

Abstract
Logic-embedded vectors (LEVs) have been introduced as a means to overcome sequential, biological barriers that prevent particle-based drug delivery systems from reaching their targets. In this chapter, we address the challenge of fabricating and optimizing LEVs to reach non-endosomal targets. We describe the general preparation, characterization, and cellular association of porous silicon-based LEVs. A specific example of LEV fabrication from start to finish, along with optimization and troubleshooting information, is presented to serve as a template for future designs.
1. Introduction
Particle-based drug delivery systems are subject to sequential physical and biological barriers that prevent them from reaching their site of activity. Logic-embedded vectors (LEVs), whose size, shape, surface, and cargo properties can be modulated for specific applications, have been proposed as a means to overcome these barriers. This concept was first introduced by Mauro Ferrari (Ferrari, 2010) and validated by Serda et al. in 2010 (Serda et al., 2010a). By definition, LEVs require the use of multiple, discrete components to achieve multiple, specific, sequential functions. This has led to the fabrication of nested LEVs, in which small, surface-tailored components are encapsulated within a porous carrier until triggered to release. The carrier may also be programmed to have specific functions, such as a long circulation time (Godin B), intravascular margination behavior (Decuzzi and Ferrari, 2008; Decuzzi et al., 2009), and controlled release of cargo including nanoparticles, bioactive molecules, imaging agents, and their combinations (Tasciotti et al.). By appropriately functionalizing the carrier surface, particles can be programmed for cell uptake (Serda et al., 2009a; Serda et al., 2009b; Serda et al., 2010b; Serda et al., 2010a; Serda et al., 2011a) and intracellular drug release (Tanaka et al., 2010). These carriers and their cargo, however, generally remain trapped within the endosomal pathway when using conventional approaches.
In this chapter, we address the challenge of fabricating and optimizing LEVs to reach targets outside of the endosomal degradation pathway. Using porous silicon (pSi) particles as a platform technology, we describe frequently used techniques for LEV preparation, characterization, and cell association. These techniques are followed by a specific example of LEV construction using pSi particles and surface-tailored iron oxide (IO) nanoparticles to achieve differential partitioning into unique cellular destinations. The procedure is described step-by-step, along with optimization and troubleshooting information.
2. Preparation of pSi carrier particles
2.1. Fabrication
Porous silicon (pSi) is generally produced by electrochemical etching, which uses hydrofluoric acid and strong electrical currents to etch pores of defined sizes into silicon substrates (Chiappini et al., 2010; Xue et al., 2011). Particles are created by lithography of the porous silicon. Quality control to produce particles of uniform size, shape, and porosity is important from a drug delivery perspective so that particle degradation and drug release can be more easily predicted. (Bimbo et al., 2010; Chiappini et al., 2010; Serda et al., 2010b; Serda et al., 2010a). Detailed fabrication protocols may be found in the papers referenced here.
2.2. Surface preparation
Silicon particles for biological applications are generally oxidized to improve their hydrophilicity and facilitate the attachment of bioactive molecules. In our laboratory, pSi particles are oxidized using Piranha etch solution to achieve a uniformly oxidized surface and remove trace organic substances. This etch solution is composed of two parts 18 M sulfuric acid (H2SO4) and one part 30% hydrogen peroxide (H2O2). Typically, we disperse the dried particles (contained in a glass vial) in H2O2 using sonication, then add a teflon stir bar and H2SO4. The solution is reacted for two hours at 100°C using a heated stir plate, after which the particles are pelleted at 4200 rpm, washed twice with Millipore water, and then resuspended in 10% nitric acid to increase the number of surface hydroxyl groups (Guhathakurta and Subramanian, 2007; Serda et al., 2011a). After 30 minutes, the particles are pelleted, washed twice with Millipore water, once with 50% 2-propanol (IPA), then resuspended in 100% IPA. At this point, Fourier Transform Infrared Spectroscopy (FTIR) and zeta potential measurements (described in further detail below) are performed for quality control purposes. The pSi concentration is assessed using a Beckman Coulter Multisizer. Particles that will be directly loaded using the incipient wetness method (described further below) are aliquoted (108 – 109 particles/tube), pelleted, and dried overnight in a vacuum desiccator.
2.3. Surface functionalization
Oxidized particles may be functionalized with different molecules to achieve specific functions in vitro and in vivo. For pSi particles, this is generally done via silanization or esterification. We use a variety of silianes with different terminal groups, including 3-(aminopropyl)triethoxysilane (APTES, Sigma-Aldrich, St. Louis, MO) and 2-(carbomethoxy)ethyltrimethoxysilane (CETES), which feature terminal amine and carboxyl groups, respectively. To modify oxidized pSi, silanes are diluted to a concentration of 0.5 – 2.0% in 95% IPA (made freshly by adding 5% water to 100% IPA). Particles are suspended in the silane solution by sonication, and allowed to react for 0.5 – 2.0 h at 35°C, while vortexing at 1400 rpm using a Thermomixer. The silane concentration and reaction time are chosen based upon the desired amount of silanization, with higher concentrations and longer reaction times yielding a more highly polymerized layer (Moon et al., 1996). An advantage of silane-based surface modification is that it can be performed before or after loading of the particle pores, and is useful for nanoparticle (NP) entrapment and as a barrier to NP release (Serda et al., 2010b; Serda et al., 2010a). After silanization, particles are centrifuged at 4200 rpm, washed three times with 100% IPA, pelleted, and dried overnight in a vacuum desiccator to induce silane cross-linking.
Non-polymerizing functional groups may be used to minimize blockage of the nanoporous substrate. Molecules such as 3-(aminopropyl)(ethoxy)dimethylsilane and ethanolamine yield a monolayer of surface modification. This approach is useful when one would like to load negatively charged cargo into the pores after surface modification. Both molecules feature a terminal amine group for further functionalization, but unlike APTES, cause little to no clogging of the nanoporous substrate (Moon et al., 1996).
Bioactive ligands, such as antibodies and peptides, may be covalently linked to modified pSi surfaces using standard N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) chemistry. Briefly, carboxyl groups on the ligand surface are activated by the addition of a 10mM MES solution (pH 6.0) containing freshly dissolved 0.4mg/ml EDC and 1.1mg/ml N-hydroxysulfosuccinimide ester (Sulfo-NHS). Ligands are reacted for 15 min and then purified using a Zeba desalting column. To raise the ligand pH to 7.2 for the next chemical reaction, the Zeba column is primed with 10 mM phosphate buffer (PB, pH 7.2) prior to use. The activated ligand solution is added to APTES-modified pSi, sonicated, and allowed to react for up to 2 hours at room temperature with intermittent vortexing. After functionalization, the particles are washed twice with 10 mM PB and used immediately or lyophilized after pelleting for long-term storage. Detailed protocols for this and other common cross-linking reactions may be found in (Hermanson, 2008).
2.4. Surface characterization
Fourier Transform Infrared Spectroscopy (FTIR) is the primary method used to verify surface functionalization (Ferraro and Basile, 1985; Teo et al., 1997). FTIR measures infrared absorption resulting from vibration of the bonds between atoms. The technique uses an interferometer to isolate infrared wavelengths based on destructive interference, and then a Fourier transform is applied at each wavelength to produce an absorbance spectrum. Individual spectral peaks correlate with the presence of specific bonds and can be used to identify material composition. Peak height can be used to quantify the amount of material present. Chemical modification of a particle surface (via either covalent conjugation or electrostatic interactions) is generally verified by looking for a reduction or shift in one or more absorbance peaks compared to an unmodified control. This technique uses very little sample (~106 – 107 particles per test) and does not require any chemical dissociation.
Dynamic light scattering (DLS) measurements are used to determine particle size in solution (Goldburg, 1999). This technique relies on photon correlation spectroscopy and is suitable for spherical particles 10 nm – 3 μm in size. When a laser is passed through a dilute solution of particles, Brownian motion of the particles causes a Doppler shift in the laser scattering. This wavelength shift directly correlates with particle size. DLS can distinguish between particles of different sizes within a single sample, and is therefore useful for assessing particle dispersity and aggregation. Since DLS is easy and requires little sample (~106 particles per test), we use it for both particle optimization and quality control.
Zeta potential measurements can be used to collect information about the surface charge of particles in solution (Serda et al., 2011b). Like DLS, zeta potential measurements rely on light scattering detection of particle movement and electron field analysis. Devices such as the Brookhaven Instrument Corporation (Holtsville, NY) Zeta-PALS instrument have both functionalities and thus allow a single sample to be used for both assays. Zeta potential is measured by applying an electric field through a dilute particle solution and measuring the resultant particle migration velocity. We typically suspend a small amount of particles (<106 particles per test) in 10 mM phosphate buffer (PB) for analysis. This solution mimics physiological pH while allowing electrical conductance across the solution. PB can be easily titrated to a variety of pHs by mixing 10 mM sodium phosphate monobasic buffer (pH 4.8) and 10 mM sodium phosphate dibasic buffer (pH 9.3). It should be noted that zeta potential is an average bulk measurement, and thus cannot distinguish between particles of different surface charges within a single sample.
A variety of colorimetric assays may be used to measure for the presence of specific functional groups on the pSi surface. This is particularly useful when optimizing or troubleshooting multi-step conjugation reactions, such as the covalent attachment of ligands via a silane intermediary. Our group favors the use of APTES for ligand addition, thus we commonly use a nitrobenzaldehyde assay to quantify the presence of amine groups (Moon et al., 1996; Moon et al., 1997; Xiao et al., 1998). Briefly, particles are washed once in 99.2% ethanol mixed with 0.8% glacial acetic acid (hereafter called the coupling solution), pelleted at 4500g, and then resuspended in coupling solution containing 0.7 mg/ml 4-nitrobenzaldehyde (4-NBZ, Sigma Aldrich). The 4-NBZ is allowed to react with the terminal amine groups for 3 hours at 50°C while mixing at 900 rpm. Particles are then pelleted, washed 3 times in coupling solution, and then resuspended in a hydrolysis solution containing 0.133% glacial acetic acid, 49.933% ethanol and 49.933% Millipore water. This step cleaves the bound 4-NBZ, which absorbs light at 268 nm. After pelleting the hydrolyzed particles, the supernatant is transferred to a UV-transparent 96-well plate, measured on a spectrophotometer, and compared to the titration curve of known 4-NBZ concentrations. Similar protocols are available in the literature for measuring other reactive terminal groups commonly used for protein conjugation including carboxyl groups (Everaerts et al., 2007). Generally, it is important to select an assay that is highly sensitive and quantitative, allowing the same assay to be used for both optimization and quality control purposes.
3. LEV Assembly
3.1. Loading via the incipient wetness method
To load nanoparticles (NPs) into pSi carriers, in our laboratories we use a technique termed incipient wetness loading (Serda et al., 2010b). Here, dried pSi particles are treated with a high concentration (1.0 – 5.0 mg/ml) of NPs contained within an aqueous or organic solution. Bath sonication is initially used to disperse the mixture and repeated several times over the course of 20–30 minutes. To separate the pSi from the unloaded NPs, it is important to centrifuge the mixture at a speed and time that selectively pellets only the pSi. The supernatant is removed and the pSi particles are washed 2 or 3 times in the buffer of choice.
To increase carrying capacity of LEV, charge-based loading methods may be employed alongside incipient wetness loading (Tasciotti et al., 2008; Serda et al.; Serda et al., 2010a). Particle charge plays an important role in the retention of NPs within porous structures. Oxidized pSi is highly negatively charged (pKa ~2.2) as a result of the hydroxyl groups present on the particle surface. For efficient loading, one should choose a cargo of sufficiently different pKa and loading buffer with a pH between that of the pSi and NPs. For samples of unknown pKa, this value can be determined experimentally by measuring the particle zeta potential across a range of pH. To load aminated iron oxide NPs (pKa ~8), for example, a borate buffer at pH 5.0 is used. When the pKa of the NPs is too close to that of the pSi, such as for carboxylated iron oxide NPs (pKa ~4), NP retention can be improved by modifying the NPs with positively charged molecules like chitosan (pKa ~12, described in further detail below).
3.2. Loading characterization
Scanning electron microscopy (SEM) can be used to characterize LEVs after loading. This technique uses a narrow electron beam to collect high-resolution, high-magnification images of backscattered electrons emitted from sample surfaces. Due to the narrowness of the excitation beam, the resultant images have a high depth-of-field that can be used to understand particle topography. Since an SEM of sufficient resolution can distinguish individual NPs within the pores of pSi particles, we use SEM on a regular basis to confirm pSi loading, evaluate the nature of NP loading, and qualitatively determine loading efficiency. Sample preparation is simple and requires only a small amount of LEVs (~106) to be dried on a SEM stub, and generally uses much less sample than destructive, time-consuming, chemical-based assays.
4. LEV uptake and trafficking by cells
4.1. Transmission electron microscopy (TEM)
The gold standard for investigating the intracellular uptake and localization of nanoparticles, including LEVs, is transmission electron microscopy (TEM). TEM uses an electron beam to shine through ultra-thin sample preparations and produce an image with nanometer or sub-nanometer resolution. Cellular structures including lipid membranes, organelles, and chromatin can be imaged in cross-section when samples are sliced appropriately thin (<100 nm). This technique is especially useful for visualizing individual LEV components that are beyond the resolution limit of optical microscopes. An additional advantage of this approach is that LEV components are not required to have intrinsic optical properties. We use this technique to analyze the subcellular localization of LEV formulations at different time-points after internalization by cells. The association of LEV components with specific organelles can give insights into the mechanisms that regulate LEV uptake and trafficking, as well as their influence on cell structure and function.
5. Case study: Fabrication and characterization of chitosan-coated iron oxide LEVs
LEVs capable of releasing nanoscale cargo from the endosome may be fabricated using chitosan as a permeation enhancer. Chitosan is a biocompatible, biodegradable, cationic polysaccharide comprised of repeating units of glucosamine and N-acetyl glucosamine. The cationic properties of this polymer make it uniquely suited for enhancing nanoparticle transport through tissues (Ghosn et al., 2009) and cells (Serda et al., 2010a). Below we describe how chitosan can be electrostatically wrapped around anionic nanoparticles and then loaded into pSi particles to protect its bioactivity. For each step, optimization and troubleshooting information is provided.
5.1. Step 1: Chitosan preparation
Chitosan may be purchased in a variety of molecular weights and with different amounts of deacetylation. Varying these parameters affects the polymer packing density, charge, and toxicity. Chitosan with a molecular weight of 130 kDa and 86% deacetylation (Protasan, UP CL 113; Novamatrix, Norway) works well with silicon LEVs, being sufficiently small and positively charged while demonstrating minimal toxicity. Since chitosan is not soluble at physiological pH, it must be dissolved in an acidic solution. We dissolve chitosan at 30 mg/ml in 50 mM borate buffer (pH 5.0, Sigma-Aldrich).
5.2. Step 2: Chitosan coating of anionic iron oxide
Chitosan readily coats nanoparticles with a negative surface charge, such as carboxylated superparamagnetic iron oxide nanoparticles (SPIONs; Ocean NanoTech, Springdale, AZ), via electrostatic interactions. For SPIONs of 30 nm diameter, chitosan is added at a ratio of 1:0.6 (w/w) at room temperature for 1 h. The ideal ratio is dependent on SPION size: if too little chitosan is added, the SPIONs cross-link and aggregate; if too much chitosan is added, it becomes difficult to remove the excess chitosan. To find the ideal ratio for specific SPIONs, the mean zeta potential and DLS diameter should be monitored across multiple ratiometric dilutions. Note that nanoparticles should be purified (Step 3) prior to analysis to avoid interference from unbound chitosan. The reaction also requires a sufficient volume to coat uniformly the nanoparticles without cross-linking them; thus it is recommended that the stock nanoparticles be suspended at the concentration of 0.1% (w/v).
5.3. Step 3: Removal of unbound chitosan
To remove unbound chitosan, purification columns may be used. For SPIONs, we can take advantage of their magnetic properties to hold the nanoparticles in place while washing away free chitosan. Unbound chitosan is removed using a MACS magnetic purification column (Miltenyi Biotec, Inc., Auburn, CA) and washing twice with 1.0 ml of borate buffer (pH 5.0) while the sample is held next to the magnet. The sample is then removed from the magnet and released into a fresh tube using an additional 0.2 ml borate buffer. Alternatively, a SuperMag Multitube Separator (Ocean NanoTechwith two 0.5 ml washes may be used.
5.4. Step 4. Confirming the presence of chitosan coating
Several assays are useful for confirming and optimizing the chitosan coating procedure. Chitosan-coated SPIONs have several unique spectral peaks that can be detected by FTIR (Fig. 1). Twenty microliter samples (~5 μg) are dried on a glass slide in a vacuum desiccator, mixed with potassium bromide (KBr) (Sigma-Aldrich), and pressed into pellets for analysis on a Nicolet FTIR Spectrophotometer (Thermo Scientific, Pittsburgh, PA). The infrared peaks contributed by the chitosan become more distinctive as the chitosan concentration is increased; therefore, this approach may be used to quantify chitosan binding.
Figure 1.

FTIR spectra of chitosan, iron oxide (IO), and IO-chitosan before and after filtering. Successful chitosan coating is characterized by the presence of distinctive peaks.
The number of chitosan molecules per SPION can be quantified directly using a colorimetric assay adapted from (Genta et al.). Aliquots of purified samples (20 μl, ~5 μg) are raised to 150 μl using a glycine–HCl buffer (980 mM glycine, 80 mM NaCl, and 20 mM HCl) and then diluted 1:1 using freshly prepared 0.3% (w/v) Congo red dye in water. Chitosan-coated SPIONs have a distinctive absorbance peak centered at 520 nm, whereas uncoated SPIONs peak at ~560 nm. The height of the 520 nm peak can be measured and compared to chitosan standards (spiked with SPIONs) to quantify the chitosan coating.
DLS and zeta potential analysis can be used to measure the mean particle diameter and surface charge, respectively. An acidic buffer, such as borate buffer, pH 5.0, should be used rather than physiological buffers to avoid chitosan precipitation. It is important that the samples appear monodisperse (i.e. only showing one peak when using DLS) and have a negative surface charge prior to loading.
5.5. Step 5: Loading of SPIONs
Oxidized pSi particles (5 × 106) are dried overnight in a vacuum desiccator. SPIONs are added to oxidized pSi at high concentration (0.5–1.0 mg/ml) and loaded via the incipient wetness method for 30 min at room temperature. Samples are centrifuged at 1000g to pellet selectively the loaded pSi, washed twice in borate buffer, and suspended in fresh borate buffer (for storage) or serum-free cell culture media (for immediate use). One advantage of this loading approach is that it preferentially selects for chitosan-coated particles. At pH 5.0, adequately coated SPIONs have a positive zeta potential (+26 mV), whereas uncoated SPIONs have a negative zeta potential (−27 mV); thus, only the coated particles load into oxidized silicon particles (−23 mV). It should be noted that chitosan removal (Step 3) is critical for successful loading, since free chitosan (+71 mV) will compete with coated SPIONs for entry into the pores (Fig. 2). Loading works best with freshly purified solutions, since the chitosan-coated SPIONs tend to aggregate with storage.
Figure 2.
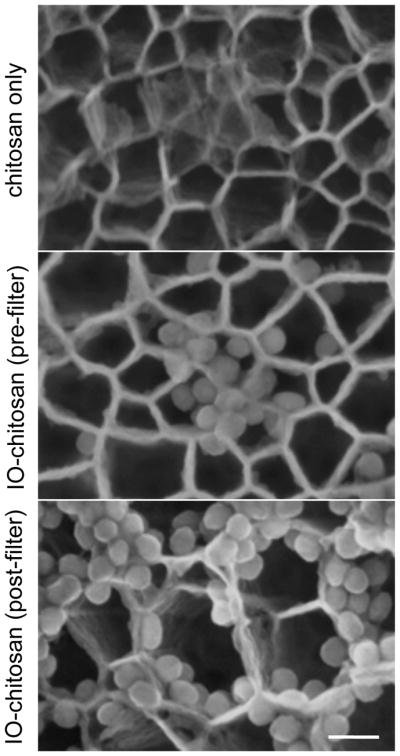
SEM images of pSi loading with IO-chitosan. The importance of removing excess chitosan is highlighted here: Free chitosan competes with IO and results in poor IO loading. Scale bar is 50nm.
5.6. Step 6: Verification of LEV assembly
A small aliquot of assembled LEVs is dried overnight on a SEM stub in a vacuum desiccator. Images of LEV surface topography are collected using an SEM, such as a Hitachi S-5500, under high vacuum at 20–30 kV with a spot size of 3.0–5.0. Fig. 3 shows examples of LEVs loaded with aminated SPIONs (+21 mV) and chitosan-coated SPIONs (+26 mV) at multiple magnifications.
Figure 3.

SEM images of pSi loaded with IO-amine and IO-chitosan via the incipient wetness method. The positively charged NPs appear to preferentially line the walls of the pores. The IO-chitosan shows some minor aggregation on the particle surface. Scale bars are 500nm, 80nm, and 40nm.
5.7. Step 7: Incubation of LEVs with live cells
Cells are seeded at a concentration of 1 × 105 cells per well in 6-well plates. Twenty four hours after seeding, 10–20 LEVs per cell (normalized to the number of seeded cells) are added directly to the cell culture media. Particles are sonicated immediately before addition to ensure that they are monodisperse. Cells and LEVs are co-incubated at 37 °C for 2–24 hours, after which the cells are washed twice with phosphate buffered saline (PBS) to remove non-internalized LEVs.
5.8. Step 8: TEM preparation
Cells are fixed for 1 h using freshly prepared 0.1 M cacodylate buffer (pH 7.3) containing 3% glutaraldehyde and 2% paraformaldehyde. After fixation, the samples are washed 3 times, treated with 0.1% tannic acid (in 0.1 M cacodylate buffer) for 15 minutes, washed 3 times, and post-fixed with 1% osmium tetroxide (in 0.1 M cacodylate buffer) for 30 min. All washing steps up to the osmium tetroxide step use 0.1 M cacodylate buffer; thereafter, the samples are washed 3 times in distilled water. After washing, the samples are stained en-bloc with 1% uranyl acetate for 30 min, and then dehydrated in increasing concentrations of ethanol (50%, 70%, 80%, 90%, 95%, 100%, 100%, 100%) for 5 min each. Polymer infiltration is performed by the sequential addition of 2 ml of a 1:1 mixture of LX-112 resin and ethanol for 1 h, followed by a 2:1 LX-112:ethanol mixture for 1 h, and followed by 100% LX-112 applied twice for 1 h each. The samples are then embedded in 1.0 ml of 100% LX-112 resin and allowed to polymerize for 48 h at 70 °C. After polymerization, the resin is released from the 6-well plate using liquid nitrogen. Areas of interest are cut out using a jeweler’s saw and trimmed with a razor blade. Ultrathin sections (~70 nm) are cut from trimmed blocks using a microtome, such as the Leica Ultracut (Leica, Deerfield, IL). Sections are floated onto TEM grids and counterstained using a Leica EM stainer. Samples are sequentially treated with 1% uranyl acetate for 4 min, a water wash, and 1% lead citrate for 1 min. Samples are then examined using a TEM, such as the JEM 1010 (JEOL, USA, Inc., Peabody, MA) with an accelerating voltage of 80 kV. Digital images are obtained using the AMT Imaging System (Advanced Microscopy Techniques Corp, Danvers, MA).
5.9. Step 9: Evaluation of cell-particle associations
TEM images of early LEV uptake provide insights into the mechanisms of particle uptake. Fig. 4 shows cells 2 h after co-incubation with IO-chitosan or IO-chitosan-loaded LEVs. Ruffles in the cell membrane appear to fold-over, loosely capturing IO-chitosan and pSi via what is probably pinocytosis. Internalized LEVs appear in large vacuoles. At 2 h, the endosomal membrane is loosely associated with the LEVs, providing plenty of space for unloading of the SPIONs.
Figure 4.
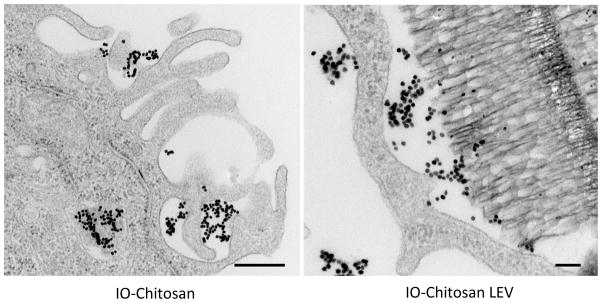
TEM images of IO-chitosan (left) and LEVs loaded with IO-chitosan (right) 2 hours after co-incubation with cells. IO nanoparticles appear as dark dots while pSi appears as porous sponge-like structure. At this time-point, internalized IO and LEVs are surrounded by a well-defined endosomal membrane. This membrane is relatively loose, allowing for the unloading of pSi cargo. Scale bar is 500nm.
The trafficking of individual LEV components may be monitored using TEM. Fig. 5 shows the cells 24 h after co-incubation with empty pSi (left), IO-chitosan (middle), or IO-chitosan loaded LEVs (right). We find that pSi particles are retained within the endo-lysosomal pathway, even when their cargo trafficks to other intracellular destinations. Freely administered IO-chitosan appears to remain trapped in the endosomes. By contrast, IO-chitosan that was previously protected within pSi particles is now seen free in the cytoplasm, in close proximity to mitochondria. It is known that protonation of the chitosan backbone during endosome maturation leads to localized membrane destabilization (Bowman and Leong, 2006), which in turn may facilitate IO escape. Differential trafficking is observed for IO-amine of similar surface charge (Serda et al., 2010a). When released from the pSi after endosomal uptake, these cationic particles accumulate in multi-vesicular bodies and are exported out of the cell. The differential trafficking of IO-amine and IO-chitosan LEVs suggests that: (i) endosomal escape of IO is mediated directly by the presence of chitosan, and (ii) loaded IO-chitosan is protected from exposure to physiological pH, and thereby better retains its bioactivity than freely administered IO-chitosan.
Figure 5.
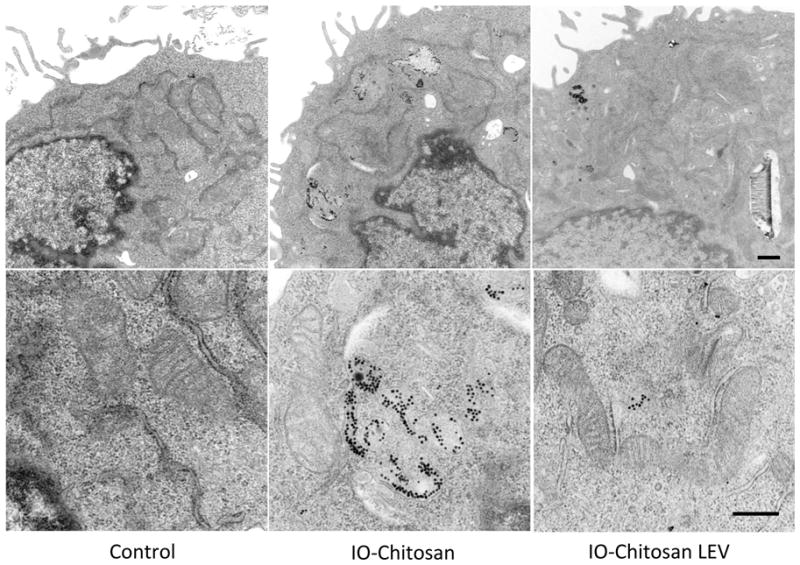
TEM images of cells treated for 24 hours. Freely administered IO-chitosan appears in multi-vesicular bodies, whereas protected IO-chitosan freely associates with mitochondria. An ultra-structural analysis reveals no associated cell toxicity. Indicators of healthy cell function include the presence of an intact cell membrane and the absence of nuclear condensation, membrane blebbing, and vacuoles. Scale bars are 500nm.
6. Conclusions
A major advantage of LEVs is their ability to deliver components to multiple intracellular sites, creating an opportunity for simultaneously targeting independent or synergistic pathways. In this chapter, we provide an example of how pSi carriers may be fabricated, loaded with bioactive particles, and differentially partitioned upon cellular uptake. Examples of characterization techniques are presented together with optimization and troubleshooting information, in order to serve as a template for the design and fabrication of new LEVs. It is exciting to speculate that future designs will be programmed to perform coordinated and synergistic modulation of intracellular functions.
Acknowledgments
We thank Hitachi for training and use of the S-5500 high-resolution scanning electron microscope. This research was supported by the Department of Defense grants DODW81XWH-07–1-0596 and DODW81XWH-09–1-0212; NASA NNJ06HE06A; NIH RO1CA128797, U54CA143837, MH58920, and RC2GM092599; MDACC Institutional Core Grant CA016672; and The State of Texas, Emerging Technology Fund.
Contributor Information
Anne L. van de Ven, Email: avandeven@tmhs.org, Department of Nanomedicine, Methodist Hospital Research Institute, Houston, TX 77030
Aaron Mack, Email: amack@tmhs.org, Department of Nanomedicine, Methodist Hospital Research Institute, Houston, TX 77030.
Kenneth Dunner, Jr., Email: jdunner@mdanderson.org, High Resolution Microscopy Imaging Facility, MD Anderson Cancer Center, Houston, TX 77030
Mauro Ferrari, Email: mferrari@tmhs.org, President and CEO, Methodist Hospital Research Institute, Houston, TX 77030
Rita Serda, Email: reserda@tmhs.org, Department of Nanomedicine, Methodist Hospital Research Institute, Houston, TX 77030
References
- 1.Ferrari M. Frontiers in cancer nanomedicine: directing mass transport through biological barriers. Trends Biotechnol. 2010;28:181–188. doi: 10.1016/j.tibtech.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serda RE, Mack A, van de Ven AL, Ferrati S, Dunner K, Jr, Godin B, Chiappini C, Landry M, Brousseau L, Liu X, Bean AJ, Ferrari M. Logic-embedded vectors for intracellular partitioning, endosomal escape, and exocytosis of nanoparticles. Small. 2010a;6:2691–2700. doi: 10.1002/smll.201000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Godin B, GJ, Serda RE, Ferrati S, Liu X. Multistage mesoporous silicon-based nanocarriers: biocompatibility and controlled degradation in physiological fluids. 35th Annual Meeting & Exposition of the Controlled Release Society; 2008. p. 575. [PMC free article] [PubMed] [Google Scholar]
- 4.Decuzzi P, Ferrari M. Design maps for nanoparticles targeting the diseased microvasculature. Biomaterials. 2008;29:377–384. doi: 10.1016/j.biomaterials.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 5.Decuzzi P, Pasqualini R, Arap W, Ferrari M. Intravascular delivery of particulate systems: does geometry really matter? Pharm Res. 2009;26:235–243. doi: 10.1007/s11095-008-9697-x. [DOI] [PubMed] [Google Scholar]
- 6.Tasciotti E, Liu X, Bhavane R, Plant K, Leonard AD, Price BK, Cheng MM, Decuzzi P, Tour JM, Robertson F, Ferrari M. Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nat Nanotechnol. 2008;3:151–157. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- 7.Serda RE, Gu J, Bhavane RC, Liu X, Chiappini C, Decuzzi P, Ferrari M. The association of silicon microparticles with endothelial cells in drug delivery to the vasculature. Biomaterials. 2009a;30:2440–2448. doi: 10.1016/j.biomaterials.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 8.Serda RE, Gu J, Burks JK, Ferrari K, Ferrari C, Ferrari M. Quantitative mechanics of endothelial phagocytosis of silicon microparticles. Cytometry A. 2009b;75:752–760. doi: 10.1002/cyto.a.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serda RE, Mack A, Pulikkathara M, Zaske AM, Chiappini C, Fakhoury J, Webb D, Godin B, Conyers JL, Liu XW, Bankson JA, Ferrari M. Cellular Association and Assembly of a Multistage Delivery System. Small. 2010b doi: 10.1002/smll.201000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serda RE, Blanco E, Mack A, Stafford SJ, Amra S, Li Q, van de Ven A, Tanaka T, Torchilin VP, Wiktorowicz JE, Ferrari M. Proteomic analysis of serum opsonins impacting biodistribution and cellular association of porous silicon microparticles. Molecular imaging. 2011a;10:43–55. [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka T, Mangala LS, Vivas-Mejia PE, Nieves-Alicea R, Mann AP, Mora E, Han HD, Shahzad MM, Liu X, Bhavane R, Gu J, Fakhoury JR, Chiappini C, Lu C, Matsuo K, Godin B, Stone RL, Nick AM, Lopez-Berestein G, Sood AK, Ferrari M. Sustained small interfering RNA delivery by mesoporous silicon particles. Cancer Res. 2010;70:3687–3696. doi: 10.1158/0008-5472.CAN-09-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiappini C, Tasciotti E, Fakhoury JR, Fine D, Pullan L, Wang YC, Fu L, Liu X, Ferrari M. Tailored porous silicon microparticles: fabrication and properties. Chemphyschem. 2010;11:1029–1035. doi: 10.1002/cphc.200900914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xue M, Zhong X, Shaposhnik Z, Qu Y, Tamanoi F, Duan X, Zink JI. pH-Operated mechanized porous silicon nanoparticles. Journal of the American Chemical Society. 2011;133:8798–8801. doi: 10.1021/ja201252e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bimbo LM, Sarparanta M, Santos HA, Airaksinen AJ, Makila E, Laaksonen T, Peltonen L, Lehto VP, Hirvonen J, Salonen J. Biocompatibility of Thermally Hydrocarbonized Porous Silicon Nanoparticles and their Biodistribution in Rats. ACS Nano. 2010;4:3023–3032. doi: 10.1021/nn901657w. [DOI] [PubMed] [Google Scholar]
- 15.Guhathakurta S, Subramanian A. Effect of Hydrofluoric Acid in Oxidizing Acid Mixtures on the Hydroxylation of Silicon Surface. Journal of The Electrochemical Society. 2007;154:136–146. [Google Scholar]
- 16.Moon JH, Shin JW, Kim SY, Park JW. Formation of Uniform Aminosilane Thin Layers: An Imine Formation To Measure Relative Surface Density of the Amine Group. Langmuir. 1996;12:4621–4624. [Google Scholar]
- 17.Hermanson GT. Bioconjugate Techniques. Academic Press; San Diego, CA: 2008. [Google Scholar]
- 18.Ferraro JR, Basile LJ. Fourier transform infrared spectroscopy. 1985. [Google Scholar]
- 19.Teo LS, Chen CY, Kuo JF. Fourier Transform Infrared Spectroscopy Study on Effects of Temperature on Hydrogen Bonding in Amine-Containing Polyurethanes and Poly(urethane‚àíurea)s. Macromolecules. 1997;30:1793–1799. [Google Scholar]
- 20.Goldburg WI. Dynamic light scattering. AAPT; 1999. [Google Scholar]
- 21.Serda RE, Godin B, Blanco E, Chiappini C, Ferrari M. Multi-stage delivery nano-particle systems for therapeutic applications. Biochimica et biophysica acta. 2011b;1810:317–329. doi: 10.1016/j.bbagen.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moon JH, Kim JH, Kim K-j, Kang TH, Kim B, Kim CH, Hahn JH, Park JW. Absolute Surface Density of the Amine Group of the Aminosilylated Thin Layers: Ultraviolet Visible Spectroscopy, Second Harmonic Generation, and Synchrotron-Radiation Photoelectron Spectroscopy Study. Langmuir. 1997;13:4305–4310. [Google Scholar]
- 23.Xiao SJ, Textor M, Spencer ND, Sigrist H. Covalent Attachment of Cell-Adhesive, (Arg-Gly-Asp)-Containing Peptides to Titanium Surfaces. Langmuir. 1998;14:5507–5516. [Google Scholar]
- 24.Everaerts F, Torrianni M, Hendriks M, Feijen J. Quantification of carboxyl groups in carbodiimide cross-linked collagen sponges. Journal of biomedical materials research Part A. 2007;83:1176–1183. doi: 10.1002/jbm.a.31398. [DOI] [PubMed] [Google Scholar]
- 25.Ghosn B, van de Ven A, Tam J, Sokolov K, Gillenwater A, Richards-Kortum R, Roy K. Reversible mucosal permeation enhancement with imidazole-modified chitosan for optical contrast agent delivery. Molecular Imaging and Biology. 2009 doi: 10.1117/1.3309739. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genta I, Perugini P, Pavanetto F, Modena T, Conti B, Muzzarelli RA. Microparticulate drug delivery systems. Exs. 1999;87:305–313. doi: 10.1007/978-3-0348-8757-1_22. [DOI] [PubMed] [Google Scholar]
- 27.Bowman K, Leong KW. Chitosan nanoparticles for oral drug and gene delivery. Int J Nanomedicine. 2006;1:117–128. doi: 10.2147/nano.2006.1.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]