

Abstract
Investigations of the molecular mechanisms underlying major depressive disorder (MDD) have been hampered by the complexity of brain tissue and sensitivity of gene expression profiling approaches. To address these issues, we used discrete microdissections of postmortem dorsolateral prefrontal cortex (DLPFC) (area 9) and an oligonucleotide (60mer) microarray hybridization procedure that increases sensitivity without RNA amplification. Mixed-effects statistical methods were used to rigorously control for medication usage in the subset of medicated depressed subjects. These analyses yielded a rich profile of dysregulated genes. Two of the most highly dysregulated genes of interest were stresscopin, a neuropeptide involved in stress responses, and Forkhead box D3 (FOXD3), a transcription factor. Secondary cell-based analysis demonstrated that stresscopin and FoxD3 are increased in neurons of DLPFC gray matter of MDD subjects. These findings identify abnormal gene expression in a discrete region of MDD subjects and contribute to further elucidation of the molecular alterations of this complex mood disorder.
Keywords: microarray, stresscopin, urocortin III, stress, corticotrophin releasing hormone, FoxD3, fibroblast growth factor
Introduction
Depression is a devastating neurobiological illness that afflicts an estimated 12–15% of the population at some point in life and is a leading cause of disability worldwide (Kessler et al., 1994). Although medications are available and effective for ∼65% of patients, a therapeutic response requires weeks to months, and, in many cases, it may take up to 1 year or more to identify the most effective drug or combination of drugs for a particular individual. This is partly attributable to the fact that the pathophysiology underlying depression has been difficult to access.
Brain imaging studies demonstrate altered blood flow, metabolism, and morphology in limbic structures of depressed patients (Drevets, 2001; Manji et al., 2001; Nestler et al., 2002a; Warner-Schmidt and Duman, 2006). Structural brain imaging studies demonstrate atrophy of specific limbic structures, including prefrontal cortex (Drevets et al., 1997; Bremner et al., 2002), hippocampus (Sheline et al., 1996, 1998, 1999; Bremner et al., 2000), and amygdala (Sheline et al., 1998; Hastings et al., 2004). Postmortem morphometric studies from a number of research groups have also reported a reduction in the density and size of neurons and the number and density of glia in the prefrontal and anterior cingulate cortex, amygdala, and hippocampus, which could contribute to the reduced volume of these regions (Ongur et al., 1998; Rajkowska et al., 1999; Cotter et al., 2001).
Although these brain imaging and postmortem studies provide important information, the molecular determinants that underlie mood disorders have not been identified. This is attributable in part to the inability to directly examine and analyze brain tissue in living humans. Postmortem studies have been conducted to address this issue, although the cellular heterogeneity of complex brain structures poses a challenge. In addition, it has been difficult to examine all of the genes involved in neuronal and glial function that could influence mood.
To address these issues, we used gene expression microarray analysis of a well defined structure, the dorsolateral prefrontal cortex (DLPFC) gray matter. The DLPFC tissue was obtained from the same major depressive disorder (MDD) brains that were subjected to morphometric analysis of this region, providing further cellular characterization of the samples (Rajkowska et al., 1999). Using a human 20,000 oligo array chip combined with a sensitive indirect labeling procedure that does not require RNA amplification, we examined gene expression changes in 14 pairs of MDD patients and matched nonpsychiatric controls. Data were analyzed using a mixed-model approach to allow us to statistically control for medication usage. A variety of pathways previously implicated in mood disorders were dysregulated, as well as two novel genes of particular interest to us given their potential role in mood disorders: stresscopin and Forkhead box D3 (FOXD3). We confirmed dysregulation of expression by in situ hybridization in human tissue sections. In addition, we confirm and extend recent studies identifying altered expression of fibroblast growth factor receptor-3 (FGFR3). These findings identify abnormal gene expression in a discrete region of MDD subjects and contribute to further elucidation of the molecular alterations of this complex mood disorder.
Materials and Methods
Human subjects
Brain samples were collected at autopsy at the Cuyahoga County Coroner's Office (Cleveland, OH). Informed written consent was obtained from the legal next-of-kin of all subjects. Next-of-kin for all subjects were interviewed using the Structured Clinical Interview for Diagnostic and Statistical Manual of Mental Disorders IV (SCID) (First et al., 2001), and retrospective psychiatric assessments were conducted in accordance with Institutional Review Board policies. The use of retrospective informant-based Axis I diagnoses was validated by Kelly and Mann (1996) and reviewed by Lewis (2002). Fifteen subjects met diagnostic criteria for MDD according to the Diagnostic and Statistical Manual of Mental Disorders IV (American Psychiatric Association, 2000). All subjects with MDD were experiencing a depressed episode within the last month of life, and, as such, the results should be interpreted as reflecting on the “state” rather than the “trait” of depression when the subjects were asymptomatic. Fifteen psychiatrically normal control subjects were matched with the 15 depressed subjects according to age, ethnicity, gender, and postmortem delay (Table 1). There was no evidence of a neurological disorder in any of the subjects. Among the 15 depressed subjects, nine had prescriptions for antidepressant medication and one also for an antipsychotic medication during the last month of life. An antidepressant medication (sertraline, venlafaxine, or nortriptyline) was present in the blood of four depressed subjects, and amitriptyline and chlorpromazine were present in a fifth subject. Ethanol was detected postmortem in the blood of two subjects and urine of one subject. None of these three depressed subjects met criteria for an alcohol use disorder.
Table 1.
Case demographics of the matched controls and MDD subjects
| Array | Control |
MDD |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | Sex/race | Age | PMI | pH average | ID | Sex/race | Age | PMI | pH average | |
| 1 | Ctrl 1 | M/C | 71 | 24 | 6.82 | MDD 1 | M/C | 74 | 25 | 6.67 |
| 2 | Ctrl 2 | M/AAm | 55 | 18 | 6.50 | MDD 2 | M/C | 55 | 29 | 6.64 |
| 3 | Ctrl 3 | M/C | 42 | 20 | 6.82 | MDD 3 | M/C | 42 | 20 | 6.64 |
| 4 | Ctrl 4 | M/C | 38 | 30 | 6.74 | MDD 4 | M/C | 33 | 18 | 6.79 |
| 5 | Ctrl 5 | M/C | 35 | 25 | 6.74 | MDD 5 | M/C | 45 | 29 | 6.86 |
| 6 | Ctrl 6 | M/C | 33 | 23 | 6.86 | MDD 6 | M/C | 36 | 11 | 6.97 |
| 7 | Ctrl 7 | M/C | 54 | 17 | 6.87 | MDD 7 | M/C | 54 | 23 | 6.24 |
| 8 | Ctrl 8 | M/AAm | 30 | 19 | 6.98 | MDD 8 | M/AAm | 30 | 18 | 6.91 |
| 9 | Ctrl 9 | F/C | 50 | 27 | 6.74 | MDD 9 | F/C | 50 | 23 | 6.83 |
| 10 | Ctrl10 | M/C | 69 | 18 | 6.70 | MDD10 | F/C | 73 | 17 | 6.57 |
| 11 | Ctrl11 | M/C | 70 | 20 | 6.81 | MDD11 | M/C | 73 | 10 | 6.57 |
| 12 | Ctrl12 | M/AAm | 46 | 11 | 6.95 | MDD12 | M/AAm | 46 | 17 | 6.26 |
| 13 | Ctrl13 | F/C | 37 | 13 | MDD13 | F/C | 40 | 25 | 6.32 | |
| 14 | Ctrl14 | F/AAm | 60 | 19 | 6.74 | MDD14 | F/C | 63 | 24 | 6.32 |
| Average | 49.29 | 20.29 | 6.79 | Average | 51.00 | 20.64 | 6.61 | |||
| SD | 14.26 | 5.17 | 0.12 | SD | 14.99 | 5.87 | 0.25 | |||
| SEM | 1.02 | 0.37 | 0.01 | SEM | 1.07 | 0.42 | 0.02 | |||
M, Male; F, female; C, Caucasian; AAm, African American; PMI, postmortem interval (hours).
Human brain tissue preparation
Blocks of tissue from the DLPFC containing Brodmann's area 9 were frozen at autopsy using isopentane cooled in dry ice and stored at −80°C. Frozen sections were cut on a cryostat (50 μm), and the first two sections collected were stained for Nissl substance to distinguish area 9. The immediately adjacent sections were used to collect punches of tissue. The diameter of the punches was adjusted to include all six cortical layers of the gray matter of area 9, and the underlying white matter was excluded (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Fifteen control samples were matched with MDD samples on the basis of sex, age, and race. In addition, control and MDD groups did not significantly differ with regard to agonal state, sample pH, or postmortem interval (Table 1).
Microarray hybridization
Total RNA was isolated from pooled punches (40 punches of 2–3 mm in diameter per subject) from 50 μm sections of DLPFC (Brodmann's area 9) by tissue punch. Total RNA was isolated using a nonphenolic procedure (RNA Aqueous; Ambion, Austin, TX). Optical density values at 260/280 were consistently above 1.9, and the quality of the RNA was also confirmed by Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). Two micrograms of total RNA from MDD samples (n = 15) and matched controls (n = 15) were reverse transcribed into cDNA and indirectly labeled using a sensitive fluorescent labeling procedure (Genisphere, Hatfield, PA). A two-step hybridization and labeling protocol was used (Genisphere array 350 protocol).
The Agilent Human 1A Oligo chip (Agilent Technologies) was hybridized overnight to cDNA in Agilent buffer, washed stringently to remove nonspecifically bound probe, and then poststained with fluorescent dendrimers using Genisphere 2× SDS phosphate buffer. After posthybridization washes, slides were scanned using a GenePix scanner (Molecular Devices, Sunnyvale, CA). Image analysis was performed using GenePix Pro 4.0 software (Molecular Devices).
Microarray data analysis
Filtering and missing data imputation.
The raw dataset consisted of 15 Agilent Human 1A array scans involving comparative hybridizations of a patient labeled with cyanine-3 (Cy3) against a matched control sample labeled with Cy5. A total of 20,173 probes were scanned. All probe sets that did not correspond to a gene in GenBank were dropped, and probe sets with more than six no calls were dropped. This left 11,351 probe sets. The dataset was then analyzed in R/Bioconductor using the MAANOVA package (Cui and Churchill, 2003). Intensity data were transformed with the linear-log transformation method (Cui et al., 2003), and scatter plots were examined before and after transformation. This transformation method yielded results that were superior to the other methods tested (e.g., LOWESS). Array number 15 was dropped from additional analysis because of an ill conditioned scatter plot that could not be adequately normalized. Missing data were imputed from the dataset using the k-nearest neighbor method with the impute.knn function from the impute library in R (Troyanskaya et al., 2001) using the nearest 20 neighbors method.
Mixed-model analysis.
Although samples were matched, matching was not perfect, in particular with respect to antidepressant usage. Eight of the 14 depressed subjects were prescribed antidepressants. A statistical approach that was capable of assessing the impact of this imperfect matching was therefore required. Toward this end, we used a mixed-model approach as implemented in the MAANOVA package in R to allow all sources of variation to be rigorously assessed.
Because of limited degrees of freedom, all of the covariates of interest could not be entered into a single model. Instead, a series of models were tested with the array effect entered as a mixed effect, Depression Status (DepStatus) as a fixed effect, and a single covariate. The results suggested that illicit drug use, smoking, pH, postmortem interval, age, gender, and race had little impact on the results as determined by a lack of significant gene effects, and these models were not explored further. However, the potential confound of medication usage was deemed sufficiently important that it was analyzed in detail.
A variety of mixed-effects models were fit to the data using the MAANOVA package in R/Bioconductor. In every case, the “array” effect was fit as a random effect. Our primary analysis included DepStatus (depressed vs not depressed) and whether the subject had been treated in the last month with an antidepressant medication as a covariate. This model can be written as yas = γa + β1 × DepStatus + β2 × Med + εs, where ya,s is the log2 intensity value for subject s hybridized on array a, γa is the (random) effect of array, DepStatus is a binary depression status variable with values 0 (not depressed) or 1 (depressed), Med is a binary variable with values 0 (no antidepressant in the last month) or 1 (antidepressant taken in the last month), and ε is a (random) error term. The β parameters are the regression coefficients for the two fixed effects DepStatus and Med. It is important to note that the medication status term has significance only for the depressed subjects, because none of the control subjects were taking antidepressants.
The analysis was run using the restricted maximum likelihood method with 1000 permutation tests to yield correct significance values for the mixed model in a manner that is robust to the non-normality of most microarray data. Analyses were run on a computing cluster at the Yale Keck Center with 130 compute nodes each containing two 3.2 GHz EM64T Xeon processors. The best linear unbiased expression estimates from these models were used to calculate expression ratios adjusted for covariate effects (“corrected” expression ratios) (supplemental Table 1, available at www.jneurosci.org as supplemental material).
False discovery rate estimation.
After runs were complete, probability values corresponding to the Fs statistic were extracted, which are based on the James–Stein shrinkage estimates of the error variance (Cui et al., 2005). False discovery rate (FDR) adjusted p values were then calculated using the pava.fdr function from the SAGx library in R (Broberg, 2005). We used a 5% cutoff value for the false discovery rate. These FDR-controlled p values [FDR(p)] are shown in Table 2.
Table 2.
Most significantly regulated gene categories
| GO category | p valuea |
|---|---|
| Second-messenger-mediated signaling | 1.62E-16 |
| Intracellular signaling cascade | 3.89E-16 |
| Protein kinase C activation | 1.45E-15 |
| Muscarinic acetylcholine receptor, adenylate cyclase inhibiting pathway | 2.19E-15 |
| Learning and/or memory | 1.91E-14 |
| Positive regulation of mismatch repair | 6.61E-13 |
| Negative regulation of protein ubiquitination | 6.61E-13 |
| Negative regulation of protein modification | 6.61E-13 |
| Regulation of mismatch repair | 6.61E-13 |
| Dopamine transport | 8.71E-13 |
| Wnt receptor signaling pathway | 1.41E-12 |
| Regulation of cell differentiation | 1.91E-12 |
| Negative regulation of enzyme activity | 2.45E-12 |
| Negative regulation of biological process | 3.80E-12 |
| Signal transduction | 1.10E-11 |
| Cell communication | 2.14E-11 |
| Negative regulation of protein amino acid phosphorylation | 2.82E-11 |
| Negative regulation of amino acid metabolic process | 2.82E-11 |
| Cell–cell signaling | 3.63E-11 |
| Cell surface receptor-linked signal transduction | 3.89E-11 |
| Regulation of developmental process | 4.37E-11 |
| Regulation of peptidyl-tyrosine phosphorylation | 4.57E-11 |
| Negative regulation of cellular process | 7.76E-11 |
| Tissue remodeling | 9.12E-11 |
| Regulation of signal transduction | 1.05E-10 |
ap values were calculated using Gene Go software with a false discovery rate cutoff of 0.01. Overall, 113 GO categories showed p values <1E-6, and 271 had p values <1E-4.
Functional annotation.
Functional classification was performed as follows. Gene symbols for the significant genes were subjected to pathway analysis with the Gene Go program (http://www.genego.com/) as well as additional analysis with the Tree Machine (http://bioinfo.vanderbilt.edu/gotm/frame.php) and DAVID (http://niaid.abcc.ncifcrf.gov/) programs. In addition, each gene symbol was analyzed using the Bioinformatics Harvester program (http://harvester.embl.de/), and PubMed searches were performed to supplement this information. Then, for each gene, a scientifically informative summary of molecular function was arrived at that best summarized the collected information. Disease associations were determined using the Medgene program (http://medgene.med.harvard.edu/MEDGENE/)
Quantitative real-time PCR
An aliquot of the total RNA that was previously extracted from prefrontal cortex punches was used for secondary validation using real-time PCR. Five hundred nanograms of total RNA was used for cDNA synthesis using oligo-dT primers and SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA) and subsequently diluted with nuclease-free water to 25 ng/μl cDNA. Gene-specific high-melt temperature primers for genes of interest were designed using Primer 3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi; Whitehead Institute for Biomedical Research, Cambridge, MA) and expressed sequence information obtained from GenBank (National Center for Biotechnology Information). PCR reactions were conducted on an ABI 7900 Sequence Detection System (Applied Biosystems, Foster City, CA) using a hot-start SYBR-green based method (Quantitect; Qiagen, Valencia, CA), followed by melt curve analysis to verify specificity of product. The CT value (cycle number at threshold) was used for calculations of relative amount of mRNA molecules. The CT value of each target gene was normalized by subtraction of the CT value from multiple housekeeping genes. This value is the ΔCT. The difference in ΔCT between control and MDD represents the ΔΔCT, and the relative quantitative change was showed as 2-ΔΔCT. All of the genes of interest were normalized to the housekeeping gene cyclophilin.
In situ hybridization
Sections of human brain were cut at 14 μm from blocked DLPFC. Rat brains were also sectioned at 14 μm after animals were killed, and brains were stored at −80°C. Brains were mounted onto “probe-on plus” glass slides (Fisher Scientific, Houston, TX). To prepare cRNA probe for in situ hybridization histochemistry, the fragments of FOXD3 cDNA (human FOXD3; GenBank accession number AF197560.1; position 1094–1255) and UCN3 (also referred to as stresscopin) cDNA (human UCN3; GenBank accession number NM_053049.2; position 511–624) were amplified by PCR using T7 promoter attached primer, and PCR products were used to generate 35S-radiolabeled riboprobes using T7 RNA polymerase in vitro transcription (T7-MEGAshortscript; Ambion). The rat sequences of FoxD3 and stresscopin are 96 and 93% identical, respectively, to the human sequences, and the same probes were used for in situ hybridization analysis of sections from both species.
After fixation and acetylation, human or rat sections were hybridized with 35S-CTP incorporated RNA probes at 60°C for 16 h in the hybridization solution. Sections were then washed at 60°C, dehydrated with graded alcohols, air dried, and exposed to film. Control experiments were conducted with sense strand riboprobes for both stresscopin and FoxD3, and, in both cases, no significant levels of labeling was detected.
Combined immunohistochemistry and in situ hybridization
The immuno-in situ method used in this study was performed as described previously (Newton et al., 2002). Briefly, fresh-frozen brains were sectioned at 14 μm thickness and mounted onto probe-on plus glass slides (Fisher Scientific). Immunohistochemical identification was performed under RNase-free conditions. Sections were fixed in 4% paraformaldehyde–PBS for 10 min, followed by 0.6% hydrogen peroxide treatment for 5 min. Sections were incubated in a blocking solution (2.5% BSA and 3000 U/ml heparin in PBS) for 30 min, reacted with mouse monoclonal antibody recognizing neuronal-specific nuclear protein (NeuN) (1:200) in RNase free solution (0.25% Triton X-100, 1% BSA, 3000 U/ml heparin, and 10 U/ml anti-RNase in PBS), and then reacted with secondary antibody for 1 h at room temperature. Sections were then incubated in a preformed avidin and biotinylated horseradish peroxidase macromolecular complex (ABC reagent). After this incubation, sections were rinsed in phosphate buffer and then stained with DAB staining kit (Vector Laboratories, Burlingame, CA). Slides were air dried before by in situ hybridization analysis or stored at −80°C. After in situ hybridization was conducted as described above, sections were dipped in nuclear track emulsion (NT2B, Kodak IB1654433; Eastman Kodak, Rochester, NY) for visualization at the single-cell level.
Stress and antidepressant models
For restraint stress, adult Sprague Dawley male rats (300–350 g) were subjected to immobilization stress in Disposable Rodent Restrainers (Braintree Scientific, Braintree, MA) for 1 h daily over a period of 15 consecutive days. For chronic unpredictable stress, rats were subjected to chronic unpredictable stress for 15 d exactly as described previously (Ortiz et al., 1996). Control rats were either not handled or were handled daily but not subjected to stress. For antidepressant studies, vehicle, fluoxetine (5 mg/kg), venlafaxine (15 mg/kg), or citalopram (15 mg/kg) were administered twice daily for 21 d, and brains were harvested for in situ hybridization analysis 4 h after the last vehicle or drug treatment.
Results
We began with 15 matched pairs of postmortem samples that were hybridized to Agilent Human 1A expression arrays. One array did not meet our quality control standards and was discarded (see Materials and Methods). Of the remaining 14 arrays, 20,173 probes were scanned. Probes that did not correspond to an annotated gene were eliminated as were those for which there were more than six “no calls” across the 14 arrays and two channels scanned. This left 11,351 probe sets for analysis.
One difficulty associated with postmortem analysis is that many variables are not under experimental control, such as patient demographics and medication usage. In an effort to minimize the effects of these extraneous variables, samples were matched on the basis of age, gender, and race (Table 1). However, because antidepressant usage was specific for patients with depression, it was not possible to match on the basis of this potential confound. We therefore analyzed the data using a mixed-model methodology that allowed us to model the effects of covariates such as medication usage while adjusting for any dependencies between samples hybridized on the same array. This methodology has the added advantage of allowing for permutation tests that rigorously compute p values that are robust to the effects of non-normality.
Because of a limitation in the degrees of freedom of our dataset, covariates were tested individually. Age, gender, and race were found to have no significant effects on the results, as expected based on the adequate matching on these variables, and these variables were not examined further. However, because we could not match subjects on the basis of antidepressant usage, we felt it necessary to enter this variable as a covariate. After correction for antidepressant usage in the month before death, a total of 249 probes were significant at a false discovery rate of 5% (supplemental Table 1, available at www.jneurosci.org as supplemental material). In comparison, >1000 probes were significant at a false discovery rate of 5% without correction for antidepressant usage. We did not have adequate power to test antidepressant effects on gene expression with this dataset. However, expression ratios adjusted for the effects of antidepressants were calculated from the mixed-model equations and are presented in Table 2 for comparison with the unadjusted expression ratios. The five genes most strongly related to antidepressant usage in the entire dataset of 11,351 probes were LGI4, NT5C1A, IFNAR1, C9orf115, and KIAA1641 and had uncorrected p values in the range 0.00045–0.0010 but did not survive false discovery rate control.
The significant gene list was then subjected to functional annotation. Although a diverse array of molecular pathways were altered by depression status, a smaller number of important cellular functions and molecular pathways showed multiple affected genes. Here we discuss the results in terms of broad categories of genes with multiple members that showed regulation (Table 2) and relate these results to previously published findings from the depression literature. We then explore two novel genes, stresscopin and FoxD3, as well as FGF-related genes, in more detail.
Intracellular signaling
A number of major signal transduction pathways showed multiple genes with significant expression changes. These include the β-catenin/WNT pathway, cAMP signaling pathways, and protein kinase C (PKC)/calcium signaling pathways.
We observed downregulation of the WNT receptors FZD8 and FZD9 and upregulation of the WNT receptor ligand WNT5A. The β-catenin/WNT pathway is regulated by antidepressants and antipsychotics (Beasley et al., 2002; Coyle and Duman, 2003). Opposing regulation of the FZD receptors and Wnt ligand makes it difficult to determine the functional consequences on Wnt signaling in postmortem tissue. However, the possible significance of altered Wnt signaling is highlighted by studies demonstrating that a major downstream effector of this pathway, glycogen synthase kinase-3, is inhibited by lithium as well as antidepressant drugs (Beasley et al., 2002; Coyle and Duman, 2003).
We observed increased phosphodiesterase type 4A (PDE4A) expression in depressed brains. cAMP and PDE4 isoforms have been implicated in mood regulation (O'Donnell and Zhang, 2004; Carlezon et al., 2005). For example, PDE4 inhibitors such as rolipram have antidepressant efficacy in animal models and in clinical trials (O'Donnell and Zhang, 2004). In addition, recent studies demonstrate that deletion of PDE4A (D'Sa et al., 2006) or PDE4D (Zhang et al., 2002) produces an antidepressant response in behavioral models of depression. Based on these findings, increased PDE4A in depressed brain would be expected to decrease cAMP levels and could thereby contribute to depressive behavior.
A number of other expression changes that we observed would be expected to decrease phosphorylation of the cAMP response element binding protein (CREB), a major transcription factor target of cAMP and calcium signaling, and could have a prodepressive effect (Carlezon et al., 2005). The transcriptional activity of CREB is regulated by phosphorylation, which is in turn controlled by protein kinases and phosphatases. Previous studies have demonstrated that expression of CREB can produce antidepressant effects in certain brain regions (Chen et al., 2001a; Wallace et al., 2004), although opposite effects are observed when CREB is expressed in the nucleus accumbens (Pliakas et al., 2001; Nestler and Carlezon, 2006). It is known that calmodulin activates calcium/calmodulin-dependent protein kinase IV, which in turn phosphorylates and inactivates PDE4 (O'Donnell and Zhang, 2004) and phosphorylates CREB. PKC can also lead to indirect phosphorylation of CREB. Decreased CALM2, decreased PKC (PRKCG), and increased protein phosphatase 2CA (PPP2CA) that we observed in MDD would be expected to decrease CREB phosphorylation and decrease CREB activity. An association between CREB activation and MDD has been reported previously, in which cAMP binding in cerebral cortex of depressed subjects was found to be decreased (Dwivedi et al., 2004) and CREB expression was found to be increased by antidepressant treatment (Chen et al., 2001b).
The decreased PKCγ (PRKCG) expression that we observed is interesting in light of evidence that this neuron-specific PKC isoform is required for certain forms of learning and memory (Douma et al., 1998; Colombo and Gallagher, 2002; Alvarez-Jaimes et al., 2004; Dwivedi et al., 2004; Rossi et al., 2005). Depression is associated with impaired prefrontal cognitive function, including impaired working memory (Harvey et al., 2004). We also observed increased diacylglycerol (DAG) kinase α expression, which competes with PKC for DAG and is a substrate for PKC. We observed upregulation of phospholipase 2 (PLA2G6), which would be expected to increase DAG levels and increase PKC activation, perhaps in compensation for the decreased levels of PKC.
Transduction of extracellular signals
A number of categories of genes involved in transducing extracellular signals were altered by depression status. These include genes involved in extracellular matrix and cellular adhesion, signaling and metabolism of GABA and glutamate, the major inhibitory and excitatory neurotransmitters, ion channel function, G-protein-coupled receptors, genes involved in growth factor signaling, steroid hormone signaling, and neurotransmitter transport or metabolism.
GABA and glutamate signaling have received considerable attention with respect to depression and other mood disorders. Compared with controls, depressed patients have elevated cortical glutamate levels (Sanacora et al., 2004) and decreased cortical GABA levels (Sanacora et al., 1999). Glutamate-ammonia ligase [glutamine synthetase (GLUL)] is an enzyme that is involved in the clearance of glutamate from synaptic sites by conversion to glutamine. The decreased levels of GLUL in depressed brains could account, in part, for the increased glutamate seen in depressed brains in this study. ALDH9A1, an enzyme that catalyzes the dehydrogenation of γ-aminobutyraldehyde to GABA, was increased in depressed brain. GABAB receptor agonists have antidepressant and anxiolytic effects in rodents (Pilc and Nowak, 2005; Nowak et al., 2006). The decreased GABBR1 expression noted in our study would be expected to have a prodepressive effect. A recent microarray study has reported altered expression of several GABA and glutamate transporter and receptor genes (Choudary et al., 2005), but there were no significant effects on these genes in the present study.
Another receptor that was altered in MDD is the cannabinoid 2 receptor (CNR2). Levels of CNR2 expression were increased by more than twofold, and this was confirmed by real-time (RT)-PCR (Table 3). CNR2 is expressed primarily in glia and has been implicated in immune and inflammation responses (Fernandez-Ruiz and Gonzales, 2005; Fernandez-Ruiz et al., 2007). CNR2 has been implicated in neuroprotective mechanisms, and levels of CNR2 expression in activated microglia and astrocytes are increased in response to treatments that produce local inflammatory responses.
Table 3.
Confirmation of microarray identified genes by real time-PCR (RT-PCR)
| Gene | GenBank | Description | Array fold change | Array p value* | RT-PCR fold change | RT-PCR p value |
|---|---|---|---|---|---|---|
| UCN3 | NM_053049 | Stresscopin (urocortin 3) | 1.99 | 4.75E-03 | 2.11 | <0.05 |
| FOXD3 | AF197560 | Forkhead box D3 | 2.09 | 2.35E-02 | 2.52 | <0.05 |
| CNR2 | X74328 | Cannabinoid receptor 2 | 2.66 | 5.35E-03 | 2.63 | <0.05 |
| CCIN | NM_005893 | Calicin, a basic cytoskeletal protein | 1.59 | 1.36E-02 | 2.05 | <0.05 |
| PRODH2 | U80018 | Proline dehydrogenase | 1.57 | 3.28E-02 | 1.96 | 0.10 |
| NELF | NM_015537 | Nasal embryonic LHRH factor | 0.69 | 3.57E-02 | 0.82 | <0.05 |
| MDFI | BC007836 | MyoD family inhibitor | 0.68 | 3.37E-04 | 0.66 | <0.05 |
Total RNA (12.5 ng) was reverse transcribed for a single-well reaction of real-time PCR (RT-PCR). All of the genes of interest were normalized to cyclophilin. The results are the mean ± SEM, n = 4 or more pairs. *p < 0.05 compared with controls (unpaired t test).
Stresscopin (UCN3) is one of several neuropeptides in the corticotrophin-releasing hormone (CRH) family. CRH is a critical factor in the response of the brain to stress and has been implicated in mood disorders (Holsboer, 2000). Stresscopin acts primarily at CRH type 2 receptors, and intraperitoneal injection of stresscopin in rats was found to impair memory trace formation and avoidance during a passive avoidance learning task (Klenerova et al., 2003). The upregulation that we observed in depressed brains could represent a homeostatic response to stress and/or other pathology associated with depression. Additional studies of stresscopin expression and regulation in MDD are presented below.
Control of cell cycle, cell death, and differentiation
A number of genes involved in cell cycle control, mitosis, or DNA replication, development and/or differentiation, and cell survival were altered. Previous studies have demonstrated that stress causes atrophy, decreased glia, and decreased proliferation of neurons (hippocampus) and non-neuronal cell types in hippocampus and/or prefrontal cortex, respectively, whereas antidepressant treatments have the opposite effect (Manji and Duman, 2001; Nestler et al., 2002b; Duman and Monteggia, 2006). Alterations in the expression level of genes involved in cellular proliferation and survival are therefore of interest.
FAS is a key player in apoptosis. The increased expression of FAS and TNR6, a receptor for TNFSF6, that we observed in depressed brains would be expected to lead to increased caspase activation and apoptosis and could play a role in the glial loss that is observed in postmortem depressed brain (Ongur et al., 1998; Rajkowska et al., 1999; Cotter et al., 2001). YWHAH inhibits BAD, which in turn increases cell death. The decreased YWHAH expression that we observed would be expected to disinhibit BAD and increase cell death. BBC3 is a negative regulator of the antiapoptotic BCL2 and was downregulated. Likewise, protein kinase, interferon-inducible double-stranded RNA dependent activator is activated by stress signals (Ito et al., 1999) and is thought to be proapoptotic and was also found to be upregulated. Overall, these results may indicate an increased apoptotic tone in depressed brains.
Inflammation
A number of genes involved in immunity and/or inflammation were altered. We observed increased expression of a tumor necrosis factor (TNF) family receptor (TNFRSF11B), and TNF receptor signaling has been implicated in depression-like behavior in mice (Simen et al., 2006). We observed increased expression of interferon α 6 (IFNA6) as well as interferon receptor 1 (IFNAR1). Interferons have been associated with MDD and in particular with depression associated with multiple sclerosis (Kagaya et al., 1997; Patten and Metz, 2001; Feinstein et al., 2002; Pandya and Patten, 2002).
Regulation of chromatin state and transcription factors
A number of genes involved in chromatin binding, regulation, or repair and a large number of transcription factors and genes involved in transcriptional regulation were affected.
YWHAH was substantially downregulated in depressed brain. This molecule negatively regulates histone deacetylase 9 (HDAC9). HDAC9, as well as HDAC5, are decreased by antidepressant treatment, and overexpression of HDAC5 blocks the effects of antidepressants in the social defeat model (Tsankova et al., 2006). FBXL10 is a histone demethylase for lysine 36 of histone H3 and was upregulated in depressed brain. Increased histone H3 dimethylation binding to the BDNF promoter regions 3 and 4 in response to social defeat stress has been reported (Tsankova et al., 2006). These data suggest that H3 methylation accounts in part for the sustained decrease in BDNF gene expression in response to stress.
FOXD3 is a transcription factor that plays an important role in vertebrate nervous system development, including the determination, migration, and differentiation of neural crest lineages (Dottori et al., 2001; Cheung et al., 2005). FOXD3 is expressed in a variety of other progenitor cells, including embryonic stem cells (Pan et al., 2006; Pan and Thomson, 2007), and appears to play a key role in regulating development.
Secondary confirmation
We chose seven genes for secondary confirmation by quantitative RT-PCR (Table 3), including five that are upregulated (UCN3, CNR2, FOXD3, PRODH2, and CCIN) and two that are downregulated (MDFI and NELF). The purpose of these confirmations was to obtain a general sense of the reliability of our array results using an independent method. We selected genes because of their potential relevance to MDD, most notably stresscopin, and because of their high degree of regulation. Quantitative PCR was consistent with the microarray results and significant changes (p < 0.05 by t test) for six of the seven genes. There was a tendency for the other gene, PRODH2, to replicate the array results, but the effect did not reach significance (p = 0.10). These results demonstrate good reproducibility of the expression changes using an independent method.
Altered expression of stresscopin and CRH-related genes
Altered expression of stresscopin is potentially relevant to depression pathophysiology because hyperactivity of hypothalamic–pituitary–adrenal (HPA) axis, including increased CRH, is one of the most consistent abnormalities associated with mood disorders (Holsboer, 2000). Fifty to 60% of depressed patients exhibit increased levels of cortisol and loss of feedback inhibition of the HPA axis. Moreover, previous studies have demonstrated that levels of CRH immunoreactivity are increased in the CSF of depressed subjects (Nemeroff et al., 1984), and postmortem studies report decreased levels of CRH receptor binding in cerebral cortex of suicide subjects (Nemeroff et al., 1988). The highly significant upregulation of stresscopin identified in the current study by microarray and confirmed by RT-PCR (1.99- and 2.11-fold, respectively) provides additional evidence that the CRH system is dysregulated in depression. Additional studies were conducted to examine the regulation and regional and cellular distribution of stresscopin, as well as other members of the CRH family.
Stresscopin in situ hybridization analysis was conducted using a radiolabeled human stresscopin riboprobe and frozen sections of DLPFC from MDD subjects and matched controls. The results demonstrate that stresscopin mRNA expression is enriched in gray matter relative to white matter in the DLPFC (Fig. 1). In addition, stresscopin mRNA was significantly increased in the gray matter (143.9 ± 10%) but not white matter of sections from MDD subjects (Fig. 1). Emulsion analysis of the radiolabeled stresscopin riboprobe suggests that the number of grains over individual cells is increased in the MDD subjects rather than an increase in number of cells expressing stresscopin, although this is based on a qualitative analysis of sections. This indicates that the increase is not attributable to a recruitment of new cells expressing stresscopin and suggests that the function of stresscopin may be increased within this population of neurons or possibly the surrounding cells. This possibility must be directly addressed by quantitative analysis of stresscopin expression in these neurons. This is the first demonstration of stresscopin expression in human brain, as well as increased expression of this gene in the DLPFC of depressed subjects.
Figure 1.
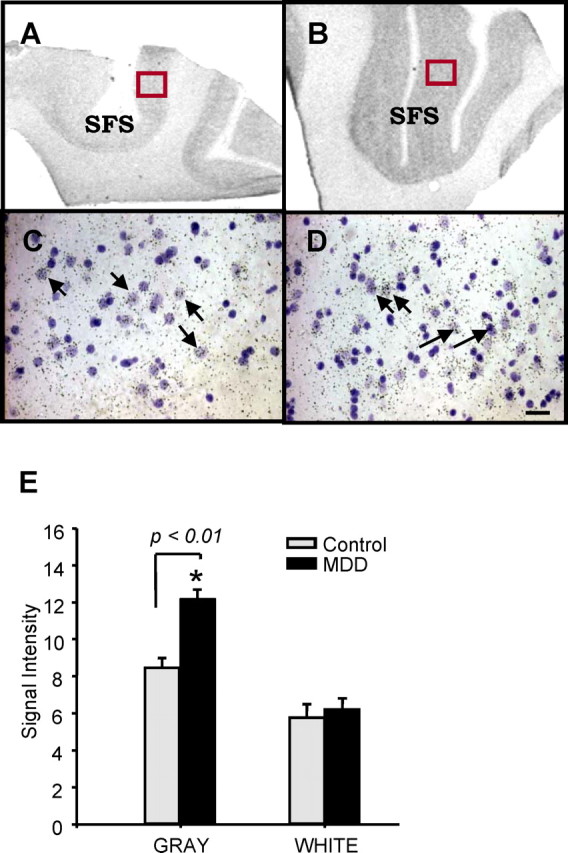
A,B, Expression of stresscopin in human DLPFC. In situ hybridization analysis of stresscopin comparing sections from matched control and MDD subject was conducted using a 35S-labeled antisense riboprobe. Expression of stresscopin mRNA was increased in the gray matter of DLPFC of MDD compared with the matched control. C and D show emulsion of stresscopin in the DLPFC of matched control and MDD subject (400×). Cresyl violet counterstained cells that do not express stresscopin mRNA appear violet. SFS, Superior frontal sulcus. Scale bar, 50 μm. E, The results are presented as the mean ± SEM (n = 5). *p < 0.01 compared with controls (unpaired t test).
Additional studies were conducted to characterize the expression and regulation of stresscopin. Because of the limited number of sections available from MDD and controls, these studies were first conducted in rat brain PFC and retrosplenial cortex, as well as human DLPFC sections. Double-labeling studies clearly demonstrate clustering of stresscopin mRNA labeled grains over neurons (NeuN-immunopositive cells), many of which appear to be pyramidal cells, in both rat and human cortical sections (Fig. 2D–F). In contrast, very few grains appear over GFAP-positive cells (Fig. 2G,H).
Figure 2.
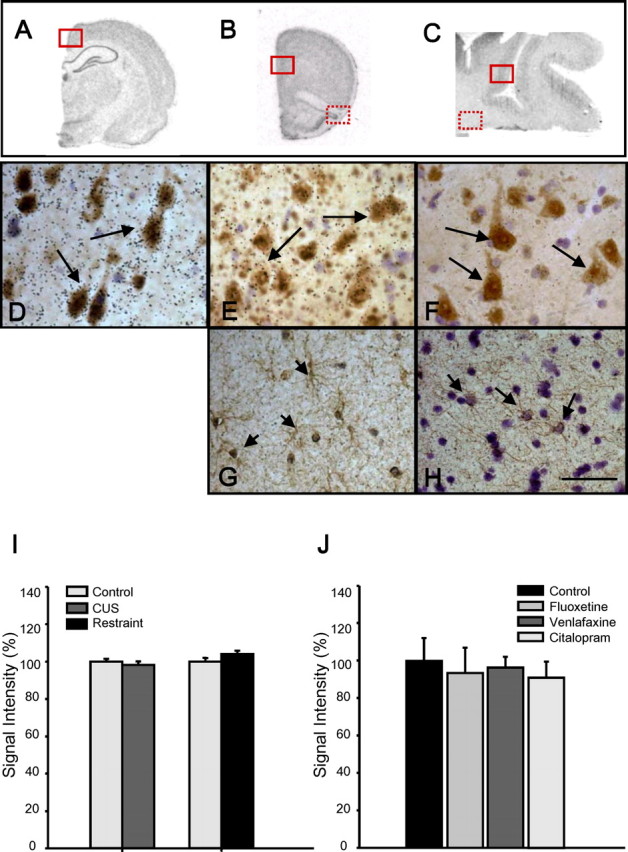
A–H, Stresscopin colocalization and regulation by stress. Combined stresscopin in situ hybridization and immunohistochemistry for a marker of neurons (NeuN) or glia was conducted on rat retrosplenial (A, D) and PFC (B, E, G) or human PFC (C, F, H). Black emulsion grains can be clearly seen over brown DAB-stained NeuN-positive cells (arrow). I, J, The influence of chronic stress, either restraint or unpredictable stress (I), or antidepressant administration (3 weeks) on stresscopin expression (J) was determined using a 35S-labeled antisense riboprobe. The results are expressed as the mean ± SEM (n = 5 for stress, n = 4 for antidepressants).
To examine the factors that could influence the regulation of stresscopin in humans, we examined the influence of chronic stress in two rodent models, repeated restraint or unpredictable stress, each for 15 d. The results demonstrate that there is no significant effect of either chronic restraint or chronic unpredictable stress on levels of stresscopin expression (Fig. 2I), suggesting that other factors account for the altered expression of this gene in depressed subjects. In addition, there was no effect of chronic antidepressant administration on the expression of stresscopin (Fig. 2J).
In addition to CRH and stresscopin, the other members of this family include urocortin and urocortin II (Muller and Wurst, 2004; Nemeroff and Vale, 2005). Based on receptor affinity, urocortin, as well as corticotrophin-releasing factor (CRF), are ligands for CRF receptor type 1 (CRFR1), whereas urocortin, urocortin II, and stresscopin are ligands for CRFR2 (Muller and Wurst, 2004; Nemeroff and Vale, 2005). In contrast to the highly significant increase in the expression of stresscopin (Tables 3, 4), levels of CRF, urocortin, and urocortin II were not significantly altered in the MDD subjects (Table 4). Expression of CRF binding protein, which binds and inactivates CRF, was decreased, although this effect did not reach significance based on rigorous FDR statistics (Table 4). In addition, levels of CRF receptor 1 (CRHR1) mRNA, but not CRHR2, were decreased but again did not reach significance based on FDR statistics (Table 4). There were no changes in the expression levels of the glucocorticoid receptor subtypes or related genes (i.e., modulatory and DNA binding proteins) (Table 4).
Table 4.
Expression of stresscopin and CRF-related genes in MDD
| Category | Gene | GenBank ID | Description | Fold changes | p value |
|---|---|---|---|---|---|
| CRH system | CRH | BC011031 | Corticotropin-releasing hormone | 1.00 | NS |
| UCN | NM_003353 | Urocortin | 1.10 | NS | |
| SRP | NM_033199 | Stresscopin-related peptide (urocortin 2) | 1.13 | NS | |
| SPC | NM_053049 | Stresscopin (urocortin 3) | 1.99 | <0.05* | |
| CRHR1 | NM_004382 | Corticotropin-releasing hormone receptor 1 | 0.81 | NS | |
| CRHR2 | NM_001883 | Corticotropin-releasing hormone receptor 2 | 0.95 | NS | |
| CRHBP | BC018038 | Corticotropin-releasing hormone-binding protein | 0.85 | NS | |
| Glucocorticoid | NR3C1 | NM_000176 | Glucocorticoid receptor | 1.11 | NS |
| NR3C2 | NM_000901 | Mineralocorticoid receptor | 1.16 | NS | |
| GMEB1 | AF099013 | Glucocorticoid modulatory element binding protein 1 | 1.08 | NS | |
| GMEB2 | AF173867 | Glucocorticoid modulatory element binding protein 2 | 1.05 | NS | |
| GRLF1 | NM_004491 | Glucocorticoid receptor DNA binding factor 1 | 1.16 | NS |
Expression of stresscopin and CRF-related genes were examined by microarray analysis. The results are expressed as mean ± SEM. An asterisk indicates significant at p value indicated (unpaired t test). NS, Not significant.
Altered expression of FoxD3
Although there is no a priori reason to predict a role for FoxD3 in MDD, the high degree of dysregulation (Table 3) prompted studies to further examine the regulation and distribution of this transcription factor (Weigel et al., 1989). The name forkhead family derives from two spiked-head structures in embryos of the Drosophila forkhead mutant, which are defective in formation of the anterior and posterior gut (Weigel et al., 1989). Family members have a characteristic winged-helix DNA binding domain and are required for a variety of developmental processes, including the maintenance of differentiated cell states, tissue-specific gene expression, embryogenesis, and tumorigenesis (Kaufmann and Knochel, 1996; Zhou et al., 2002).
In situ hybridization analysis was performed using a human radiolabeled FoxD3 riboprobe and demonstrated that FoxD3 mRNA is enriched in gray matter, with a laminar distribution in the middle and deep layers of DLPFC (Fig. 3). Levels of radiolabeled FoxD3 riboprobe were significantly increased (125.5 ± 12%; n = 10) in the gray matter but not white matter of sections from MDD subjects (Fig. 3). Emulsion analysis of radiolabeled FoxD3 demonstrates intense clustering of grains over cells and suggests that the number of grains over individual cells is increased in the MDD subjects rather than an increase in number of cells expressing FoxD3. This possibility must be directly addressed by quantitative analysis of FoxD3 expression in these neurons. This is also the first demonstration of FoxD3 expression in human brain, as well as increased expression of this gene in depressed subjects.
Figure 3.
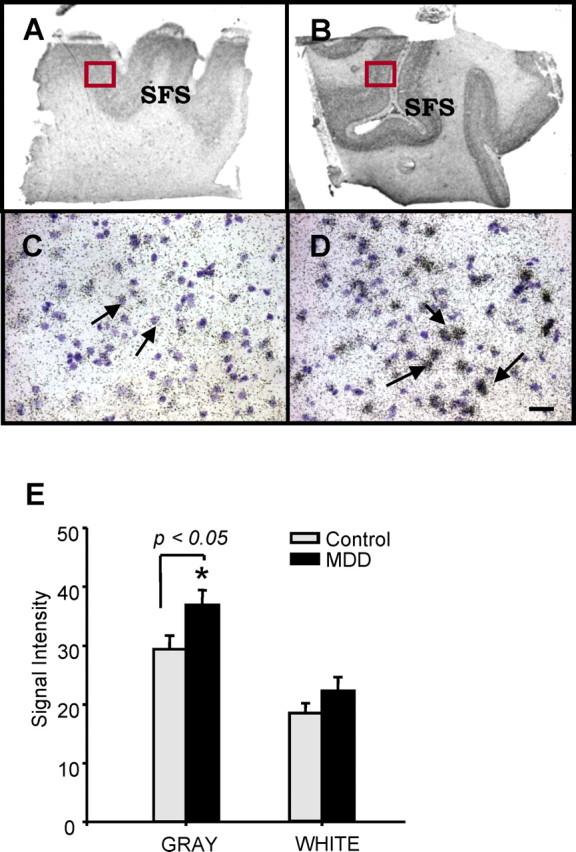
A,B, Expression of FoxD3 in human DLPFC. In situ hybridization analysis of FoxD3 comparing sections from matched control and MDD subject was conducted using a 35S-labeled antisense riboprobe. Expression of FoxD3 mRNA was increased in the gray matter of DLPFC of MDD compared with the matched control. SFS, Superior frontal sulcus. C and D show emulsion of FoxD3 in the DLPFC of matched control and MDD subject (400×). Cresyl violet counterstained cells that do not express FoxD3 mRNA appear violet. E, The results are presented as the mean ± SEM (n = 10). *p < 0.05 compared with controls (unpaired t test).
As described for stresscopin, rodent brain sections were used first because of the limited number of human sections. Double-labeling studies demonstrate clustering of grains of FoxD3 over NeuN-immunopositive cells in both rodent PFC and human DLPFC (Fig. 4D–F) but not over GFAP-positive cells (Fig. 4G,H). We also examined the influence of chronic restraint and unpredictable stress on the expression of FoxD3. There is a small but significant increase in the expression of FoxD3 after exposure to chronic unpredictable stress (115.2 ± 4.6%; n = 9) but not restraint stress (106 ± 3.8%; n = 9) (Fig. 4I). There was no effect of chronic antidepressant administration on the expression of FoxD3 (Fig. 4J).
Figure 4.
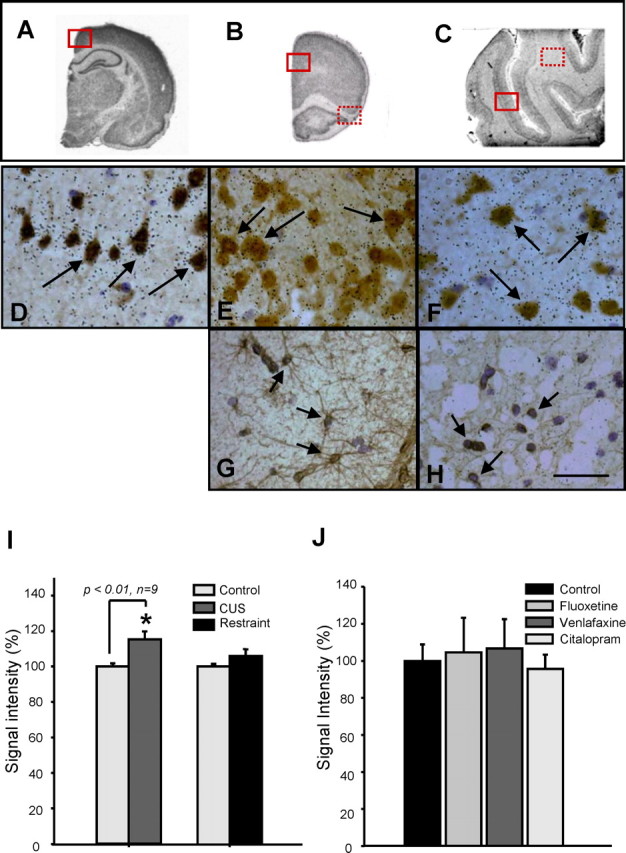
A–H, FoxD3 colocalization and regulation by stress. Combined FoxD3 in situ hybridization and immunohistochemistry for a marker of neurons (NeuN) or glia was conducted on rat retrosplenial (A, D) or PFC (B, E, G) or human PFC (C, F, H). Black emulsion grains can be clearly seen over brown DAB-stained NeuN-positive cells (arrow). I, J, The influence of chronic stress, either restraint or unpredictable stress (I), or antidepressant administration (3 weeks) on FoxD3 expression (J) was determined using a 35S-labeled antisense riboprobe. Chronic unpredictable stress, but not restraint stress, increased the expression of FoxD3 by ∼15% in the prefrontal cortex. The results are expressed as the mean ± SEM (n = 9 for stress, n = 3 or 4 for antidepressants). *p < 0.01 compared with control (unpaired t test).
Altered expression of FGF-related genes
A recent postmortem study has demonstrated altered expression of specific genes in the FGF family of growth factors in depressed subjects (Evans et al., 2004). This includes decreased levels of FGF1, increased levels of FGF9, and decreased levels of FGFR2 and FGFR3 in DLPFC. In the current study, we confirm the decreased expression of FGFR3 in DLPFC of MDD subjects but found no change in levels of FGF1 and FGF9 or FGFR2. We also detected downregulation of fibroblast growth factor binding protein 1 in depressed subjects.
Discussion
Depression is a complex disorder attributable to an interaction between genetics and the environment. Alterations in the expression of genes, either as a result of environmental factors or mutation of promoter elements that control gene expression, contribute to the behavioral phenotype and biological state that defines depression. Gene expression studies using microarrays allows for a broad analysis of alterations in human gene expression. Microarrays can be used to identify novel genes and gene expression patterns that contribute, in part, to the pathophysiology of MDD and could potentially identify unique therapeutic drug targets.
In the current study, we examined the gene expression profile of a well defined region of the DLPFC in postmortem brains of MDD subjects, using a sensitive hybridization approach that does not require amplification. The sections were taken from the same control and MDD subjects and the same part of the DLPFC that were used for morphological analysis, a study that demonstrates decreased number of glia and size of neurons (Rajkowska et al., 1999) and that is consistent with other reports (Ongur et al., 1998; Cotter et al., 2001, 2002). Although the focus of the current study is the gray matter of the DLPFC, it will be important to also examine gene expression changes in white matter to fully characterize the alterations underlying MDD. In addition, a matched-pair design was used in which controls were carefully selected to match MDD samples in a variety of factors, including pH and agonal state. Medication usage was controlled for statistically using a mixed-effects model. Together, this approach allows for gene expression profiling of gray matter of a discrete region of the DLPFC of MDD subjects.
Some of the identified genes are new, and others extend and confirm previous gene expression alterations that have been identified by traditional approaches or more recent microarray studies. The relationship of these gene changes to the morphological alterations reported previously in the DLPFC of these MDD patients is currently unknown, although it is notable that the highest expression of stresscopin and FoxD3 is observed in the same DLPFC layers (III, V, and VI). Additional analysis of these genes, as well as multiple genes involved in signal transduction, metabolic, synthetic, and repair pathways identified in the current study will be required to understand this relationship. Together, these studies begin to identify trends for MDD gene profiles, as well as identify novel gene profiles that could contribute to depression. Brief descriptions of relevant gene families and pathways are provided in Results, and additional studies will be needed to provide secondary validation, replication in other MDD cohorts, and to characterize the function of these genes in MDD. Here we will focus on three gene families that were chosen for additional analysis because of their relevance based on previous studies (stresscopin), novelty (FoxD3), and previous microarray analysis (FGFR3).
Altered expression of stresscopin
One of the most highly regulated genes identified in the current study is stresscopin or UCN3, a CRF-related peptide that is an agonist for CRFR2. This novel discovery extends previous work demonstrating elevation of CRF and dysregulation of the HPA axis in mood disorders (Holsboer, 2000). In situ hybridization analysis demonstrates specific expression of stresscopin in gray matter of human DLPFC, and double-labeling with a neuronal marker demonstrates that stresscopin mRNA is expressed predominantly in neurons in rodent PFC and human DLPFC. Stresscopin mRNA levels are not altered in rodent stress models (chronic restraint or unpredictable stress), suggesting that other environmental factors, promoter polymorphisms, or epigenetic factors contribute to the elevation in levels of stresscopin observed in MDD subjects. Alternatively, it is possible that the rodent models, which use normal animals, do not accurately reflect the human condition. Levels of stresscopin in the hypothalamus are reported to be upregulated in response to acute stress (Venihaki et al., 2004).
Fifty to 60% of depressed patients exhibit increased levels of cortisol and loss of feedback inhibition of the HPA axis. Moreover, previous studies have demonstrated that levels of CRF immunoreactivity are increased in the CSF of depressed subjects(Nemeroff et al., 1984), and postmortem studies report decreased levels of CRF receptor binding in cerebral cortex of suicide subjects (Nemeroff et al., 1988). The highly significant upregulation of stresscopin identified in the current study by microarray and confirmed by RT-PCR (1.99- and 2.11-fold, respectively) provides additional evidence that the CRF system is dysregulated in depression.
Rodent studies suggest that the function of stresscopin in the brain may be related, as the name implies, to coping mechanisms in response to aversive or stressful stimuli (Muller and Wurst, 2004; Nemeroff and Vale, 2005). Other functions assigned to stresscopin include the effects of stress on appetite (Muller and Wurst, 2004). Although the function of the CRFR1 receptor has been clearly linked to anxiety and depression, leading to the development of selective antagonists for the treatment of these disorders, the function of CRFR2 and related peptides has been less clear. Studies of CRFR2 null mutant mice have reported an increase in anxiety (Bale et al., 2000; Kishimoto et al., 2000) and depression-like behavior (Bale and Vale, 2003), suggesting that stresscopin and other ligands for this receptor may function to decrease anxiety. This is supported by studies of stresscopin infusions (Venihaki et al., 2004). Other studies demonstrate that activation of either CRFR1 or CRFR2 enhance defensive startle but that the two receptor subtypes function in opposition to regulate the flexibility of startle (Risbrough et al., 2004). Other studies have demonstrated an interaction of CRFR1 and CRFR2, suggesting that they are independent but related systems (Muller and Wurst, 2004).
Based on these findings, one interpretation of the current results is that the dramatic increase of stresscopin expression represents an attempt to compensate for the stress and depression experienced by the MDD subjects. In this context, stresscopin could represent a marker of depression and other mood disorders. Additional studies will be required to further identify the factors that control the expression of stresscopin and to determine the function of this neuropeptide in humans.
In contrast to stresscopin, levels of CRF, urocortin, urocortin II, and CRFR2 were not significantly different in the MDD subjects. The finding that CRF expression was not altered suggests that the elevated CRF immunoreactivity reported in the CSF of depressed subjects (Nemeroff et al., 1984; Gillespie and Nemeroff, 2005) may not be a result of increased CRF mRNA expression. Alternatively, increased expression of CRF in other brain regions, most notably the hypothalamus and amygdala, could contribute to the elevation of CRF levels in CSF. There was a tendency for decreased expression of CRF binding protein and CRFR1 in MDD subjects. Although these effects did not reach significance, a reduction in CRF binding protein could result in an increase in free CRF, and decreased CRFR1 expression is consistent with reports of decreased CRF receptor binding in suicide subjects (Nemeroff et al., 1988).
Altered expression of FoxD3 and transcription factors
Another one of the most highly regulated genes is FoxD3, a member of the forkhead family of transcriptional regulators. In situ hybridization and colocalization studies demonstrate that FoxD3 is expressed primarily in neurons of human DLPFC. Studies of rodent stress models also demonstrate a small but significant induction of FoxD3, although the magnitude of this effect indicates that other factors may also contribute to the large elevation observed in MDD subjects.
Previous reports suggest that FoxD3 promotes differentiation of neural crest cells and regulates differentiation of crest-derived cell types by inhibiting melanoblast development (Dottori et al., 2001; Kos et al., 2001; Sasai et al., 2001). However, the specific function of this gene in the adult brain has not yet been determined.
Altered expression of FGF-related genes
Basic and clinical research studies have reported that the expression of certain growth and/or neurotrophic factors are decreased by stress and depression and that antidepressant treatment blocks or reverses these effects (Duman, 2004a,b). In addition, brain imaging studies have demonstrated that the volume of certain limbic structures, including the prefrontal cortex and hippocampus, are reduced in depressed subjects (Sheline et al., 1996; Drevets et al., 1997; Duman, 2004a). Together, these findings have contributed to a neurotrophic hypothesis of depression (Duman et al., 1997; Duman, 2004b). Although much of this work has focused on BDNF and related neurotrophic factors, recent basic and clinical studies have also demonstrated a role for FGF family members. The results of the current study confirm the findings of a recent postmortem study (Evans et al., 2004), demonstrating decreased expression of FGFR3 in the DLPFC. There were also several differences between the previous and current studies as discussed in Results. Studies of additional MDD subjects will be needed to confirm these findings, to determine what accounts for the differences observed between these two studies, and to examine the functional consequences of altered expression of specific FGF and receptor subtypes. Nevertheless, confirmation of decreased levels of FGFR3 suggests that the regulation of this gene could be a marker of depression and possibly contribute to the underlying pathophysiology of mood disorders.
Conclusions
The results of this study identify new genes not shown previously to be altered in MDD as well as genes implicated previously in mood disorders. The role of these novel genes in depression, and in some cases in CNS function, will require additional studies but open new avenues of research. Altered expression of stresscopin and CRF-related genes further demonstrates that the HPA axis and genes involved in mediating the actions of stress are dysregulated in mood disorders. It is also interesting to speculate that altered HPA axis activity could contribute to altered expression of other genes implicated in MDD. Together, these effects could contribute to the morphological, as well as functional, changes that have been observed in MDD subjects. Additional postmortem studies will be needed to confirm that these genes are consistently altered in other MDD subjects. It is likely that many of these genes are altered as a consequence of long-term stress and depression, but it is also possible that some are also involved in the etiology of mood disorders. Additional studies are necessary to determine whether these changes represent trait or state markers of mood disorders, as well as studies to determine the influence of psychotropic drugs on the expression of these genes. This work contributes to ongoing studies of the identification and characterization of gene expression profiles underlying stress-related neurobiological illnesses.
Footnotes
This work was supported by National Institutes of Health Grants MH25642, MH45481 (R.S.D.), MH67996 (C.A.S.), RR17701 (C.A.S., G.R.), MH61578 and MH60451 (G.R.) and RR19895-02, a National Alliance for Research on Schizophrenia and Depression Young Investigator Award, National Center for Research Resources Grant 1KL2RR024138 (A.S.), and by the postdoctoral fellowship program of Korea Science and Engineering Foundation (H.J.K.). We thank Nicholas Carriero and Robert Bjornson from the Yale Center for High Performance Computation in Biology and Biomedicine.
References
- Alvarez-Jaimes L, Betancourt E, Centeno-Gonzalez M, Feliciano-Rivera MZ, Rodriguez D, de Ortiz SP, Maldonado-Vlaar CS. Spatial learning in rats is impaired by microinfusions of protein kinase C-gamma antisense oligodeoxynucleotide within the nucleus accumbens. Neurobiol Learn Mem. 2004;81:120–136. doi: 10.1016/j.nlm.2003.12.001. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Ed 4. Washington, DC: American Psychiatric Association; 2000. Diagnostic and statistical manual of mental disorders. [Google Scholar]
- Bale TL, Vale WW. Increased depression-like behaviors in corticotropin-releasing factor receptor-2-deficient mice: sexually dichotomous responses. J Neurosci. 2003;23:5295–5301. doi: 10.1523/JNEUROSCI.23-12-05295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, Contarino A, Smith GW, Chan R, Gold LH, Sawchenko PE, Koob GF, Vale WW, Lee KF. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat Genet. 2000;24:410–414. doi: 10.1038/74263. [DOI] [PubMed] [Google Scholar]
- Beasley C, Cotter D, Everall I. An investigation of the Wnt-signalling pathway in the prefrontal cortex in schizophrenia, bipolar disorder and major depressive disorder. Schizophr Res. 2002;58:63–67. doi: 10.1016/s0920-9964(01)00376-0. [DOI] [PubMed] [Google Scholar]
- Bremner J, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS. Hippocampal volume reduction in major depression. Am J Psychiatry. 2000;157:115–118. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- Bremner J, Vythilingam M, Vermetten E, Nazeer A, Adil J, Khan S, Staib LH, Charney DS. Reduced volume of orbitofrontal cortex in major depression. Biol Psychiatry. 2002;51:273–279. doi: 10.1016/s0006-3223(01)01336-1. [DOI] [PubMed] [Google Scholar]
- Broberg P. A comparative review of estimates of the proportion unchanged genes and the false discovery rate. BMC Bioinformatics. 2005;6:199. doi: 10.1186/1471-2105-6-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon W, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Chen AC, Shirayama Y, Shin KH, Neve RL, Duman RS. Expression of the cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effect. Biol Psychiatry. 2001a;49:753–762. doi: 10.1016/s0006-3223(00)01114-8. [DOI] [PubMed] [Google Scholar]
- Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry. 2001b;50:260–265. doi: 10.1016/s0006-3223(01)01083-6. [DOI] [PubMed] [Google Scholar]
- Cheung M, Chaboissier MC, Mynett A, Hirst E, Schedl A, Briscoe J. The transcriptional control of trunk neural crest induction, survival, and delamination. Dev Cell. 2005;8:179–192. doi: 10.1016/j.devcel.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Choudary P, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, Myers RM, Bunney WE, Akil H, Watson SJ, Jones EG. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci USA. 2005;102:15653–15658. doi: 10.1073/pnas.0507901102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo PJ, Gallagher M. Individual differences in spatial memory among aged rats are related to hippocampal PKCgamma immunoreactivity. Hippocampus. 2002;12:285–289. doi: 10.1002/hipo.10016. [DOI] [PubMed] [Google Scholar]
- Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry. 2001;58:545–553. doi: 10.1001/archpsyc.58.6.545. [DOI] [PubMed] [Google Scholar]
- Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb Cortex. 2002;12:386–394. doi: 10.1093/cercor/12.4.386. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Duman RS. Finding the intracellular signaling pathways affected by mood disorder treatments. Neuron. 2003;38:157–160. doi: 10.1016/s0896-6273(03)00195-8. [DOI] [PubMed] [Google Scholar]
- Cui X, Churchill GA. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003;4:210. doi: 10.1186/gb-2003-4-4-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Kerr MK, Churchill GA. Transformations for cDNA microarray data. Stat Appl Genet Mol Biol. 2003;2 doi: 10.2202/1544-6115.1009. Article4. [DOI] [PubMed] [Google Scholar]
- Cui X, Hwang JT, Qiu J, Blades NJ, Churchill GA. Improved statistical tests for differential gene expression by shrinking variance components estimates. Biostatistics. 2005;6:59–75. doi: 10.1093/biostatistics/kxh018. [DOI] [PubMed] [Google Scholar]
- D'Sa C, Terwilliger R, Simen B, Edwards F, Schultz H, Russell RS, Duman RS. Role of the rolipram-sensitive cAMP-dependent phosphodiesterase type 4A in the mechanism of antidepressant action: insights form constitutive and conditional PDE4A-null mutant mice. Soc Neurosci Abstr. 2006;32:191–3. [Google Scholar]
- Dottori M, Gross MK, Labosky P, Goulding M. The winged-helix transcription factor Foxd3 suppresses interneuron differentiation and promotes neural crest cell fate. Development. 2001;128:4127–4138. doi: 10.1242/dev.128.21.4127. [DOI] [PubMed] [Google Scholar]
- Douma BR, Van der Zee EA, Luiten PG. Translocation of protein kinase Cgamma occurs during the early phase of acquisition of food rewarded spatial learning. Behav Neurosci. 1998;112:496–501. doi: 10.1037//0735-7044.112.3.496. [DOI] [PubMed] [Google Scholar]
- Drevets W. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr Opin Neurobiol. 2001;11:240–249. doi: 10.1016/s0959-4388(00)00203-8. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Simpson JR, Todd RD, Reich T, Vannier M, Raichle ME. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- Duman RS. Depression: a case of neuronal life and death? Biol Psychiatry. 2004a;56:140–145. doi: 10.1016/j.biopsych.2004.02.033. [DOI] [PubMed] [Google Scholar]
- Duman RS. Role of neurotrophic factors in the etiology and treatment of mood disorders. Neuromolecular Med. 2004b;5:11–25. doi: 10.1385/NMM:5:1:011. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Shukla PK, Lyons J, Faludi G, Palkovits M, Sarosi A, Conley RR, Roberts RC, Tamminga CA, Pandey GN. Protein kinase A in postmortem brain of depressed suicide victims: altered expression of specific regulatory and catalytic subunits. Biol Psychiatry. 2004;55:234–243. doi: 10.1016/j.biopsych.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Evans SJ, Choudary PV, Neal CR, Li JZ, Vawter MP, Tomita H, Lopez JF, Thompson RC, Meng F, Stead JD, Walsh DM, Myers RM, Bunney WE, Watson SJ, Jones EG, Akil H. Dysregulation of the fibroblast growth factor system in major depression. Proc Natl Acad Sci USA. 2004;101:15506–15511. doi: 10.1073/pnas.0406788101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein A, O'Connor P, Feinstein K. Multiple sclerosis, interferon beta-1b and depression A prospective investigation. J Neurol. 2002;249:815–820. doi: 10.1007/s00415-002-0725-0. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ruiz J, Gonzales S. Cannabinoid control of motor function at the basal ganglia. Handb Exp Pharmacol. 2005:479–507. doi: 10.1007/3-540-26573-2_16. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ruiz J, Romero J, Velasco G, Tolon RM, Ramos JA, Guzman M. Cannabinoid CB2 receptor: a new target for controlling neural cell survival? Trends Pharmacol Sci. 2007;28:39–45. doi: 10.1016/j.tips.2006.11.001. [DOI] [PubMed] [Google Scholar]
- First M, Spitzer RL, Gibbon M, Williams JBW. New York: Biometrics Research, New York State Psychiatric Institute; 2001. Structured clinical interview for DSM-IV-TR axis I disorders, Research version, Patient Edition (SCID-I/NP) [Google Scholar]
- Gillespie CF, Nemeroff CB. Hypercortisolemia and depression. Psychosom Med. 2005;67(Suppl 1):S26–S28. doi: 10.1097/01.psy.0000163456.22154.d2. [DOI] [PubMed] [Google Scholar]
- Harvey PO, Le Bastard G, Pochon JB, Levy R, Allilaire JF, Dubois B, Fossati P. Executive functions and updating of the contents of working memory in unipolar depression. J Psychiatr Res. 2004;38:567–576. doi: 10.1016/j.jpsychires.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Hastings R, Parsey RV, Oquendo MA, Arango V, Mann JJ. Volumetric analysis of the prefrontal cortex, amygdala, and hippocampus in major depression. Neuropsychopharmacology. 2004;29:952–959. doi: 10.1038/sj.npp.1300371. [DOI] [PubMed] [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Ito T, Yang M, May WS. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem. 1999;274:15427–15432. doi: 10.1074/jbc.274.22.15427. [DOI] [PubMed] [Google Scholar]
- Kagaya A, Uchitomi Y, Takezaki E, Fukue M, Tsukano K, Kugaya A, Minagawa H, Takebayashi M, Zensho H, Oyamada T, Yamawaki S. Plasma levels of cyclic GMP, immune parameters and depressive status during interferon therapy. A prospective study in Japan. Neuropsychobiology. 1997;35:128–131. doi: 10.1159/000119333. [DOI] [PubMed] [Google Scholar]
- Kaufmann E, Knochel W. Five years on the wings of fork head. Mech Dev. 1996;57:3–20. doi: 10.1016/0925-4773(96)00539-4. [DOI] [PubMed] [Google Scholar]
- Kelly TM, Mann JJ. Validity of DSM-III-R diagnosis by psychological autopsy: a comparison with clinician ante-mortem diagnosis. Acta Psychiatr Scand. 1996;94:337–343. doi: 10.1111/j.1600-0447.1996.tb09869.x. [DOI] [PubMed] [Google Scholar]
- Kessler R, McGonagle KA, Zhao S, Nelson CB, Hughes M, Eshleman S, Wittchen H, Kendler KS. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States: results from the national comorbidity survey. Arch Gen Psychiatry. 1994;51:8–19. doi: 10.1001/archpsyc.1994.03950010008002. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Radulovic J, Radulovic M, Lin CR, Schrick C, Hooshmand F, Hermanson O, Rosenfeld MG, Spiess J. Deletion of crhr2 reveals an anxiolytic role for corticotropin-releasing hormone receptor-2. Nat Genet. 2000;24:415–419. doi: 10.1038/74271. [DOI] [PubMed] [Google Scholar]
- Klenerova V, Kaminsky O, Sida P, Hlinak Z, Krejci I, Hynie S. Impaired passive avoidance acquisition in Wistar rats after restraint/cold stress and/or stresscopin administration. Gen Physiol Biophys. 2003;22:115–120. [PubMed] [Google Scholar]
- Kos R, Reedy MV, Johnson RL, Erickson CA. The winged-helix transcription factor FoxD3 is important for establishing the neural crest lineage and repressing melanogenesis in avian embryos. Development. 2001;128:1467–1479. doi: 10.1242/dev.128.8.1467. [DOI] [PubMed] [Google Scholar]
- Lewis DA. The human brain revisited: opportunities and challenges in postmortem studies of psychiatric disorders. Neuropsychopharmacology. 2002;26:143–154. doi: 10.1016/S0893-133X(01)00393-1. [DOI] [PubMed] [Google Scholar]
- Manji HK, Duman RS. Impairments of neuroplasticity and cellular resilience in severe mood disorders: implications for the development of novel therapeutics. Psychopharmacol Bull. 2001;35:5–49. [PubMed] [Google Scholar]
- Manji H, Drevets WC, Charney DS. The cellular neurobiology of depression. Nat Med. 2001;7:541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- Muller MB, Wurst W. Getting closer to affective disorders: the role of CRH receptor systems. Trends Mol Med. 2004;10:409–415. doi: 10.1016/j.molmed.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Vale WW. The neurobiology of depression: inroads to treatment and new drug discovery. J Clin Psychiatry. 2005;66(Suppl 7):5–13. [PubMed] [Google Scholar]
- Nemeroff CB, Widerlov E, Bissette G, Walleus H, Karlsson I, Eklund K, Kilts CD, Loosen PT, Vale W. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. 1984;226:1342–1344. doi: 10.1126/science.6334362. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M. Reduced corticotropin releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry. 1988;45:577–579. doi: 10.1001/archpsyc.1988.01800300075009. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Nestler E, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002a;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Gould E, Manji H, Buncan M, Duman RS, Greshenfeld HK, Hen R, Koester S, Lederhendler I, Meaney M, Robbins T, Winsky L, Zalcman S. Preclinical models: status of basic research in depression. Biol Psychiatry. 2002b;52:503–528. doi: 10.1016/s0006-3223(02)01405-1. [DOI] [PubMed] [Google Scholar]
- Newton SS, Dow A, Terwilliger R, Duman R. A simplified method for combined immunohistochemistry and in-situ hybridization in fresh-frozen, cryocut mouse brain sections. Brain Res Brain Res Protoc. 2002;9:214–219. doi: 10.1016/s1385-299x(02)00148-4. [DOI] [PubMed] [Google Scholar]
- Nowak G, Partyka A, Palucha A, Szewczyk B, Wieronska JM, Dybala M, Metz M, Librowski T, Froestl W, Papp M, Pilc A. Antidepressant-like activity of CGP 36742 and CGP 51176, selective GABAB receptor antagonists, in rodents. Br J Pharmacol. 2006;149:581–590. doi: 10.1038/sj.bjp.0706845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell J, Zhang H-T. Antidepressant effects of inhibitors of cAMP phosphodiesterase (PDE4) Trends Pharm Sci. 2004;25:158–163. doi: 10.1016/j.tips.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA. 1998;95:13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz J, Fitzgerald LW, Lane S, Terwilliger R, Nestler EJ. Biochemical adaptations in the mesolimbic dopamine system in response to repeated stress. Neuropsychopharmacology. 1996;14:443–452. doi: 10.1016/0893-133X(95)00152-4. [DOI] [PubMed] [Google Scholar]
- Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007;17:42–49. doi: 10.1038/sj.cr.7310125. [DOI] [PubMed] [Google Scholar]
- Pan G, Li J, Zhou Y, Zheng H, Pei D. A negative feedback loop of transcription factors that controls stem cell pluripotency and self-renewal. FASEB J. 2006;20:1730–1732. doi: 10.1096/fj.05-5543fje. [DOI] [PubMed] [Google Scholar]
- Pandya R, Patten S. Depression in multiple sclerosis associated with interferon beta-1a (Rebif) Can J Psychiatry. 2002;47:686. doi: 10.1177/070674370204700720. [DOI] [PubMed] [Google Scholar]
- Patten SB, Metz LM. Interferon beta-1 a and depression in relapsing-remitting multiple sclerosis: an analysis of depression data from the PRISMS clinical trial. Mult Scler. 2001;7:243–248. doi: 10.1177/135245850100700406. [DOI] [PubMed] [Google Scholar]
- Pilc A, Nowak G. GABAergic hypotheses of anxiety and depression: focus on GABA-B receptors. Drugs Today (Barc) 2005;41:755–766. doi: 10.1358/dot.2005.41.11.904728. [DOI] [PubMed] [Google Scholar]
- Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA., Jr Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci. 2001;21:7397–7403. doi: 10.1523/JNEUROSCI.21-18-07397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, Overholser JC, Roth BL, Stockmeier CA. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–1098. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- Risbrough VB, Hauger RL, Roberts AL, Vale WW, Geyer MA. Corticotropin-releasing factor receptors CRF1 and CRF2 exert both additive and opposing influences on defensive startle behavior. J Neurosci. 2004;24:6545–6552. doi: 10.1523/JNEUROSCI.5760-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi MA, Mash DC, DeToledo-Morrell L. Spatial memory in aged rats is related to PKCgamma-dependent G-protein coupling of the M1 receptor. Neurobiol Aging. 2005;26:53–68. doi: 10.1016/j.neurobiolaging.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, Petroff OA, Berman RM, Charney DS, Krystal JH. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 1999;56:1043–1047. doi: 10.1001/archpsyc.56.11.1043. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, Krystal JH, Mason GF. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- Sasai N, Mizuseki K, Sasai Y. Requirement of FoxD3-class signaling for neural crest determination in Xenopus. Development. 2001;128:2525–2536. doi: 10.1242/dev.128.13.2525. [DOI] [PubMed] [Google Scholar]
- Sheline Y, Wany P, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA. 1996;93:3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline Y, Gado MH, Price JL. Amygdala core nuclei volumes are decreased in recurrent major depression. NeuroReport. 1998;9:2023–2028. doi: 10.1097/00001756-199806220-00021. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Sanghavi M, Mintun MA, Gado MH. Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci. 1999;19:5034–5043. doi: 10.1523/JNEUROSCI.19-12-05034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simen BB, Duman CH, Simen AA, Duman RS. TNFalpha signaling in depression and anxiety: behavioral consequences of individual receptor targeting. Biol Psychiatry. 2006;59:775–785. doi: 10.1016/j.biopsych.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, Botstein D, Altman RB. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17:520–525. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Venihaki M, Sakihara S, Subramanian S, Dikkes P, Weninger SC, Liapakis G, Graf T, Majzoub JA. Urocortin III, a brain neuropeptide of the corticotropin-releasing hormone family: modulation by stress and attenuation of some anxiety-like behaviours. J Neuroendocrinol. 2004;16:411–422. doi: 10.1111/j.1365-2826.2004.01170.x. [DOI] [PubMed] [Google Scholar]
- Wallace TL, Stellitano KE, Neve RL, Duman RS. Effects of cyclic adenosine monophosphate response element binding protein overexpression in the basolateral amygdala on behavioral models of depression and anxiety. Biol Psychiatry. 2004;56:151–160. doi: 10.1016/j.biopsych.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Warner-Schmidt J, Duman RS. Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus. 2006;16:239–249. doi: 10.1002/hipo.20156. [DOI] [PubMed] [Google Scholar]
- Weigel D, Jurgens G, Kuttner F, Seifert E, Jackle H. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell. 1989;57:645–658. doi: 10.1016/0092-8674(89)90133-5. [DOI] [PubMed] [Google Scholar]
- Zhang HT, Huang Y, Jin SJC, Frith SA, Nuvarna N, Conti M, O'Donnell JM. Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmacology. 2002;27:587–595. doi: 10.1016/S0893-133X(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Kato H, Asanoma K, Kondo H, Arima T, Kato K, Matsuda T, Wake N. Identification of FOXC1 as a TGF-beta1 responsive gene and its involvement in negative regulation of cell growth. Genomics. 2002;80:465–472. [PubMed] [Google Scholar]