

Abstract
Aim
To investigate the effect of increases in extracellular Ca2+ entry produced by the L-type Ca2+ channel agonist FPL-64176 (FPL) upon acute atrial arrhythmogenesis in intact Langendorff-perfused mouse hearts and its dependence upon diastolic Ca2+ release from sarcoplasmic reticular Ca2+ stores.
Methods
Confocal microscope studies of Fluo-3 fluorescence in isolated atrial myocytes were performed in parallel with electrophysiological examination of Langendorff-perfused mouse hearts.
Results
Atrial myocytes stimulated at 1 Hz and exposed to FPL (0.1 μm) initially showed (<10 min) frequent, often multiple, diastolic peaks following the evoked Ca2+ transients whose amplitudes remained close to control values. With continued pacing (>10 min) this reverted to a regular pattern of evoked transients with increased amplitudes but in which diastolic peaks were absent. Higher FPL concentrations (1.0 μm) produced sustained and irregular patterns of cytosolic Ca2+ activity, independent of pacing. Nifedipine (0.5 μm), and caffeine (1.0 mm) and cyclopiazonic acid (CPA) (0.15 μm) pre-treatments respectively produced immediate and gradual reductions in the F/F0 peaks. Such nifedipine and caffeine, or CPA pre-treatments, abolished, or reduced, the effects of 0.1 and 1.0 μm FPL on cytosolic Ca2+ signals. FPL (1.0 μm) increased the incidence of atrial tachycardia and fibrillation in intact Langendorff-perfused hearts without altering atrial effective refractory periods. These effects were inhibited by nifedipine and caffeine, and reduced by CPA.
Conclusion
Enhanced extracellular Ca2+ entry exerts acute atrial arrhythmogenic effects that is nevertheless dependent upon diastolic Ca2+ release. These findings complement reports that associate established, chronic, atrial arrhythmogenesis with decreased overall inward Ca2+ current.
Keywords: atrial arrhythmogenesis, Ca2+ homeostasis, calcium entry, murine hearts, store calcium
Atrial arrhythmias constitute the most common sustained disorders of cardiac rhythm encountered in clinical practice and result in substantial mortality and morbidity. For example, atrial fibrillation (AF) is associated with stroke, thromboembolism, heart failure and impaired quality of life; treatment strategies have largely proved inadequate (Thrall et al. 2006). It is attributed to irregular and rapid atrial electrical activity due to ectopic activity, single circuit or multiple wavelets of re-entry throughout the atria (Moe et al. 1964, Nattel 2002).
Persistent atrial arrhythmia appears to lead to changes in electrical properties of the cardiac tissue. This electrical remodelling may include alterations in cellular Ca2+ homeostasis including overload (Ryu et al. 2005, Yeh et al. 2008). Studies in both animal models and human atrial cardiomyocytes from patients in established AF report reduced current densities through L-type Ca2+ channels (ICa,L) (Yue et al. 1997, Bosch et al. 1999, Van Wagoner et al. 1999, Skasa et al. 2001, Yagi et al. 2002). The balance between inward currents through L-type Ca2+ channels (LTCCs) and outward K+ currents are responsible for the plateau phase that extends the duration of action potentials in human cardiomyocytes. They are also responsible for Ca2+ entry that in turn regulates the contractile force (Fabiato & Fabiato 1975, Falk 1998). Their inhibition results in a shortening of both atrial action potential duration and effective refractory period (Morillo et al. 1995, Wijffels et al. 1995, Li & Nattel 1997). This in turn favours re-entry and persistence of the AF condition. Both human hearts with chronic AF (Christ et al. 2004) and a rabbit model of rapid atrial pacing (Bosch et al. 2003) showed parallel reductions in β-2 subunit expression and ICa,L followed by changes in the α1C subunit mRNA levels. This supports the concept that reduced α subunit expression underlies this reduction in voltage-dependent (ICa,L) despite increased single ICa,L channel open probability as reported in hearts of patients with established AF (Klein et al. 2003). Conversely, it is well established that AF is associated with an abnormal sarcoplasmic reticular (SR) Ca2+ release reflecting increased open probabilities in the relevant RyR2 release channels resulting from their hyperphosphorylation (Nattel et al. 2007).
However, such earlier studies described results from models of established AF or chronic AF patients. There are relatively few reports on the features of acute atrial arrhythmogenesis that can include either atrial tachycardia (AT) or AF, and the possible involvement of LTCC activity. Reductions in ICa,L form a characteristic and pathophysiologically important part of myocardial remodelling observed during long-lasting AF. However, recent studies indicate that they are not present in patients with non-persistent AF (Skasa et al. 2001). Nevertheless, acute atrial arrhythmias (both fibrillation and flutter) occur with a 20–50% incidence following cardiothoracic surgery, particularly coronary artery bypass grafting. They most frequently occur 2 days after surgery and are rare after 7–15 postoperative days. However such episodes are usually short-lived. This incidence is significantly reduced by treatment with β-blockers and/or Ca2+ channel antagonists (Podesser et al. 1995, Yilmaz et al. 1996, Kim et al. 2002, Dobrilovic et al. 2005, Baker & White 2007, Iwamoto & Inoue 2007). Conversely, activation of β-adrenergic signalling leads to threefold to fourfold increases in calcium current. The latter results from protein kinase A phosphorylation of calcium channels (Hulme et al. 2006).
The present study accordingly goes on to investigate the possible roles of altered Ca2+ homeostasis on the initiation of acute atrial arrhythmias at both the cellular and the whole organ levels in intact murine hearts, through enhancing ICa,L. Murine hearts were used in view of their potential utility in future atrial studies of the effects of genetic modifications. Despite inevitable differences from human hearts, such genetically modified models have been successfully used to model mechanisms of ventricular arrhythmogenesis in LQTS3 (Thomas et al. 2008), Brugada syndrome (Stokoe et al. 2007) and catecholaminergic polymorphic ventricular tachycardia (Priori et al. 2001, Cerrone et al. 2005, Goddard et al. 2008). We provide experimental evidence that enhanced ICa,L-mediated extracellular Ca2+ entry exerts acute atrial arrhythmogenic effects that nevertheless depend upon diastolic release of intracellularly stored SR Ca2+. We compared the effects of FPL-64176 (methyl 2,5-dimethyl-4-[2-(phenylmethyl) benzoyl]-1H-pyrrole-3-carboxylate; FPL) on regularly stimulated isolated murine atrial myocytes and atrial arrhythmogenesis in isolated Langendorff-perfused murine hearts. FPL is known to prolong opening of single LTCCs during depolarization and slow channel closing upon repolarization (Rampe & Lacerda 1991, Baxter et al. 1993, Lauven et al. 1999, Fan et al. 2000). FPL was used both alone and in combination with three agents with known effects upon cellular Ca2+ homeostasis. Of these, nifedipine acts as a competitive LTCC blocker (Triggle 2003) inhibiting inward Ca2+ current (Shen et al. 2000, Thomas et al. 2007). Caffeine is thought to increase the release of intracellularly stored Ca2+, thereby ultimately depleting such stores, whether by sensitizing cardiac SR Ca2+ release-ryanodine receptors (RyR2s) to cytosolic Ca2+ or inhibiting phosphodiesterase activity, thereby increasing cellular cyclic adenosine monophosphate (cAMP) (Daly 2007). Finally, cyclopiazonic acid (CPA), is known to inhibit SR Ca2+-ATPase activity (Seidler et al. 1989, Du et al. 1996) by blocking its Ca2+ access channel (Moncoq et al. 2007, Palomeque et al. 2007), thereby altering levels of SR Ca.
Materials and methods
Inbred male and female 129/Sv wild-type mice (Harlan, Bicester, UK) kept in an animal house at room temperature were subjected to 12 h light–dark cycles, and fed sterile rodent chow with constant access to water. All procedures conformed to the U.K. Animals (Scientific Procedures) Act (1986). The following solutions were used in the course of the study: Solution A was prepared for electrophysiological experiments on Langendorff-perfused hearts. It consisted of normal bicarbonate-buffered Krebs-Henseleit (KH) solution containing (in mm): 119 NaCl, 25 NaHCO3, 4.0 KCl, 1.2 KH2PO4, 1.0 MgCl2, 1.8 CaCl2, 10 glucose and 2.0 sodium pyruvate, and at pH 7.4 maintained by bubbling with 95% O2–5% CO2 (British Oxygen, Manchester, UK). Solution B was the basic stock solution from which other solutions (C-H) used in myocyte isolation were derived (in mm): 125 NaCl, 4.75 KCl, 1.2 MgSO4, 1.2 KH2PO4, 30 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid (Hepes), 10 glucose, 50 taurine and titrated to pH 7.4 with NaOH. It was filtered with a 0.2 μm filter (Millipore, Billerica, MA, USA) to remove microbes and small particles. Solution C: 750 μm Ca2+-containing perfusion solution was prepared by further adding 750 μm CaCl2. Solution D: Ca2+-free nitrilotriacetic acid (NTA)-containing solution was prepared adding 5 mm NTA to solution B, and titrating the resulting solution to pH 6.95 with NaOH. Solution E: Enzyme solution was prepared by adding 1.5 mg mL−1 collagenase (Worthington Type II), 2.0 mg mL−1 hyaluronidase (Sigma, Gillingham Dorset, UK), and 100 μm CaCl2 to solution B. Solution F: a further digestion buffer made by adding 1.0 mg mL−1 collagenase and 1.0 mg mL−1 BSA to solution B. Solution G: Enzyme washout solution was made by adding 1.0 mg mL−1 BSA and 250 μm CaCl2 to solution B. Solution H: Ca2+-containing solution was made by adding 1.2 mm CaCl2 to solution B. After preparation, solutions D, E, F and G were always filtered using a 0.2 μm filter to remove microbes and small particles.
Measurement of cytosolic Ca2+ transients
Mice, aged 10–12 weeks at which viable atrial myocyte isolation could most readily be accomplished, were killed by cervical dislocation. A total of nine mice were used. Hearts were rapidly excised and cannulated in ice-cold KH solution (solution A). They were then mounted onto a Langendorff perfusion system and successively perfused with solution C (for 4 min), solution D (4 min) and solution E (10–12 min) at 37 °C. The heart was then removed from the perfusion apparatus. The atrial appendages were then excised and chopped into several pieces in solution F. These were further incubated for another 5–10 min with gentle manual agitation using a 1 mL tip transfer pipette. All these latter steps were performed at 36–37 °C. Cells were then separated from the enzymic solution by centrifuging at 243 g for 3 min. The resulting isolated cells were then washed using solution G followed after 5 min, by centrifugation at 30 g for 2 min. The cells were then resuspended in solution H and after a 5 min interval, centrifuged again at 30 g for 2 min. The cells were then maintained at room temperature in solution H for the experiments that followed. The cells were then placed on a grade 1 circular laminin-coated coverslip (Menzel, Glasbearbeitungswerk, Germany) that formed the floor of a 1.5 mL perfusion chamber to which it was fixed with vacuum grease. They were then loaded with the acetoxymethyl (AM) ester of Fluo-3 (Molecular Probes, Leiden, The Netherlands) by incubation with 5 μm Fluo-3 AM in solution H (1.2 mm CaCl2) for 10–20 min in the dark before washout of the Fluo-3 containing solution. They were then transferred onto the stage of a Zeiss LSM-510 laser scanning confocal system (Zeiss, Oberkochen, Germany) with a ×20 air objective on a Zeiss Axiovert 100 M inverted microscope. Fluo-3 fluorescence emission was excited with a 488 nm argon laser and measured at wavelengths between 505 and 550 nm. Images were then analysed using in-house custom-made software. Series of 500 frames (128 × 64 pixels per frame) were collected at a scanning frequency of 25 ms per frame to monitor fluorescence changes over time. Fluorescence (F) measurements were corrected for background signal in regions outside the cells. They were made within defined regions of interest (ROI; F) and were normalized to their baseline fluorescence (F0) values. For each of the myocytes studied, peak F/F0 values were calculated throughout each time series acquired. This typically contained around 12–13 peaks from which the mean peak F/F0 could be obtained. A mean peak F/F0 was then calculated for that series. Cells were paced at 1 Hz (5 V above excitation threshold of 30–60 V for 2 ms) with two field electrodes. All fluorescence studies were carried out at room temperature. Ca2+ transients were measured from ROIs covering entire cells.
Atrial electrophysiological experiments
Male and female mice (age 3–6 months), at which their aortic size permitted reproducible in situ cannulation, were randomly selected, heparinized with 50 IU heparin (IP) 10–15 min before being killed by cervical dislocation (Schedule I: Animals Scientific Procedures Act 1986). A total of 72 mice were used. The heart was cannulated in situ using a straight-cut and smoothed 21-gauge needle previously filled with solution A, dissected and then fixed securely with a straight 60 g pressure micro-aneurysm clip (Harvard Apparatus, Edenbridge, UK). The cannulated heart was perfused with solution A at room temperature and then mounted onto a Langendorff system (Balasubramaniam et al. 2004) then perfused at a constant flow rate of 2–2.5 mL min−1 (Watson-Marlow Bredel Peristaltic pumps, model 505S; Falmouth, Cornwall, UK) with solution A. The perfusate was first filtered through 200 μm and 5 μm membranes (Millipore UK, Watford, UK), and warmed to 37 °C by a water-jacketed heat exchange coil (Model C-58A; Techne, Cambridge, UK) before entering the coronary arterial network. Viable hearts regained a pink appearance and spontaneous rhythmic contractions upon warming. The hearts were perfused retrogradely for not less than 10 min in the absence of stimulation. Experiments were only performed in intact Langendorff preparations showing clear-cut 1 : 1 atrioventricular (AV) conduction during the intrinsic activity following cannulation.
The electrophysiological studies involved comparisons of records from simultaneous recordings made at two sites. Thus, in addition to the paired platinum-stimulating electrodes placed on the right atrium, two bipolar recording electrodes of 1 mm interpole spacing were placed on the left atrium and left ventricle respectively. Hearts were initially paced for not less than 5 min at 10 Hz to permit them to regain their physiological steady state. Three types of pacing protocols were then used. First, hearts were studied at their intrinsic rates in the absence of stimulation. Second, hearts were subject to regular pacing at 10 Hz using 2 ms square-wave stimuli set at two times the excitation threshold (Grass S48 stimulator; Grass-Telefactor, Slough, UK). Third, hearts were studied using a programmed electrical stimulation (PES) procedure adopted from clinical techniques previously used in ventricular studies (Saumarez & Grace 2000, Balasubramaniam et al. 2004) but recently introduced in clinical studies of atrial electrophysiology (Wellens 2003, Brembilla-Perrot et al. 2005). These began using standard baseline pacing stimuli at a frequency of 10 Hz for 20 s. Drive trains of eight paced beats (S1) were each followed by an extra stimulus (S2) every ninth beat, initially at an S1–S2 interval equal to the pacing interval. Each subsequent cycle reduced the S1–S2 interval by 1 ms until atrial refractoriness was reached. The resulting electrogram signals were amplified, band-pass filtered (30 Hz to 1 kHz; Gould 2400S; Gould-Nicolet Technologies, Ilford, Essex, UK) and digitized at a sampling frequency of 5 kHz (Spike2 software; CED1401plus; Cambridge Electronic Design, Cambridge, UK). The present protocols thus differed from our previous studies on ventricular arrhythmogenesis (Balasubramaniam et al. 2004, Stokoe et al. 2007, Thomas et al. 2008) in pacing hearts from the atria rather than the ventricles. Furthermore, use of the previous 8 Hz pacing rate permitted atrial escape phenomena that precluded regular activation by the stimulus train. Accordingly we used a higher pacing rate of 10 Hz to ensure regular atrial stimulation under all the pharmacological conditions. In some hearts, this resulted in a gradual development of an AV block. Nevertheless, withdrawal of the regular pacing permitting resumption of normal intrinsic activity led to restoration of normal 1 : 1 AV conduction once pharmacological agents were withdrawn. These findings likely relate to AV node refractoriness at high pacing rates: we observed that the atria had shorter refractory periods (24 ± 7 ms, n = 33) than the AV node (61 ± 12 ms, n = 5).
Statistical analysis
Statistical analysis was carried out using a repeated-measures one-way anova to compare data using SPSS software. Results from individual hearts acquired during pharmacological intervention were compared with their respective untreated controls using a one-way anova for correlated samples (SPSS software). A probability of P < 0.05 was taken as statistically significant. Cross-tabulations with chi-squared or Fisher’s exact test were used as appropriate for categorical variables. Data are expressed as means ± standard errors of the mean (SEM). The numbers, n, denote either numbers of whole hearts or of peak F/F0 values.
Agents
All drugs and other chemical agents were purchased from Sigma-Aldrich (Poole, UK) except where otherwise indicated. FPL-64176 was initially made up with dimethyl sulfoxide to make a stock concentration of 5.0 mm, and kept wrapped in foil to prevent light degradation, and was stored at −20 °C. Nifedipine was dissolved in 96% ethanol to make a 1.0 mm stock concentration, kept wrapped in foil to prevent light degradation and was kept refrigerated at 4 °C. Caffeine was dissolved directly in solution A or H as appropriate and kept at room temperature. CPA was prepared in 96% ethanol to make a 10 mm stock concentration, and stored at −20 °C. Final drug concentrations were achieved by dilution with solution A for electrophysiological experiments and solution H for experiments in single cells.
Results
FPL modifies Ca2+ homeostasis in atrial myocytes
The murine atrial myocytes that were obtained by our isolation procedure appeared as elongated cells with rounded but tapered ends with well-defined striations. They were viable for up to 6–8 h prior to fluorophore loading. We studied a total of 108 cells from nine hearts using confocal microscopy following loading with Fluo-3-AM. These were then subjected to regular pacing stimuli at a frequency of 1 Hz; higher pacing rates led to loss of capture in such isolated cells. The resulting fluorescence signals, F, were expressed normalized to their baseline values F0. Such values were obtained over ROIs that covered the entire cell area. In the absence of added pharmacological agents, this resulted in regular successions of entrained Ca2+ transients with stable amplitudes of mean F/F0 5.36 ± 0.89 (n = 192 peaks from 16 cells) that decayed to stable baselines with no evidence of diastolic events (Fig. 1Aa,B; Table 1). Such Ca2+ transients would be the result of cycles of depolarization-induced SR Ca2+ release into the cytosol and its subsequent return from cytosol to stores whose magnitudes and time courses would be sensitive to manoeuvres affecting either process. The isolated atrial myocytes thus provided stable preparations in which the effect of pharmacological agents on the overall magnitudes and transients of the resulting Fluo-3 Ca2+ signals could be investigated over prolonged periods.
Figure 1.
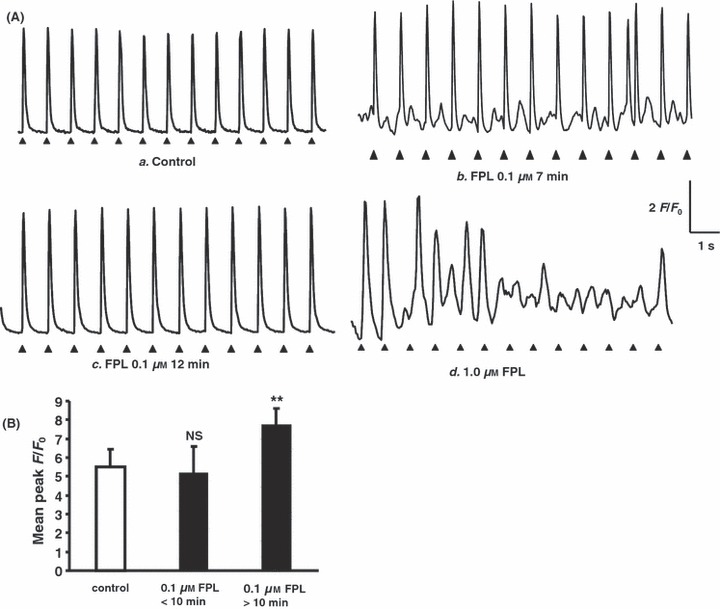
Effects of FPL on Ca2+ transients from regularly stimulated atrial myocytes. (A) Ca2+ transients obtained in a typical control myocyte (a) and cells 7 (b) and 12 min following addition of FPL (0.1 μm) (c), and immediately following addition of 1.0 μm FPL (d), showing diastolic transients (b) that eventually disappear (c), and sustained irregular patterns of cytosolic Ca2+ activity independent of the continued pacing (d). (B) Peak F/F0 values (mean ± SEM) obtained from control conditions (clear bar) and corresponding values obtained <10 min and >10 min following addition of 0.1 μm FPL (filled bars) (vs. control, NS: no significant difference, **P < 0.01).
Table 1.
Occurrence of diastolic calcium release in atrial myocytes
| Agents | Number of cells showing diastolic release/total no. of cells | Number of evoked peaks examined | Number of diastolic release events |
|---|---|---|---|
| Control | 0/16 | 192 | 0 |
| FPL (0.1 μm) | 4/4** | 50 | 55*** |
| FPL (1 μm) | 8/8** | Sustained irregular patterns of cytosolic Ca2+ activity | |
| Nifedipine | 0/8 | 96 | 0 |
| CPA pre-treated 20 min | 0/16 | 192 | 0 |
| Caffeine pre-treated 15 min | 0/12 | 150 | 0 |
| Nifedipine + 0.1 μm FPL | 0/9 | 117 | 0 |
| Nifedipine + 1 μm FPL | 0/6 | 78 | 0 |
| CPA + 0.1 μm FPL<10 min | 3/3** | 47 | 46*** |
| CPA + 0.1 μm FPL>10 min | 4/11* | 143 | 46***,§§§ |
| CPA + 1.0 μm FPL | 1/3 | 39 | 5***,§§§ |
| Caffeine + 0.1 μm FPL | 0/6 | 78 | 0 |
| Caffeine + 1 μm FPL | 0/6 | 78 | 0 |
CPA, cyclopiazonic acid.
Test agents vs. control ***P < 0.001 on chi-squared testing; test agents vs. control, *P < 0.05, **P < 0.01 on Fisher exact testing; test agents vs. FPL, §§§P <0.001 on chi-squared testing against results obtained with 0.1 μM FPL.
The addition of FPL then resulted in significant disruptions of this regular pattern of Ca2+ transients. The regular series of evoked deflections persisted with the introduction of low concentrations (0.1 μm) of FPL (Fig. 1Ab,B). The amplitudes of such evoked Ca2+ transients then initially remained close to control values, at 5.14 ± 1.46 (P > 0.05; n = 50 peaks from four cells). However, at early times (<10 min) following addition of FPL, there were additional smaller diastolic Ca2+ deflections that similarly extended over the entire cell area (Fig. 1Ab). These consisted of frequent, sometimes multiple diastolic peaks (55 diastolic peaks observed over 50 transients in four of four cells: see Table 1). However, with continued pacing beyond 10 min these subsidiary peaks disappeared, consistent with a reduction in available SR Ca upon which such events might depend. This left a sustained pattern of large evoked transients with a peak F/F0 of 7.73 ± 0.86 (P < 0.001; n = 50 peaks from four cells) (Fig. 1Ac,B). Higher FPL concentrations (1.0 μm) initially resulted in an increased peak F/F0, but this was then often followed by a loss of the regular pattern of evoked activity. This was replaced by sustained irregular patterns of cytosolic Ca2+ activity that was independent of continued pacing (Fig. 1Ad, eight cells).
The effects of FPL are modified by agents known to modify Ca2+ homeostasis
The above findings demonstrated that FPL alters cellular Ca2+ homeostasis, consistent with previous reports. FPL is thought to modify LTCCs through prolonging their opening during depolarization and their inactivation upon repolarization, enhancing the duration of the consequent tail currents (Rampe & Lacerda 1991). The FPL actions observed here accordingly could take place entirely through an increased entry of extracellular Ca2+, the effect of such an entry on Ca2+-induced Ca2+ release (CICR) from the SR Ca2+ store or both these effects. These possibilities were investigated using agents known to modify such Ca2+ entry, RyR2-mediated Ca2+ release or SR Ca2+ reuptake. The latter two manoeuvres would be expected to alter SR store Ca2+ through separate and therefore independent mechanisms.
First, nifedipine was used as a dihydropyridine-LTCC blocker to diminish extracellular Ca2+ entry. In the present experiments on atrial myocytes, nifedipine pre-treatment lasting 10 min promptly reduced peak F/F0, to 2.38 ± 0.19 (n = 96 peaks from eight cells) consistent with this known effect in reducing extracellular Ca2+ entry (Fig. 2Aa,b). It then abolished the diastolic Ca2+ transients produced by a subsequent addition of 0.1 μm FPL (Fig. 2Ac; Table 1). It also abolished the irregular activity produced by 1.0 μm FPL (Fig. 2Ad) leaving regular sequences of F/F0 peaks with mean amplitudes of 3.08 ± 0.70 (n = 117 peaks) (Fig. 2B) and 3.78 ± 0.83 (n = 78 peaks) (Fig. 2B) respectively. These observations are consistent with a dependence of these altered patterns of Ca2+ homeostasis induced by FPL upon Ca2+ entry, whether directly through LTCC-mediated Ca2+ entry or indirectly through SR-mediated Ca2+ release. These latter possibilities were then resolved using the agents CPA and caffeine, as described below.
Figure 2.
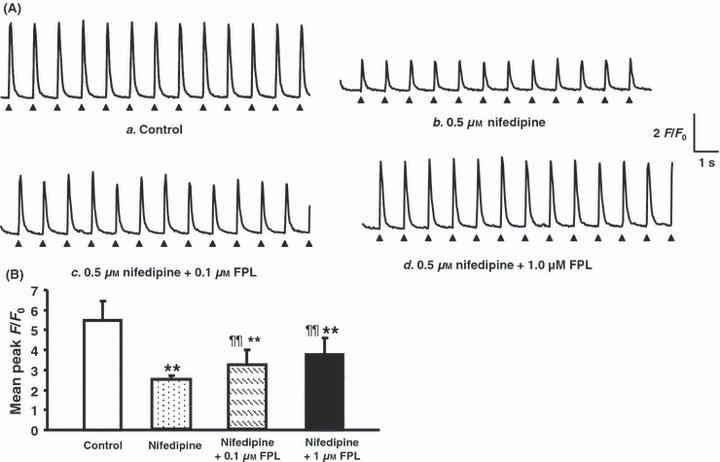
Nifedipine pre-treatment following addition of FPL on Ca2+ transients from regularly stimulated atrial myocytes. (A) Ca2+ transients obtained before (a) and immediately following introduction of 0.5 μm nifedipine (b), their partial recovery following the further addition of 0.1 and 1.0 μm FPL (c, d respectively): none of these records showed diastolic events. (B) Peak F/F0 values (mean ± SEM) obtained under control conditions (clear bar) compared with (vs. control, **P < 0.01; vs. nifedipine ¶¶P < 0.01) corresponding values obtained following addition of nifedipine (0.5 μm) (second bar from left) and a further addition of FPL (0.1 and 1 μm) (third and fourth bars from left respectively).
Second, caffeine was used as an agent to increase the release of intracellularly stored Ca2+ with a resulting gradual depletion of SR Ca2+. The present experiments in atrial cells demonstrated that a 15 min pre-treatment with caffeine (1.0 mm) similarly reduced the peak F/F0 of the Ca2+ transients to 2.75 ± 0.21 (n = 150 peaks from 12 cells) consistent with such a hypothesis (Fig. 3Aa,b,B). Such transients showed prolonged kinetics, relative to those shown under control conditions consistent with our previously reported actions on the time course of CICR (Zhang et al. 2009). These were not observed with the remaining pharmacological interventions. This is illustrated in the full-width half-maximum values in Table 2. Following such pre-treatment, the addition of FPL (0.1 and 1.0 μm) resulted in a persistent, regular pattern of stimulation-induced responses, with reduced amplitudes of 3.04 ± 0.78 (n = 78 peaks from six cells) and 2.89 ± 0.28 (n = 78 peaks from six cells) (Fig. 3Ac,d,B) respectively but an absence of diastolic Ca2+ release. Caffeine pre-treatment thus rescued the cells from the effects of FPL alone.
Figure 3.
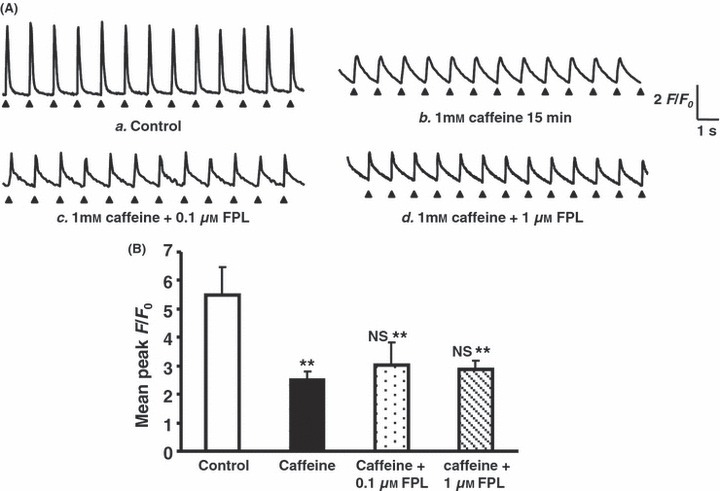
Caffeine pre-treatment following addition of FPL on Ca2+ transients from regularly stimulated atrial myocytes. (A) Ca2+ transients obtained before (a) and pre-treatment after 15 min following introduction of 1 mm caffeine (b), following introduction of 0.1 μm FPL (c, d): none of these records showed diastolic events. (B) Peak F/F0 values (mean ± SEM) obtained under control conditions (clear bar) compared with (vs. control **P < 0.01; vs. caffeine NS: no significant difference) corresponding values obtained following addition of caffeine (0.5 μm) (filled bar) and a further addition of FPL (0.1 and 1 μm) (third and fourth bars from left respectively).
Table 2.
Full-width half-maximum (FWHM) values for Ca2+ transients obtained from regularly stimulated atrial myocytes under different conditions
| Agents | No. of peaks | FWHM (ms, mean ± SEM) |
|---|---|---|
| Control | 12 | 118 ± 7 |
| 0.1 μm FPL | 12 | 117 ± 13 |
| Nifedipine | 12 | 114 ± 21 |
| Caffeine 15 min | 12 | 336 ± 38* |
| CPA 20 min | 12 | 125 ± 11 |
| Nifedipine + 0.1 μm FPL | 12 | 123 ± 21 |
| Nifedipine + 1.0 μm FPL | 12 | 121 ± 17 |
| Caffeine + 0.1 μm FPL | 12 | 178 ± 15* |
| Caffeine + 1.0 μm FPL | 12 | 185 ± 8* |
| CPA + 0.1 μm FPL | 12 | 102 ± 14 |
| CPA + 1.0 μm FPL | 12 | 117 ± 8 |
CPA, cyclopiazonic acid.
P < 0.05 when tested against control.
Third, CPA was used as a further, independent means of depleting SR store Ca2+ through its inhibition of SR Ca2+−ATPase activity. In the present experiments, CPA (0.15 μm) resulted in a progressive reduction of the amplitude of the evoked transients to a peak F/F0 of 3.64 ± 1.06 (n = 168 peaks). (Fig. 4Aa,b,B; Table 1). CPA pre-treatment also markedly reduced the incidence of cells that showed diastolic phenomena following further addition of 0.1 μm FPL. Thus, of 14 cells studied, only three cells which were examined at a period <10 min following addition of 0.1 μm FPL showed extensive diastolic Ca2+ release phenomena (following 46 of 47 examined peaks) (Fig. 4Ac,B; Table 1). The remaining 11 cells studied >10 min following addition of FPL showed a lower incidence of such transients. These followed 46 of 143 peaks and only occurred in 4 of the 11 cells studied (Fig. 4Ad). They similarly disappeared >10 min following addition of FPL, consistent with their dependence upon a limited Ca store.
Figure 4.
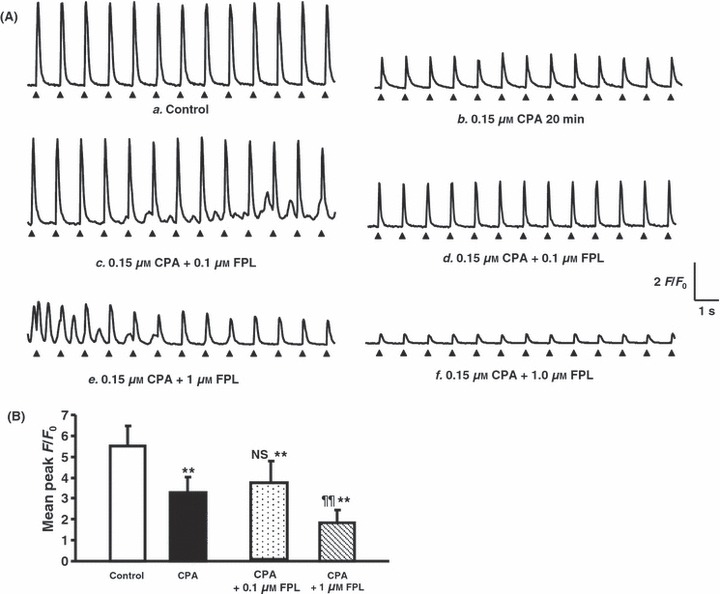
Cyclopiazonic acid (CPA) pre-treatment following addition of FPL on Ca2+ transients from regularly stimulated atrial myocytes. (A) Results obtained during control conditions (a) and 20 min following the addition of CPA (0.15 μm) (b) and further introduction of FPL at concentrations of 0.1 μm (c, d) and 1.0 μm (e, f) showing an initial appearance of diastolic signals (c, e) followed by their disappearance and a decline in the amplitude of the evoked Ca2+ transients (d, f). (B) Peak F/F0 values (mean ± SEM) obtained before (open bar) and following addition of CPA (0.15 μm) (filled bar) and a further addition of FPL (vs. control **P < 0.01; vs. CPA NS: no significant difference, ¶¶P < 0.01) (third and fourth bars from left).
Pre-treatment with CPA also reduced the incidence of cells that showed an irregular pattern of Ca2+ activity in response to the addition of 1.0 μm FPL (Fig. 4Ae). Of the five such cells studied, two showed a persistent irregular pattern but three continued to show a regular pattern of activity in response to the regular pattern of stimulation. However, the latter consisted of a series of Ca2+ transients that declined to a peak F/F0 of 1.82 ± 0.62 (n = 39 peaks; Fig. 4Af,B) with continued pacing beyond 10 min (Fig. 4Af,B; Table 1). Only one cell showed diastolic Ca2+ release in 5 of 13 peaks even 8–10 min following addition of FPL that nevertheless disappeared after continued pacing.
Cellular Ca2+ changes correlate directly with arrhythmogenic tendencies in intact perfused hearts
The above results directly correlated with findings from dual ventricular and atrial bipolar electrogram (BEG) recordings from Langendorff-perfused hearts implicating such alterations in Ca2+ homeostasis in atrial arrhythmogenic phenomena. The atrial recordings in Figs 5 and 6 show both atrial and ventricular deflections whereas atrial deflections were absent from ventricular BEG traces. Atrial pacing at 10 rather than 8 Hz avoided atrial escape phenomena and ensured regular activation by the stimulus train.
Figure 6.
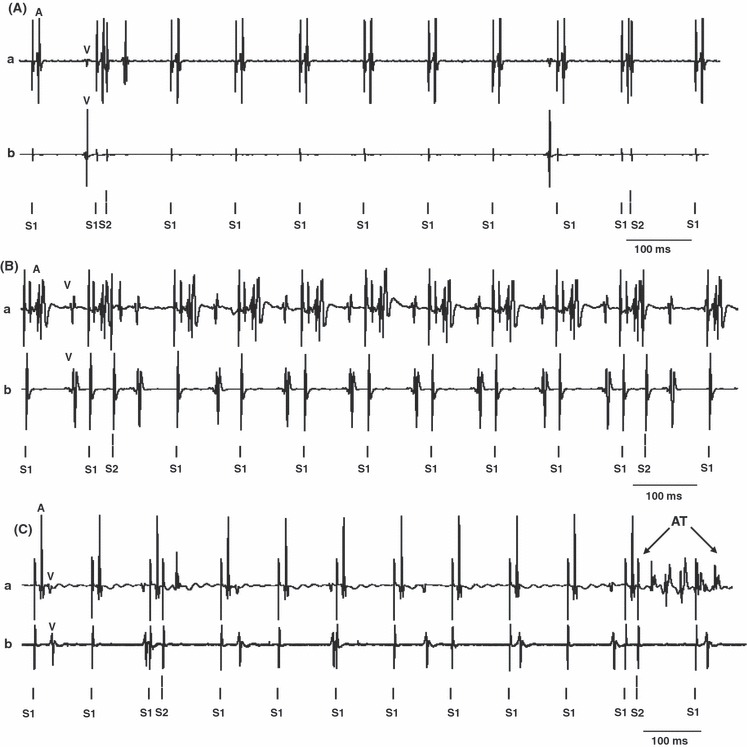
Nifedipine, caffeine and cyclopiazonic acid (CPA) pre-treatment abolish FPL-induced arrhythmogenic effects. Traces of atrial (a) and ventricular (b) bipolar electrogram records obtained during programmed electrical stimulation (PES) (timing of regular (S1) and extrasystolic stimulation (S2) indicated below each trace) under the following conditions: (A) No atrial tachycardia (AT) and atrial fibrillation (AF) after nifedipine (0.5 μm). (B) No atrial tachycardia (AT) and atrial fibrillation (AF) after 1 mm caffeine pre-treatment. (C) A brief AT after CPA (0.15 μm) pre-treatment. In the individual traces, ventricular deflections are marked ‘V’ and atrial deflections are marked ‘A’.
In the absence of test agents, none of the hearts showed arrhythmic activity, whether in the form of regular AT or irregular AF, during either intrinsic activity or regular pacing (Table 3). Three showed AT lasting 0.5, 1.0 and 5.0 s during PES. However, addition of FPL (1.0 μm) produced strong arrhythmic effects in all seven hearts studied. This was accompanied by evidence of third-degree AV block in all seven cases during intrinsic activity. We observed a total of 12 episodes of sinus pause, in two of seven hearts treated with FPL as also illustrated in Fig. 5A, with durations in the range of 0.4–1.6 s. These were not seen in untreated hearts. Thus, following addition of FPL, three hearts showed AT lasting 5.6, 5.7 and 14.0 s during intrinsic activity (Fig. 5A), two showed AT lasting 0.8 and 6.0 s (Fig. 5B) and two showed AF lasting 2.1 and 5.5 s respectively during regular pacing. During PES, five hearts showed AT lasting 3.8, 7.7, 0.4, 2.0 and 6.8 s and four showed AF lasting 3.4, 3.1, 0.6 and 18.7 s respectively (Fig. 5C,D). These arrhythmogenic phenomena took place in an absence of the altered refractoriness that has been associated with re-entrant phenomena occurring during atrial arrhythmogenesis in the experimental system used here. Tissue refractoriness has previously been closely associated with the inducibility or otherwise of ventricular arrhythmogenesis in murine hearts (Maguire et al. 2003). We assessed atrial refractoriness in intact hearts using a decremental pacing protocol both before and following FPL activation. The atrial effective refractory period (AERP) was determined from the shortest S1–S2 interval that failed to elicit an atrial deflection (Thomas et al. 2007). The addition of FPL (1.0 μm) did not produce significant changes in AERP (control: 22.10 ± 7.80 ms, n = 22 hearts; FPL: 26.47 ± 6.50 n = 7 hearts; P > 0.05).
Table 3.
Occurrence of arrhythmogenesis (AT or AF) under different protocols
| Agents | Intrinsic (n) | Regular pacing (10 Hz) (n) | PES (n) |
|---|---|---|---|
| Control | 0 (36) | 0 (36) | 3 (36) |
| FPL | 3 (7)** | 4 (7)** | 7 (7)** |
| Nifedipine pre-treated | 0 (12)* | 0 (12)* | 3 (12)## |
| CPA pre-treated 20 min | 0 (11)* | 0 (11)* | 1 (11)*** |
| Caffeine pre-treated >15 min | 0 (6)* | 0 (6)* | 0 (6)*** |
| Nifedipine + FPL | 0 (6)* | 0 (6)* | 5 (6)** |
| CPA + FPL | 0 (6)* | 0 (6)* | 4 (6)** |
| Caffeine + FPL | 0 (6)* | 0 (6)* | 1 (6)## |
AT, atrial tachycardia; AF, atrial fibrillation; CPA, cyclopiazonic acid; PES, programmed electrical stimulation.
Test agents vs. control P < 0.01**; test agents vs. FPL P < 0.05*; test agents vs. FPL P < 0.01## and P < 0.001*** on Fisher exact testing. Each entry provides both the incidence of arrhythmogenesis and the number of hearts studied in brackets.
Figure 5.
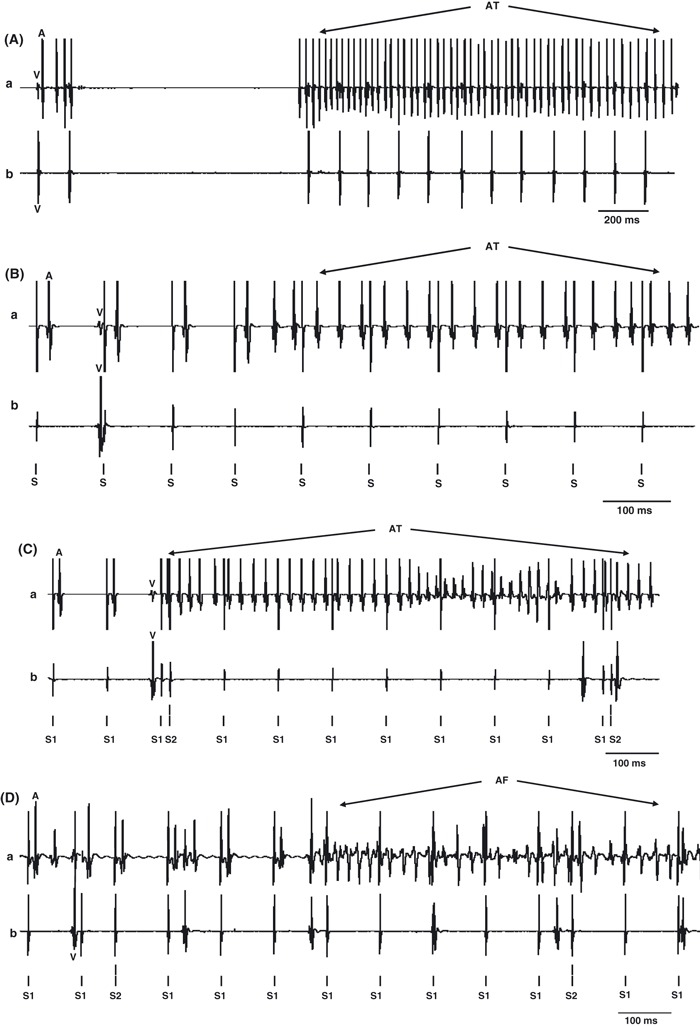
Examples of atrial arrhythmogenic effects of FPL. Traces of atrial (a) and ventricular (b) bipolar electrogram records obtained under the following conditions: (A) Spontaneous atrial tachycardia (AT) during intrinsic pacing. (B) AT during regular pacing. (C, D) AT (C) and atrial fibrillation (AF; D) respectively during programmed electrical stimulation (PES); timing of regular (S1) and extrasystolic stimulation (S2) indicated below each trace. In the individual traces, ventricular deflections are marked ‘V’ and atrial deflections are marked ‘A’.
Nifedipine, caffeine and CPA treatments reduced these arrhythmic effects of FPL. Nifedipine pre-treatment combined with a further addition of FPL in six hearts did not result in arrhythmogenesis during either intrinsic or regular pacing but five hearts showed AT (lasting 1.1, 1.1, 0.3, 2.6 and 29.2 s) during PES (Fig. 6A). Caffeine pre-treatment abolished FPL-mediated arrhythmic effects during intrinsic, regular pacing in six hearts. One heart showed AT during PES lasting 5.2 s (Fig. 6B). Finally, with pre-treatment with CPA combined with a further addition of FPL in five hearts, none of the hearts showed arrhythmogenesis during either intrinsic or regular pacing. During PES two hearts showed AT (lasting 0.6 and 5.4 s) and two showed sustained AF both lasting 5.0 s (Fig. 6C).
Discussion
The present study investigated the effect of enhancing LTCC opening on the initiation of acute atrial arrhythmias at both the cellular and the whole organ levels in intact murine hearts. We provide an independent confirmation of the finding that enhanced extracellular Ca2+ entry through using the LTCC agonist FPL exerts acute atrial arrhythmogenic effects that nevertheless depend upon diastolic release of intracellularly stored SR Ca2+. Thus treatment with FPL resulted in an appearance both of diastolic Ca2+ events at the cellular level, and atrial arrhythmogenesis despite an absence of changes in AERP at the level of intact perfused hearts. These phenomena – whether at the cellular or the whole organ level – were inhibited if the addition of FPL followed pre-treatment with the LTCC antagonist nifedipine, suggesting their dependence upon voltage-activated entry of extracellular Ca2+. They were also abolished by pre-treatments with either caffeine or CPA. This provided two independent methods of reducing SR Ca2+. The first would increase RyR2-mediated release of intracellularly stored Ca2+. The second would inhibit Ca2+-ATPase activity. The latter findings are consistent with a dependence of the effects of FPL upon a finite SR Ca2+ store. Together these findings thus attribute acute atrial arrhythmogenesis produced by FPL in an otherwise normal heart to an increased CICR that is dependent upon both Ca entry and SR Ca2+.
There have been extensive studies on chronic or established AF (Yue et al. 1997, Bosch et al. 1999, Van Wagoner et al. 1999, Skasa et al. 2001, Yagi et al. 2002). Thus, persistent AF has been associated with a reduction in atrial action potential duration (Li & Nattel 1997). This has been attributed to reductions in current densities through L-type Ca2+ channels (ICa,L). Recent studies have correspondingly shown reduced expression in mRNA coding for the α1C subunit of L-type channels in atria from patients with persistent AF (Brundel et al. 1999, Lai et al. 1999). Experimental studies that applied chronic atrial pacing at rates mimicking AF demonstrated depressed Ca2+ currents and numbers of dihydropyridine binding sites (Gaspo et al. 1999). There is also recent evidence for reduced L-type Ca2+ currents in the atrial myocardium of patients with persistent AF (Bosch et al. 1999, Van Wagoner et al. 1999). These changes in turn have been implicated in the electrical remodelling associated with AF (Yue et al. 1997, Bosch et al. 1999, Van Wagoner et al. 1999, Skasa et al. 2001, Yagi et al. 2002). These remodelling changes are not present in patients with non-persistent AF (Skasa et al. 2001). Conversely, AF is accompanied by abnormal SR Ca2+ release that likely reflects increased open probability in hyperphosphorylated RyR2-release channels (Hulme et al. 2006). In contrast, a recent report described transgenic (TG) mice with a cardiac over-expression of junctin and an accompanying 2.4 fold ICa,L over-expression. This described the electrophysiological features of significantly lower heart rates in TG compared with WT litters. There were also signs of AF on examination using limb-lead electrocardiography. The hearts also showed accompanying fibrotic and anatomical alterations. However, the precise causal relationships between these observations were not tested in detail (Hong et al. 2002).
However, there have been relatively few reports on factors bearing on the acute initiation of AF in otherwise structurally normal hearts despite its accepted clinical importance (see introduction for references). Previous studies had examined the relationship between altered Ca2+ homeostasis at the cellular level (Venetucci et al. 2007), and acute ventricular arrhythmogenic tendency in intact hearts (Balasubramaniam et al. 2004, 2005, Goddard et al. 2008). At the cellular level, the specific LTCC modulator FPL applied at a concentration of 1.0 μm increases the amplitude of both Ca2+ currents and Ca2+ transients more than it increased the rate of rise of Ca2+ transients in rat ventricular myocytes. This would potentially increase the release of intracellularly stored SR Ca2+ through enhancing CICR (Fan & Palade 2002). Correspondingly, FPL produced a Ca2+-mediated ventricular arrhythmogenecity at the level of intact Langendorff-perfused murine hearts. However, these relationships could potentially differ for atrial arrhythmogenesis. Atrial myocytes show important differences from ventricular myocytes particularly in their tubular and SR membrane systems that might reflect functional differences in their Ca2+ homeostatic processes. They do not possess extensive T-tubular systems (Mackenzie et al. 2001, 2004) but instead have prominent transversely oriented SR, Z-tubular, elements. Instead, atrial cells show an abundant corbular SR containing non-junctional RyR2s (Jorgensen et al. 1993). Junctional RyR2- ICa,L clusters are confined to the cell peripheries (Mackenzie et al. 2001). This may reflect atrial activation normally involving CICR in a pattern of centripetal propagation into the cell interior from superficial T-SR junctions (Mackenzie et al. 2001, 2004, Bootman et al. 2006).
The present experiments explored for both direct and indirect effects of an enhanced entry of extracellular Ca2+ on both Ca2+ homeostasis at both the cellular level and possible arrhythmogenic consequences at the level of whole hearts. They used the following pharmacological agents known to modify Ca2+ homeostasis: (1) FPL may modify LTCCs by prolonging channel opening during depolarization and channel closing upon repolarization enhancing the duration of the consequent tail currents (Rampe & Lacerda 1991) with possible actions upon SR Ca2+ release (Katoh et al. 2000, Copello et al. 2007). In rat ventricular myocytes, it increases both Ca2+ currents and Ca2+ transients more than it increases the rate of rise of Ca2+ transients, potentially increasing CICR. (2) Nifedipine is a known competitive dihydropyridine-LTCC blocker in ventricular cells with a KD of 40 nm (Shen et al. 2000, Balasubramaniam et al. 2004, Thomas et al. 2007). (3) Caffeine increases the release of intracellularly stored Ca2+ either by sensitizing RyR2s to cytosolic Ca2+ or inhibiting phosphodiesterase activity, thereby increasing cellular cAMP (Daly 2007), consequently increasing their open probabilities (Trafford et al. 2000). This would produce a gradual depletion of SR Ca2+ as reported previously for ventricular cells (Gaburjakova & Gaburjakova 2006, Venetucci et al. 2007). (4) CPA acts through inhibition of SERCA activity. At 100–200 nm concentrations it produces a ∼50% reduction of such activity (Schwinger et al. 1997) whilst sparing Ca2+ sensitivity in the contractile myofilaments (Takahashi et al. 1995), Ca2+ currents (Bonnet et al. 1994), and Na+-Ca2+ exchange activity (Goeger et al. 1988, Yard et al. 1994).
The experiments demonstrated that the addition of FPL disrupted the regular pattern of Ca2+ transients observed in regularly stimulated isolated Fluo-3-loaded atrial myocytes studied under confocal microscopy. These findings correlated with an appearance of atrial arrhythmogenesis at the level of intact isolated hearts. Such effects took place in atria at lower FPL concentrations than were needed to produce the corresponding ventricular effects. These changes occurred despite an absence of alterations in AERP.
These pro-arrhythmic actions of FPL whether at the cellular or the whole-heart level were dependent not only upon entry of extracellular Ca2+ but also on availability of releasable SR store Ca2+. Thus, nifedipine pre-treatment abolished the changes in Ca2+ homeostasis produced by a subsequent introduction of FPL at the level of single cells, on the one hand, and reduced FPL-induced arrhythmogenesis in whole hearts on the other. This directly correlates with the known clinical actions of Ca2+ channel antagonists in preventing acute postoperative AF (Podesser et al. 1995, Yilmaz et al. 1996, Kim et al. 2002, Dobrilovic et al. 2005, Baker & White 2007, Iwamoto & Inoue 2007). It is consistent with the potential arrhythmogenic effects of extracellular Ca2+ entry. Pre-treatments with CPA or caffeine would both be expected to deplete SR Ca2+ either through an inhibition of Ca2+-ATPase activity or an increase in RyR2-mediated release of intracellularly stored Ca2+. These manoeuvres similarly blocked FPL-mediated alterations in Ca2+ signalling at the level of isolated atrial myocytes as well as its arrhythmogenic effects in intact hearts.
These findings together are consistent with a model for the acute atrial arrhythmogenesis in an otherwise normal heart to a process of CICR, dependent upon both Ca entry and SR Ca2+. These would act within a model for an acute atrial arrhythmogenesis in an otherwise normal heart that involves triggered activity. This thus only resulted in transient rather than sustained arrhythmogenic episodes in an absence of alterations in the refractory period. The arrhythmogenic phenomena parallel the diastolic Ca2+ events produced by FPL observed at the cellular level. Furthermore, they were rescued by pharmacological manoeuvres demonstrated to reduce either SR Ca2+ or extracellular Ca2+ entry at the cellular level. These manoeuvres correspondingly rescued the diastolic Ca2+ events at the cellular level brought about by FPL.
Acknowledgments
This study was supported by grants from the British Heart Foundation, the Medical Research Council, the Wellcome Trust and the Biotechnology and Biological Sciences Research Council, UK and the National Natural Science Foundation of China (NSFC 30371571 and 30672209), PRC and the Helen Kirkland Fund for Cardiac Research. Y.H.Z. thanks the Department for Education and Skills of the United Kingdom and the China Scholarship Council for support and M.J.K. thanks the Physiological Laboratory for the award of an Avrith Studentship.
Conflict of interest
We report no conflict of interest.
References
- Baker WL, White CM. Post-cardiothoracic surgery atrial fibrillation: a review of preventive strategies. Ann Pharmacother. 2007;41:587–598. doi: 10.1345/aph.1H594. [DOI] [PubMed] [Google Scholar]
- Balasubramaniam R, Chawla S, Mackenzie L, Schwiening CJ, Grace AA, Huang CL. Nifedipine and diltiazem suppress ventricular arrhythmogenesis and calcium release in mouse hearts. Pflugers Arch. 2004;449:150–158. doi: 10.1007/s00424-004-1321-2. [DOI] [PubMed] [Google Scholar]
- Balasubramaniam R, Chawla S, Grace AA, Huang CL. Caffeine-induced arrhythmias in murine hearts parallel changes in cellular Ca2+ homeostasis. Am J Physiol Heart Circ Physiol. 2005;289:H1584–H1593. doi: 10.1152/ajpheart.01250.2004. [DOI] [PubMed] [Google Scholar]
- Baxter AJ, Dixon J, Ince F, Manners CN, Teague SJ. Discovery and synthesis of methyl 2,5-dimethyl-4-[2-(phenylmethyl)benzoyl]-1H-pyrrole-3-carboxylate (FPL 64176) and analogues: the first examples of a new class of calcium channel activator. J Med Chem. 1993;36:2739–2744. doi: 10.1021/jm00071a004. [DOI] [PubMed] [Google Scholar]
- Bonnet V, Badaoui A, Huchet-Cadiou C, Leoty C. Potentiation of the twitch responses by inhibitors of sarcoplasmic reticulum Ca2+-ATPase in frog atrial fibres. Eur J Pharmacol. 1994;264:69–76. doi: 10.1016/0014-2999(94)90637-8. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Higazi DR, Coombes S, Roderick HL. Calcium signalling during excitation-contraction coupling in mammalian atrial myocytes. J Cell Sci. 2006;119:3915–3925. doi: 10.1242/jcs.03223. [DOI] [PubMed] [Google Scholar]
- Bosch RF, Zeng X, Grammer JB, Popovic K, Mewis C, Kuhlkamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res. 1999;44:121–131. doi: 10.1016/s0008-6363(99)00178-9. [DOI] [PubMed] [Google Scholar]
- Bosch RF, Scherer CR, Rub N, Wohrl S, Steinmeyer K, Haase H, Busch AE, Seipel L, Kuhlkamp V. Molecular mechanisms of early electrical remodeling: transcriptional downregulation of ion channel subunits reduces I(Ca,L) and I(to) in rapid atrial pacing in rabbits. J Am Coll Cardiol. 2003;41:858–869. doi: 10.1016/s0735-1097(02)02922-4. [DOI] [PubMed] [Google Scholar]
- Brembilla-Perrot B, Beurrier D, Houriez P, Suty-Selton C, Nippert M, Claudon O, Andronache M, Ernst Y, Khaldi E, Belhakem H, Popovic B, Terrier de la Chaise A, Louis P. Electrophysiologic characteristics of atria in patients without heart disease. Pacing Clin Electrophysiol. 2005;28:1066–1072. doi: 10.1111/j.1540-8159.2005.00240.x. [DOI] [PubMed] [Google Scholar]
- Brundel BJ, Van Gelder IC, Henning RH, Tuinenburg AE, Deelman LE, Tieleman RG, Grandjean JG, Van Gilst WH, Crijns HJ. Gene expression of proteins influencing the calcium homeostasis in patients with persistent and paroxysmal atrial fibrillation. Cardiovasc Res. 1999;42:443–454. doi: 10.1016/s0008-6363(99)00045-0. [DOI] [PubMed] [Google Scholar]
- Cerrone M, Colombi B, Santoro M, Di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–e82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- Christ T, Boknik P, Wohrl S, Wettwer E, Graf EM, Bosch RF, Knaut M, Schmitz W, Ravens U, Dobrev D. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation. 2004;110:2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A. [DOI] [PubMed] [Google Scholar]
- Copello JA, Zima AV, Diaz-Sylvester PL, Fill M, Blatter LA. Ca2+ entry-independent effects of L-type Ca2+ channel modulators on Ca2+ sparks in ventricular myocytes. Am J Physiol Cell Physiol. 2007;292:C2129–C2140. doi: 10.1152/ajpcell.00437.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly JW. Caffeine analogs: biomedical impact. Cell Mol Life Sci. 2007;64:2153–2169. doi: 10.1007/s00018-007-7051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrilovic N, Vadlamani L, Buchert B, Wright CB. Diltiazem prophylaxis reduces incidence of atrial fibrillation after coronary artery bypass grafting. J Cardiovasc Surg (Torino) 2005;46:457–461. [PubMed] [Google Scholar]
- Du GG, Ashley CC, Lea TJ. Ca2+ effluxes from the sarcoplasmic reticulum vesicles of frog muscle: effects of cyclopiazonic acid and thapsigargin. Cell Calcium. 1996;20:355–359. doi: 10.1016/s0143-4160(96)90041-x. [DOI] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol. 1975;249:469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk RH. Etiology and complications of atrial fibrillation: insights from pathology studies. Am J Cardiol. 1998;82:10N–17N. doi: 10.1016/s0002-9149(98)00735-8. [DOI] [PubMed] [Google Scholar]
- Fan JS, Palade P. Effects of FPL 64176 on Ca transients in voltage-clamped rat ventricular myocytes. Br J Pharmacol. 2002;135:1495–1504. doi: 10.1038/sj.bjp.0704598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JS, Yuan Y, Palade P. Kinetic effects of FPL 64176 on L-type Ca2+ channels in cardiac myocytes. Naunyn Schmiedebergs Arch Pharmacol. 2000;361:465–476. doi: 10.1007/s002100000219. [DOI] [PubMed] [Google Scholar]
- Gaburjakova J, Gaburjakova M. Comparison of the effects exerted by luminal Ca2+ on the sensitivity of the cardiac ryanodine receptor to caffeine and cytosolic Ca2+ J Membr Biol. 2006;212:17–28. doi: 10.1007/s00232-006-7018-z. [DOI] [PubMed] [Google Scholar]
- Gaspo R, Sun H, Fareh S, Levi M, Yue L, Allen BG, Hebert TE, Nattel S. Dihydropyridine and beta adrenergic receptor binding in dogs with tachycardia-induced atrial fibrillation. Cardiovasc Res. 1999;42:434–442. doi: 10.1016/s0008-6363(99)00036-x. [DOI] [PubMed] [Google Scholar]
- Goddard CA, Ghais NS, Zhang Y, Williams AJ, Colledge WH, Grace AA, Huang CL. Physiological consequences of the P2328S mutation in the ryanodine receptor (RyR2) gene in genetically modified murine hearts. Acta Physiol (Oxf) 2008;194:123–140. doi: 10.1111/j.1748-1716.2008.01865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeger DE, Riley RT, Dorner JW, Cole RJ. Cyclopiazonic acid inhibition of the Ca2+-transport ATPase in rat skeletal muscle sarcoplasmic reticulum vesicles. Biochem Pharmacol. 1988;37:978–981. doi: 10.1016/0006-2952(88)90195-5. [DOI] [PubMed] [Google Scholar]
- Hong CS, Cho MC, Kwak YG, Song CH, Lee YH, Lim JS, Kwon YK, Chae SW, Kim DH. Cardiac remodeling and atrial fibrillation in transgenic mice overexpressing junctin. FASEB J. 2002;16:1310–1312. doi: 10.1096/fj.01-0908fje. [DOI] [PubMed] [Google Scholar]
- Hulme JT, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during beta1-adrenergic regulation. Proc Natl Acad Sci USA. 2006;103:16574–16579. doi: 10.1073/pnas.0607294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto J, Inoue H. Pharmacological therapy for atrial fibrillation after cardiac surgery. Kyobu Geka. 2007;60:559–564. [PubMed] [Google Scholar]
- Jorgensen AO, Shen AC, Arnold W, McPherson PS, Campbell KP. The Ca2+-release channel/ryanodine receptor is localized in junctional and corbular sarcoplasmic reticulum in cardiac muscle. J Cell Biol. 1993;120:969–980. doi: 10.1083/jcb.120.4.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh H, Schlotthauer K, Bers DM. Transmission of information from cardiac dihydropyridine receptor to ryanodine receptor: evidence from BayK 8644 effects on resting Ca2+ sparks. Circ Res. 2000;87:106–111. doi: 10.1161/01.res.87.2.106. [DOI] [PubMed] [Google Scholar]
- Kim MH, Rachwal W, McHale C, Bruckman D, Decena BF, Russman P, Morady F, Eagle KA. Effect of amiodarone +/- diltiazem +/- beta blocker on frequency of atrial fibrillation, length of hospitalization, and hospital costs after coronary artery bypass grafting. Am J Cardiol. 2002;89:1126–1128. doi: 10.1016/s0002-9149(02)02287-7. [DOI] [PubMed] [Google Scholar]
- Klein G, Schroder F, Vogler D, Schaefer A, Haverich A, Schieffer B, Korte T, Drexler H. Increased open probability of single cardiac L-type calcium channels in patients with chronic atrial fibrillation. role of phosphatase 2A. Cardiovasc Res. 2003;59:37–45. doi: 10.1016/s0008-6363(03)00357-2. [DOI] [PubMed] [Google Scholar]
- Lai LP, Su MJ, Lin JL, Lin FY, Tsai CH, Chen YS, Huang SK, Tseng YZ, Lien WP. Down-regulation of L-type calcium channel and sarcoplasmic reticular Ca2+-ATPase mRNA in human atrial fibrillation without significant change in the mRNA of ryanodine receptor, calsequestrin and phospholamban: an insight into the mechanism of atrial electrical remodeling. J Am Coll Cardiol. 1999;33:1231–1237. doi: 10.1016/s0735-1097(99)00008-x. [DOI] [PubMed] [Google Scholar]
- Lauven M, Handrock R, Muller A, Hofmann F, Herzig S. Interaction of three structurally distinct Ca2+ channel activators with single L-type Ca2+ channels. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:122–128. doi: 10.1007/s002109900059. [DOI] [PubMed] [Google Scholar]
- Li GR, Nattel S. Properties of human atrial ICa at physiological temperatures and relevance to action potential. Am J Physiol. 1997;272:H227–H235. doi: 10.1152/ajpheart.1997.272.1.H227. [DOI] [PubMed] [Google Scholar]
- Mackenzie L, Bootman MD, Berridge MJ, Lipp P. Predetermined recruitment of calcium release sites underlies excitation-contraction coupling in rat atrial myocytes. J Physiol. 2001;530:417–429. doi: 10.1111/j.1469-7793.2001.0417k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie L, Roderick HL, Berridge MJ, Conway SJ, Bootman MD. The spatial pattern of atrial cardiomyocyte calcium signalling modulates contraction. J Cell Sci. 2004;117:6327–6337. doi: 10.1242/jcs.01559. [DOI] [PubMed] [Google Scholar]
- Maguire CT, Wakimoto H, Patel VV, Hammer PE, Gauvreau K, Berul CI. Implications of ventricular arrhythmia vulnerability during murine electrophysiology studies. Physiol Genomics. 2003;15:84–91. doi: 10.1152/physiolgenomics.00034.2003. [DOI] [PubMed] [Google Scholar]
- Moe GK, Rheinboldt WC, Abildskov JA. A computer model of atrial fibrillation. Am Heart J. 1964;67:200–220. doi: 10.1016/0002-8703(64)90371-0. [DOI] [PubMed] [Google Scholar]
- Moncoq K, Trieber CA, Young HS. The molecular basis for cyclopiazonic acid inhibition of the sarcoplasmic reticulum calcium pump. J Biol Chem. 2007;282:9748–9757. doi: 10.1074/jbc.M611653200. [DOI] [PubMed] [Google Scholar]
- Morillo CA, Klein GJ, Jones DL, Guiraudon CM. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation. 1995;91:1588–1595. doi: 10.1161/01.cir.91.5.1588. [DOI] [PubMed] [Google Scholar]
- Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415:219–226. doi: 10.1038/415219a. [DOI] [PubMed] [Google Scholar]
- Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- Palomeque J, Petroff MV, Sapia L, Gende OA, Mundina-Weilenmann C, Mattiazzi A. Multiple alterations in Ca2+ handling determine the negative staircase in a cellular heart failure model. J Card Fail. 2007;13:143–154. doi: 10.1016/j.cardfail.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Podesser BK, Schwarzacher S, Zwoelfer W, Binder TM, Wolner E, Seitelberger R. Comparison of perioperative myocardial protection with nifedipine versus nifedipine and metoprolol in patients undergoing elective coronary artery bypass grafting. J Thorac Cardiovasc Surg. 1995;110:1461–1469. doi: 10.1016/S0022-5223(95)70069-2. [DOI] [PubMed] [Google Scholar]
- Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- Rampe D, Lacerda AE. A new site for the activation of cardiac calcium channels defined by the nondihydropyridine FPL 64176. J Pharmacol Exp Ther. 1991;259:982–987. [PubMed] [Google Scholar]
- Ryu K, Shroff SC, Sahadevan J, Martovitz NL, Khrestian CM, Stambler BS. Mapping of atrial activation during sustained atrial fibrillation in dogs with rapid ventricular pacing induced heart failure: evidence for a role of driver regions. J Cardiovasc Electrophysiol. 2005;16:1348–1358. doi: 10.1111/j.1540-8167.2005.00266.x. [DOI] [PubMed] [Google Scholar]
- Saumarez RC, Grace AA. Paced ventricular electrogram fractionation and sudden death in hypertrophic cardiomyopathy and other non-coronary heart diseases. Cardiovasc Res. 2000;47:11–22. doi: 10.1016/s0008-6363(00)00096-1. [DOI] [PubMed] [Google Scholar]
- Schwinger RH, Brixius K, Bavendiek U, Hoischen S, Muller-Ehmsen J, Bolck B, Erdmann E. Effect of cyclopiazonic acid on the force-frequency relationship in human nonfailing myocardium. J Pharmacol Exp Ther. 1997;283:286–292. [PubMed] [Google Scholar]
- Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem. 1989;264:17816–17823. [PubMed] [Google Scholar]
- Shen JB, Jiang B, Pappano AJ. Comparison of L-type calcium channel blockade by nifedipine and/or cadmium in guinea pig ventricular myocytes. J Pharmacol Exp Ther. 2000;294:562–570. [PubMed] [Google Scholar]
- Skasa M, Jungling E, Picht E, Schondube F, Luckhoff A. L-type calcium currents in atrial myocytes from patients with persistent and non-persistent atrial fibrillation. Basic Res Cardiol. 2001;96:151–159. doi: 10.1007/s003950170065. [DOI] [PubMed] [Google Scholar]
- Stokoe KS, Thomas G, Goddard CA, Colledge WH, Grace AA, Huang CL. Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/Delta murine hearts modelling long QT syndrome 3. J Physiol. 2007;578:69–84. doi: 10.1113/jphysiol.2006.117945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Kato Y, Adachi M, Agata N, Tanaka H, Shigenobu K. Effects of cyclopiazonic acid on rat myocardium: inhibition of calcium uptake into sarcoplasmic reticulum. J Pharmacol Exp Ther. 1995;272:1095–1100. [PubMed] [Google Scholar]
- Thomas G, Gurung IS, Killeen MJ, Hakim P, Goddard CA, Mahaut-Smith MP, Colledge WH, Grace AA, Huang CL. Effects of L-type Ca2+ channel antagonism on ventricular arrhythmogenesis in murine hearts containing a modification in the Scn5a gene modelling human long QT syndrome 3. J Physiol. 2007;578:85–97. doi: 10.1113/jphysiol.2006.121921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G, Killeen MJ, Grace AA, Huang CL. Pharmacological separation of early afterdepolarizations from arrhythmogenic substrate in ΔKPQ Scn5a murine hearts modelling human long QT 3 syndrome. Acta Physiol (Oxf) 2008;192:505–517. doi: 10.1111/j.1748-1716.2007.01770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrall G, Lane D, Carroll D, Lip GY. Quality of life in patients with atrial fibrillation: a systematic review. Am J Med. 2006;119:448 e1–448 e19. doi: 10.1016/j.amjmed.2005.10.057. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Sibbring GC, Diaz ME, Eisner DA. The effects of low concentrations of caffeine on spontaneous Ca release in isolated rat ventricular myocytes. Cell Calcium. 2000;28:269–276. doi: 10.1054/ceca.2000.0156. [DOI] [PubMed] [Google Scholar]
- Triggle DJ. 1,4-Dihydropyridines as calcium channel ligands and privileged structures. Cell Mol Neurobiol. 2003;23:293–303. doi: 10.1023/A:1023632419813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wagoner DR, Pond AL, Lamorgese M, Rossie SS, McCarthy PM, Nerbonne JM. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ Res. 1999;85:428–436. doi: 10.1161/01.res.85.5.428. [DOI] [PubMed] [Google Scholar]
- Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res. 2007;100:105–111. doi: 10.1161/01.RES.0000252828.17939.00. [DOI] [PubMed] [Google Scholar]
- Wellens HJ. 25 years of insights into the mechanisms of supraventricular arrhythmias. Pacing Clin Electrophysiol. 2003;26:1916–1922. doi: 10.1046/j.1460-9592.2003.00295.x. [DOI] [PubMed] [Google Scholar]
- Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–1968. doi: 10.1161/01.cir.92.7.1954. [DOI] [PubMed] [Google Scholar]
- Yagi T, Pu J, Chandra P, Hara M, Danilo P, Jr, Rosen MR, Boyden PA. Density and function of inward currents in right atrial cells from chronically fibrillating canine atria. Cardiovasc Res. 2002;54:405–415. doi: 10.1016/s0008-6363(02)00279-1. [DOI] [PubMed] [Google Scholar]
- Yard NJ, Chiesi M, Ball HA. Effect of cyclopiazonic acid, an inhibitor of sarcoplasmic reticulum Ca2+-ATPase, on the frequency-dependence of the contraction–relaxation cycle of the guinea-pig isolated atrium. Br J Pharmacol. 1994;113:1001–1007. doi: 10.1111/j.1476-5381.1994.tb17092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh Y-H, Wakili R, Qi XY, Chartier D, Boknik P, Kääb S, Ravens U, Coutu P, Dobrev D, Nattel S. Calcium handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol. 2008;1:93–102. doi: 10.1161/CIRCEP.107.754788. [DOI] [PubMed] [Google Scholar]
- Yilmaz AT, Demirkilic U, Arslan M, Kurulay E, Ozal E, Tatar H, Ozturk O. Long-term prevention of atrial fibrillation after coronary artery bypass surgery: comparison of quinidine, verapamil, and amiodarone in maintaining sinus rhythm. J Card Surg. 1996;11:61–64. doi: 10.1111/j.1540-8191.1996.tb00010.x. [DOI] [PubMed] [Google Scholar]
- Yue L, Feng J, Gaspo R, Li GR, Wang Z, Nattel S. Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ Res. 1997;81:512–525. doi: 10.1161/01.res.81.4.512. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Schwiening C, Killeen MJ, Zhang Y, Ma A, Lei M, Grace AA, Huang CL. Pharmacological alterations of cellular Ca homeostasis parallel initiation of atrial arrhythmogenesis in Langendorff perfused murine hearts. Clin Exp Pharmacol Physiol. 2009;36:969–980. doi: 10.1111/j.1440-1681.2009.05170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]