

Abstract
Background
Animal studies suggest that the arginine vasopressin (AVP) system may play a role in glucose metabolism, but data from humans are limited.
Methods and Results
We analysed plasma copeptin (copeptin), a stable C-terminal fragment of the AVP pro-hormone. Using baseline and longitudinal data from a Swedish population-based sample (n=4742, mean age 58 years, 60% women), we examined the association of increasing quartiles of copeptin (lowest quartile as reference) with prevalent diabetes at baseline, insulin resistance (top quartile of fasting plasma insulin among non-diabetic subjects), and incident diabetes on long-term follow up using multivariable logistic regression. New-onset diabetes was ascertained through 3 national and regional registers. All models were adjusted for clinical and anthropometric risk factors, cystatin C, and C-reactive protein. In cross-sectional analyses, increasing copeptin was associated with prevalent diabetes (P=0.04) and insulin resistance (P<0.001). During 12.6 years of follow up 174 subjects (4%) developed new-onset diabetes. The odds of developing diabetes increased across increasing quartiles of copeptin, even after additional adjustment for baseline fasting glucose and insulin (adjusted odds ratios 1.0, 1.37, 1.79, and 2.09; P for trend =0.004). The association with incident diabetes remained significant in analyses restricted to subjects with fasting whole blood glucose <5.4 mmol/L at baseline (adjusted odds ratios 1.0, 1.80, 1.92, and 3.48; P=0.001).
Conclusions
Elevated copeptin predicts increased risk for diabetes, independent of established clinical risk factors, including fasting glucose and insulin. These findings could have implications for risk assessment, novel anti-diabetic treatments, and metabolic side effects from AVP system modulation.
Keywords: arginine vasopressin, copeptin, diabetes mellitus, risk factors, epidemiology
INTRODUCTION
Diabetes is one of the major risk factors for coronary heart disease and congestive heart failure, but the causes of this interaction are incompletely understood. One possible link is that hormones implicated in cardiac diseases may play a role in diabetes development. Arginine Vasopressin (AVP), also known as antidiuretic hormone (ADH), is released from the pituitary gland in conditions of high plasma osmolality, low plasma volume and low blood pressure. Plasma copeptin, a marker of vasopressin level, is elevated in myocardial infarction1 and predict prognosis in patients who developed heart failure after myocardial infarction.2 Interestingly, recent findings also indicate a cross-sectional association between plasma vasopressin and insulin resistance.3
AVP binds to three different receptors (V1aR, V1bR and V2R). The V1aR is widely expressed,4 whereas V1bR is more specifically expressed in the pituitary gland and pancreas, and V2R in the renal collecting ducts.5–7 The antidiuretic effect of AVP is mediated through V2R and pharmacological blockade of V2R has been used in the treatment of hyponatremia8 and heart failure.9 The prothrombotic and vasoconstrictor effects of AVP are primarily mediated through the V1aR,10, 11 and AVP agonists have been used in the treatment of bleeding and hypotensive disorders.12, 13 In addition, AVP action has been linked to liver glycogenolysis (V1aR) and insulin and glucagon secretion (V1bR).14, 15 Further, AVP stimulates pituitary ACTH release (V1bR).16 This effect has been reported to be resistant to glucocorticoid feedback, suggesting cross talk between the AVP and HPA systems which could be of relevance for diabetes development.17
Previous studies in humans and animals have suggested a role for the AVP system in glucose homeostasis, insulin resistance and diabetes mellitus. In patients with poorly controlled diabetes mellitus, plasma AVP is markedly elevated18 and in healthy subjects AVP infusion leads to increased blood glucose levels.19 Mice lacking the V1aR display impaired glucose tolerance, insulin resistance and elevated AVP levels20 while mice who lack the V1bR have the opposite phenotype of lower fasting plasma glucose and increased insulin sensitivity.21 These findings suggest a model in which impaired signalling through the V1aR leads to elevated levels of AVP which in turn stimulates the V1bR and contributes to insulin resistance and diabetes development.
There are major concerns regarding the reliability of AVP measurements in plasma, as AVP is an unstable molecule both in vivo and ex vivo, is rapidly cleared from plasma and is largely attached to platelets in the circulation. Copeptin is a cleavage product of the C-terminal part of the AVP precursor that is produced in equimolar amounts with AVP, a process similar to the generation of insulin and C-peptide. In contrast to AVP, copeptin is stable, has a long half-life, and is not bound to platelets. Therefore, copeptin is found in considerably higher concentrations in plasma than AVP.22
Thus, using a validated sandwich assay for measurement of copeptin in plasma (copeptin), we tested the hypothesis that elevated copeptin is related cross-sectionally with prevalent diabetes and insulin resistance and longitudinally with increased risk of new-onset diabetes mellitus at the population level.
METHODS
Subjects
The Malmö Diet and Cancer study (MDC) is a population-based prospective cohort consisting of 28,449 persons surveyed in 1991–1996.23 From this cohort, 6,103 persons were randomly selected to be studied for the epidemiology of carotid artery disease, and this sample is referred to as the MDC cardiovascular cohort (MDC-CC). Fasting plasma samples were obtained in 5,405 subjects in the MDC-CC.24 Of those, complete data on covariates, including risk factors for diabetes, potential confounders and copeptin, were available in 4,742 individuals.
All analyses in plasma and whole blood were performed in overnight fasting samples. Analyses of fasting whole blood glucose (FBG), plasma lipids and insulin were carried out at the time of baseline examination at the Department of Clinical Chemistry, Malmö University Hospital, which is attached to a national standardization and quality control system. The limit of detection for insulin was 3mIU L −1, and the intra- and interassay coefficients of variation were 5 and 8%, respectively.25 FBG was measured in whole blood glucose by a hexokinase-glucose-6-phosphate dehydrogenase method. In fasting plasma samples we measured copeptin using a commercially available assay in the chemiluminescence/coated tube format (B.R.A.H.M.S AG, Hennigsdorf, Germany) as described previously.26 C-reactive protein (CRP) was measured by a high-sensitivity assay (Tina-quant CRP, Roche Diagnostics, Basel, Switzerland). Cystatin C was measured using a particle-enhanced immuno-nephelometric assay (N Latex Cystatin C, Dade Behring, IL).
Blood pressure was measured using a mercury-column sphygmomanometer after ten minutes of rest in the supine position. Cigarette smoking was elicited by a self-administered questionnaire, with current cigarette smoking defined as any smoking within the past year. Prevalent cardiovascular disease was defined as occurrence of myocardial infarction or stroke prior to the baseline examination obtained through national registers as described previously.24 Family history of diabetes was obtained by a questionnaire and defined as known diabetes in at least one first degree relative.
Diabetes mellitus at baseline was defined as self report of a physician diagnosis or use of diabetes medication or FBG of ≥6.1 mmol/L (corresponding to fasting plasma glucose concentration of ≥7.0 mmol/L). Subjects who belonged to the top quartile of fasting insulin concentration in the segment of the population without diabetes were defined as having insulin resistance, as proposed by the European Group for the study of Insulin Resistance.27 New-onset diabetes diagnosed after the baseline examination until December 2005 (mean follow-up time 12.6 years) was assessed in subjects free from diabetes at baseline by three registers; the Malmö HbA1c register (MHR), the nationwide Swedish National Diabetes Register (NDR)28 and the regional Diabetes 2000 register of the Scania region of which Malmö is the largest city.29 NDR and Diabetes 2000 registers required a physician diagnosis according to established diagnostic criteria (fasting plasma glucose concentration of ≥7.0 mmol/L, which corresponds to fasting whole blood glucose concentration of ≥6.1 mmol/L, measured on two different occasions).
The MHR at the Department of Clinical Chemistry, Malmö University Hospital, analysed and catalogued all HbA1c samples taken in institutional and non-institutional care in the greater Malmö area from 1988 onwards. Individuals who had at least two HbA1c recordings ≥6.0% in the MHR using the Swedish Mono-S standardization system (corresponding to 7.0% according to the US National Glycohemoglobin Standardization Program [NGSP]), after the MDC-CC baseline examination were defined as incident diabetes cases. Furthermore, subjects who were registered in the NDR or Diabetes 2000 register after the MDC-CC baseline examination were defined as incident diabetes cases.
Subjects who were free of diabetes at the baseline examination in the MDC-CC, defined by lack of self-reported history of physician-diagnosed diabetes, use of diabetes medication and FBG at the baseline examination of ≥6.1 mmol/L, but were registered as diabetes cases any time after their baseline examination in the MDC-CC until December 2005, were classified as having new-onset diabetes. Using these criteria, 174 subjects had new-onset diabetes during follow-up. Of these, more than half (52%) were identified as new-onset diabetes cases via at least two of the three registers.
A subset (n=887) of the MDC-CC was reinvestigated after a mean of 6.6 years.30 Of subjects free of diabetes or without IFG at baseline, complete data on covariates, including risk factors for diabetes, potential confounders and copeptin, were available in 745 and 620 individuals, respectively. Diabetes development was defined as FBG ≥6.1 mmol/L at the time of follow up.
The study protocols were approved by the ethics committee of Lund University. All participants provided written informed consent.
Statistical analyses
SPSS statistical software (version 14.0) was used for all analyses but calculation of the C-statistics, which was performed using Stata software version 8.0 (stata corp). Group-wise differences in continuous variables at baseline were tested using student’s t-test and reported as means ± SD if normally distributed and with Mann-Whitney test and reported as medians and interquartile ranges if not normally distributed. Variables which were not normally distributed were transformed using the natural logarithm when analysed as continuous variables. Differences in dichotomous variables were tested using chi-square tests. The P-value for linear trend in FBG and insulin levels over quartiles of copeptin in subjects without diabetes was assessed using linear regression. We used crude and multivariable adjusted logistic regression to test if increasing quartiles of copeptin (Q2–Q4, compared to Q1) were related to prevalent diabetes at baseline in the entire cohort (n=4742). In subjects without diabetes at baseline (n=4377), crude and multivariable logistic regression were used to test whether increasing quartiles of copeptin were related to insulin resistance and to new-onset diabetes. In addition, the relationship between increasing quartiles of copeptin and risk of new-onset diabetes was tested in subjects without impaired fasting glucose (IFG) at the baseline examination (fasting plasma glucose <6.1 mmol/L corresponding to FBG <5.4 mmol/L) (n=3702). Finally, multivariable logistic regression was used to test the relationship between increasing quartiles of copeptin and incident diabetes, based on FBG thresholds, in the subset of MDC-CC that was reinvestigated. Data from logistic regression analyses were expressed as odds ratio (OR) and 95 % confidence intervals (CI). A two sided P-value of <0.05 was considered statistically significant.
To assess sensitivity and specificity of copeptin in predicting new-onset diabetes in addition to classical diabetes predictors, we compared the area under the Receiver Operating Characteristic (ROC) curves using both a personal model (age, gender, BMI and family history of diabetes) and a clinical model (personal model + systolic blood pressure, triglycerides, HDL, waist circumference and FBG) as previously proposed31 with and without copeptin in each of the two models. The Integrated Discrimination Improvement (IDI) was calculated as described previously.32
We chose logistic regression in our primary analyses rather than Cox regression (time-to-event), because the diagnosis of diabetes commonly occurs years after the actual diabetes onset.33. In secondary analyses, we performed Cox regression models with the time of the diabetes event defined as the date of either the first HbA1c value ≥ 6% in MHR, registration in DM 2000, or registration in NDR. C-statistics based on the Cox regressions were calculated as described previously.34
RESULTS
Baseline characteristics according to diabetes status are shown in Table 1. A total of 365 individuals had diabetes at the baseline exam, 29% of whom had a history of a physician-diagnosis of diabetes or diabetes treatment, and 71% of whom had a FBG ≥6.1 mmol/L without an accompanying physician diagnosis.
Table 1.
Baseline characteristics of subjects with normal fasting glucose (NFG), with impaired fasting glucose (IFG) and with diabetes mellitus (DM)
| NFG subjects (n=3702) | IFG subjects (excl DM) (n=675) | DM subjects (n=365) | P-value NFG vs DM | P-value IFG vs DM | |
|---|---|---|---|---|---|
| Age (years) | 57.3 ± 5.9 | 57.9 ± 5.8 | 59.5 ± 5.5 | < 0.001 | < 0.001 |
| Men (%) | 38.3 | 53.9 | 56.2 | < 0.001 | 0.49 |
| FBG (mmol/l) | 4.8 ± 0.34 | 5.6 ± 0.19 | 8.1 ± 3.0 | < 0.001 | < 0.001 |
| Triglycerides (mmol/l)* | 1.14 (0.86–1.58) | 1.34 (1.00–1.86) | 1.64 (1.13–2.33) | < 0.001 | < 0.001 |
| Systolic BP (mmHg) | 139 ± 19 | 146 ± 19 | 150 ± 20 | < 0.001 | 0.001 |
| Diastolic BP (mmHg) | 86.1 ± 9.3 | 89 ± 9.6 | 90 ± 9.5 | < 0.001 | 0.036 |
| AHT (%) | 15.7 | 20.9 | 37.3 | < 0.001 | < 0.001 |
| BMI (kg/m2) | 25.2 ± 3.6 | 27.0 ± 3.9 | 28.7 ± 4.5 | < 0.001 | < 0.001 |
| Waist (cm) | 81.5 ± 12 | 88.8 ± 12 | 94.4 ± 13 | < 0.001 | < 0.001 |
| Waist-hip-ratio | 0.84 ± 0.09 | 0.88 ± 0.09 | 0.91 ± 0.09 | < 0.001 | < 0.001 |
| HDL (mmol/l) | 1.42 ± 0.37 | 1.31 ± 0.35 | 1.23 ± 0.35 | < 0.001 | 0.001 |
| LDL (mmol/l) | 4.1 ± 0.98 | 4.3 ± 1.0 | 4.2 ± 1.0 | 0.078 | 0.23 |
| Cystatin C (mg/l) | 0.771 ± 0.144 | 0.787 ± 0.137 | 0.809 ± 0.193 | < 0.001 | 0.038 |
| Copeptin (pmol/l)* | 5.14 (3.20–8.14) | 5.77 (3.54–9.32) | 6.90 (4.32–10.7) | < 0.001 | 0.011 |
| Insulin (mU/L)* | 6.0 (4.0–9.0) | 8.0 (6.0–12) | 12 (7.0–18) | < 0.001 | < 0.001 |
| CRP (mg/l)* | 1.3 (0.7–2.8) | 1.7 (0.8–3.4) | 2.3 (1.3–4.5) | < 0.001 | < 0.001 |
| Current smoker (%) | 25.9 | 29.1 | 24.4 | 0.50 | 0.12 |
| Family history of diabetes (%) | 3.1 | 2.1 | 3.0 | 0.90 | 0.35 |
| Previous CVD (%) | 2.1 | 2.8 | 3.8 | 0.01 | 0.37 |
Continuous variables are given as means ± SD unless otherwise specified
median (interquartile range).
AHT, anti-hypertensive treatment; BMI, Body-mass-index; FBG, fasting blood glucose; previous CVD, cardiovascular disease present before baseline examination.
At the baseline examination, concentrations of copeptin were higher among individuals with diabetes compared with subjects without diabetes (Table 1). Among non-diabetic subjects at baseline, the Pearson correlation between copeptin and potential confounders, i.e. BMI, insulin and glucose, was 0.091, 0.12 and 0.10, respectively (P<0.001 for all). Increasing quartile of copeptin was associated with an increased odds of diabetes in a crude model and after multivariable adjustment for all baseline covariates that differed between diabetes patients and subjects without diabetes (Table 1) with the exception of FBG and fasting plasma insulin concentration (model 1 adjustment) (Table 2). In the segment of the population free of diabetes, i.e. in subjects with a range of glucose values that should not affect copeptin through osmolality, copeptin was positively associated with FBG (Figure 1). Additionally, copeptin was positively associated with plasma concentration of insulin (Figure 1). The odds of insulin resistance increased with increasing copeptin in both crude analysis and after adjustment for model 1 covariates and FBG (Table 2). The association between increasing quartiles of copeptin and the HOMA-insulin resistance index (the product of fasting insulin and FBG) was similar (data not shown).
Table 2.
Prevalent diabetes and insulin resistance in relation to quartiles of baseline copeptin at baseline.
| Dependent variable | OR* (95% CI) Q2copeptin vs Q1 |
OR* (95% CI) Q3copeptin vs Q1copeptin |
OR* (95% CI) Q4copeptin vs Q1copeptin |
P (test for linear trend) | |
|---|---|---|---|---|---|
| Diabetes† | crude | 1.44 (1.00–2.07) | 1.92 (1.36–2.71) | 2.83 (2.04–3.93) | <0.001 |
| Adjusted§ | 1.19 (0.81–1.75) | 1.39 (0.96–2.01) | 1.45 (1.00–2.11) | 0.04 | |
| Hyperinsulinemia‡ | crude | 1.30 (1.06–1.60) | 1.53 (1.25–1.87) | 2.34 (1.93–2.85) | <0.001 |
| Adjusted‖ | 1.19 (0.94–1.51) | 1.25 (0.99–1.59) | 1.61 (1.26–2.06) | <0.001 | |
Odds Ratio.
Analysis of diabetes prevalence (n=365) in the entire cohort (n=4742).
Analysis of hyperinsulinemia (highest quartile of fasting plasma insulin concentration among non-diabetic subjects) among non-diabetic subjects (n=4377).
Adjusted for age, sex, HDL, triglycerides, blood pressure, antihypertensive treatment, body mass index, waist, waist/hip ratio, cystatin C, CRP and previous cardiovascular disease (model 1).
Adjusted for model 1 and FBG.
Figure 1.
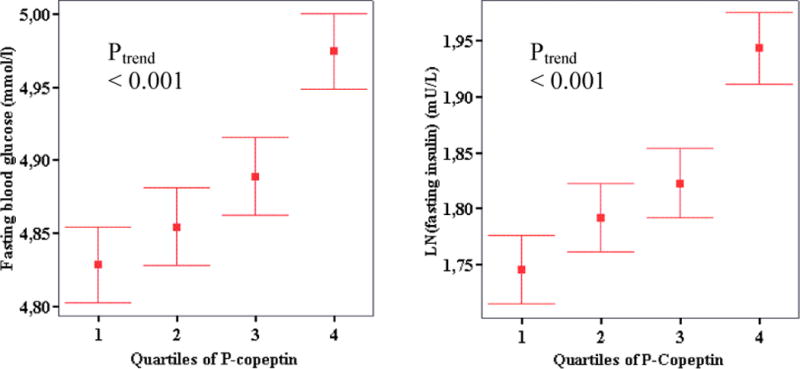
Fasting concentrations of glucose in whole blood and insulin in plasma (the latter transformed using the natural logarithm, LN), expressed as mean with 95 % confidence intervals, in non diabetic subjects belonging to quartiles of increasing copeptin (n=4377). P for trend <0.001 in both analyses.
Among participants free of diabetes at baseline (n=4377), 174 subjects developed new-onset diabetes (Table 3) according to the three different registers. MHR captured 106 subjects, whereas the Diabetes 2000 registry and the NDR captured 110 and 76 subjects, respectively. Of the 106 diabetes cases diagnosed by MHR, 71% (n=75) were also diagnosed in Diabetes 2000 and/or in NDR.
Table 3.
Baseline characteristics in subjects who did and did not convert to diabetes during follow-up
| Subjects without diabetes at baseline (n=4377) | |||
|---|---|---|---|
| Non-converters (n=4203) |
Incident diabetes (n=174) |
P | |
| Age (years) | 57.3 ± 5.9 | 57.9 ± 5.7 | 0.26 |
| Men (%) | 38.9 | 45.4 | 0.09 |
| FBG (mmol/l) | 4.9 ± 0.44 | 5.4 ± 0.44 | < 0.001 |
| Triglycerides (mmol/l)* | 1.11 (0.84–1.51) | 1.46 (1.05–1.96) | < 0.001 |
| Systolic BP (mmHg) | 140 ± 19 | 146 ± 19 | < 0.001 |
| Diastolic BP (mmHg) | 86 ± 9.3 | 90 ± 10 | < 0.001 |
| AHT (%) | 14.2 | 27.6 | < 0.001 |
| BMI (kg/m2) | 25.4 ± 3.6 | 28.2 ± 4.7 | < 0.001 |
| Waist (cm) | 82.2 ± 12 | 91.5 ± 14 | < 0.001 |
| Waist-hip-ratio | 0.84 ± 0.09 | 0.89 ± 0.10 | < 0.001 |
| HDL (mmol/l) | 1.41 ± 0.37 | 1.25 ± 0.33 | < 0.001 |
| LDL (mmol/l) | 4.2 ± 0.98 | 4.3 ± 1.0 | 0.06 |
| Cystatin C (mg/l) | 0.77 ± 0.14 | 0.82 ± 0.21 | < 0.001 |
| Copeptin (pmol/l)* | 4.98 (3.09–7.84) | 6.35 (4.05–9.88) | < 0.001 |
| Insulin (mU/L)* | 6.0 (4.0–9.0) | 9.0 (6.0–13) | < 0.001 |
| CRP (mg/l)* | 1.2 (0.6–2.6) | 2.1 (0.9–4.1) | < 0.001 |
| Current smoker (%) | 26.4 | 29.5 | 0.37 |
| Family history of diabetes (%) | 2.8 | 6.9 | 0.002 |
| Previous CVD (%) | 2.0 | 2.3 | 0.80 |
| Subjects without impaired fasting glucose at baseline (n=3702) | |||
|---|---|---|---|
| Non-converters (n=3623) |
Incident diabetes (n=79) |
P | |
| Age (years) | 57.2 ± 5.9 | 58.1 ± 5.4 | 0.20 |
| Men (%) | 36.5 | 38.0 | 0.79 |
| FBG (mmol/l) | 4.7 ± 0.34 | 5.0 ± 0.28 | < 0.001 |
| Triglycerides (mmol/l)* | 1.08 (0.82–1.47) | 1.34 (1.06–1.92) | < 0.001 |
| Systolic BP (mmHg) | 139 ± 19 | 146 ± 19 | 0.001 |
| Diastolic BP (mmHg) | 86 ± 9.2 | 89 ± 9.5 | 0.004 |
| AHT (%) | 13.4 | 24.1 | 0.006 |
| BMI (kg/m2) | 25.2 ± 3.6 | 27.5 ± 4.9 | < 0.001 |
| Waist (cm) | 81.3 ± 12 | 88.7 ± 14 | < 0.001 |
| Waist-hip-ratio | 0.84 ± 0.09 | 0.87 ± 0.09 | < 0.001 |
| HDL (mmol/l) | 1.43 ± 0.37 | 1.27 ± 0.32 | < 0.001 |
| LDL (mmol/l) | 4.1 ± 0.98 | 4.1 ± 0.93 | 0.99 |
| Cystatin C (mg/l) | 0.77 ± 0.14 | 0.83 ± 0.27 | < 0.001 |
| Copeptin (pmol/l)* | 4.90 (3.03–7.65) | 6.74 (4.44–10.9) | 0.001 |
| Insulin (mU/L)* | 6.0 (4.0–8.0) | 8.0 (6.0–11) | < 0.001 |
| CRP (mg/l)* | 1.2 (0.6–2.4) | 2.4 (1.0–4.1) | < 0.001 |
| Current smoker (%) | 26.0 | 30.4 | 0.38 |
| Family history of diabetes (%) | 3.0 | 7.6 | 0.02 |
| Previous CVD (%) | 1.9 | 2.5 | 0.67 |
Continuous variables are given as means ± SD unless otherwise specified
median (interquartile range).
AHT, anti-hypertensive treatment; BMI, Body-mass-index; FBG, fasting blood glucose; previous CVD, cardiovascular disease present before baseline examination.
When the baseline sample was further restricted to subjects without IFG (n=3702), 79 subjects developed new-onset diabetes during follow-up. Continuous copeptin concentration at baseline was significantly higher in subjects who subsequently developed new-onset diabetes compared with those who did not, regardless of whether subjects with IFG at baseline were included (Table 3). The likelihood of developing new-onset diabetes increased with increasing copeptin quartiles in crude logistic regression analysis as well as after multivariate adjustment (model 1 covariates and fasting insulin, FBG, smoking, diabetes heredity and LDL), in both subjects free of diabetes at baseline (P for trend <0.001 crude, P=0.004 multivariable-adjusted) and in subjects free of IFG at baseline (P<0.001 crude, P=0.001 multivariable-adjusted, Table 4).
Table 4.
New-onset diabetes in relation to quartiles of baseline copeptin.
| Dependent variable | OR* (95% CI) Q2copeptin vs Q1copeptin | OR* (95% CI) Q3copeptin vs Q1copeptin | OR* (95% CI) Q4copeptin vs Q1copeptin | P (test for linear trend) | |
|---|---|---|---|---|---|
| Incident diabetes among non-DM† | Crude | 1.28 (0.76–2.15) | 1.94 (1.19–3.14) | 2.64 (1.66–4.19) | <0.001 |
| adjusted§ | 1.37 (0.78–2.39) | 1.79 (1.06–3.05) | 2.09 (1.23–3.56) | 0.004 | |
| Incident diabetes among non-IFG‡ | Crude | 1.85 (0.81–4.20) | 2.12 (0.95–4.73) | 4.56 (2.18–9.52) | <0.001 |
| adjusted§ | 1.80 (0.78–4.16) | 1.92 (0.84–4.38) | 3.48 (1.58–7.65) | 0.001 | |
Odds Ratio.
Subjects who developed diabetes during follow-up (n=174) among subjects without diabetes at baseline (n=4377).
Subjects who developed diabetes during follow-up (n=79) among subjects without impaired fasting glucose at baseline (n=3702).
Adjusted for age, sex, HDL, triglycerides, blood pressure, antihypertensive treatment, body mass index, waist, waist/hip ratio, cystatin C, CRP and prevalent cardiovascular disease, smoking, family history of diabetes, LDL, FBG and fasting insulin.
In subjects free of diabetes at baseline, the area under the ROC curve increased from 0.694 to 0.710 (P=0.08) when copeptin was added to the personal model, and from 0.832 to 0.841 (P=0.007) when copeptin was added to the clinical model of diabetes prediction. In subjects free of IFG at baseline, the area under the ROC curve increased from 0.663 to 0.713 (P=0.03) and from 0.783 to 0.805 (P=0.04) when copeptin was added to the personal model and clinical model for diabetes prediction, respectively. IDI was significantly improved by adding copeptin to the personal model for diabetes prediction in both non-diabetic subjects (P=0.01) and in non-IFG subjects (P=0.02) but non-significant when added to the clinical model (P=0.35 in non-diabetic individuals, P=0.09 in non-IFG individuals).
In order to validate the association between copeptin at baseline and the register-based diabetes endpoint, we assessed the multivariate-adjusted relationship between copeptin and diabetes development based on elevated FBG, in a subset of the MDC-CC that had been reinvestigated after 6.6 years. Among those free of diabetes at baseline, 63 subjects had an elevated FBG during follow up. Increasing quartile of copeptin was associated with new-onset diabetes using this criterion both in subjects free of diabetes at baseline (OR=1.42 95% CI=1.04–1.94; P=0.029) and in subjects free of IFG at baseline (OR=1.67 95% CI=1.07–2.63; P=0.025) after full adjustment (model 1 covariates and fasting insulin, FBG, smoking, diabetes heredity and LDL) comparable to findings reported in Table 4.
Finally, Cox regression models were performed in order to exclude potential bias related to inter-individual differences in follow-up time in the logistic regressions models and the results were unchanged (Supplemental Table 1). C-statistics derived from Cox regression models were similar to those from logistic regression models. With addition of copeptin, the C-statistic from Cox regression models increased in subjects without diabetes at baseline from 0.702 to 0.718 in the personal model and from 0.835 to 0.844 in the clinical model. When analyses were restricted to subjects without IFG at baseline, the C-statistic increased from 0.671 to 0.721 in the personal model and from 0.790 to 0.811 in the clinical model.
DISCUSSION
The key finding of our study is that copeptin predicts diabetes development independently of renal function and a broad range of diabetes risk factors at baseline, including FBG and fasting insulin. The association between baseline copeptin and incident diabetes was particularly strong in those individuals free of IFG at baseline.
As expected, FBG was the strongest risk factor for new-onset diabetes. Each 1 mmol/L increase of FBG at baseline increased the risk of future diabetes with an OR of 11.4 (95% CI 7.4–17.5) in the fully-adjusted model. Nonetheless, after accounting for FBG and all other available diabetes risk factors, individuals in the top quartile of copeptin had a two- to three-fold excess risk of developing diabetes compared with those in the lowest quartile of copeptin. In subjects without IFG, there was a significant improvement in the area under the ROC curve when copeptin was added to classical diabetes risk factors, consistent with an improvement in discrimination. Additionally, inclusion of copeptin was associated with a significant IDI when compared to the personal model, and a borderline significant IDI when compared with the clinical model. The improvement in the area under the ROC curve was smaller when the entire sample, including those with IFG, was considered. This finding may result from FBG being a more powerful predictor of diabetes at FBG levels near the diagnostic limit of 6.1mmol/L. Markers that are not part of the diagnostic criteria for diabetes, such as copeptin, may better signal diabetes susceptibility earlier in the pre-diabetes state. Novel risk markers as screening tools for future diabetes risk could be particularly useful in individuals with normal FBG, who are likely to be less closely monitored than individuals with IFG. Thus, our findings suggest that copeptin could provide incremental information for prediction of diabetes.
Apart from any implications for diabetes prediction, our data support a role for the AVP system in the pathophysiology of diabetes. Animal studies have shown that mice lacking the V1aR display elevated levels of AVP, glucose intolerance and insulin resistance, whereas mice lacking the V1bR have the opposite phenotype of lower FBG and improved insulin sensitivity.20, 21 Based on these animal data and our clinical findings, it can be speculated that elevated AVP, as a consequence of AVP resistance at the level of V1aR or elsewhere, could contribute to insulin resistance and diabetes through stimulation of the V1bR. In fact, pharmacological blockade of the V2R, a potent stimulus of increased AVP secretion,35 was associated with a 5% incidence in hyperglycemic events during 30 days of tolvaptan treatment in patients with hyponatremia compared to 1% in controls, although numbers were small (12/223 vs 2/220).8 Further studies on the potential metabolic effects of pharmacological manipulation of the AVP system may be warranted.
The diabetes incidence in our study may appear low but was comparable with that in a previous Swedish study.36 Still, there are several potential factors that could result in a lower than expected number of new onset diabetes cases in our study. 1) As the participation rate was only 40%, the MDC population is healthier than the background population.37 2) The diabetes incidence we observe in the three registers is based on people who actively seek health care, leading to a lower incidence than that observed in studies that directly screen for diabetes by measuring FBG. 3) At baseline, the classification of prevalent diabetes was based solely on FBG ≥6.1 mmol/L in as many as 71% of diabetes cases. Excluding these previously unrecognized diabetes cases from follow-up minimizes the risk that we have incorrectly classified some subjects as incident, instead of prevalent, cases of diabetes. 4) Finally, due to incomplete register coverage, our study may have missed new-onset cases who were in fact diagnosed with diabetes within the health care system. These factors would be expected to bias our results towards the null. On the other hand, given the strict HbA1c criteria for new-onset diabetes in the MHR and the fact that the NDR and Diabetes 2000 registers require a physician diagnosis according to established diagnostic criteria,28, 29 those classified as new-onset diabetes cases in our study are unlikely to be misclassified. The validity of the register based new-onset diabetes diagnosis is further supported by most of the well-known risk factors for diabetes being markedly elevated at the MDC-CC baseline in these subjects as compared to those who did not develop diabetes according to the three registers (Table 3). Importantly, these differences were equally, or more, pronounced when the study population was restricted to subjects free from IFG at baseline, excluding the possibility that our findings regarding established diabetes risk factors and copeptin in relation to new-onset diabetes were solely driven by subjects who had IFG at the MDC-CC baseline exam.
The fact that we were able to reproduce the association between copeptin and the register-based diabetes endpoint, using a FBG-based endpoint in a subset of MDC-CC, strongly supports the validity of our findings.
In conclusion, copeptin predicts diabetes independently of a broad range of established diabetes risk factors, including fasting levels of glucose and insulin. Our findings suggest a role of the AVP system in diabetes development and may have implications for risk assessment and the development of novel diabetes pharmacotherapy. In addition, our results highlight the possibility that existing therapies that selectively antagonize AVP receptors could have metabolic side effects.
Supplementary Material
Acknowledgments
We thank Dr Jan-Olof Jeppsson for his valuable work with the MHR.
FUNDING SOURCES
Drs. Enhörning and Melander were supported by grants from the Swedish Medical Research Council, the Swedish Heart and Lung Foundation, the Medical Faculty of Lund University, Malmö University Hospital, the Albert Påhlsson Research Foundation, the Crafoord foundation, the Ernhold Lundströms Research Foundation, the Region Skane, the Hulda and Conrad Mossfelt Foundation, the King Gustaf V and Queen Victoria Foundation, the Lennart Hanssons Memorial Fund, and the Wallenberg Foundation. Dr. Newton-Cheh was supported by NIH K23-HL-080025, a Doris Duke Charitable Foundation Clinical Scientist Development Award, and a Burroughs Wellcome Fund Career Award for Medical Scientists. Dr. Wang was supported by NIH grants R01-HL-086875, R01-HL-083197, and R01- DK-081572, and a grant from the American Heart Association. The authors also wish to thank Brahms and Dade-Behring for their support of assay measurements.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST DISCLOSURES
Drs. Struck, Morgenthaler and Bergmann are employees of B.R.A.H.M.S AG. B.R.A.H.M.S AG holds patent rights on the copeptin assay. There are no other conflicts of interest in connection with this manuscript. All researchers have been independent from funders.
References
- 1.Reichlin T, Hochholzer W, Stelzig C, Laule K, Freidank H, Morgenthaler NG, Bergmann A, Potocki M, Noveanu M, Breidthardt T, Christ A, Boldanova T, Merki R, Schaub N, Bingisser R, Christ M, Mueller C. Incremental value of copeptin for rapid rule out of acute myocardial infarction. J Am Coll Cardiol. 2009;54:60–68. doi: 10.1016/j.jacc.2009.01.076. [DOI] [PubMed] [Google Scholar]
- 2.Voors AA, von Haehling S, Anker SD, Hillege HL, Struck J, Hartmann O, Bergmann A, Squire I, van Veldhuisen DJ, Dickstein K. C-terminal provasopressin (copeptin) is a strong prognostic marker in patients with heart failure after an acute myocardial infarction: results from the OPTIMAAL study. Eur Heart J. 2009;30:1187–1194. doi: 10.1093/eurheartj/ehp098. [DOI] [PubMed] [Google Scholar]
- 3.Saleem U, Khaleghi M, Morgenthaler NG, Bergmann A, Struck J, Mosley TH, Jr, Kullo IJ. Plasma Carboxy-terminal Pro-Vasopressin (Copeptin): A Novel Marker of Insulin Resistance and Metabolic Syndrome. J Clin Endocrinol Metab. 2009;94:2558–2564. doi: 10.1210/jc.2008-2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morel A, O’Carroll AM, Brownstein MJ, Lolait SJ. Molecular cloning and expression of a rat V1a arginine vasopressin receptor. Nature. 1992;356:523–526. doi: 10.1038/356523a0. [DOI] [PubMed] [Google Scholar]
- 5.Folny V, Raufaste D, Lukovic L, Pouzet B, Rochard P, Pascal M, Serradeil-Le Gal C. Pancreatic vasopressin V1b receptors: characterization in In-R1-G9 cells and localization in human pancreas. Am J Physiol Endocrinol Metab. 2003;285:E566–576. doi: 10.1152/ajpendo.00148.2003. [DOI] [PubMed] [Google Scholar]
- 6.de Keyzer Y, Auzan C, Lenne F, Beldjord C, Thibonnier M, Bertagna X, Clauser E. Cloning and characterization of the human V3 pituitary vasopressin receptor. FEBS Lett. 1994;356:215–220. doi: 10.1016/0014-5793(94)01268-7. [DOI] [PubMed] [Google Scholar]
- 7.Lolait SJ, O’Carroll AM, McBride OW, Konig M, Morel A, Brownstein MJ. Cloning and characterization of a vasopressin V2 receptor and possible link to nephrogenic diabetes insipidus. Nature. 1992;357:336–339. doi: 10.1038/357336a0. [DOI] [PubMed] [Google Scholar]
- 8.Schrier RW, Gross P, Gheorghiade M, Berl T, Verbalis JG, Czerwiec FS, Orlandi C. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355:2099–2112. doi: 10.1056/NEJMoa065181. [DOI] [PubMed] [Google Scholar]
- 9.Gheorghiade M, Konstam MA, Burnett JC, Jr, Grinfeld L, Maggioni AP, Swedberg K, Udelson JE, Zannad F, Cook T, Ouyang J, Zimmer C, Orlandi C. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST Clinical Status Trials. Jama. 2007;297:1332–1343. doi: 10.1001/jama.297.12.1332. [DOI] [PubMed] [Google Scholar]
- 10.Filep J, Rosenkranz B. Mechanism of vasopressin-induced platelet aggregation. Thromb Res. 1987;45:7–15. doi: 10.1016/0049-3848(87)90252-0. [DOI] [PubMed] [Google Scholar]
- 11.Ohlstein EH, Berkowitz BA. Human vascular vasopressin receptors: analysis with selective vasopressin receptor antagonists. J Pharmacol Exp Ther. 1986;239:737–741. [PubMed] [Google Scholar]
- 12.Federici AB, Mannucci PM. Management of inherited von Willebrand disease in 2007. Ann Med. 2007;39:346–358. doi: 10.1080/07853890701513738. [DOI] [PubMed] [Google Scholar]
- 13.Dunser MW, Mayr AJ, Ulmer H, Knotzer H, Sumann G, Pajk W, Friesenecker B, Hasibeder WR. Arginine vasopressin in advanced vasodilatory shock: a prospective, randomized, controlled study. Circulation. 2003;107:2313–2319. doi: 10.1161/01.CIR.0000066692.71008.BB. [DOI] [PubMed] [Google Scholar]
- 14.Keppens S, de Wulf H. The nature of the hepatic receptors involved in vasopressin-induced glycogenolysis. Biochim Biophys Acta. 1979;588:63–69. doi: 10.1016/0304-4165(79)90371-4. [DOI] [PubMed] [Google Scholar]
- 15.Abu-Basha EA, Yibchok-Anun S, Hsu WH. Glucose dependency of arginine vasopressin-induced insulin and glucagon release from the perfused rat pancreas. Metabolism. 2002;51:1184–1190. doi: 10.1053/meta.2002.34052. [DOI] [PubMed] [Google Scholar]
- 16.Holmes CL, Landry DW, Granton JT. Science review: Vasopressin and the cardiovascular system part 1–receptor physiology. Crit care. 2003;7:427–434. doi: 10.1186/cc2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabadan-Diehl C, Aguilera G. Glucocorticoids increase vasopressin V1b receptor coupling to phospholipase C. Endocrinology. 1998;139:3220–3226. doi: 10.1210/endo.139.7.6121. [DOI] [PubMed] [Google Scholar]
- 18.Zerbe RL, Vinicor F, Robertson GL. Plasma vasopressin in uncontrolled diabetes mellitus. Diabetes. 1979;28:503–508. doi: 10.2337/diab.28.5.503. [DOI] [PubMed] [Google Scholar]
- 19.Spruce BA, McCulloch AJ, Burd J, Orskov H, Heaton A, Baylis PH, Alberti KG. The effect of vasopressin infusion on glucose metabolism in man. Clin Endocrinol. 1985;22:463–468. doi: 10.1111/j.1365-2265.1985.tb00145.x. [DOI] [PubMed] [Google Scholar]
- 20.Aoyagi T, Birumachi J, Hiroyama M, Fujiwara Y, Sanbe A, Yamauchi J, Tanoue A. Alteration of glucose homeostasis in V1a vasopressin receptor-deficient mice. Endocrinology. 2007;148:2075–2084. doi: 10.1210/en.2006-1315. [DOI] [PubMed] [Google Scholar]
- 21.Fujiwara Y, Hiroyama M, Sanbe A, Aoyagi T, Birumachi J, Yamauchi J, Tsujimoto G, Tanoue A. Insulin hypersensitivity in mice lacking the V1b vasopressin receptor. J Physiol. 2007;584:235–244. doi: 10.1113/jphysiol.2007.136481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Struck J, Morgenthaler NG, Bergmann A. Copeptin, a stable peptide derived from the vasopressin precursor, is elevated in serum of sepsis patients. Peptides. 2005;26:2500–2504. doi: 10.1016/j.peptides.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 23.Berglund G, Elmstahl S, Janzon L, Larsson SA. The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med. 1993;233:45–51. doi: 10.1111/j.1365-2796.1993.tb00647.x. [DOI] [PubMed] [Google Scholar]
- 24.Persson M, Hedblad B, Nelson JJ, Berglund G. Elevated Lp-PLA2 levels add prognostic information to the metabolic syndrome on incidence of cardiovascular events among middle-aged nondiabetic subjects. Arterioscler Thromb Vasc Biol. 2007;27:1411–1416. doi: 10.1161/ATVBAHA.107.142679. [DOI] [PubMed] [Google Scholar]
- 25.Engstrom G, Hedblad B, Nilsson P, Wollmer P, Berglund G, Janzon L. Lung function, insulin resistance and incidence of cardiovascular disease: a longitudinal cohort study. J Int Med. 2003;253:574–581. doi: 10.1046/j.1365-2796.2003.01138.x. [DOI] [PubMed] [Google Scholar]
- 26.Fenske W, Stork S, Blechschmidt A, Maier SG, Morgenthaler NG, Allolio B. Copeptin in the differential diagnosis of hyponatremia. J Clin Endocrinol Metab. 2009;94:123–129. doi: 10.1210/jc.2008-1426. [DOI] [PubMed] [Google Scholar]
- 27.Balkau B, Charles MA. Comment on the provisional report from the WHO consultation. European Group for the Study of Insulin Resistance (EGIR) Diabet Med. 1999;16:442–443. [Google Scholar]
- 28.Cederholm J, Eeg-Olofsson K, Eliasson B, Zethelius B, Nilsson PM, Gudbjornsdottir S. Risk prediction of cardiovascular disease in type 2 diabetes: a risk equation from the Swedish National Diabetes Register. Diabetes Care. 2008;31:2038–2043. doi: 10.2337/dc08-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindholm E, Agardh E, Tuomi T, Groop L, Agardh CD. Classifying diabetes according to the new WHO clinical stages. Eur J Epidemiol. 2001;17:983–989. doi: 10.1023/a:1020036805655. [DOI] [PubMed] [Google Scholar]
- 30.Nilsson PM, Engstrom G, Hedblad B, Frystyk J, Persson MM, Berglund G, Flyvbjerg A. Plasma adiponectin levels in relation to carotid intima media thickness and markers of insulin resistance. Arterioscler Thromb Vasc Biol. 2006;26:2758–2762. doi: 10.1161/01.ATV.0000249638.01416.4b. [DOI] [PubMed] [Google Scholar]
- 31.Wilson PW, Meigs JB, Sullivan L, Fox CS, Nathan DM, D’Agostino RB., Sr Prediction of incident diabetes mellitus in middle-aged adults: the Framingham Offspring Study. Arch Intern Med. 2007;167:1068–1074. doi: 10.1001/archinte.167.10.1068. [DOI] [PubMed] [Google Scholar]
- 32.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172. doi: 10.1002/sim.2929. discussion 207–112. [DOI] [PubMed] [Google Scholar]
- 33.Harris MI, Klein R, Welborn TA, Knuiman MW. Onset of NIDDM occurs at least 4–7 yr before clinical diagnosis. Diabetes Care. 1992;15:815–819. doi: 10.2337/diacare.15.7.815. [DOI] [PubMed] [Google Scholar]
- 34.Pencina MJ, D’Agostino RB. Overall C as a measure of discrimination in survival analysis: model specific population value and confidence interval estimation. Stat Med. 2004;23:2109–2123. doi: 10.1002/sim.1802. [DOI] [PubMed] [Google Scholar]
- 35.Gines P, Wong F, Watson H, Milutinovic S, del Arbol LR, Olteanu D. Effects of satavaptan, a selective vasopressin V(2) receptor antagonist, on ascites and serum sodium in cirrhosis with hyponatremia: a randomized trial. Hepatology. 2008;48:204–213. doi: 10.1002/hep.22293. [DOI] [PubMed] [Google Scholar]
- 36.Ohlson LO, Larsson B, Bjorntorp P, Eriksson H, Svardsudd K, Welin L, Tibblin G, Wilhelmsen L. Risk factors for type 2 (non-insulin-dependent) diabetes mellitus. Thirteen and one-half years of follow-up of the participants in a study of Swedish men born in 1913. Diabetologia. 1988;31:798–805. doi: 10.1007/BF00277480. [DOI] [PubMed] [Google Scholar]
- 37.Manjer J, Carlsson S, Elmstahl S, Gullberg B, Janzon L, Lindstrom M, Mattisson I, Berglund G. The Malmo Diet and Cancer Study: representativity, cancer incidence and mortality in participants and non-participants. Eur J Cancer Prev. 2001;10:489–499. doi: 10.1097/00008469-200112000-00003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.