

Abstract
B-cell lymphoma 2 (Bcl-2) family proteins regulate survival, mitochondria morphology dynamics and metabolism in many cell types including neurons. Huntington's disease (HD) is a neurodegenerative disorder caused by an expanded CAG repeat tract in the IT15 gene that encodes for the protein huntingtin (htt). In vitro and in vivo models of HD and HD patients' tissues show abnormal mitochondrial function and increased cell death rates associated with alterations in Bcl-2 family protein expression and localization. This review aims to draw together the information related to Bcl-2 family protein alterations in HD to decipher their potential role in mutated htt-related cell death and mitochondrial dysfunction.
Keywords: mutated huntingtin, Huntington's disease models, Bcl-2 family proteins
Facts
The B-cell lymphoma 2 (Bcl-2) family proteins regulate the outer mitochondrial membrane (OMM) permeability thus controlling the mitochondrial potential, mitochondrial morphology dynamics and the balance between survival and death in many cell types including neurons.
HD is a neurodegenerative disease characterized by progressive neuronal dysfunction and death. In patients with HD and in in vitro and in vivo HD models, mutated htt expression leads to OMM permeabilization, loss of mitochondrial potential and mitochondrial fragmentation.
HD cell culture models and HD mouse brain models show alterations in Bcl-2 family protein expression and localization. Autopsy studies in patients' brain have detected Bcl-2 family protein alterations in HD.
Open Questions
We still need more precise evidence on the role of Bcl-2 family proteins in HD-related cell death.
We do not know the role of Bcl-2 family proteins in HD-related mitochondrial dysfunction.
Nor do we know through which molecular pathway Bcl-2 family proteins can cause mitochondrial dysfunctions and cell death in HD.
Introduction
Proteins belonging to the Bcl-2 family are key regulators of the apoptotic mitochondrial pathway. Growing evidence shows that Bcl-2 family proteins also modulate mitochondrial morphology1, 2, 3 and cellular metabolism4 independently from cell death mechanisms. Given that HD typically leads to progressive neuronal death causing neuronal cells to display several mitochondrial dysfunctions (including decreased mitochondrial potential (Δψ), abnormal calcium handling, decreased ATP synthesis capacity and increased mitochondrial fragmentation), research has for long investigated Bcl-2 family proteins in HD models and tissues from patients with HD. These studies highlighted evidence that mutated htt dysregulates Bcl-2 family protein levels and localization. Considering the complex Bcl-2 network, a literature review could help to elucidate these proteins' role in mutated htt-related mitochondrial dysfunction and cell death.
What Evidence Exists for Dysregulated Bcl-2 Family Proteins in HD?
Research has to date identified over 20 Bcl-2 family members classified, according to their function, in three Bcl-2 family subgroups: the pro-survival proteins, the pro-apoptotic BH3-only proteins and the pore-forming proteins. Here, we review the three subgroups, focusing on the Bcl-2 family members relevant for HD and gathering data on the cell culture model SThdH,5 cell culture models obtained by transient htt transfection, in genetic mouse models R6/1, R6/2,6 Tet/HD94,7 N171-82Q,8 YAC1289 and human cells from patients with HD.
The Pro-survival Bcl-2 Proteins: Bcl-2 and Bcl-xL
Bcl-2 and B-cell lymphoma-extra large (Bcl-xL) function to repress programmed cell death.10, 11 They are stably inserted in intracellular membranes (OMM and the endoplasmic reticulum (ER) membrane) and inhibit cell death by sequestering the pro-apoptotic Bcl-2 family proteins into inactive complexes.12 In the normal central nervous system (CNS), Bcl-2 is widespread expressed during the embryonic stage, whereas after birth it progressively declines with aging. Studies in adult monkey and adult human CNS disclosed, apart from rare Bcl-2 positive neurons, that most neurons and astrocytes express no Bcl-2 and that most Bcl-2 protein in adult brain results from microglial expression.13 Bcl-xL is encoded by the Bcl-x gene and, differently from Bcl-2, retains its expression in adult neurons at a relatively high level, predominantly in Purkinje cells in the cerebellum, in cerebral cortical neurons and in hippocampal neurons.14, 15
Bcl-2 level alterations in HD remain controversial. Bcl-2 levels were first investigated in HD cell culture models: mutated htt expression significantly decreased Bcl-2 protein levels in the neuroblastoma cell line Neuro2A16 and significantly decreased Bcl-2 transcript and Bcl-2 protein levels in immortalized striatal cells.17, 18 Although these results strongly suggest that mutated htt expression decreases Bcl-2 levels, they provide no definitive proof given that the proto-oncogene Bcl-2 is highly dysregulated in many tumor cell lines including those of neural origin.19, 20 Hence, data from immortalized cell cultures might not correspond to Bcl-2 levels in post-mitotic neurons.
Others have investigated Bcl-2 levels also in brains from HD mouse models. A first study detected no Bcl-2 protein level changes in the total brain or in mitochondrial fractions from R6/2 mice as compared with littermate controls.21 Accordingly, two independent studies reported no differences in Bcl-2 mRNA and protein levels between R6/1 and control mice.22, 23 Conversely, more recent studies disclosed decreased Bcl-2 mRNA and protein levels in R6/2 mouse brains17, 24 and N171-82Q mice.25
After reappraising these seemingly contradictory findings, we find it difficult to provide a definitive answer about whether Bcl-2 levels are dysregulated in HD mouse models. To understand the potential protective role of Bcl-2 in HD, Zhang et al.21 crossed R6/2 mice with transgenic mice selectively overexpressing Bcl-2 in neurons: double transgenic R6/2-Bcl-2 mice survived longer than R6/2 littermates and their motor deficits had a significantly later onset. Notwithstanding contradictory data about Bcl-2 expression in HD mouse models, this result suggests that Bcl-2 overexpression can protect neurons from toxicity elicited by mutated htt.
Bcl-2 expression was analyzed by immunostaining in post-mortem controls and the HD patients' caudate nucleus. Control neurons had low or negative Bcl-2 signals, in agreement with a previous report,13 whereas Bcl-2 labeling was stronger in HD neurons, and Bcl-2 expression reached maximum in the brains of HD patients with longer disease duration.26 In accordance with a potential neuroprotective role of Bcl-2 in HD, the investigators suggested that HD patients' neurons may increase Bcl-2 expression in an attempt to survive. An alternative hypothesis is that Bcl-2 protein in HD brains is cleaved by caspase proteases activated by mutated htt expression.27 If so, the caspase-dependent cleavage inactivates Bcl-2 antiapoptotic function and converts Bcl-2 into a Bcl-2 associated X-protein (Bax)-like protein that enhances cell death.28 According to this hypothesis, rather than inhibiting cell death, high Bcl-2 levels in HD cells could worsen neurodegeneration. The Bcl-2 upregulation in HD patients seems to be brain-specific given that no differences were found between Bcl-2 protein levels in peripheral blood cells from patients with HD and from controls.29
Few data are available about the other pro-survival protein, Bcl-xL in HD: Bcl-xL protein levels were investigated in the brain of HD mice, but no significant difference was found in total-brain lysates, striatum lysates or mitochondrial fractions in HD mice and controls.21, 22, 30 To our knowledge, only one report suggests a role of Bcl-xL in HD. The authors showed that Bcl-xL expression prevented the htt proteolysis induced by DNA damage and hypothesized that blocking Bcl-xL may activate the caspases that cause htt proteolysis.31
The Pro-apoptotic BH3-only Proteins
The pro-apoptotic BH3-only proteins, by responding to specific death and survival signals, function as sensors of cellular damage. Activating one or more among the BH3-only proteins culminates in allosteric activation of the pore-forming proteins Bax and Bcl-2 antagonist/killer (Bak).
BH3-only proteins could activate Bax and Bak through two mechanisms. The ‘direct activation' model posits that certain BH3-only proteins termed ‘activators' (eg, Bcl-2 interacting mediator of cell death (Bim), Bcl-2 interacting domain death agonist (Bid)/truncated Bid (tBid) and p53 upregulated modulator of apoptosis (Puma)) bind to Bax and Bak directly triggering their oligomerization in the OMM, whereas other BH3-only proteins, termed ‘sensitizers' (eg, Bcl-2 antagonist of cell death (Bad), Bcl-2/adenovirus E1B 19 kDa interacting protein 3 (Bnip3) and Puma) would bind only to pro-survival Bcl-2 family members, thus liberating ‘activator' proteins. Instead, the ‘indirect activation' model posits that all the BH3-only proteins bind to pro-survival Bcl-2 family members, thereby preventing them from binding to and neutralizing Bax or Bak.32
Bcl-2 Antagonist of Cell Death
Bad has a double function: it separately regulates apoptosis and glucose metabolism in multiple cell types including neurons.33, 34 Investigating in vitro HD models, Rigamonti et al.35 showed that Bad overexpression in immortalized striatal cells induces apoptosis only in clones co-expressing mutated htt, thus suggesting that mutated htt toxicity induces Bad-dependent cell death. For HD mouse models, no differences in striatal Bad protein levels were found between R6/1 and littermate controls.22 Also in the R6/2 mouse, no change was found in the brain Bad protein levels. Conversely, the investigators found a significant decrease in phosphorylated Bad (pBad) during the late-HD stages in R6/2 mice.21 Because pBad promotes mitochondrial respiration and ATP production, whereas non-pBad binds the anti-apoptotic partners Bcl-2 and Bcl-xL eliciting Bax and Bak activation,36 the low pBad/Bad ratio found in R6/2 brain lysates suggests that Bad activity shifts from positive metabolic to negative apoptotic function in HD cells.
Bcl-2 Interacting Mediator of Cell Death
The BH3-only pro-apoptotic protein Bim is expressed in three major isoforms generated by alternative splicing: Bim short (BimS), Bim long (BimL) and Bim extra-long (BimEL). All these isoforms neutralize activity in pro-survival Bcl-2- like proteins, but they differ in pro-apoptotic potency, BimS being the most effective and BimEL the least effective killer.37 In the mouse and human CNS, Bim is expressed primarily in neurons, and the most expressed isoform in the brain is BimEL.38, 39
Several observations show that mutated htt causes BimEL accumulation in various cell types. In particular, mutated htt expression induces BimEL accumulation in HEK293T cells, in Neuro-2a cells,40 in sympathetic superior cervical ganglion neurons41 and in immortalized striatal cells.42 BimEL silencing in Neuro-2a cells significantly reduces cell death elicited by mutated htt.40
In the mouse model R6/2, high BimEL levels were found in total-brain lysates and in mitochondrial fractions specifically at the late stages of disease.21, 40 These data found further confirmation in two other HD mouse models showing increased striatal BimEL in R6/1 mice at the late stages of disease and in the conditional model Tet/HD94.22 Collectively, these results demonstrate that mutated htt causes BimEL accumulation and translocation to mitochondria.
Bcl-2 Interacting Domain Death Agonist
The BH3-only pro-apoptotic protein Bid is involved in neuronal cell death in many neurological disorders such as stroke,43 ischemia44 and Alzheimer's disease.45 Full-length Bid has extremely weak pro-apoptotic activity but reaches its strongest pro-apoptotic activity after proteolytic cleavage by several proteases that produce truncated Bid (tBid).46 Bid protein is widely expressed in embryonic and postnatal brain, and its expression in post-mitotic neurons in the limbic system, basal ganglia, mesencephalic tectum and cerebellum persists at a high level into adulthood.15
Several lines of evidence show that mutated htt causes Bid and tBid accumulation in neurons and in mitochondrial neuronal fractions. In particular, western blot analysis for Bid and tBid detection in HeLa cells and Neuro2A cells disclosed that mutated htt transfection causes Bid cleavage.16
Increased Bid cleavage was also detected in brain lysates from R6/2 mice at the middle stage of HD.21 Different results come from the R6/1 model in which Garcìa-Martìnez et al.22 found no tBid accumulation, but instead found full-length Bid at high levels in the striatum and in striatal mitochondrial fractions. The Bid protein increase correlated with enhanced Bid mRNA expression. Increased full-length Bid protein levels were also found in the striatum in the conditional mouse model of HD, Tet/HD94: Bid accumulation specifically depended on mutated htt expression, given that transgene suppression completely reverted Bid protein to wild-type levels.22
Bcl-2/adenovirus E1B 19 kDa Interacting Protein 3
Bnip3 has had an emerging role in human health, as convincing evidence implicated its death, inducing activity in heart diseases, whereas Bnip3 loss of function was associated with tumor growth.47, 48 Current knowledge shows that Bnip3 is involved in cell death, autophagy and programmed mitochondrial clearance.49 Bnip3 induces cell death through at least two distinct mechanisms: Bnip3 can engage anti-apoptotic Bcl-2 family members to trigger Bax–Bak-dependent OMM permeability50 or can induce a novel mitochondrial leak pathway by interacting with the optic atrophy-1 protein (OPA1).51, 52
Because Bnip3 is expressed in the brain and skeletal muscle,53, 54 in recent years, we undertook a study aimed to investigate the potential role of Bnip3 in HD by assessing Bnip3 level and localization in htt-transfected neuronal cells, brain in HD mouse models and in muscle cells from HD patients.55 We observed that mutated htt expression causes Bnip3 accumulation in whole-cell lysates and in the mitochondrial fraction of SHSY5Y and HEK293T cells. We also found higher Bnip3 protein levels in mitochondrial fractions from R6/2 mice and in striatum from YAC128 mice than in littermate controls. Finally, we observed that Bnip3 mainly co-localized with the mitochondria in HD patients' muscle cells, whereas in control cells it mainly localized in the cytosol and nucleus. We also observed that the expression of a dominant-negative Bnip3 named Bnip3ΔTM56, 57 rescued the mitochondrial membrane potential loss in HD muscle cells.55 Overall, these data suggest that mutated htt enhances Bnip3 activity in neuronal and muscular cells.
p53-upregulated Modulator of Apoptosis/Bcl-2 Binding Component 3 (Puma/Bbc3)
Puma/Bbc3 is a BH3-only protein identified in 2001.58, 59 Puma/Bbc3 appears not to be expressed in the normal adult brain, but its expression is strongly induced in some brain diseases such as status epilepticus60 and cerebral ischemia.61 Originally identified as a transcriptional target of p53,58, 59 Puma/Bbc3 is also transcriptionally induced in neuronal cells undergoing ER stress-induced unfolded protein responses (UPR).62 Few data are available about Puma/Bbc3 expression in HD models: in SCG neurons, mutated htt expression leaves Puma/Bbc3 mRNA unchanged,41 whereas in PC12 cells it increases Puma/Bbc3 protein sixfold. Although Puma/Bbc3 protein accumulation in HD may depend on mutated htt-induced ER stress, because this datum remains unconfirmed in other HD models, the possible pathogenetic role of Puma/Bbc3 in HD needs further clarification.
The Pore-forming Proteins Bax and Bcl-2 Antagonist/Killer (Bak)
Bax and Bak act downstream of the pro-survival and BH3-only members and have a key role in the mitochondrial apoptotic pathway. Bak is normally inserted in the OMM; Bax is predominantly cytosolic, but, once activated, it translocates from the cytosol into the OMM where together with Bak it forms the apoptotic pore, the point of no return in mitochondrial apoptosis.63 Besides inducing apoptosis, Bax and Bak influence mitochondrial fission–fusion dynamics in healthy cells.1, 2 Both Bax and Bak are expressed in neurons: Bax is expressed in many regions in the human CNS64, 65 including the human caudate nucleus;26 and Bak is expressed in the human brain at the fetal, adult and elder stages.66
Ample evidence underlining the link between Bax activation and HD pathogenesis comes from research assessing Bax protein levels and localization in neuronal cell culture models of HD and in the brains of HD mouse models. Mutated htt transfection in PC12 cells increased Bax protein levels fourfold as compared with wt htt transfection;67 in Neuro2A cells, mutated htt expression induced Bax translocation from the cytosol to the mitochondria.40 Only one in vitro study argues against a Bax role in mutated htt-induced cell death: King et al.21 showed that in sympathetic superior cervical ganglion (SCG) neurons mutated htt induced Bax-independent cell death.
Studies in HD mouse models showed increased Bax levels in the brain mitochondrial fractions from R6/1 and R6/2 mice21, 22 and increased Bax mRNA in the cortex and cerebellum from R6/1 mice, brain areas in which Bax mRNA correlated with the number of apoptotic nuclei.23 Bax expression was also analyzed in the caudate nucleus from HD patients. The cytoplasmic Bax signal was stronger in caudate neurons from HD patients than from controls. Bax expression was already maximal in HD brains at disease onset and the highest in the most severely compromised shrunken and dark neurons.26 Hence, data from HD mouse models and human tissues converge to indicate that mutated htt expression causes Bax protein accumulation and Bax translocation to the OMM in neurons. Bax dysregulation in HD seems not to be brain specific insofar as Bax protein levels were also higher in lymphocytes and monocytes from HD patients than from controls.29 This finding probably depends on the fact that htt is ubiquitously expressed in human tissues.68
Little is known about Bak levels in HD cells: Garcìa-Martìnez et al.22 analyzed the R6/1 mouse striatum and found equal Bak levels in R6/1 mice and littermate controls. Although these data may indicate that Bak has no role in HD cell death and mitochondrial dysfunction, others observed that immortalized striatal cells overexpressing wt htt are completely protected from Bak-induced death.35 Whether and, if so, how Bak intervenes in the pathogenic pathway induced by mutated htt therefore remains an intriguing topic for further investigation.
Through Which Molecular Pathway Could Bcl-2 Family Proteins Cause Mitochondrial Dysfunctions and Cell Death in HD?
By summarizing the data about Bcl-2 family proteins in cell culture models of HD (Table 1), mouse models of HD (Table 2) and cells and tissues from HD patients (Table 3), we formulated a hypothetical model for explaining Bcl-2 family protein activation in HD (Figure 1).
Table 1. Evidence for Bcl-2 family protein dysregulation in cell culture models of HD (Significant differences between HD and control cells are highlighted in bold).
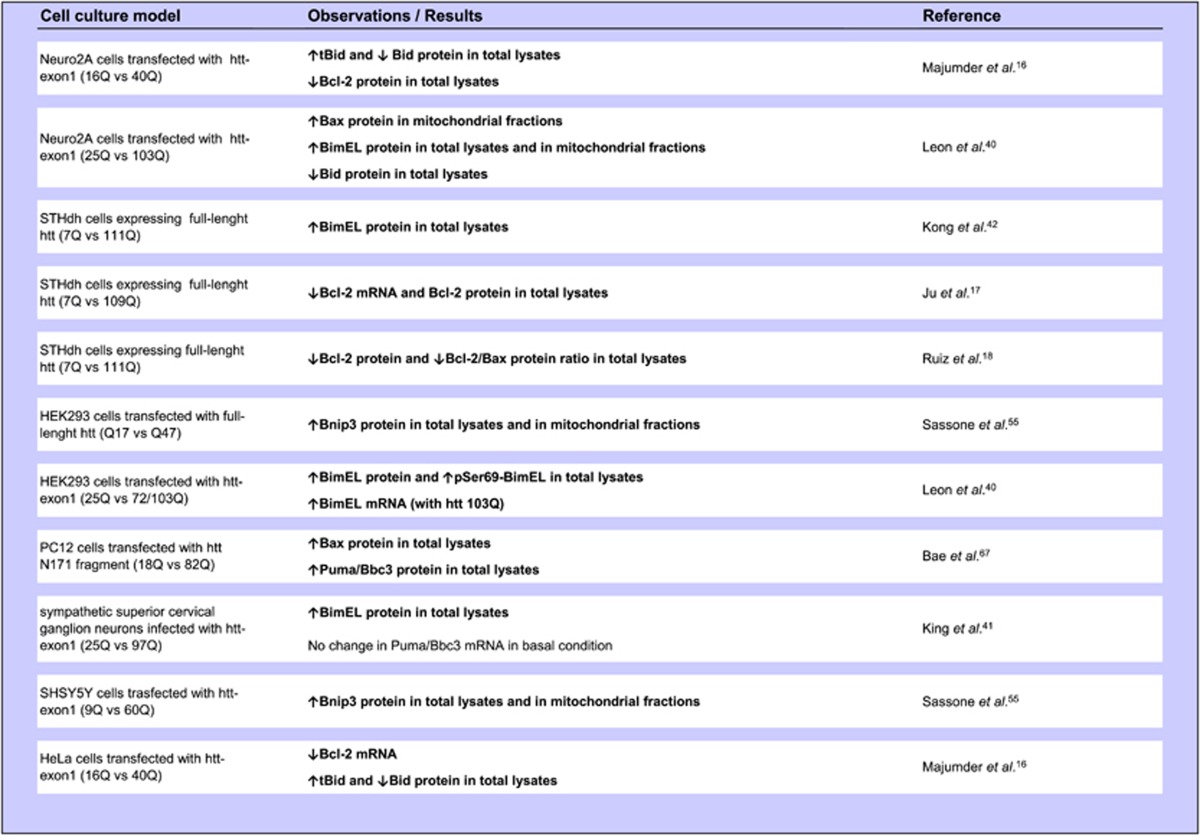
Table 2. Evidence for Bcl-2 family protein dysregulation in mouse models of HD (Significant differences between HD and control cells are highlighted in bold).

Table 3. Evidence for Bcl-2 family protein dysregulation in HD patients' cells (Significant differences between HD and control cells are highlighted in bold).

Figure 1.

Hypothetical model explaining the pathway through which mutated htt (mhtt) causes OMM permeabilization. Bax activation in HD may depend on the ‘activators' BH3-only proteins and the ‘sensitizers' that bind only to pro-survival proteins Bcl-2 and Bcl-xL. Mutated htt may interact with the Bcl-2 protein network at multiple levels by modulating Bcl-2 expression, inducing Bid accumulation and Bid cleavage, promoting BNip3 activation and increasing non-pBad levels. BimEL accumulation/activation may depend on htt control over BDNF expression or ER stress-induced UPR or both mechanisms
Because many results converge to demonstrate that mutated htt causes BimEL activation (Tables 1 and 2), we hypothesize that BimEL, a protein that in healthy cells associates with cellular microtubule complexes,69 in HD neurons localizes to mitochondria where, according to the ‘direct activation' model (paragraph 2.2), it would trigger Bax activation. This hypothesis receives support from evidence that both BimEL and Bax accumulate in the mitochondrial fractions from R6/1 and R6/2 mouse models. Alternatively, according to the ‘indirect activation' model, BimEL in HD cells may bind to Bcl-2, thereby preventing it from neutralizing Bax. In this context, Bcl-2 may have a neuroprotective role in HD. This hypothesis receives support from evidence that Bcl-2 overexpression in neurons slows down neurodegeneration in the R6/2 model.21 What remains unclear is how mutated htt activates BimEL. An interesting clue comes from evidence that BimEL expression is negatively regulated by the brain-derived neurotrophic factor (BDNF).70, 71 By wide consensus, the HD mutation results in lower BDNF levels in the brain72 because htt is indirectly involved in transcriptional control over the BDNF gene.73 Mutated huntingtin might therefore elicit BimEL accumulation/activation by inhibiting BDNF expression. BimEL is also a well-known downstream target of ER stress.74 Insofar, as proteins with an abnormally long polyglutamine expansion cause ER stress,75 BimEL activation in HD may also depend on mutated htt induced-UPR. A question awaiting further research is whether Puma/Bbc3, the other potent BH3-only protein activated in response to ER stress, has a role in HD.
Another BH3-only protein potentially involved in HD pathogenesis is Bid. In HD models, mutated htt elicits Bid cleavage or full-length Bid accumulation or both events (Tables 1 and 2). Bid activation was suggested as a key event in many neurodegenerative diseases because Bid is highly expressed in neurons.76 In HD cells, full-length Bid and tBid, by migrating to mitochondria, could sustain Bax activation,77 thus amplifying the mitochondrial damage.
The molecular mechanism by which mutated htt causes full-length Bid to accumulate is undefined. Because full-length Bid pro-apoptotic activity is extremely weak, high Bid expression levels are nevertheless unlikely to cause cell death in HD. Conversely, because tBid exhibits strong pro-apoptotic activity, it may have a key role in HD cell death. This possibility receives support also from evidence that mutated htt enhances activity of caspase-878, 79 and calpain80 that both enzymes are able to cleave full-length Bid.46
The most recently BH3-only protein potentially implicated in HD pathogenesis is Bnip3. Our results provide evidence that Bnip3, an apoptotic regulator that in healthy cells localizes to the cytosol or the nuclei, in neuronal and non-neuronal cells expressing mutated htt mainly localizes to the mitochondria.55 Because Bnip3 activation causes Δψ loss, mitochondrial fragmentation and mitophagy,51, 81 Bnip3 could be implicated in the mitochondrial dysfunction in HD. Precisely, how mutated htt induces Bnip3 activation remains an unanswered question. Previous reports show that the Bnip3–mitochondria association is strongly stabilized by acidosis82, 83 or by an increased cytosolic calcium concentration,84 conditions that may both take place in HD cells owing to mitochondrial respiration inhibition. An alternative possibility is that htt binds directly or indirectly to Bnip3, but this issue awaits elucidation in future studies.
Summary and Perspective
Current evidence, derived from research efforts by many investigators over a lengthy time-span, increasingly implies that mutated htt adversely influences Bcl-2 family protein levels and localization. Studies conducted in vitro and in vivo provide convincing evidence that mutated htt expression activates at least four BH3-only proteins. Although mutated htt may upregulate each of these four proteins through different mechanisms, their activation culminates in an identical consequence, namely Bax activation. Bax activation, by promoting cytochrome c release, may underpin the progressive neuronal apoptosis in HD patients' brains. Although this hypothesis receives support from early studies that identified apoptotic-like cells in the HD striatum,85, 86 evidence for HD cell death arising through apoptosis alone is controversial.87 An alternative possibility is that Bcl-2 protein family dysregulation alters HD mitochondrial dynamics in an apoptosis-independent manner. Especially interesting in this context is a report that Bax-induced mitochondrial fission and Bax-initiated cytochrome c release are separable events and that Bcl-2 family proteins can influence mitochondrial fission–fusion dynamics in HD cells independently of apoptosis.1, 2 If true, Bcl-2 family proteins in HD may be responsible for the fragmented mitochondrial morphology, changes in mitochondrial ultrastructure and impaired mitochondrial trafficking demonstrated in in vitro and in vivo models of HD.88, 89, 90
Future studies should aim to go beyond analyzing Bcl-2 family protein levels because their expression level often poorly indicates protein activation, especially given that most BH3-only members undergo strong regulation by post-translational mechanisms. Future studies aimed to elucidate the molecular mechanisms underlying the Bcl-2 protein interactions documented in in vitro and in vivo HD models will exploit innovative fluorescence techniques91 that may more clearly illustrate the Bcl-2 role in HD. Equally important are the genetic approaches such as crossing HD mouse models onto mice knockout for Bcl-2 genes that will provide definitive proof that Bcl-2 family members are pathogenetically involved in HD.
Finally, future studies involving innovative human cellular models such as iPS from patients with HD, will clarify whether Bcl-2 family proteins, already molecular targets in cancer therapy,92 may also be a therapeutic target for HD.
Acknowledgments
JS, AM and AC are supported by Cariplo Foundation (grant number 2012-0593). JS is also supported by Italian Health Minister (grant number GR-2010-2318394). FS is supported by Associazione Mauro Emolo O.N.L.U.S and AICH Milano O.N.L.U.S. We thank Huntington's patients and their families for their precious support. We also thank the anonymous reviewer for helpful comments on the manuscript.
Glossary
- HD
Huntington's disease
- htt
huntingtin
- OMM
outer mitochondrial membrane
- ER
endoplasmic reticulum
- Δψ
mitochondrial potential
- BH3
Bcl-2 Homology 3
- CNS
central nervous system
- OPA1
optic atrophy-1 protein
- UPR
unfolded protein response
- SGC
superior cervical ganglion
- BDNF
brain-derived neurotrophic factor
- Bcl-2
B-cell lymphoma 2
- Bcl-xL
B-cell lymphoma-extra large
- Bad
Bcl-2 antagonist of cell death
- Bim
Bcl-2 interacting mediator of cell death
- Bid
Bcl-2 interacting domain death agonist
- Bnip3
Bcl-2/adenovirus E1B 19 kDa interacting protein 3
- Puma/Bbc3
p53-upregulated modulator of apoptosis/Bcl-2 binding component 3
- Bax
Bcl-2-associated X-protein
- Bak
Bcl-2 antagonist/killer
The authors declare no conflict of interest.
Footnotes
Edited by A Verkhratsky
References
- Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- Sheridan C, Delivani P, Cullen SP, Martin SJ. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell. 2008;31:570–585. doi: 10.1016/j.molcel.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB, Goebbels S, et al. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009;184:707–719. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–956. doi: 10.1038/nature01825. [DOI] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Allsopp TE, Wyatt S, Paterson HF, Davies AM. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 1993;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Merry DE, Veis DJ, Hickey WF, Korsmeyer SJ. bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development. 1994;120:301–311. doi: 10.1242/dev.120.2.301. [DOI] [PubMed] [Google Scholar]
- González-García M, García I, Ding L, O'Shea S, Boise LH, Thompson CB, et al. Bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA. 1995;92:4304–4308. doi: 10.1073/pnas.92.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewska M, Mai JK, Zapata JM, Ashwell KW, Schendel SL, Reed JC, et al. Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ. 2002;9:145–157. doi: 10.1038/sj.cdd.4400934. [DOI] [PubMed] [Google Scholar]
- Majumder P, Raychaudhuri S, Chattopadhyay B, Bhattacharyya NP. Increased caspase-2, calpain activations and decreased mitochondrial complex II activity in cells expressing exogenous huntingtin exon 1 containing CAG repeat in the pathogenic range. Cell Mol Neurobiol. 2007;27:1127–1145. doi: 10.1007/s10571-007-9220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju TC, Chen HM, Lin JT, Chang CP, Chang WC, Kang JJ, et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington's disease. J Cell Biol. 2011;194:209–227. doi: 10.1083/jcb.201105010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz C, Casarejos MJ, Rubio I, Gines S, Puigdellivol M, Alberch J, et al. The dopaminergic stabilizer, (-)-OSU6162, rescues striatal neurons with normal and expanded polyglutamine chains in huntingtin protein from exposure to free radicals and mitochondrial toxins. Brain Res. 2012;1459:100–112. doi: 10.1016/j.brainres.2012.04.021. [DOI] [PubMed] [Google Scholar]
- Reed JC, Meister L, Tanaka S, Cuddy M, Yum S, Geyer C, et al. Differential expression of bcl2 protooncogene in neuroblastoma and other human tumor cell lines of neural origin. Cancer Res. 1991;51:6529–6538. [PubMed] [Google Scholar]
- Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 2010;1:e40. doi: 10.1038/cddis.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ona VO, Li M, Drozda M, Dubois-Dauphin M, Przedborski S, et al. Sequential activation of individual caspases, and of alterations in Bcl-2 proapoptotic signals in a mouse model of Huntington's disease. J Neurochem. 2003;87:1184–1192. doi: 10.1046/j.1471-4159.2003.02105.x. [DOI] [PubMed] [Google Scholar]
- García-Martínez JM, Pérez-Navarro E, Xifró X, Canals JM, Díaz-Hernández M, Trioulier Y, et al. BH3-only proteins Bid and Bim(EL) are differentially involved in neuronal dysfunction in mouse models of Huntington's disease. J Neurosci Res. 2007;85:2756–2769. doi: 10.1002/jnr.21258. [DOI] [PubMed] [Google Scholar]
- Teles AV, Rosenstock TR, Okuno CS, Lopes GS, Bertoncini CR, Smaili SS. Increase in bax expression and apoptosis are associated in Huntington's disease progression. Neurosci Lett. 2008;438:59–63. doi: 10.1016/j.neulet.2008.03.062. [DOI] [PubMed] [Google Scholar]
- Chiang MC, Chen CM, Lee MR, Chen HW, Chen HM, Wu YS, et al. Modulation of energy deficiency in Huntington's disease via activation of the peroxisome proliferator-activated receptor gamma. Hum Mol Genet. 2010;19:4043–4058. doi: 10.1093/hmg/ddq322. [DOI] [PubMed] [Google Scholar]
- Duan W, Peng Q, Masuda N, Ford E, Tryggestad E, Ladenheim B, et al. Sertraline slows disease progression and increases neurogenesis in N171-82Q mouse model of Huntington's disease. Neurobiol Dis. 2008;30:312–322. doi: 10.1016/j.nbd.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vis JC, Schipper E, de Boer-van Huizen RT, Verbeek MM, de Waal RM, Wesseling P, et al. Expression pattern of apoptosis-related markers in Huntington's disease. Acta Neuropathol. 2005;109:321–328. doi: 10.1007/s00401-004-0957-5. [DOI] [PubMed] [Google Scholar]
- Sanchez Mejia RO, Friedlander RM. Caspases in Huntington's disease. Neuroscientist. 2001;7:480–489. doi: 10.1177/107385840100700604. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, et al. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- Almeida S, Sarmento-Ribeiro AB, Januário C, Rego AC, Oliveira CR. Evidence of apoptosis and mitochondrial abnormalities in peripheral blood cells of Huntington's disease patients. Biochem Biophys Res Commun. 2008;374:599–603. doi: 10.1016/j.bbrc.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Hansson O, Castilho RF, Korhonen L, Lindholm D, Bates GP, Brundin P. Partial resistance to malonate-induced striatal cell death in transgenic mouse models of Huntington's disease is dependent on age and CAG repeat length. J Neurochem. 2001;78:694–703. doi: 10.1046/j.1471-4159.2001.00482.x. [DOI] [PubMed] [Google Scholar]
- Khoshnan A, Ko J, Tescu S, Brundin P, Patterson PH. IKKalpha and IKKbeta regulation of DNA damage-induced cleavage of huntingtin. PLoS One. 2009;4:e5768. doi: 10.1371/journal.pone.0005768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happo L, Strasser A, Cory S. BH3-only proteins in apoptosis at a glance. J Cell Sci. 2012;125:1081–1087. doi: 10.1242/jcs.090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Walensky LD, Zhang CY, Choi CS, Fisher JK, Molina AJ, et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat Med. 2008;14:144–153. doi: 10.1038/nm1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giménez-Cassina A, Martínez-François JR, Fisher JK, Szlyk B, Polak K, Wiwczar J, et al. BAD-dependent regulation of fuel metabolism and K(ATP) channel activity confers resistance to epileptic seizures. Neuron. 2012;74:719–730. doi: 10.1016/j.neuron.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti D, Bauer JH, De-Fraja C, Conti L, Sipione S, Sciorati C, et al. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci. 2000;20:3705–3713. doi: 10.1523/JNEUROSCI.20-10-03705.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN. BAD: undertaker by night, candyman by day. Oncogene. 2008;27 (Suppl 1:S53–S70. doi: 10.1038/onc.2009.44. [DOI] [PubMed] [Google Scholar]
- Ley R, Hadfield K, Howes E, Cook SJ. Identification of a DEF-type docking domain for extracellular signal-regulated kinases 1/2 that directs phosphorylation and turnover of the BH3-only protein BimEL. J Biol Chem. 2005;280:17657–17663. doi: 10.1074/jbc.M412342200. [DOI] [PubMed] [Google Scholar]
- O'Reilly LA, Cullen L, Visvader J, Lindeman GJ, Print C, Bath ML, et al. The proapoptotic BH3-only protein bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am J Pathol. 2000;157:449–461. doi: 10.1016/S0002-9440(10)64557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Hattori H, Sasaki T, Gotoh J, Hamada J, Fukuuchi Y. Temporal profiles of the subcellular localization of Bim, a BH3-only protein, during middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2002;22:810–820. doi: 10.1097/00004647-200207000-00006. [DOI] [PubMed] [Google Scholar]
- Leon R, Bhagavatula N, Ulukpo O, McCollum M, Wei J. BimEL as a possible molecular link between proteasome dysfunction and cell death induced by mutant huntingtin. Eur J Neurosci. 2010;31:1915–1925. doi: 10.1111/j.1460-9568.2010.07215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MA, Goemans CG, Hafiz F, Prehn JH, Wyttenbach A, Tolkovsky AM. Cytoplasmic inclusions of Htt exon1 containing an expanded polyglutamine tract suppress execution of apoptosis in sympathetic neurons. J Neurosci. 2008;28:14401–14415. doi: 10.1523/JNEUROSCI.4751-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong PJ, Kil MO, Lee H, Kim SS, Johnson GV, Chun W. Increased expression of Bim contributes to the potentiation of serum deprivation-induced apoptotic cell death in Huntington's disease knock-in striatal cell line. Neurol Res. 2009;31:77–83. doi: 10.1179/174313208X331572. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellecchia M, et al. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci. 2005;25:10262–10272. doi: 10.1523/JNEUROSCI.2818-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T, Plesnila N, Prehn JH, Henshall DC. In vivo contributions of BH3-only proteins to neuronal death following seizures, ischemia, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31:1196–1210. doi: 10.1038/jcbfm.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhamandas JH, Mactavish DJ. β-Amyloid protein (Aβ) and human amylin regulation of apoptotic genes occurs through the amylin receptor. Apoptosis. 2012;17:37–47. doi: 10.1007/s10495-011-0656-3. [DOI] [PubMed] [Google Scholar]
- Yin XM. Bid, a BH3-only multi-functional molecule, is at the cross road of life and death. Gene. 2006;369:7–19. doi: 10.1016/j.gene.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Chinnadurai G, Vijayalingam S, Gibson SB. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene. 2008;27 (Suppl 1:S114–S127. doi: 10.1038/onc.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton TR, Gibson SB. The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. 2009;16:515–523. doi: 10.1038/cdd.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ney PA. Mechanisms and biology of B-cell leukemia/lymphoma 2/adenovirus E1B interacting protein 3 and Nip-like protein X. Antioxid Redox Signal. 2011;14:1959–1969. doi: 10.1089/ars.2010.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J. 2007;405:407–415. doi: 10.1042/BJ20070319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landes T, Emorine LJ, Courilleau D, Rojo M, Belenguer P, Arnauné-Pelloquin L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010;11:459–465. doi: 10.1038/embor.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinsay MN, Lee Y, Rikka S, Sayen MR, Molkentin JD, Gottlieb RA, et al. Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J Mol Cell Cardiol. 2010;48:1146–1156. doi: 10.1016/j.yjmcc.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, et al. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B, Choi SY, Park OH, Sun W, Geum D. Differential expression of BNIP family members of BH3-only proteins during the development and after axotomy in the rat. Mol Cells. 2012;33:605–610. doi: 10.1007/s10059-012-0051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassone J, Colciago C, Marchi P, Ascardi C, Alberti L, Di Pardo A, et al. Mutant Huntingtin induces activation of the Bcl-2/adenovirus E1B 19-kDa interacting protein (BNip3) Cell Death Dis. 2010;1:e7. doi: 10.1038/cddis.2009.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci USA. 2002;99:12825–12830. doi: 10.1073/pnas.202474099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–157. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- Engel T, Murphy BM, Hatazaki S, Jimenez-Mateos EM, Concannon CG, Woods I, et al. Reduced hippocampal damage and epileptic seizures after status epilepticus in mice lacking proapoptotic Puma. FASEB J. 2010;24:853–861. doi: 10.1096/fj.09-145870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niizuma K, Endo H, Nito C, Myer DJ, Chan PH. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke. 2009;40:618–625. doi: 10.1161/STROKEAHA.108.524447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimertz C, Kögel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–597. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara A, Hirose Y, Wang A, Yoshimi N, Tanaka T, Mori H. Localization of Bax and Bcl-2 proteins, regulators of programmed cell death, in the human central nervous system. Virchows Arch. 1996;429:249–253. doi: 10.1007/BF00198341. [DOI] [PubMed] [Google Scholar]
- MacGibbon GA, Lawlor PA, Sirimanne ES, Walton MR, Connor B, Young D, et al. Bax expression in mammalian neurons undergoing apoptosis, and in Alzheimer's disease hippocampus. Brain Res. 1997;750:223–234. doi: 10.1016/s0006-8993(96)01351-0. [DOI] [PubMed] [Google Scholar]
- Obonai T, Mizuguchi M, Takashima S. Developmental and aging changes of Bak expression in the human brain. Brain Res. 1998;783:167–170. doi: 10.1016/s0006-8993(97)01361-9. [DOI] [PubMed] [Google Scholar]
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Sassone J, Colciago C, Cislaghi G, Silani V, Ciammola A. Huntington's disease: the current state of research with peripheral tissues. Exp Neurol. 2009;219:385–397. doi: 10.1016/j.expneurol.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3:287–296. doi: 10.1016/s1097-2765(00)80456-6. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang J, Liu Z, Woo CW, Thiele CJ. Downregulation of Bim by brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from paclitaxel but not etoposide or cisplatin-induced cell death. Cell Death Differ. 2007;14:318–326. doi: 10.1038/sj.cdd.4401983. [DOI] [PubMed] [Google Scholar]
- Almeida S, Laço M, Cunha-Oliveira T, Oliveira CR, Rego AC. BDNF regulates BIM expression levels in 3-nitropropionic acid-treated cortical neurons. Neurobiol Dis. 2009;35:448–456. doi: 10.1016/j.nbd.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Duennwald ML, Lindquist S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 2008;22:3308–3319. doi: 10.1101/gad.1673408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Plesnila N. Targeting Bid to prevent programmed cell death in neurons. Biochem Soc Trans. 2006;34:1334–1340. doi: 10.1042/BST0341334. [DOI] [PubMed] [Google Scholar]
- Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20:929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez I, Xu CJ, Juo P, Kakizaka A, Blenis J, Yuan J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron. 1999;22:623–633. doi: 10.1016/s0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Singaraja R, Xanthoudakis S, Gutekunst CA, Leavitt BR, Metzler M, et al. Recruitment and activation of caspase-8 by the Huntingtin-interacting protein Hip-1 and a novel partner Hippi. Nat Cell Biol. 2002;4:95–105. doi: 10.1038/ncb735. [DOI] [PubMed] [Google Scholar]
- Gafni J, Ellerby LM. Calpain activation in Huntington's disease. J Neurosci. 2002;22:4842–4849. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier DP, Wilson A, Graham RM, Thompson JW, Bishopric NH, Webster KA. Acidosis regulates the stability, hydrophobicity, and activity of the BH3-only protein Bnip3. Antioxid Redox Signal. 2006;8:1625–1634. doi: 10.1089/ars.2006.8.1625. [DOI] [PubMed] [Google Scholar]
- Graham RM, Frazier DP, Thompson JW, Haliko S, Li H, Wasserlauf BJ, et al. A unique pathway of cardiac myocyte death caused by hypoxia-acidosis. J Exp Biol. 2004;207:3189–3200. doi: 10.1242/jeb.01109. [DOI] [PubMed] [Google Scholar]
- Graham RM, Thompson JW, Wei J, Bishopric NH, Webster KA. Regulation of Bnip3 death pathways by calcium, phosphorylation, and hypoxia-reoxygenation. Antioxid Redox Signal. 2007;9:1309–1315. doi: 10.1089/ars.2007.1726. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Faull RL, Lawlor P, Beilharz EJ, Singleton K, Walker EB, et al. In situ evidence for DNA fragmentation in Huntington's disease striatum and Alzheimer's disease temporal lobes. Neuro Report. 1995;6:1053–1057. doi: 10.1097/00001756-199505090-00026. [DOI] [PubMed] [Google Scholar]
- Thomas LB, Gates DJ, Richfield EK, O'Brien TF, Schweitzer JB, Steindler DA. DNA end labeling (TUNEL) in Huntington's disease and other neuropathological conditions. Exp Neurol. 1995;133:265–272. doi: 10.1006/exnr.1995.1029. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lim PJ, Karbowski M, Monteiro MJ. Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum Mol Genet. 2009;18:737–752. doi: 10.1093/hmg/ddn404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, Giacomello M, Hudec R, Lopreiato R, Ermak G, Lim D, et al. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington's disease to apoptotic stimuli. EMBO Mol Med. 2010;2:490–503. doi: 10.1002/emmm.201000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med. 2011;17:377–382. doi: 10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kale J, Liu Q, Leber B, Andrews DW. Shedding light on apoptosis at subcellular membranes. Cell. 2012;151:1179–1184. doi: 10.1016/j.cell.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Davids MS, Letai A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J Clin Oncol. 2012;30:3127–3135. doi: 10.1200/JCO.2011.37.0981. [DOI] [PMC free article] [PubMed] [Google Scholar]