

Abstract
The conclusive evidence supporting a role for NK cells in defense against viruses has been obtained under conditions of NK cell deficiencies prior to infections. NK cell proliferation can be induced during infections, but the advantages of resulting expansion have been unclear because NK cell basal frequency is already high. However, NK cell decreases are also observed during certain conditions of viral infection. Given the range of potent antiviral and immunoregulatory functions of NK cells, such “disappearance” dramatically changes the resources available to the host. New studies demonstrate that proliferation dependent on activating receptors for virus-induced ligands is key for NK cell maintenance, and allows their continued availability for control of adaptive immune responses and immunopathology. This pathway for sustaining NK cells may represent a system used generally to select subsets for rescue during homeostatic purging. In the case of NK cells, though, nonselection limits continued access to the many beneficial functions of NK cells. The observations resolve the long-standing conundrum of reported NK cell increases and decreases during viral infections. Moreover, they demonstrate a previously unappreciated role for activating receptors, i.e. to keep NK cells here today and also tomorrow.
Keywords: Activating receptors, NK cells, Proliferation, Viral infections
Introduction
The demonstration in the 1970s, that viral infections, and the type 1 IFN molecules produced during viral infections, induce NK cell-mediated killing, stimulated a frenzy of activity aimed at evaluating the role of these cells in defense. Correlations between low NK cell functions after different viral infections in humans and increased susceptibility to certain viral infections in mice with genetic deficiencies in cytotoxic function, provided support for the hypothesis that NK cells, by killing virus-infected cells, are important innate mediators of resistance [1, 2]. The conclusive evidence defining a protective role for classic, non-T, NK cells was provided by documenting increased susceptibility when NK cell numbers are lacking prior to infection. Sensitivity to herpesgroup viruses, particularly murine CMV (MCMV) in mice [1, 2] and human CMV (HCMV) in humans, is increased when NK cells are depleted or NK cell deficiencies are present before infection [1, 3]. It is clear, however, that not all viral infections are directly regulated by NK cells [1].
Since the original observations, there have been dramatic advances in several important related areas including characterization of: (i) the natural killer receptor complexes carrying genes for multiple activating and inhibiting receptors, as well as the roles played by these receptors in regulating NK cell responses [4, 5]; (ii) NK cell subsets based on the expression of activating and inhibiting receptors [6, 7]; (iii) the wide range of direct antimicrobial and immunoregulatory functions mediated by NK cells [1]; and (iv) innate cytokine regulation of NK cell responses [1, 2, 8-11]. In addition, a variety of ligands for NK activating receptors are being identified and being shown to be induced during viral infections. The picture emerging in vivo is that during a wide range of infections, innate cytokines quickly induce proliferation of many NK cells for short period of time, and that at later times during certain viral infections, there can also be increases in the numbers and proportions of NK cell subsets expressing particular activating receptors. The benefits of NK cell proliferation have, however, remained elusive. NK cells are at relatively high frequencies in the blood, spleen, and certain tissues and in contrast to the extremely low frequency of antigen-specific T cells prior to first infections (<104), high proportions (20–100%) of NK cells basally express particular activating receptors and/or combinations of activating receptors. Thus, a requirement to increase their numbers for early defense is unlikely to explain the advantage of inducing NK cell proliferation during an infection. Moreover, paradoxical NK cell decreases have been reported under different conditions of infections such as infection with different viruses and/or infections of different individuals with the same virus. Hence, NK cells appear either to increase or to decrease in numbers during viral infections. The mechanisms and the biological consequences of these contrasting responses are poorly understood. If NK cells are “disappearing”, it must be remembered that they are no longer available to contribute their potential antiviral or immunoregulatory functions to aid an infected host.
The pathways promoting NK cell responses, as well as the key contributions made by individual responses, are being defined during viral infections. Individual innate cytokines stimulate subset NK cell responses without direct NK cell recognition of target cells [1, 8-11]. Cytotoxic function, however, ultimately requires stimulation of activating receptors on NK cells by ligands on target cell surfaces, and these activating receptors trigger additional NK cell responses [5, 12]. Given that a common pathway of stimulation is used as a result of activating receptor engagement, it has been difficult to separate the biological advantages of killing as compared with the other NK cell responses. New approaches have proven that proliferation driven through an activating receptor for a viral ligand is key in sustaining NK cells, permitting the cells to carry out their functions under conditions of prolonged viral infection, and that this is beneficial to the host even in the absence of NK cell cytotoxicity [13]. The results provide novel insights into the advantages of NK cell proliferation to an infected host and help explain a wide range of observations made regarding the presence or absence of NK cells during different conditions of viral infections. The data and the implications are presented in this mini review.
NK receptors
NK cells enhance resistance to some but not all viral infections [1]. The factors contributing to the NK cell ability or failure to mediate protection are incompletely understood. Deficiencies in cytotoxic function can increase susceptibility to infection, and delivery of this cytotoxic function is dependent on activating receptors [2]. CD16, also known as FcγRIII (the receptor recognizing the Fc portion of antibody molecules bound to the surface of infected target cells), is the first of the activating receptors to have been identified on NK cells. Engagement of these molecules stimulates NK cells to mediate antibody-dependent cellular cytotoxicity against virus-infected cells with the specificity directed by the antigen-binding site on the antibody.
Since characterization of the Fc receptor, an extensive list of NK receptors directly recognizing ligands expressed on target cells has been identified [2] (Table 1). NK cells have an assortment of receptors, encoded by germline-genes, and these can be functionally distinguished as either activating or inhibiting. Some of the genes for these molecules are highly polygenic, and several are polymorphic among populations, as highlighted in the case of the C-type lectin-like Ly49 receptor family in rodents and the immunoglobulin-like receptor (KIR) family in primates. Because of these genetic differences, there are variations between strains and individuals in regard to the type and number of genes inherited [44, 45]. Moreover, expression of these receptors is stochastically regulated, such that there are random combinations of receptors on the surface of a given NK cell. The activating receptors signal through the ITAM present in the cytoplasmic tails of associated adaptor molecules such as the Fc receptor g or DAP12. Tyrosine phosphorylation of ITAM initiates signaling cascades that induce increased NK cytotoxicity, cytokine production, and proliferation [46, 47]. Inhibitory receptors signal through the ITIM in their cytoplasmic tails. Upon ligand binding, protein tyrosine phosphatases are recruited to the ITIM and mediate inhibition of NK cell effector functions by dephosphorylating the activated signaling molecules in the vicinity. For virtually all of the activating receptors, basal expression is on a high proportion of NK cells. The balance between signaling through activating and inhibitory receptors modifies NK cell responses.
Table 1.
NK cells in viral infection: evidence for activating receptors in defense and proliferation
| Species | Virus family | Virus | Phasea) | Receptor involvement in antiviral function and NK proliferation | Ligand for receptor | Evidence for NK cell in defenseb) | References |
|---|---|---|---|---|---|---|---|
| Increases in NK cell numbers or subset frequencies | |||||||
| Human | Retroviridae | HIV | Acute | KIR3DS1 | HLA-B (Bw4-80I)c) | +++ | [14, 15] |
| Human | Flaviviridae | HCV | Chronic | NKp30, NKp46 | ++ | [16] | |
| Human | Herpesviridae | HCMV | NDd) | CD94/NKG2C | ++ | [17] | |
| Human | Herpesviridae | EBV | Acute | ++ | [18] | ||
| Mouse (Ly49H+) | Herpesviridae | MCMV | Acute | Ly49H | m157 | ++++ | [12, 13, 19-22] |
| Decreases in NK cell numbers or subset frequencies | |||||||
| Human | Retroviridae | HIV | Chronic | ++ | [23-26] | ||
| Human | Flaviviridae | HCV | Chronic | ++ | [25, 27, 28] | ||
| Human | Herpesviridae | VZV | Acute | ++ | [29] | ||
| Monkey | Rhabdoviridae | Ebola | Acute | ++ | [30] | ||
| Mouse (Ly49H−) | Herpesviridae | MCMV | Acute | ++ | [13] | ||
| Unknown | |||||||
| Human | Orthomyxoviridae | Influenza | ND | NKp46 | HA | + | [31] |
| Human | Herpesviridae | HCMV | Acute | NKG2D | Stress molecules | + | [32] |
| Human | Rhabdoviridae | Ebola | ND | NKp30 | ? | + | [33] |
| Human | Herpesviridae | HSV | ND | NCRe) | ? | + | [34] |
| Human | Herpesviridae | EBV | ND | KIR2DS1 | HLA-C | + | [35] |
| Human | Poxviridae | Vaccinia | ND | NCR | ? | + | [36] |
| Mouse | Orthomyxoviridae | Influenza | Acute | NKp46 | ? | ++++ | [37] |
| Mouse | Herpesviridae | MCMV | Acute | Ly49P | H-2Dk+m04 | ++++ | [38] |
| Mouse | Herpesviridae | MCMV | Acute | NKG2D | Stress molecules | ++++ | [39, 40] |
| Monkey | Retroviridae | SIV | Acute | Minimal | [41] | ||
| Mouse | Arenaviridae | LCMV | Acute and chronic | − | [1, 42] | ||
| Mouse | Herpesviridae | MHV-68 | Acute | − | [43] | ||
Acute or chronic.
Results in publications summarized as follows: ++++: in vivo and in vitro demonstration; +++: in vivo correlation and in vitro data; ++: in vivo correlation; +: in vitro demonstration only; −: evidence for no role.
Putative ligand.
Not defined.
NKp30, NKp40, and NKp46.
NK activating receptors and ligand pairs during viral infections
Much progress has been made in identifying combinations of activating receptors and ligands for their stimulation, and the conditions for having both during viral infections. The different activating receptors appear to be in place to respond to different ligands induced on target cells through different types of pathways. A well-studied example is the natural killer group 2, member D (NKG2D) activating receptor that recognizes molecules on stressed cells [48]. The gene for this receptor is maintained across different species and individuals. Its ligands can be elicited through a number of pathways under stress conditions and by certain innate cytokines produced during viral infections. The ligands include RAE1, MULT1, and H60 in mice and the ULBP and MIC molecules in humans. Given that the activating NKG2D receptor is broadly expressed, has limited polymorphisms, and recognizes host gene products that are induced in infected cells, this receptor and its stress ligands are likely to have applied powerful unidirectional evolutionary pressure causing viruses to incorporate detection–avoidance mechanisms. In fact, the importance of the NKG2D interactions with stress ligands is supported by the observations that human and mouse CMV contain genes that interfere with the expression of the ligands on infected cell surfaces [48]. Furthermore, downregulation of NKG2D ligand expression is observed following HSV-1 infection in human cells [49].
Other well-characterized NK-activating receptor/ligand pairs studied in viral systems are the products of highly polygenic and polymorphic genes in both the host and the virus. These conditions allow for “holes” in the genetic repertoires for certain activating receptors, providing viruses with opportunities to pass through some members of a diverse population while a subset of infected host populations is equipped to mount an effective defense. The balance is likely to have provided evolutionary pressure on both sides. The polygenic/polymorphic combinations were first suggested by epidemiological genetic studies that demonstrated strong associations between certain NK receptors (and their HLA class I ligands) and disease outcomes in several virus infections such as HCV, HCMV, and HIV [45]. One example of an identified activating receptor/MHC class I pair is the epistatic association between KIR3DS1 and HLA-Bw4-80I, the putative HLA class I ligand for the receptor. Here, there is population diversity in the inheritance of the receptor gene, as well as extensive allelic diversity in HLA-B, and the KIR3DS1/HLA-Bw4-801 combination is strongly associated with slower HIV-1 disease progression [45]. This receptor/ligand pair has been recently shown to interact in mediating killing of infected cells to lead to the inhibition of HIV-replication [14]. In the mouse, a representative activating receptor/MHC class I pair with diversity in both the receptor and the ligand genes, which is important for defense, is Ly49P recognition of MCMV-infected cells expressing H2-Dk. In this case, a requirement for the MCMV-encoded m04 gene for effective defense against MCMV has been established [38]. The virus-induced molecular changes promoting interactions between activating receptors and MHC I molecules are, however, undefined.
The list of activating receptors directly recognizing viral gene products on infected cells is still small. One viral product binding to the NKp46-activating receptor is HA of influenza, and this combination has potential general importance for defense because of the expression of the receptor and sensitivity to virus strains across species [31]. The best characterized example of an activating receptor/ligand pair during a viral infection, however, is Ly49H, the receptor conferring resistance to MCMV infection in certain mouse strains, and m157, its viral ligand [19, 20]. The evidence supporting the importance of Ly49H recognition of m157 in defense against infection is definitive and includes studies evaluating the effects of deleting either the receptor or the ligand [21, 22]. Although many other receptor/ligand pairs are likely to be identified in different viral infections that have a documented contribution of NK cells to defense, it is important to remember that not all viral infections are controlled by NK cells. For example, there is no evidence for an NK cell role in the regulation of lymphocytic choriomeningitis virus (LCMV) replication or for any identified LCMV-induced ligands for NK cell-activating receptors [1, 42]. Although less thoroughly studied, NK cell contributions to defense against infection with the γ-herpesvirus MHV-68 [43] and the SIV are reported to be minimal [41]. There are several known mechanisms regulating NK cell functions during viral infections and more are likely to be identified, but the presence of an activating receptor/infection-induced ligand pair may be key for a strong NK cell contribution to defense.
NK cell responses
Killing, cytokine production, and proliferation
NK cells mediate killing, following engagement of their activating receptors by ligands on target cell surfaces, by triggering the lytic machinery. In the mouse, the tight correlation between increased sensitivity to MCMV infection and deficiencies in either cytotoxic factors [2] or the NK activating receptor Ly49H [22] indicates that NK cell killing of virus-infected cells as a result of stimulation through Ly49H is important in early antiviral defense; however, NK cells can also be stimulated to produce a variety of cytokines, such as IFN-γ and TNF, and chemokines to subsequently deliver antiviral effects [1]. Furthermore, NK cells have been reported to delivery immunoregulatory functions through either cytokine production or killing. For example, NK cell-produced IFN-γ has effects on downstream chemokine induction for enhancing inflammation in tissues [50] and on promoting the differentiation of Th1 over Th2 cells [51]; and NK cell cytotoxicity can facilitate downstream T-cell responses [52]. More recently, NK cells have also been shown to be producers of IL-10 [13, 16, 53-56], and this response can play a role in controlling the magnitude of adaptive immune responses by mediating effects on antigen-presenting cells and/or directly on T cells. Finally, NK cell proliferation can be induced in response to cytokines and activating receptors, but the importance of this response has been difficult to define. Thus, NK cells have the potential to respond to an infection with proliferation and activation of a number of different mechanisms, including those delivered through cell-to-cell contact or through soluble intermediaries, to provide antiviral and immunoregulatory functions.
Innate cytokine driven
Innate cytokines produced at early times after a wide range of viral infections potently induce NK cell responses. These have been best characterized during MCMV and LCMV infections of mice [8-11, 57-62]. In both systems, type 1 IFN production stimulates NK cell cytotoxicity and results in the induction of NK cell blastogenesis and proliferation [1, 10, 11, 57-62]. Remarkably, the first demonstration of NK cell proliferation in vivo was at times of type 1 IFN production during LCMV infections in a strain of mice now known to be lacking Ly49h [60]. The role for type 1 IFN in the induction of NK cell proliferation has been definitively established by examining responses during infections under conditions of blocks in cytokine functions and following administration of IFN [1, 10, 11, 61]. Type 1 IFN moleclues induce IL-15, and this cytokine contributes to NK cell division following viral infection or type 1 IFN treatment [11]. A strong IL-12 response is induced during MCMV but not LCMV infection, and NK cell IFN-γ production is elicited when IL-12 is present [9, 11, 57]. Interestingly, type 1 IFN exposure negatively regulates both IFN-γ production and IL-12-induced IFN-γ production [58, 59]. Given the pleiotropic and overlapping effects assigned to different cytokines, the ability to distinctly define dominant roles for individual cytokines in individual NK cell responses, i.e. elevated killing, cytokine production, or proliferation, is somewhat surprising. The infection-induced innate cytokines use different receptors, however, and preferentially use different signaling components [11]. Thus, the in vivo evidence indicates that despite a potential to stimulate overlapping responses, NK cell responses are differentially regulated by the composition of innate cytokines elicited during a particular viral infection. It is important to note, however, that the innate cytokine-driven increases in total NK cell yields are moderate or difficult to detect and declines are often observed over narrow periods [11]. Thus, their contribution to NK cell proliferation appears to be limited or controlled.
Activating receptor driven
Activating receptors are ultimately required for the delivery of cytotoxicity in vitro and in vivo, and stimulation through these receptors is also reported to induce proliferation in vitro and in vivo as well as NK cell IFN-γ production in vitro [2, 46]. Although their role in killing for antiviral effects is strongly supported in vivo, it has been difficult to assign an importance for activating receptor-mediated stimulation for NK cell cytokine responses during viral infections. There is more evidence that activating receptors are important in overall NK cell proliferation. The most definitive case is for the contribution made by activating receptors to NK cell proliferation following MCMV challenge in mice. If the mouse strain examined has the Ly49h gene for the Ly49H receptor, and the challenge virus expresses the Ly49H ligand m157, there is an increase in the proportion of Ly49H+ NK cells several days after infection [12]. In naïve mice, Ly49H is expressed on about 55% of NK cells, but the subset of cells expressing this molecule is increased during virus infection of immunocompetent mice to approximately 80%, and is accompanied by a modest increase in total NK cell numbers. In vivo [12] and in vitro [63] studies have shown that stimulation through Ly49H can directly elicit proliferation of NK cells, and it has recently been suggested that induction of NK cell proliferation through this activating receptor can lead to the development of “memory” NK cells [64]. New studies suggest that IL-12 production may help support activating receptor-stimulated proliferation under conditions limiting availability of an IL-15 contribution [65], and that the same receptors can function to support proliferation in the absence of type 1 IFN function [66], but the relative importance of the different pathways remains to be determined. Given that the preferential expansion of an NK cell subset is apparent after viral control under the conditions of acute infections in immunocompetent mice, the biological relevance of the proliferative response is not established by any of these particular observations.
To address the importance of the activating receptor in overall NK cell proliferation in isolation from its function in killing, mice mutated in a gene critical for the killing function, i.e. perforin 1 (Prf1−/−), have been examined [13]. In comparison with immunocompetent mice, MCMV infection of Ly49h−/− (Ly49h-deficient) or Prf1−/− (Prf1-deficient) mice results in similar early elevated MCMV levels. There are profound differences, however, in the NK cell responses of the mutated mice. Although basal NK cell proportions and numbers are similar, NK cells disappear in infected Ly49h-deficient, but expand dramatically in infected Prf1-deficient, mice, resulting in greater than tenfold higher NK cell yields in Prf1−/− mice with virtually all of the NK cells expressing Ly49H. In comparison to either the immunocompetent or the Prf1−/− conditions, NK cell proliferation is reduced at intermediate times after infection in the absence of Ly49h, and the remaining NK cells have an immature phenotype. Responses under conditions of double-deficiencies in both genes prove that the elevated responses in the absence of Prf1 are dependent on Ly49H. Thus, Ly49H is required for the proliferation and maintenance of NK cells under the conditions of prolonged infection.
Although it is not yet clear whether or not all of the NK activating receptors and virus-induced ligand pairs described in the section NK receptors promote NK cell proliferation, there is accumulating evidence for NK cell proliferation mediated by activating receptors in humans. Preferential expansion of KIR+ NK cells can be detected during acute HIV infection. Under these conditions, the KIR-expressing population is increased twofold in comparison with levels in uninfected individuals, and NK cells expressing the activating receptor KIR3DS1 are specifically expanded in the context of host HLA-Bw4-80I [15]. Another activating receptor-specific NK expansion has been observed during HCMV infection in humans with the proportion of NK cells expressing CD94/NKG2C being >20-fold higher in HCMV-seropositive as compared with HCMV-seronegative individuals [17]. NK cell expansion has also been reported following EBV [18] and HCV infections [16]. Thus, studies in mice and humans show that NK cells can be expanded during viral infections and that the presence of an activating receptor for a virus-induced ligand results in the selection of NK cells for expansion. Furthermore, the results in mice prove that the activating receptor-dependent response is also required for maintenance.
NK cell loss
The possibility of loss of NK cells during viral infections was first suggested in studies demonstrating a correlation between low NK cell function and viral infections in humans (reviewed in [1]), but this observation remained undeveloped for a number of years. With the extended characterization of NK cell surface markers, reductions in NK cell numbers and/or subsets are now being reported during viral infections of humans and primates (Table 1). Comparing the studies is difficult because of the range of infection conditions examined and because of the different phenotypes used to define the cells; however, it is clear that the percentages and total numbers of NK cells are significantly decreased in some HIV-infected individuals [23-26]. Here, the losses are identified by a marked reduction in CD56dim NK cells, the major population mediating NK cell cytotoxicity. Interestingly, this early severe loss of CD56dim is followed by an increase in CD3−CD56−CD16+ NK cells that have low perforin and elevated SHIP-1 expression [67]. Therefore, decreases in expected NK cell subsets and increases in anergic populations may contribute to the general deficit of NK cell functions during chronic HIV-1 infection. Other examples of NK cell loss include decreases of CD3−CD16+CD56+ NK cells in chronic HCV infection in humans [25, 27], of CD56+CD3− NK cells in varicella zoster virus (VZV) infections in humans [29], and of CD16+CD3− NK cells during simian Ebola infections [30]. The possibility of avoiding these population shifts by an accompanying stimulation through activating receptors remains to be tested during HCV, VZV, and Ebola infections.
As for MCMV infection in mice, however, both increases and decreases in NK cell populations have been observed in human HIV infection [15, 23-25, 68]. Here, the reported NK cell subset increases are associated with expression of a particular activating receptor, thereby providing support for the importance of stimulation through an activating receptor/ligand pair in the maintenance of NK cell numbers [15]. In most of the cases reporting NK cell decreases, the populations have been defined by a pan-NK cell marker such as CD56 or CD16 in combination with exclusion of the T-cell marker CD3, without further characterization of KIR receptor expression. As noted, there is high polygenic and polymorphic variation in KIR receptor genes. KIR3DS1, a highly polymorphic allelic form of the inhibitory receptor KIR3DL1, is the activating receptor expressed in expanded NK cells during acute HIV infection [45]. Thus, defining a critical role for KIR3DS1 in NK cell maintenance during HIV infections requires the identification of the absence of the KIR3DS1 activating receptor expression in individuals that have decreased NK cell numbers during HIV infection, as well as the evaluation of the confounding signals that might be delivered by an inhibitory receptor that could potentially be cross-recognized by the ligand of the activating receptor.
Biological consequences of NK cell maintenance or loss
The different responses to viral infections indicate that NK cells can be stimulated for proliferation through their activating receptors for maintenance if the appropriate receptors are present on the NK cells and if the viruses induce expression of the receptor ligands on the virus-infected cells. Without such a pathway, NK cell numbers decrease during sustained or overwhelming viral infections. What are the advantages and/or disadvantages of the two conditions? Proliferation might be a part of the response required to change the assortment of functions that NK cells can mediate, such as production of additional cytokines. Maintaining NK cells, however, clearly provides the host with an opportunity to use different pathways of stimulation to preserve these populations and their wide range of antiviral and immunoregulatory functions (Fig. 1). Interestingly, different regulatory mechanisms are in place to control the different pathways of NK cell activation, i.e. cytokine as compared with activating receptor stimulation, and these are delivered at different locations within the cell [2]. For example, feedback inhibition of cytokine-activated intracellular JAK-STAT signaling is shaped by STAT levels and protein/enzymatic activities in the cytoplasm at the sites of cytokine receptor activation and/or in the nucleus at the site of delivery of the effects on gene expression. Conversely, activating receptor-mediated signaling is negatively regulated by the recruitment of inhibitory receptors and stimulation of phosphatases that overcome kinase activation at the sites of NK cell interactions with target cells. Thus, even if the conditions lead to one stimulatory pathway being negatively regulated, maintaining NK cell populations provides an opportunity to access these NK cell functions that are stimulated through alternative pathways. Moreover, if the TRAIL is induced, as it can be in response to type 1 IFN exposure, there is an opportunity to kill those target cells that express the death receptors for TRAIL. Under these conditions, the responsiveness of NK cells to direct activating-receptor stimulation can be virtually turned-off but the NK cells are still effective because the stimulatory signal is delivered to the target rather than the effector (NK) cell.
Figure 1.
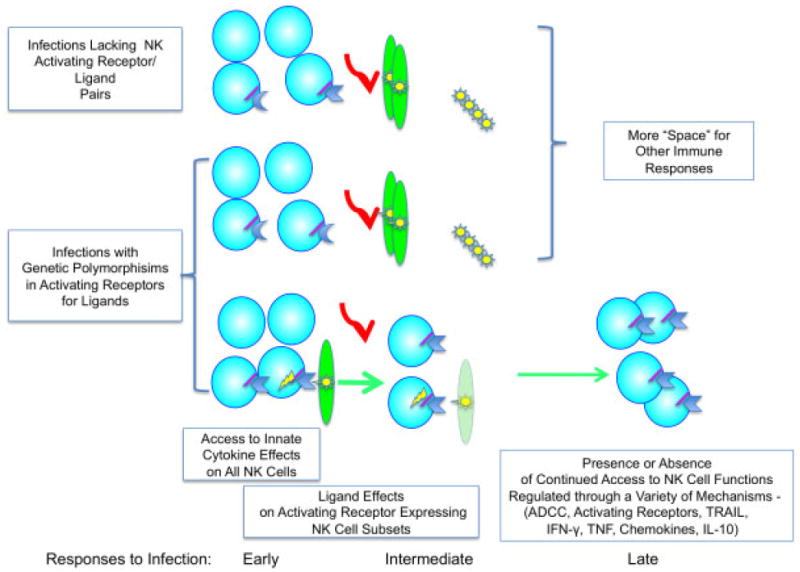
Model for contributions made to sustaining NK cells and hence their NK cell functions by activating receptors and virus-induced ligand pairs. Genetic variability in the expression of particular activating receptors in different individuals, as well as differences in virus induction of ligands, helps explain the paradoxical reports of increases in NK cell subsets and decreases in overall NK cells under different conditions of infection. If the conditions of infection fail to result in the induction of an activating receptor and virus-induced pair, NK cells will be decreasing as the infection advances. If a receptor ligand pair is present, the NK cell subset expressing the activating receptor for the virus-induced ligand will be preferentially stimulated and marked for rescue from purging. Thus, proliferation of NK cells through activating receptors provides a mechanism to allow their continued access and contributions to a variety of responses during infection, including documented antiviral and immunoregulatory effects mediated through killing or cytokine/chemokine production. (Red arrows represent decreases in subsets failing to be stimulated, and green arrows represent induction of proliferation and maintenance.)
Maintaining NK cells also has the potential to preserve their various immunoregulatory functions, including shaping adaptive responses via NK cell production of cytokines, including IFN-γ and IL-10 [1, 13, 16, 53-56], under different conditions of need. The IFN-γ response can mediate a number of immune enhancing or shaping functions. IL-10 can have a variety of effects but is a potent negative regulator of adaptive immunity. In the context of chronic HCV infections in humans, the production of IL-10 by NK cells has been suggested to interfere with viral clearance [16]. However, in other systems IL-10 can have an important role in protecting against immunopathology. The system of MCMV infection in Prf1-deficient mice has clearly shown that the Ly49H activating receptor maintains the NK cell population permitting NK cell IL-10 production to play a critical role in regulating CD8+ T-cell responses and CD8+ T-cell-dependent disease [13]. In light of this new understanding, it is interesting to consider CD8+ T-cell responses during infections of mice with LCMV, an agent with no evidence of an NK cell contribution to defense or of a virus-induced ligand/activating receptor pair. Here, Prf1-deficient mice fail to control LCMV infections, and CD8+ T-cell expansion is sustained with dramatic consequences for immunopathology [69]. Thus, the opportunity to access NK cell responses negatively regulating adaptive immunity can be extremely beneficial to the host and maintaining NK cells through the later periods of infection might dramatically increase the opportunities for the delivery of these functions.
If, as the evidence suggests, keeping NK cells around is helpful but losing them is problematic, why then has the system evolved to allow NK cell loss? Certainly, NK cells have their own potential to mediate pathology, and it is likely that balancing the magnitudes of NK and T-cell responses when they overlap is very important. An alternative or additional possibility, however, is that the NK cell “disappearance” in the absence of activating receptor function during viral infections represents a pathway generally used to purge cells if they are not specifically contributing to the challenge at hand. T-regulatory cells have recently been reported to “disappear” from the liver during parasitic infections [70], and the question of whether NKT cells in the liver are “disappearing” or simply downregulating their receptors either during viral infections or after cytokine treatment has been a difficult controversy to resolve [71]. A common mechanism to remove lymphocytes not receiving a stimulatory signal through an activating receptor would provide room for other cell proliferative responses, facilitate homeostasis, and contribute to T-cell “contraction” after their initial expansion once viral infections had been cleared [72]. In the case of NK cells, the disappearance might be unusually problematic for the host because maintaining the cells provides continued access to the other beneficial effects mediated by NK cells. Furthermore, bone marrow is compromised during viral infections and NK cell populations basally turn over from bone marrow precursors at a relatively slow rate. Thus, NK cell depletions during viral infections result in the loss of cells, under conditions that hinder recovery, when the cells could still be of use. A better understanding of the advantages of losing NK and T-cell populations during infections will require careful characterization of the mechanisms contributing to their “disappearance”. It is clear, however, that linking the maintenance of cells to stimulation through an activating receptor whose ligand is induced during infection is a powerful mechanism to select cells proven beneficial for rescue.
Concluding remarks
In summary, much remains to be learned about the regulation of NK cell numbers during viral infections, but the studies discussed in this review argue that an important function of NK cell activating receptors is to stimulate proliferation for cell maintenance. The studies also suggest that this pathway is in place to protect cells from the mechanisms that purge those populations failing to sufficiently demonstrate immediate value. Given the polygenic and polymorphic diversity of many of these receptors between both individuals and strains, the observations provide an explanation for the reported increases and decreases in NK cell numbers under different conditions of viral infection. If a virus does not induce a ligand for an NK cell activating receptor, proliferation will not be induced. On the other hand, if an individual does not have a receptor for the virus-induced ligand, proliferation will also be lacking although it might be induced in another individual. By linking the activating receptor to proliferation and maintenance, the immune system has evolved a strong test for selecting cell subsets that clearly contribute to defense; however, losing cells that do not pass the test is an expensive way of making space because decreasing NK cell numbers limits the availability of their many other functions that do not depend on engagement of a particular activating receptor. It may be possible to promote the maintenance of NK cells using purposeful stimulation through known activating receptors, suggesting novel therapeutic interventions that would benefit infected individuals. In addition, it now seems that NK cells, if present, are intimately involved in both innate and adaptive immunity, and hence challenging the narrow assignment of NK cell functions to innate immunity.
Acknowledgments
The work in the authors’ laboratory is supported by the National Institutes of Health, USA. C. A. Biron thanks R. Ahmed for a lively discussion on homeostasis and T-cell contraction.
Abbreviations
- HCMV
human CMV
- LCMV
lymphocytic choriomeningitis virus
- MCMV
murine CMV
- NKG2D
natural killer group 2, member D
- VZV
varicella zoster virus
Footnotes
Conflict of Interest: The authors declare no financial or commercial conflict of interest.
References
- 1.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 2.Lee SH, Miyagi T, Biron CA. Keeping NK cells in highly regulated antiviral warfare. Trends Immunol. 2007;28:252–259. doi: 10.1016/j.it.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 1989;320:1731–1735. doi: 10.1056/NEJM198906293202605. [DOI] [PubMed] [Google Scholar]
- 4.Yokoyama WM, Plougastel BF. Immune functions encoded by the natural killer gene complex. Nat Rev Immunol. 2003;3:304–316. doi: 10.1038/nri1055. [DOI] [PubMed] [Google Scholar]
- 5.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caligiuri MA. Human natural killer cells. Blood. 2008;112:461–469. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayakawa Y, Huntington ND, Nutt SL, Smyth MJ. Functional subsets of mouse natural killer cells. Immunol Rev. 2006;214:47–55. doi: 10.1111/j.1600-065X.2006.00454.x. [DOI] [PubMed] [Google Scholar]
- 8.Orange JS, Wang B, Terhorst C, Biron CA. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J Exp Med. 1995;182:1045–1056. doi: 10.1084/jem.182.4.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orange JS, Biron CA. An absolute and restricted requirement for IL-12 in natural killer cell IFN-gamma production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J Immunol. 1996;156:1138–1142. [PubMed] [Google Scholar]
- 10.Orange JS, Biron CA. Characterization of early IL-12, IFN-alphabeta, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J Immunol. 1996;156:4746–4756. [PubMed] [Google Scholar]
- 11.Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, Caligiuri MA, et al. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol. 2002;169:4279–4287. doi: 10.4049/jimmunol.169.8.4279. [DOI] [PubMed] [Google Scholar]
- 12.Dokun AO, Kim S, Smith HR, Kang HS, Chu DT, Yokoyama WM. Specific and nonspecific NK cell activation during virus infection. Nat Immunol. 2001;2:951–956. doi: 10.1038/ni714. [DOI] [PubMed] [Google Scholar]
- 13.Lee SH, Kim KS, Fodil-Cornu N, Vidal SM, Biron CA. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J Exp Med. 2009;206:2235–2251. doi: 10.1084/jem.20082387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alter G, Martin MP, Teigen N, Carr WH, Suscovich TJ, Schneidewind A, Streeck H, et al. Differential natural killer cell-mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J Exp Med. 2007;204:3027–3036. doi: 10.1084/jem.20070695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alter G, Rihn S, Walter K, Nolting A, Martin M, Rosenberg ES, Miller JS, et al. HLA class I subtype-dependent expansion of KIR3DS1+ and KIR3DL1+ NK cells during acute human immunodeficiency virus type 1 infection. J Virol. 2009;83:6798–6805. doi: 10.1128/JVI.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Maria A, Fogli M, Mazza S, Basso M, Picciotto A, Costa P, Congia S, et al. Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur J Immunol. 2007;37:445–455. doi: 10.1002/eji.200635989. [DOI] [PubMed] [Google Scholar]
- 17.Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, Lopez-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood. 2004;104:3664–3671. doi: 10.1182/blood-2004-05-2058. [DOI] [PubMed] [Google Scholar]
- 18.Williams H, McAulay K, Macsween KF, Gallacher NJ, Higgins CD, Harrison N, Swerdlow AJ, et al. The immune response to primary EBV infection: a role for natural killer cells. Br J Haematol. 2005;129:266–274. doi: 10.1111/j.1365-2141.2005.05452.x. [DOI] [PubMed] [Google Scholar]
- 19.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 20.Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci USA. 2002;99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bubic I, Wagner M, Krmpotic A, Saulig T, Kim S, Yokoyama WM, Jonjic S, et al. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J Virol. 2004;78:7536–7544. doi: 10.1128/JVI.78.14.7536-7544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fodil-Cornu N, Lee SH, Belanger S, Makrigiannis AP, Biron CA, Buller RM, Vidal SM. Ly49h-deficient C57BL/6 mice: a new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J Immunol. 2008;181:6394–6405. doi: 10.4049/jimmunol.181.9.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarazona R, Casado JG, Delarosa O, Torre-Cisneros J, Villanueva JL, Sanchez B, Galiani MD, et al. Selective depletion of CD56(dim) NK cell subsets and maintenance of CD56(bright) NK cells in treatment-naive HIV-1-seropositive individuals. J Clin Immunol. 2002;22:176–183. doi: 10.1023/a:1015476114409. [DOI] [PubMed] [Google Scholar]
- 24.Azzoni L, Rutstein RM, Chehimi J, Farabaugh MA, Nowmos A, Montaner LJ. Dendritic and natural killer cell subsets associated with stable or declining CD4+ cell counts in treated HIV-1-infected children. J Infect Dis. 2005;191:1451–1459. doi: 10.1086/429300. [DOI] [PubMed] [Google Scholar]
- 25.Meier UC, Owen RE, Taylor E, Worth A, Naoumov N, Willberg C, Tang K, et al. Shared alterations in NK cell frequency, phenotype, and function in chronic human immunodeficiency virus and hepatitis C virus infections. J Virol. 2005;79:12365–12374. doi: 10.1128/JVI.79.19.12365-12374.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong HS, Eberhard JM, Keudel P, Bollmann BA, Ballmaier M, Bhatnagar N, Zielinska-Skowronek M, et al. HIV infection is associated with a preferential decline in less-differentiated CD56dim CD16+ NK cells. J Virol. 2010;84:1183–1188. doi: 10.1128/JVI.01675-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morishima C, Paschal DM, Wang CC, Yoshihara CS, Wood BL, Yeo AE, Emerson SS, et al. Decreased NK cell frequency in chronic hepatitis C does not affect ex vivo cytolytic killing. Hepatology. 2006;43:573–580. doi: 10.1002/hep.21073. [DOI] [PubMed] [Google Scholar]
- 28.Golden-Mason L, Madrigal-Estebas L, McGrath E, Conroy MJ, Ryan EJ, Hegarty JE, O’Farrelly C, et al. Altered natural killer cell subset distributions in resolved and persistent hepatitis C virus infection following single source exposure. Gut. 2008;57:1121–1128. doi: 10.1136/gut.2007.130963. [DOI] [PubMed] [Google Scholar]
- 29.Vossen MT, Biezeveld MH, de Jong MD, Gent MR, Baars PA, von Rosenstiel IA, van Lier RA, et al. Absence of circulating natural killer and primed CD8+ cells in life-threatening varicella. J Infect Dis. 2005;191:198–206. doi: 10.1086/426866. [DOI] [PubMed] [Google Scholar]
- 30.Reed DS, Hensley LE, Geisbert JB, Jahrling PB, Geisbert TW. Depletion of peripheral blood T lymphocytes and NK cells during the course of ebola hemorrhagic Fever in cynomolgus macaques. Viral Immunol. 2004;17:390–400. doi: 10.1089/vim.2004.17.390. [DOI] [PubMed] [Google Scholar]
- 31.Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, Davis DM, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature. 2001;409:1055–1060. doi: 10.1038/35059110. [DOI] [PubMed] [Google Scholar]
- 32.Dunn C, Chalupny NJ, Sutherland CL, Dosch S, Sivakumar PV, Johnson DC, Cosman D. Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J Exp Med. 2003;197:1427–1439. doi: 10.1084/jem.20022059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuller CL, Ruthel G, Warfield KL, Swenson DL, Bosio CM, Aman MJ, Bavari S. NKp30-dependent cytolysis of filovirus-infected human dendritic cells. Cell Microbiol. 2007;9:962–976. doi: 10.1111/j.1462-5822.2006.00844.x. [DOI] [PubMed] [Google Scholar]
- 34.Chisholm SE, Howard K, Gomez MV, Reyburn HT. Expression of ICP0 is sufficient to trigger natural killer cell recognition of herpes simplex virus-infected cells by natural cytotoxicity receptors. J Infect Dis. 2007;195:1160–1168. doi: 10.1086/512862. [DOI] [PubMed] [Google Scholar]
- 35.Stewart CA, Laugier-Anfossi F, Vely F, Saulquin X, Riedmuller J, Tisserant A, Gauthier L, et al. Recognition of peptide-MHC class I complexes by activating killer immunoglobulin-like receptors. Proc Natl Acad Sci USA. 2005;102:13224–13229. doi: 10.1073/pnas.0503594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chisholm SE, Reyburn HT. Recognition of vaccinia virus-infected cells by human natural killer cells depends on natural cytotoxicity receptors. J Virol. 2006;80:2225–2233. doi: 10.1128/JVI.80.5.2225-2233.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, Hanna J, et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol. 2006;7:517–523. doi: 10.1038/ni1322. [DOI] [PubMed] [Google Scholar]
- 38.Kielczewska A, Pyzik M, Sun T, Krmpotic A, Lodoen MB, Munks MW, Babic M, et al. Ly49P recognition of cytomegalovirus-infected cells expressing H2-Dk and CMV-encoded m04 correlates with the NK cell antiviral response. J Exp Med. 2009;206:515–523. doi: 10.1084/jem.20080954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krmpotic A, Busch DH, Bubic I, Gebhardt F, Hengel H, Hasan M, Scalzo AA, et al. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat Immunol. 2002;3:529–535. doi: 10.1038/ni799. [DOI] [PubMed] [Google Scholar]
- 40.Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, Lanier LL. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med. 2003;197:1245–1253. doi: 10.1084/jem.20021973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi EI, Reimann KA, Letvin NL. In vivo natural killer cell depletion during primary simian immunodeficiency virus infection in rhesus monkeys. J Virol. 2008;82:6758–6761. doi: 10.1128/JVI.02277-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bukowski JF, Woda BA, Habu S, Okumura K, Welsh RM. Natural killer cell depletion enhances virus synthesis and virus-induced hepatitis in vivo. J Immunol. 1983;131:1531–1538. [PubMed] [Google Scholar]
- 43.Usherwood EJ, Meadows SK, Crist SG, Bellfy SC, Sentman CL. Control of murine gammaherpesvirus infection is independent of NK cells. Eur J Immunol. 2005;35:2956–2961. doi: 10.1002/eji.200526245. [DOI] [PubMed] [Google Scholar]
- 44.Carlyle JR, Mesci A, Fine JH, Chen P, Belanger S, Tai LH, Makrigiannis AP. Evolution of the Ly49 and Nkrp1 recognition systems. Semin Immunol. 2008;20:321–330. doi: 10.1016/j.smim.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Kulkarni S, Martin MP, Carrington M. The Yin and Yang of HLA and KIR in human disease. Semin Immunol. 2008;20:343–352. doi: 10.1016/j.smim.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacFarlane AWT, Campbell KS. Signal transduction in natural killer cells. Curr Top Microbiol Immunol. 2006;298:23–57. doi: 10.1007/3-540-27743-9_2. [DOI] [PubMed] [Google Scholar]
- 47.Biron CA, Sen GC. Innate immune responses to viral infection. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 249–278. [Google Scholar]
- 48.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 49.Schepis D, D’Amato M, Studahl M, Bergstrom T, Karre K, Berg L. Herpes simplex virus infection downmodulates NKG2D ligand expression. Scand J Immunol. 2009;69:429–436. doi: 10.1111/j.1365-3083.2009.02241.x. [DOI] [PubMed] [Google Scholar]
- 50.Salazar-Mather TP, Hamilton TA, Biron CA. A chemokine-to-cytokine-to-chemokine cascade critical in antiviral defense. J Clin Invest. 2000;105:985–993. doi: 10.1172/JCI9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 52.Krebs P, Barnes MJ, Lampe K, Whitley K, Bahjat KS, Beutler B, Janssen E, et al. NK-cell-mediated killing of target cells triggers robust antigen-specific T-cell-mediated and humoral responses. Blood. 2009;113:6593–6602. doi: 10.1182/blood-2009-01-201467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brady J, Hayakawa Y, Smyth MJ, Nutt SL. IL-21 induces the functional maturation of murine NK cells. J Immunol. 2004;172:2048–2058. doi: 10.4049/jimmunol.172.4.2048. [DOI] [PubMed] [Google Scholar]
- 54.Maroof A, Beattie L, Zubairi S, Svensson M, Stager S, Kaye PM. Posttranscriptional regulation of II10 gene expression allows natural killer cells to express immunoregulatory function. Immunity. 2008;29:295–305. doi: 10.1016/j.immuni.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deniz G, Erten G, Kucuksezer UC, Kocacik D, Karagiannidis C, Aktas E, Akdis CA, et al. Regulatory NK cells suppress antigen-specific T cell responses. J Immunol. 2008;180:850–857. doi: 10.4049/jimmunol.180.2.850. [DOI] [PubMed] [Google Scholar]
- 56.Brockman MA, Kwon DS, Tighe DP, Pavlik DF, Rosato PC, Sela J, Porichis F, et al. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood. 2009;114:346–356. doi: 10.1182/blood-2008-12-191296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biron CA, Su HC, Orange JS. Function and regulation of natural killer (NK) cells during viral infections: characterization of responses in vivo. Methods. 1996;9:379–393. doi: 10.1006/meth.1996.0043. [DOI] [PubMed] [Google Scholar]
- 58.Nguyen KB, Cousens LP, Doughty LA, Pien GC, Durbin JE, Biron CA. Interferon alpha/beta-mediated inhibition and promotion of interferon gamma: STAT1 resolves a paradox. Nat Immunol. 2000;1:70–76. doi: 10.1038/76940. [DOI] [PubMed] [Google Scholar]
- 59.Miyagi T, Gil MP, Wang X, Louten J, Chu WM, Biron CA. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J Exp Med. 2007;204:2383–2396. doi: 10.1084/jem.20070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Biron CA, Welsh RM. Blastogenesis of natural killer cells during viral infection in vivo. J Immunol. 1982;129:2788–2795. [PubMed] [Google Scholar]
- 61.Biron CA, Young HA, Kasaian MT. Interleukin 2-induced proliferation of murine natural killer cells in vivo. J Exp Med. 1990;171:173–188. doi: 10.1084/jem.171.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Biron CA, Orange JS. IL12 in acute viral infectious disease. Res Immunol. 1995;146:590–600. doi: 10.1016/0923-2494(96)83036-7. [DOI] [PubMed] [Google Scholar]
- 63.French AR, Sjolin H, Kim S, Koka R, Yang L, Young DA, Cerboni C, et al. DAP12 signaling directly augments proproliferative cytokine stimulation of NK cells during viral infections. J Immunol. 2006;177:4981–4990. doi: 10.4049/jimmunol.177.8.4981. [DOI] [PubMed] [Google Scholar]
- 64.Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457:557–561. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun JC, Ma A, Lanier LL. Cutting edge: IL-15-independent NK cell response to mouse cytomegalovirus infection. J Immunol. 2009;183:2911–2914. doi: 10.4049/jimmunol.0901872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Geurs TL, Zhao YM, Hill EB, French AR. Ly49H engagement compensates for the absence of type I interferon signaling in stimulating NK cell proliferation during murine cytomegalovirus infection. J Immunol. 2009;183:5830–5836. doi: 10.4049/jimmunol.0901520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alter G, Suscovich TJ, Kleyman M, Teigen N, Streeck H, Zaman MT, Meier A, et al. Low perforin and elevated SHIP-1 expression is associated with functional anergy of natural killer cells in chronic HIV-1 infection. AIDS. 2006;20:1549–1551. doi: 10.1097/01.aids.0000237371.31315.48. [DOI] [PubMed] [Google Scholar]
- 68.Alter G, Teigen N, Ahern R, Streeck H, Meier A, Rosenberg ES, Altfeld M. Evolution of innate and adaptive effector cell functions during acute HIV-1 infection. J Infect Dis. 2007;195:1452–1460. doi: 10.1086/513878. [DOI] [PubMed] [Google Scholar]
- 69.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–743. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 70.Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O’Brien S, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wesley JD, Tessmer MS, Chaukos D, Brossay L. NK cell-like behavior of Valpha14i NK T cells during MCMV infection. PLoS Pathog. 2008;4:e1000106. doi: 10.1371/journal.ppat.1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]