estimated by subtracting the mean current amplitude recorded in the 5 ms before the event from the average of five data points at the peak of the event. The decay of EPSC was fit with a single exponential component to obtain an estimate of the decay time constant. Mean EPSC waveforms were obtained by averaging between 100 and 200 individual events. Due to variation in the mean amplitudes and frequencies of EPSCs, when reporting the mean values for data pooled from series of experiments, we have normalized these values to those recorded in the preconditioning period before the application of trains of depolarizing or hyperpolarizing steps. To calculate differences in the distributions of sEPSC amplitudes, we constructed cumulative probability plots. Non-stationary noise analysis (Traynelis et al., 1993; Hartveit & Veruki, 2006) was used to estimate the average number of functional receptors at the peak of the response. Briefly, the mean current (300–500 events) was scaled to each individual event (MiniAnalysis software), and the resultant variance plot as a function of the mean was fitted with a parabola.
All results are presented as mean ± SEM. Statistical analysis of EPSC and sEPSC amplitudes (and frequencies of sEPSCs) was performed with SigmaStat v3 (Aspire Software International, Ashburn, VA, USA) using Mann–Whitney U-tests, Kolmogorov–Smirnov and anova as appropriate. Statistical analysis of paired-pulse and NMDA EPSCs was carried out using the Prism 4 (Graphpad Software Inc.) unpaired Student’s t-test. Significant differences were inferred when P < 0.05.
Results
Evoked alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor-mediated excitatory postsynaptic currents
The input resistance and cell capacitance were not different between wild-type (370 ± 10.7 MΩ and 24.5 ± 2.8 pF, n = 14) and P2X4−/− (349 ± 9.7 MΩ and 29.9 ± 21.9 pF, n = 15) mice. With similar stimuli (3–4 V, 0.2 ms), the mean EPSC amplitude in wild-type mice was not different from that in P2X4−/− mice (40.8 ± 5.2 pA, n = 12 and 37.6 ± 4.3 pA, n = 14) (Fig. 1A). Evoked EPSCs were completely blocked by tetrodotoxin (300 nm) or CNQX (30 μm; n = 3) (Fig. 1A) in both groups of mice.
FIG. 1.
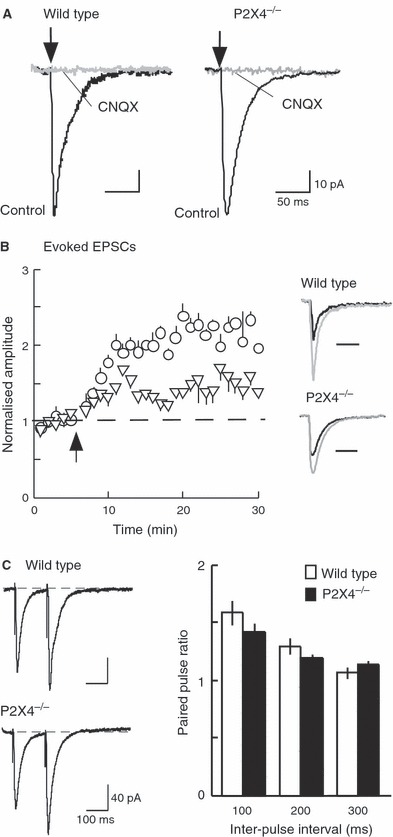
AMPA receptor-mediated evoked EPSCs. (A) CNQX (30 μm) completely blocks EPSCs in wild-type and P2X4−/− mice. (B) Potentiation of EPSC amplitude following tetanic stimulation is much less in P2X4−/− (triangles, n = 4) than wild-type (open circles, n = 4) mice. At 15 and 25 min after the tetanus, the difference is highly significant (P < 0.001). Inset: EPSCs in control (black) and after tetanus (gray) in wild-type and P2X4−/− mice. (C) Paired-pulse facilitation. Left: typical recordings from wild-type (top) and P2X4−/− (bottom) mice. Each trace is an average of five sweeps, showing two EPSCs evoked with an interpulse interval of 150 ms. Right: summary data show no difference in paired-pulse facilitation between wild-type (open bars) and P2X4−/− (filled bars) mice.
A single tetanic stimulus (100 Hz for 1 s) combined with postsynaptic depolarization (0 mV for 1 s) evoked a sustained increase in EPSC amplitude. This increase was less in P2X4−/− (approximately 0.5-fold increase) than in wild-type (approximately 2.3-fold increase) mice (Fig. 1B). The increase did not reverse over the time course of the experiment (30–60 min). There was no change in the rise time or decay time constant of the EPSC during this potentiation in either group of mice. The lesser potentiation observed in P2X4−/− mice confirms the observations reported by Sim et al. (2006) using extracellular field recording.
Figure 1C shows that the amplitude of the second EPSC was larger than the first when they were separated by 100 or 200 ms. Such paired-pulse facilitation is generally considered to result from ‘residual’ presynaptic calcium elevation enhancing release evoked by the second action potential of the pair (Katz & Miledi, 1968). However, paired-pulse facilitation was not different between wild-type and P2X4−/− mice.
Spontaneous excitatory postsynaptic currents
Very few sEPSCs were observed in the presence of tetrodotoxin, and the following experiments were therefore carried out without tetrodoxin but in the presence of picrotoxin (50 μm). sEPSCs had monophasic rise times and single exponential decays. The rise times (10–90%) and decay time constants of sEPSCs were not different between wild-type and P2X4−/− mice (wild-type: 1.4 ± 0.1 and 12.2 ± 0.4 ms; P2X4−/−: 1.6 ± 0.3 and 16.1 ± 0.3 ms; n = 600 events in each case). The mean amplitude in wild-type mice was 15.3 ± 1.3 pA (n = 600 events, Fig. 2C) and in P2X4−/− mice it was 15.4 ± 2.5 pA (n = 600 events, Fig. 2A). CNQX (30 μm) blocked all events in wild-type and P2X4−/− mice (Fig. 2C). (+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine maleate (30 μm) had no effect on their amplitude, frequency or decay time constant. Over a period of 30 min recording, sEPSCs from both groups had stable amplitudes (Fig. 2D), and their frequency progressively decreased. The frequencies did not differ between wild-type and P2X4−/− mice [at 10 min: 0.77 ± 0.15 Hz (n = 4) and 0.74 ± 0.14 Hz (n = 5), respectively; at 30 min: 0.50 ± 0.01 Hz (n = 4) and 0.54 ± 0.13 Hz (n = 5), respectively].
FIG. 2.
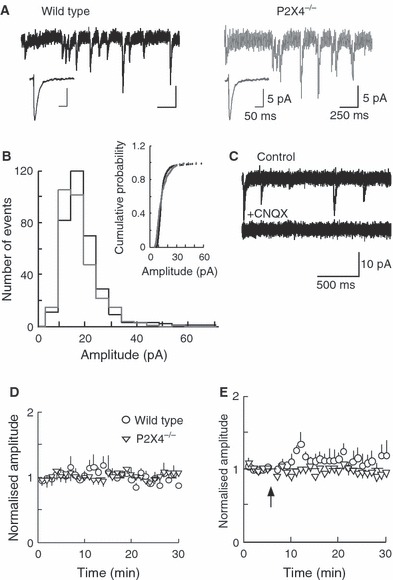
(A) Typical sEPSCs from wild-type (left) and P2X4−/− (right) mice. Inset: average event (n = 200) at higher time resolution. (B) EPSC amplitude histograms do not differ between wild-type (black) and knockout (gray) mice. Inset: cumulative probability plot. (C) CNQX (30 μm) blocks all sEPSCs. (D) Normalized average amplitude of sEPSCs of wild-type (open circles, n = 4) and P2X4−/− (triangles, n = 5) mice during 30 min recording. (E) Tetanic stimulation (arrow) does not produce long-lasting potentiation of the amplitudes of sEPSCs in wild-type (open circles, n = 5) or P2X4−/− (open triangles, n = 4) mice. Holding potentials, −60 mV.
A series of 10 depolarizing pulses from −60 to +20 mV (3 s duration, given at 6 s intervals) increased the amplitudes of sEPSCs. This effect was significantly larger in wild-type (from 13.5 ± 1.3 to 24.1 ± 1.3 pA, n = 8) than in P2X4−/− (from 13.7 ± 0.1 to 17.0 ± 0.8 pA, n = 9) mice (Fig. 3A). There was no difference between the time courses of the sEPSC between the two groups. The effect on sEPSC amplitude in wild-type mice was apparent from the rightward skew in the amplitude histograms (11.6–15.1 pA) (Fig. 3C), but this was negligible in P2X4−/− mice. (+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine maleate (30 μm; n = 5) had no effect on the potentiation of sEPSC amplitudes that followed a series of depolarizing pulses (Fig. 3B). The potentiation was blocked by nifedipine (10 μm; n = 8) (Fig. 3B), indicating its dependence on calcium entry through voltage-dependent calcium channels (Baxter & Wyllie, 2006).
FIG. 3.
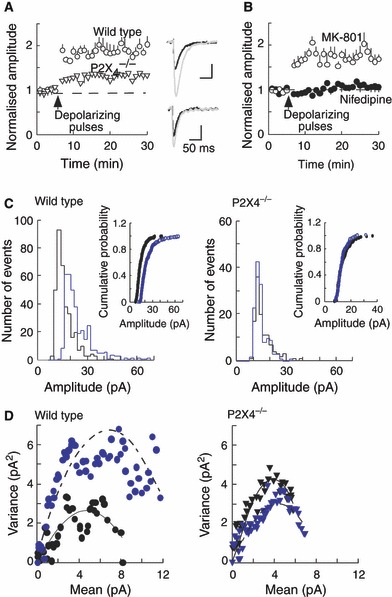
Depolarizing postsynaptic pulses cause a sustained increase in sEPSC amplitudes. (A) sEPSC amplitudes increase by about 90% in neurons from wild-type mice (open circles, n = 8) and by about 30% in neurons from P2X4−/− mice (open triangles, n = 9). (B) In wild-type mice, the increase in sEPSCs was unaffected by (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine maleate (MK-801) (10 μm, open circles, n = 5), but was blocked by nifedipine (10 μm, filled circles, n = 7). In A and B, the stimulus (upward arrowhead) was a train of 10 depolarizing steps (from −60 to 20 mV, 3 s duration) applied at 6 s intervals. (C) Amplitude histograms before (black line) and after (gray line) the depolarizing pulse stimulus, and cumulative probability plots (inset) show the increase in amplitude observed in wild-type (left) but not in P2X4−/− (right) mice. (D) For wild-type mice (left), non-stationary noise analysis for control sEPSCs (black symbols) and sEPSCs following depolarizing pulses (gray symbols) shows an increase in variance against the change in mean amplitude, suggesting an increase in receptor number. This is not seen for P2X4−/− mice (right).
Non-stationary noise analysis (Traynelis et al., 1993; Hartveit & Veruki, 2006) for wild-type mice showed a rightward skew in the parabola following depolarizing pulses (Fig. 3D), indicating that the potentiation is probably the result of an increase in receptor number at the synapses. In contrast, the results obtained from P2X4−/− mice showed overlapping amplitude histograms for control sEPSCs and sEPSCs following depolarizing pulses. We observed that the frequency of sEPSCs after depolarizing pulses did not display the rundown at 30 min that was seen in the control recordings from wild-type mice (values were 0.82 ± 0.12 and 0.96 ± 0.05 Hz at 30 min, n = 6). In knockout mice, the frequencies were 0.69 ± 0.19 and 0.70 ± 0.2 Hz, n = 6. These differences between wild-type and P2X4−/− mice suggest that the latter group has an impaired ability to deliver new AMPA receptors to the subsynaptic membrane following a rise in postsynaptic [Ca2+]i.
Hyperpolarization would increase the driving force through open P2X4 receptor channels, and reduce the current through NMDA receptors and voltage-dependent calcium currents. Figure 4A shows that a series of hyperpolarizing pulses is followed by a sustained increase in sEPSC amplitude in wild-type but not in P2X4−/− mice. The baseline amplitude of 14.4 ± 1.6 pA increased to 21.1 ± 1.7 pA (n = 5). There was no change in sEPSC frequency (0.63 ± 0.18 Hz). In P2X4−/− mice, the sEPSC amplitudes were 13.1 ± 0.7 pA (n = 6) before and 13.9 ± 0.11 pA (n = 6) after the series of hyperpolarizing pulses, and the frequency was 0.62 ± 0.12 Hz (n = 6). Non-stationary noise analysis indicated an increase in the variance at the largest change from the mean amplitude value (Fig. 4D), suggesting an increase in the number of AMPA receptors present at the synapse. This shift in amplitude was much smaller in the P2X4−/− mice (Fig. 4D). sEPSC amplitudes were not different between wild-type and P2X4−/− mice, and hyperpolarizing pulses did not elicit any sustained increase in sEPSC amplitude when recordings were made with electrodes containing BAPTA (10 mm) (n = 3) (Fig. 4B). The increase did not appear to involve L-type voltage-dependent calcium channels, because it was unaffected by nifedipine (10 μm) (n = 6) (Fig. 4C).
FIG. 4.
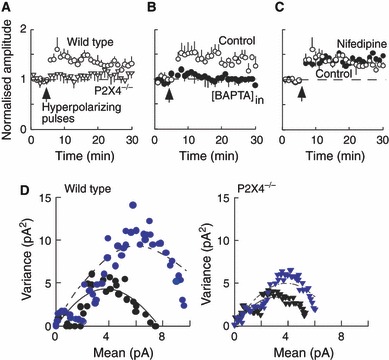
Hyperpolarizing postsynaptic pulses cause a sustained increase in sEPSC amplitude. (A) In wild-type mice (open circles, n = 5), there is an increase of about 50% in the sEPSC amplitude following a train of hyperpolarizing pulses. This is not observed in P2X4−/− mice (triangles, n = 6). (B) The increase in sEPSCs is not observed when recording electrodes contain BAPTA (10 mm) (filled circles, n = 3) compared with when they contain EGTA (0.2 mm) (Control, open circles, n = 5). (C) The increase in sEPSCs is unaffected by nifedipine (10 μm) (filled circles, n = 6) compared with control (open circles, n = 5). (D) In wild-type mice (left), non-stationary noise analysis of sEPSCs in control conditions (black circles) and following hyperpolarizing pulses (gray circles) suggests an increase in receptor number underlying this potentiation. In P2X4−/− mice (right), non-stationary noise analysis of sEPSCs in control conditions (black triangles) and following hyperpolarizing pulses (gray triangles) shows a much smaller rightward shift. Representative record from one experiment.
Evoked N-methyl-d-aspartate receptor-mediated excitatory postsynaptic currents
The NMDA receptor EPSCs were isolated by recording at +40 mV (Fig. 5A); the amplitude was measured at 100 ms from their onset to eliminate any contamination from reversed AMPA components. The decay time constants were 65.6 ± 2.5 ms (n = 13) and 63.7 ± 4.5 ms (n = 16) in wild-type and knockout mice, respectively. There was no difference in the amplitudes of NMDA EPSCs between wild-type (40.8 ± 5.0 pA, n = 13) and knockout (42.2 ± 3.8 pA, n = 16) mice (Fig. 6B), and the AMPA : NMDA ratio also showed no significant difference between the two groups of animals (P = 0.74) (Fig. 5C). When intracellular EGTA was replaced with BAPTA, the AMPA : NMDA ratio was reduced in both wild-type and P2X4−/− mice, but there remained no difference between the two groups (P = 0.17).
FIG. 5.
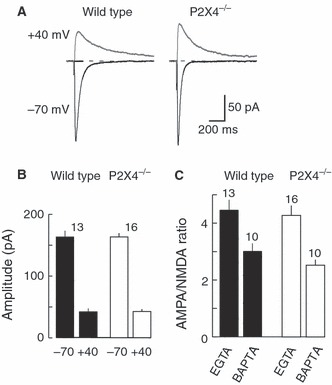
NMDA receptor-mediated evoked EPSCs at +40 mV. (A) Typical recordings of evoked EPSCs recorded at −70 and +40 mV (each average of five sweeps) from wild-type (left) and P2X4−/− (right) mice. (B) The amplitudes of the AMPA (peak amplitude, at −70 mV) and NMDA (amplitude at 100 ms, at +40 mV) components did not differ between wild-type (filled columns) and P2X4−/− (open columns) mice. (C) AMPA : NMDA ratio does not differ between wild-type (filled columns) and P2X4−/− (open columns) mice, whether the recording electrode contained EGTA or BAPTA.
FIG. 6.
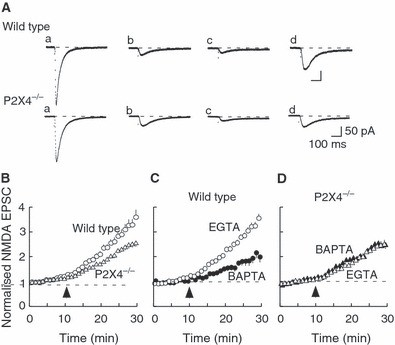
NMDA receptor-mediated EPSCs isolated pharmacologically at −100 mV. (A) Representative EPSCs at indicated times [a, before CNQX; b, in CNQX with 0.1 mM magnesium (2min); c, in CNQX and 0.1 mM Mg (10 min); d, in CNQX and 0 mM Mg (20 min)]. Traces are averages of five sweeps. (B) The increase in NMDA receptor-mediated EPSCs was less in neurons from P2X4−/− mice (triangles, n = 15) than in neurons from wild-type mice (open circles, n = 14). (C) In wild-type mice, the progressive increase in EPSC amplitude that occurred in Mg-free solution was much less when recording electrodes contained BAPTA (filled circles) rather than EGTA (open circles). (D) In P2X4−/− mice, no difference was observed whether electrodes contained BAPTA (filled triangles) or EGTA (open triangles).
The NMDA receptor EPSCs were also isolated pharmacologically by holding neurons at −70 mV in reduced MgCl2 (0.1 mm) and CNQX (30 μm); the EPSCs were evoked during a hyperpolarizing step (1 s) from −70 to −100 mV. The amplitude of the NMDA receptor-mediated EPSC was 21.3 ± 1.5 pA (n = 14) and 18.7 ± 1.3 pA (n = 15) in P2X4−/− mice (Fig. 6A) (P > 0.05). In wild-type mice, complete removal of magnesium from the external solution led to a progressive increase in the amplitude of the EPSC, by a factor of 1.75 ± 0.17 (i.e. control = 1.00; n = 14) over 20 min (Fig. 6B). However, in P2X4−/− mice, this slow increase (which we term facilitation) was substantially less (1.15 ± 0.11, n = 15, P < 0.005) (Fig. 6B). The facilitated NMDA EPSC was completely abolished by d-2-amino-5-phosphonopentanoic acid (20 μm). In control experiments, we found that there was no change in NMDA EPSCs in either wild-type or P2X4−/− mice when recordings were continued in 0.1 mm MgCl2 over the same period of 20 min; in other words, the slow increase in amplitude was elicited by the change to a totally magnesium-free solution.
We hypothesized that Ca2+ entry through P2X4 receptors may contribute to the observed difference between wild-type and P2X4−/− mice in the facilitation of NMDA receptor EPSCs in the magnesium-free solution. Figure 6C shows that, in wild-type mice, the facilitation was much less when recording electrodes contained BAPTA (10 mm) rather than EGTA (0.2 mm); after 20 min the increase was by a factor of 1.04 ± 0.14 (n = 11) (compared with control, P < 0.005) (Fig. 6C). However, in P2X4−/− mice, the facilitation following complete magnesium removal was not different (P = 0.34) whether the recording electrodes contained EGTA (1.15 ± 0.11, n = 15) or BAPTA (1.29 ± 0.05, n = 9) (Fig. 6D). These findings suggest that the slow facilitation of NMDA receptor-mediated EPSCs that follows the complete removal of magnesium was dependent on the presence of P2X4 receptors. The slow time course of the facilitation might indicate the recruitment of NMDA receptors to the synapses.
The NR2B subunits have been reported to play a role in synaptic transmission at CA1 neurones (Gardoni et al., 2009). Ifenprodil (3 μm), an antagonist selective for receptors that contain a NR2B subunit (Williams, 1993), prevented the facilitation of NMDA receptor EPSCs in wild-type mice. The normalized amplitude was 0.83 ± 0.16 (n = 14) compared with a control value of 1.75 ± 0.17 (n = 14) (Fig. 7A). Ifenprodil had no effect on the (lesser) facilitation of NMDA EPSCs observed in P2X4−/− mice (ifenprodil: 0.89 ± 0.17, n = 7; control: 1.15 ± 0.11, n = 15) (Fig. 7B). In these experiments, ifenprodil was simply added to the nominally magnesium-free solution. In separate experiments, we evoked the NMDA EPSCs at 0.05 Hz for 30 min, holding at −70 mV in the presence of CNQX (30 μm) and nominally magnesium-free solution. Ifenprodil (3 μm) reduced the amplitude of NMDA EPSCs by 30% in wild-type mice (Fig. 7B) but had no effect in P2X4−/− mice.
FIG. 7.
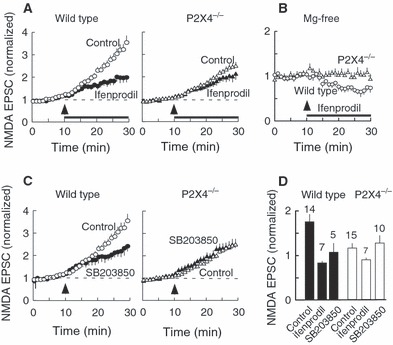
Growth in NMDA receptor-mediated EPSC in Mg-free solution (facilitation) involves NR2B subunits. (A) In wild-type mice (left), the increase in EPSC amplitude is much reduced by ifenprodil (3 μm). In control cells (open circles), the facilitation was by a factor of 1.7 ± 0.2 (n = 14); in ifenprodil the factor was 0.8 ± 0.12 (n = 14). In P2X4−/− mice (right), ifenprodil has no effect on the growth in EPSC amplitude. The application of ifenprodil (solid bar) began at the same time as the change to Mg-free solution (upward arrowhead). (B) After prolonged treatment with Mg-free solution (20 min), ifenprodil reduces the amplitude of the EPSC in neurons from wild-type mice (open circles), but not in neurons from P2X4−/− mice (triangles). (C) In wild-type mice (left), the increase in EPSC amplitude (Control, open circles) was reduced by 4-(4-fluorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)1H-imidazole (SB203850) (5 μm, 1 h pretreatment) (closed circles). This effect was not seen in neurons from P2X4−/− mice (right). (D) In wild-type mice, the degree of inhibition by ifenprodil (3 μm) was similar to that observed by pretreatment with SB203850 (5 μm).
These findings indicate that, in the wild-type mice, there is little evidence for an NR2B component in the synaptic NMDA receptors, but that an NR2B component appears in the synaptic NMDA EPSC during 20 min perfusion with magnesium-free solution; this increase in the NR2B component was not observed in P2X4−/− mice. It has been suggested that phosphorylation of NR2B subunits may stabilize them at the synaptic membrane (Goebel-Goody et al., 2009). We found that pretreatment with the p38 MAP kinase inhibitor 4-[5-(4-fluorophenyl)-2-(4-[(S)-methylsulfinyl]phenyl)-3H-imidazol-4-yl]pyridine [4-(4-fluorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)1H-imidazole] (5 μm, 1 h) also prevented the slow facilitation in magnesium-free solution; the level was 1.06 ± 0.21 (n = 10) compared with the control level of 1.75 ± 0.17 (n = 14). This level was not significantly different (P = 0.34) from that observed for P2X4−/− mice, either in the presence of 4-(4-fluorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)1H-imidazole (1.36 ± 0.17, n = 10) or in its absence (1.15 ± 0.11, n = 15) (Fig. 7C). Figure 7D summarizes the effect of both ifenprodil and 4-(4-fluorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)1H-imidazole in wild-type and P2X4−/− mice.
Discussion
We discuss five aspects of the present work. Four deal with the AMPA component of the EPSC in mouse hippocampal CA1 cells, and the fifth deals with the NMDA component. First, our results provide no evidence that any component of the evoked EPSC is directly mediated by P2X receptors. Pankratov et al. (1998) found that a component of the EPSC (up to 25% of amplitude) in CA1 neurons from rats (16–19 days old) was not blocked by CNQX (20 μm), but it was blocked by pyridoxal phosphate-6-azophenyl-2′-4′-disulphonic acid (10 μm). However, rat P2X4 receptors are known to be very insensitive to blockade by pyridoxal phosphate-6-azophenyl-2′-4′-disulphonic acid (North, 2002), and this raises the possibility that pyridoxal phosphate-6-azophenyl-2′-4′-disulphonic acid may have been blocking residual AMPA receptor currents. We have not observed any such residual component in 20–26-day-old mice using 30 μm CNQX, and we also observed no difference in the EPSCs between wild-type and P2X4−/− mice.
The second observation that merits discussion relates to LTP. When depolarization was paired with a train of stimuli applied to the Schaffer collateral, we observed approximately twofold potentiation of the AMPA EPSC (recorded at −60 mV), which lasted for at least 20 min. This potentiation was much reduced in slices from P2X4−/− mice, which is consistent with the observations using extracellular field recordings by Sim et al. (2006). The main purpose of the present work was to determine whether this reduced LTP could have a simple explanation, such as calcium entry through postsynaptic P2X4 receptors that was absent in the knockout mice. This is a reasonable hypothesis, given the high calcium permeability of P2X4 receptors (Soto et al., 1996; North, 2002).
Third, our experiments provide no evidence for any presynaptic component to the effect of P2X4 receptors. Presynaptic actions of P2X receptors have been described in other regions of the central nervous system (e.g. spinal cord, Gu & MacDermott, 1997; mesencephalic nucleus motoneurons, Khakh & Henderson, 1998) but electron microscopic studies by Rubio & Soto (2001) indicate that the predominant localization of P2X4 receptors in the CA1 region of the hippocampus is postsynaptic. The absence of any difference in paired-pulse facilitation or in sEPSC frequency between wild-type and P2X4−/− mice is consistent with the failure to observe presynaptic P2X4 receptors in CA1.
The fourth area for discussion is the sEPSCs. We found that the frequency of sEPSCs in the presence of tetrodotoxin was too low to allow useful observation on ‘miniature’ EPSCs in 20–26-day-old mice. There appears to be a species difference here, because similarly prepared slices from rat hippocampus exhibit sEPSCs in the presence of tetrodotoxin (Baxter & Wyllie, 2006). In the present experiments, the spontaneous events will also include events that are driven by presynaptic action potentials. Nonetheless, two forms of exclusively postsynaptic manipulation elicited an increase in the mean amplitude of such events.
In wild-type mice, a train of postsynaptic depolarizing pulses potentiated sEPSCs in a way that was independent of NMDA receptors [not blocked by (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine maleate; Fig. 3B] but was dependent on the activation of L-type voltage-dependent calcium channels (blocked by nifedipine, Fig. 4A). These findings replicate those of Wyllie et al. (1994) and Baxter & Wyllie (2006) in rat CA1 neurons. Previous work indicates that the steps involved in the increase in EPSC amplitude following a train of depolarizing pulses involves (i) an increase in [Ca2+]i (including calcium-induced calcium release from intracellular stores), (ii) activation of phosphatidylinositol 3 kinase, and (iii) increased delivery of AMPA receptors to the subsynaptic membrane (Baxter & Wyllie, 2006). The present experimental results are consistent with the first and third of these points (i.e. the block by intracellular BAPTA, and the rightward shift in the variance/peak amplitude curve). The absence of this potentiation of sEPSCs in P2X4−/− mice is not readily explained by a simple loss of postsynaptic membrane P2X4 receptors. However, nifedipine (3 μm) has been reported to block P2X receptors in PC12 cells (Hur et al., 2001); if it had such an action on hippocampal P2X4 receptors, then this might underlie a component of the potentiation. Alternatively, it might suggest compensatory changes to AMPA receptor recycling mechanisms resulting from the loss of P2X4 receptors.
Our experiments using hyperpolarizing pulses were prompted by the expectation that this would increase calcium entry through any open P2X4 channels, in conditions where entry through NMDA receptors and voltage-gated calcium channels would be negligible (see Pankratov et al., 2006). We found that strong hyperpolarization also elicited a sustained increase in the amplitude of sEPSCs in wild-type mice (Fig. 4); non-stationary noise analysis again indicated that this resulted from an increase in the number of AMPA receptors in the synaptic membrane. This increase was not observed with intracellular BAPTA (Fig. 4B), and was not seen in cells from P2X4−/− mice (Fig. 4A). One interpretation of these results is that there is ‘tonic’ activation of P2X4 receptors by endogenous levels of extracellular ATP, and that this effect is absent in the knockout mice. However, taken together with the results of the experiments with depolarizing pulses discussed above, it seems more parsimonious to evoke some compensatory change in AMPA receptor recycling, the mechanism of which is still not understood.
Fifth, we discuss the differences in the NMDA EPSC between wild-type and P2X4−/− mice that became evident in magnesium-free conditions. The time course of the NMDA component of the isolated NMDA EPSC in mouse CA1 cells was similar to that previous described (Kirson & Yaari, 1996), and not different between wild-type and knockout mice. In wild-type mice, complete removal of magnesium revealed an ifenprodil-sensitive component to the EPSC that developed over 10–20 min (facilitation). This facilitation was not observed with intracellular BAPTA, suggesting that it required a certain level of [Ca2+]i. It also required the activity of p38 MAP kinase, at some stage of the signaling cascade. Interpretation of the molecular sites at which the calcium and phosphorylation are operating is beyond the scope of the present work. However, the time course would be consistent with a progressive increase in the contribution of NR2B subunits to the NMDA EPSC during the facilitation, either by insertion or lateral diffusion from extrasynaptic sites (Tovar & Westbrook, 2002). Why does this occur in magnesium-free solution, and why is it absent in P2X4−/− mice?
The facilitation did not occur in 0.1 mm magnesium, but only after the complete removal of extracellular magnesium (Fig. 6). It seems unlikely that this is due to calcium entry through progressive unblocking of NMDA channels over time, given that the magnesium concentrations required for 50% inhibition of NMDA receptor currents in mouse CA1 neurons are in the millimolar range (at 0 mV) (Kirson & Yaari, 1996). However, Hsu et al. (2000) found that the removal of magnesium elicited a persistent suppression of LTP, and attributed this to increased calcium through NMDA receptors triggering a protein kinase C-dependent signaling cascade. Other possibilities are (i) an increase in calcium entry through P2X4 receptors, and (ii) altered coupling between [Ca2+]i and NR2B receptor insertion in P2X4−/− mice. The first of these possibilities could result from a changed form of extracellular ATP (i.e. an increase in ATP4−) or from relief of the magnesium block of P2X4 receptors (Negulyaev & Markwardt, 2000).
During maturation, the subunit composition of postsynaptic receptors on CA1 mouse pyramidal cells is known to change from NR1/2B to NR1/2A/2B (Kohr et al., 2003). In mature (6–10-week-old) mice, the NMDA EPSC in CA1 neurons is partially blocked by ifenprodil (Miwa et al., 2008), and this is consistent with our own results (Fig. 7A). However, the amplitude of the NMDA EPSC in zero magnesium was unaffected in P2X4−/− mice (Fig. 7A). This suggests that the absence of the P2X4 receptors has resulted in changed NMDA receptor composition, and that this is most readily revealed by conditions (e.g. magnesium removal) that drive NR2B subunits into the postsynaptic membrane.
In summary, our studies show that genetically modified mice lacking P2X4 receptors have impairments in two aspects of synaptic transmission in hippocampal CA1 neurons. First, they have reduced LTP of AMPA receptor EPSCs when Schaffer stimulation is paired with depolarization, and they do not show the increase in sEPSCs driven by postsynaptic depolarization. A possible common explanation for this might be that, in P2X4−/− mice, a rise in [Ca2+]i less effectively brings new AMPA receptors to the subsynaptic membrane. Second, prolonged exposure (20 min) to magnesium-free solution, coupled with synaptic activation of NMDA receptors, results in a progressive facilitation of the NMDA synaptic current that results from insertion of NR2B subunits; this is also not seen in P2X4−/− mice. Ifenprodil can itself partially inhibit LTP (von Engelhardt et al., 2008; Li et al., 2007; Miwa et al., 2008), and it is possible that the first aspect results from the second. However, in neither case does the phenotype support the simple hypothesis that calcium entry through activated P2X4 receptors in the postsynaptic membrane is directly responsible for effects on synaptic transmission.
Acknowledgments
This work was supported by the Wellcome Trust.
Glossary
- AMPA
alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate
- BAPTA
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid
- [Ca2+]i
concentration of intracellular calcium
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- EGTA
ethylene glycol bis(2-aminoethylether)-N,N,N′,N′-tetra-acetic acid
- EPSC
excitatory postsynaptic current
- LTP
long-term potentiation
- NMDA
N-methyl-d-aspartate
- sEPSC
spontaneous excitatory postsynaptic current
References
- Baxter AW, Wyllie DJ. Phosphatidylinositol 3 kinase activation and AMPA receptor subunit trafficking underlie the potentiation of miniature EPSC amplitudes triggered by the activation of L-type calcium channels. J. Neurosci. 2006;26:5456–5469. doi: 10.1523/JNEUROSCI.4101-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collinridge G. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;36:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan TM, Khakh BS. Contribution of calcium ions to P2X channel responses. J. Neurosci. 2004;24:3413–3420. doi: 10.1523/JNEUROSCI.5429-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Mauceri D, Malinverno M, Polli F, Costa C, Tozzi A, Silliquini S, Picconi B, Cattebeni F, Calabresi P, Di Luca M. Decreased NR2B subunits synaptic levels cause impaired long-term potentiation but not long-term depression. J. Neurosci. 2009;29:669–677. doi: 10.1523/JNEUROSCI.3921-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel-Goody SM, Davies KD, Alvestad Linger RM, Freund RK, Browning MD. Phosphoregulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience. 2009;158:1446–1459. doi: 10.1016/j.neuroscience.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Gu JG, MacDermott AB. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature. 1997;389:749–753. doi: 10.1038/39639. [DOI] [PubMed] [Google Scholar]
- Hartveit E, Veruki ML. Studying properties of neurotransmitter receptors by non-stationary noise analysis of spontaneous synaptic currents. J. Physiol. 2006;574:751–785. doi: 10.1113/jphysiol.2006.111856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu K-S, Ho W-C, Huang C-C, Tsai J-J. Transient removal of extracellular Mg2+ elicits persistent suppression of LTP at hippocampal CA1 synapses via PKC activation. J. Neurophysiol. 2000;84:1279–1288. doi: 10.1152/jn.2000.84.3.1279. [DOI] [PubMed] [Google Scholar]
- Hur E-M, Park T-J, Kim K-T. Coupling of L-type voltage-sensitive calcium channels to P2X2 purinoceptors in PC12 cells. Am. J. Physiol. Cell Physiol. 2001;280:C1121–C1129. doi: 10.1152/ajpcell.2001.280.5.C1121. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J. Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Henderson G. ATP receptor-mediated enhancement of fast excitatory neurotransmitter release in the brain. Mol. Pharmacol. 1998;54:372–378. doi: 10.1124/mol.54.2.372. [DOI] [PubMed] [Google Scholar]
- Kirson ED, Yaari Y. Synaptic NMDA receptors in developing mouse hippocampal neurons: functional properties and sensitivity to ifenprodil. J. Physiol. 1996;497:437–455. doi: 10.1113/jphysiol.1996.sp021779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr G, Jensen V, Koester HJ, Mihaljevic ALA, Utvik JK, Kvello A, Ottersen OP, Seeburgh PH, Sprengel R, Hvalby O. Intracellular domains of NMDA receptor subtypes are determinants for long-term potentation. J. Neurosci. 2003;23:10791–10799. doi: 10.1523/JNEUROSCI.23-34-10791.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Huang FS, Abbas AK, Wigström H. Role of NMDA receptor subtypes in different forms of NMDA-dependent synaptic plasticity. BMC Neurosci. 2007;8:55–64. doi: 10.1186/1471-2202-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Miwa H, Fukaya M, Watabe AM, Watanabe M, Manabe T. Functional contribution of synaptically localized NR2B subunits of the NMDA receptor to synaptic transmission and long-term potentiation in the adult mouse CNS. J. Physiol. 2008;586:2539–2550. doi: 10.1113/jphysiol.2007.147652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negulyaev YA, Markwardt F. Block by extracellular Mg2+ of single human purinergic P2X4 receptor channels expressed in human embryonic kidney cells. Neurosci. Lett. 2000;279:165–168. doi: 10.1016/s0304-3940(99)00976-3. [DOI] [PubMed] [Google Scholar]
- North RA. Molecular physiology of P2X receptors. Physiol. Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Pankratov Y, Castro E, Miras-Portugal MT, Krishtal O. A purinergic component of the excitatory postsynaptic current mediated by P2X receptors in the CA1 neurons of the rat hippocampus. Eur. J. Neurosci. 1998;10:3898–3902. doi: 10.1046/j.1460-9568.1998.00419.x. [DOI] [PubMed] [Google Scholar]
- Pankratov Y, Lalo U, Verkhratsky A, North RA. Vesicular release of ATP at central synapses. Pflugers Arch. 2006;452:589–597. doi: 10.1007/s00424-006-0061-x. [DOI] [PubMed] [Google Scholar]
- Pankratov Y, Lalo U, Verkhratsky A, North RA. Quantal release of ATP in mouse cortex. J. Gen. Physiol. 2007;129:257–265. doi: 10.1085/jgp.200609693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio ME, Soto F. Distinct localization of P2X receptors at excitatory postsynaptic specializations. J. Neurosci. 2001;21:641–653. doi: 10.1523/JNEUROSCI.21-02-00641.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim JA, Chaumont S, Jo J, Ulmann L, Young MT, Cho K, Buell G, North RA, Rassendren F. Altered hippocampal synaptic potentiation in P2X4 knock-out mice. J. Neurosci. 2006;26:9006–9009. doi: 10.1523/JNEUROSCI.2370-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Ellis-Davies GC, Magee JC. Mechanism of the distance-dependent scaling of Schaffer collateral synapses in rat CA1 pyramidal neurons. J. Physiol. 2003;548:245–258. doi: 10.1113/jphysiol.2002.036376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto F, Garcia-Guzman M, Gomez-Hernandez JM, Hollmann M, Karschin C, Stühmer W. P2X4: an ATP-activated ionotropic receptor cloned from rat brain. Proc. Natl. Acad. Sci. USA. 1996;93:3684–3688. doi: 10.1073/pnas.93.8.3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–264. doi: 10.1016/s0896-6273(02)00658-x. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Silver RA, Cull-Candy SG. Estimated conductance of glutamate receptor channels activated during EPSCs at the cerebellar mossy fiber-granule cell synapse. Neuron. 1993;11:279–289. doi: 10.1016/0896-6273(93)90184-s. [DOI] [PubMed] [Google Scholar]
- von Engelhardt J, Doganci B, Jensen V, Hvalby Ø, Göngrich C, Taylor A, Barkus C, Sanderson DJ, Rawlins JNP, Seeburg PH, Bannerman DM, Monyer H. Contribution of hippocampal and extra-hippocampal NR2B-containing NMDA receptors to performance on spatial learning tasks. Neuron. 2008;60:846–860. doi: 10.1016/j.neuron.2008.09.039. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptors: selectivity and mechanisms at recombinant heteromeric recptors. Mol. Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Wyllie DJ, Manabe T, Nicoll RA. A rise in postsynatic Ca2+ potentiates miniature excitatory postsynaptic currents and AMPA responses in hippocampal neurons. Neuron. 1994;12:127–138. doi: 10.1016/0896-6273(94)90158-9. [DOI] [PubMed] [Google Scholar]