


Abstract
The transcriptional regulator NprR controls the expression of genes essential for the adaptative response of Bacillus cereus. NprR belongs to the RNPP family of directly regulated quorum sensors from Gram-positive bacteria. It is activated by the re-imported signaling peptide NprX. To elucidate the activation mechanism of this quorum-sensing system, we analyzed the conformation changes induced on binding of NprX. We solved the crystal structure of the NprR/NprX binary complex and characterized the apo form of NprR in solution. We demonstrated that apo NprR is a dimer that switches to a tetramer in the presence of NprX. Mutagenesis, and functional analysis allowed us to identify the protein and peptide residues directly involved in the NprR activation process. Based on the comparison with the Rap proteins, we propose a model for the peptide-induced conformational change allowing the apo dimer to switch to an active tetramer specifically recognizing target DNA sequences.
INTRODUCTION
Quorum-sensing (QS) is a mode of cell–cell communication based on the secretion and recognition of diffusible biomolecules by cognate bacteria. This sensing mechanism allows bacteria to coordinate their behavior as a whole community according to population density. Bacteria use QS systems to control, in a multi-cellular manner, important bacterial processes, such as virulence, sporulation, genetic transfer and antibiotic production. QS relies on secreted signaling molecules called autoinducers or pheromones, to directly or indirectly regulate gene expression. In general, Gram-positive bacteria use processed oligopeptides as signaling molecules (1). They are subdivided in two groups according to whether they interact with receptors at the cell surface or inside the cell (2). The first group consists of peptides interacting with the external domain of membrane embedded histidine kinases that transmit the signal to cytoplasmic response regulators of two component systems (3). The second group includes peptides, which are re-imported in the bacterium where they interact with cytoplasmic receptors directly regulating the QS response (4,5). In these direct QS systems, the gene of the signaling peptide is generally in the vicinity of the gene encoding its cognate QS regulator, thus forming a characteristic regulator/peptide-signaling cassette (6). The re-imported signaling peptides mainly originate from short genes coding for small exported proteins that are ∼20–60 amino acids in length. After processing by external proteases (7,8), their mature forms composed of 5–11 residues (9–11) are actively re-imported into the cell by oligopeptide permeases (12–14). Once inside the bacterial cell, the mature signaling peptides either activate or inhibit their related quorum sensor.
The QS regulators of these direct QS systems have been grouped in a new protein family called RNPP according to the names of the first four identified members: Rap, NprR, PlcR and PrgX (15). They are characterized by a C-terminal domain containing several tetratricopeptide repeats (TPRs), a structural motif of 34 residues mediating protein–protein or protein–peptide interactions (16,17). Except the Rap proteins, the RNPP regulators display a transcriptional activity and carry an N-terminal helix-turn-helix (HTH)-type DNA-binding domain (18,19).
The crystal structures of PrgX from Enterococcus faecalis (20,21) and PlcR from Bacillus cereus (15) have been solved in complex with their cognate signaling peptides. PrgX forms a tetramer, whereas PlcR is a dimer, but they display a similar peptide-binding site. Recently, the crystal structures of two Rap proteins from Bacillus subtilis, RapH (22) and RapF (23) have been solved in the absence of their Phr signaling peptide. Instead of the HTH-type DNA-binding domain observed in the other RNPP proteins, they display an antiparallel 3-helix bundle linked to the TPR domain through a flexible region and a small α-helix. This N-terminal domain is involved in protein/protein interaction and carries a phosphatase activity in some of the Rap proteins (24,25). Up to now, there was no structure reported for the last identified member of the family, the transcription factor NprR from the B. cereus group (26). Interestingly, the NprR sequence contains between the predicted HTH and TPR domains an insertion of a hundred residues, which shares sequence similarity with the N-terminal domain of the Rap proteins. NprR was proposed to be an evolutionary intermediate between the Rap proteins and the RNPP proteins carrying transcription factor activity (15,26).
In the presence of the signaling peptide NprX, NprR activates the expression of genes encoding proteins involved in food supply, stress and antibiotic resistance, as well as in the synthesis of a lipopeptide (27). It was demonstrated that the NprR regulon is required for the necrotrophic lifestyle of Bacillus thuringiensis. The minimal active form of NprX has been identified as the heptapeptide SKPDIVG corresponding to an internal region of the carboxy-terminal part (26). This heptapeptide forms a tight complex with NprR, but the biological form predominantly present in the bacterial cell has not been characterized yet. Isothermal titration calorimetry (ITC) experiments demonstrated that the NprR/NprX7 complex specifically binds to DNA sequences centered on the −35 box of the nprA promoter, the first identified target of NprR (26). The study of NprR/NprX polymorphism within the B. cereus group revealed seven NprR groups associated with seven cognate NprX heptapeptide classes. The NprR/NprX cell–cell communication system was found to be strain specific with possible cross talk between some phylogenetic groups (26).
In this article, we present the crystal structure of a truncated form of B. cereus NprR deleted of its HTH-domain in complex with its cognate signaling peptide NprX. Together with small angle scattering experiments and biochemical analysis of apo NprR and engineered mutant proteins, this structure–function study provides new insights into the NprX-dependent activation mechanism of NprR.
MATERIALS AND METHODS
Protein preparation
The nprR gene from B. thuringiensis strain 407 has been cloned into a pQE60 plasmid and expressed as recombinant protein in Escherichia coli strain M15 [pRep4] with a C-terminal 6xHis tag, as already described (26). The gene encoding the NprR(ΔHTH) protein was constructed by deleting the N-terminal 60 residues of NprR. The double mutations (Y223A/F225A) and (N407A/Y410A) as well as the single R126A mutation were introduced in the genes of the His6-tagged NprR and NprR(ΔHTH) proteins by PCR amplification using appropriate primers (Supplementary Table S1). The recombinant plasmids used in this work are also described in Supplementary Table S1. The wild-type and mutant NprR proteins have been produced and purified as described (26). The protein samples were aliquoted, flash frozen and stored at −20°C in 20 mM Tris–HCl (pH 8) and 100 mM NaCl.
Production of SeMet-labeled NprR(ΔHTH)
Selenomethonine-labeled NprR(ΔHTH) was produced in E. coli strain M15 [pREP4, pQE60ΩnprRΔHTH] grown overnight at 28°C in the presence of 100 µg/ml of ampicilin and 25 µg/ml of kanamicin in M9 minimal medium supplemented with 100 µg/ml of Lys, Thr and Phe and 50 µg/ml of Leu, Ile, Val and Met. At mid-exponential growth phase (OD600 = 0.6), cells were harvested by centrifugation at 5000 rpm for 10 min and resuspended in fresh M9 medium supplemented as before except for methionine that was replaced by 50 µg/ml of selenomethionine. After incubation for 30 min at 37°C, expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 4 h at 37°C. The purification protocole was unchanged.
Peptide preparation
The NprX peptides (Genscript) were resuspended at 25 mM in protein storage buffer. They were aliquoted, flash frozen and stored at −20°C.
Size-exclusion chromatography multi-angle light scattering
The experiments were performed using a Viskotec TDA305 triple detector array with an integrated GPCmax VE 2001 system (Malvern, France). The protein samples were injected at a concentration of 2 mg/ml (≈40 µM) on a SuperdexTM 200 HR 10/300 (GE Healthcare) column equilibrated in 20 mM Tris–HCl (pH 7.5), 100 mM NaCl. The OmniSEC software of the manufacturer was used for acquisition and analysis of the data. Bovine serum albumin (Sigma–Aldrich) was used as standard reference protein for detector calibration.
Dynamic light scattering
Dynamic light scattering (DLS) measurements were carried out at 20°C on a Wyatt DynaPro light scattering instrument with both protein forms, NprR and NprR(ΔHTH), at 20 µM alone or in complex with 200 µM NprX7. The samples were centrifuged at 15 000g for 10 min at 4°C just before use. For each sample, 20 acquisitions (5 sec each) were recorded. The data were analyzed using the Dynamics V6 software (Wyatt Technologies Corp.).
Native polyacrylamide gel electrophoresis
The samples were loaded on 8% acrylamide gels prepared with 2 M Tris–HCl buffer at pH 8.8. The running buffer contained 0.05 M Tris–HCl (pH 8.3) and 0.5 M Glycine. Migration was performed at 100 V for 2 h 30 min at room temperature. The gels were stained with Coomassie blue.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were performed at 20°C with an ITC200 calorimeter (Microcal®). The protein concentration in the microcalorimeter cell and the peptide concentration in the syringe were set at 25 µM and 250 µM, respectively. A first injection of 0.4 µl was followed by 20 injections of 2 µl at intervals of 180 s. The data were analyzed according to the one binding-site model using the MicroCal Origin software provided by the manufacturer.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) experiments were performed with VP-DSC calorimeter (Microcal®). Samples equilibrated at 20°C were heated up to 80°C at a constant rate of 1°C/min. Data were analyzed using the MicroCal Origin software provided by the manufacturer.
Crystallization
The NprR(ΔHTH)/NprX complex crystallized at 18°C in 0.975 M Na Citrate, 0.1 M Hepes (pH 7.6). The selenomethionine labeled protein crystallized in the same conditions. The crystals were flash frozen in the crystallization solution supplemented with 30% glycerol.
Crystallographic analysis
The diffraction data sets used for the resolution of the structure were collected at 100 K on beamline Proxima-I at the synchrotron SOLEIL (Gif-sur-Yvette, France). Diffraction data from selenomethionine labeled crystals were collected at a single energy (12.6615eV) corresponding to the maximum of the anomalous dispersion factor f′′. The native data set was collected at a wavelength of 0.980 Å. Both data sets were processed using the XDS package (28). Phasing was performed by the single-wavelength anomalous diffraction (SAD) method. The sub-structure determination was performed with the SHELX program suite (29). Initial phasing was performed by the program PHASER (30), and density modification was carried out with RESOLVE (31). An initial model covering ∼80% of the NprR(ΔHTH) sequence was automatically built using BUCCANEER (32). This partial model was then used as input for phasing using the program PHASER, and completion of the sub-structure was performed by a new run of RESOLVE and BUCCANEER. Subsequent model building was done manually with COOT (33) alternating with refinement cycles using BUSTER (34). Non-crystallographic symmetry was applied in the first steps of refinement. The final refinement cycles were performed using PHENIX (35) against the 3.2 Å resolution native data set. The B-factors were refined individually. The final model was evaluated using COOT validation tools and the MolProbity software (36). Final statistics are listed in Table 2. The atomic coordinates and structures factors of the NprR(ΔHTH)/NprX8n complex have been deposited in the Protein Data Bank under the ID code 4GPK.
Table 2.
X-ray data processing and refinement statistics
| Data processing statistics | Native data set | SAD data set (SeMet) |
|---|---|---|
| Space group | P1 | P1 |
| Unit-cell parameters (Å) | a = 122.2; b = 133.3; c = 137.5 | a = 121.1; b = 133.0; c = 138.5 |
| Unit-cell angles (°) | α = 108.2; β = 104.8; γ = 103.8 | α = 108.4; β = 105.3 γ = 103.2 |
| Resolution range (Å)a | 30.0–3.2 (3.4–3.2) | 45.0–3.5 (3.7–3.5) |
| No. of unique reflections | 121 662 (19 211) | 187 722 (27 788) |
| Completeness (%) | 98.1 (96.9) | 96.2 (88.4) |
| Redundancy | 3.9 (3.8) | 3.8 (3.7) |
| Mean I/σ(I) | 18.3 (3.3) | 10.25 (2.2) |
| Rmeas (%)b | 5.0 (44.5) | 16.3 (82.8) |
| Refinement statistics | Against native data set | |
|---|---|---|
| Resolution range | 29.74–3.19 (3.23–3.19) | |
| No. of complexes/a.u. | 12 | |
| Rworkc | 26.95 (34.93) | |
| Rfreed | 29.94 (39.76) | |
| Ramachandran | ||
| Favored (%) | 93.8 | |
| Disallowed (%) | 0.8 | |
| R.M.S.D. | ||
| Bond lengths (Å) | 0.002 | |
| Bond angles (°) | 0.632 | |
| Chirality | 0.05 | |
| Mean B value (Å2) | 73.14 | |
aNumbers in parentheses represent values in the highest resolution shell.
bRmeas = Σhkl [N/N-1]1/2Σi |Ii(hkl) - <I(hkl)>| / ΣhklΣi Ii(hkl) where N is the multiplicity of a given reflection, Ii(hkl) is the integrated intensity of a given reflection, and <I(hkl)> is the mean intensity of multiple corresponding symmetry-related reflections.
cRwork = Σ ||Fobs| - |Fcalc|| /Σ |Fobs|, where |Fobs| and |Fcalc| are the observed and calculated structure factor amplitudes, respectively.
dRfree is the same as Rwork but calculated with a 20% subset of all reflections that was never used in refinement.
Structure analysis
Structure comparisons and calculations of rms distances have been performed by the protein structure comparison service FOLD (37). Calculation of contacts and interfaces have been performed with the Protein Interfaces, Surfaces and Assemblies (PISA) service (38). Multiple sequence alignment was performed using ClustalW (39) and optimized using Seaview (40). The secondary structure elements have been added at the top of the alignment using ESPript (41). Figures of 3D structures were generated using PYMOL (DeLano Scientific LLC).
SAXS analysis
SAXS data were recorded on beamline Swing at SOLEIL Synchrotron (Gif sur Yvette, France) at a wavelength of 1.003 Å on a 17 cm × 17 cm low-noise Aviex charge-coupled device detector positioned at a distance of 1850 mm from the sample, with the direct beam off-centered. The useful Q-range was 0.018–0.4 Å−1, where Q = 4πsinθ/λ is the momentum transfer, and 2θ is the scattering angle. 40 µl of NprR(ΔHTH) sample at 14 mg/ml were injected into a size-exclusion column (SHODEX KW403) using an Agilent© High Performance Liquid Chromatography system cooled at 15°C and eluted directly into the SAXS flow through capillary cell at a flow rate of 150 μl/min. SAXS data were collected online throughout the whole elution time, with a frame duration of 2 s and a dead time between frames of 1 s. The first 30 frames collected during the first minutes of the elution flow were averaged to account for buffer scattering. The 10 frames corresponding to the top of the elution peak were averaged and used for data processing after baseline subtraction.
The model of apo NprR(ΔHTH) was built using DaDiModO, a program for refining structures of multidomain proteins against small-angle scattering data (42). Starting from the type II dimer of the NprR(ΔHTH)/NprX complex, a more elongated model fitting the experimental data was obtained by introducing flexibility in the α3-α4 loop of TPR2 as well as in the N- and C-terminal extremities. Stepwise generic conformational changes were applied cyclically in a stochastic optimization algorithm that performs a search in the protein conformation space.
Functional analysis
Activation of the nprA gene expression by NprR was measured by complementing the recombinant B. thuringiensis 407 nprA’Z ΔRX strain with the plasmids described in Supplementary Table S2. The 407 nprA’Z ΔRX strain was obtained by deleting the nprR-nprX genes from a strain containing a chromosomal transcriptional fusion between the promoterless lacZ gene and the nprA promoter regulated by NprR (26). ß-galactosidase activities were measured at 37°C in sporulation-specific HCT medium as previously described (26). The onset of stationary phase (time 0) was defined as the first divergence point of mass increase from the exponential rate. The specific activities are expressed in units of ß-galactosidase per milligram of protein (Miller units). Each assay was independently repeated at least three times and a representative graph was shown for each experiment.
RESULTS
NprX binding shifts NprR from a dimer to a tetramer
During the purification procedure, full-length NprR and the HTH-truncated form NprR(ΔHTH) eluted from the calibrated size exclusion chromatography column as dimers. This was confirmed by Size-Exclusion Chromatography Multi-Angle Light Scattering (SEC-MALS) analysis. The molecular weight corresponding to the elution peak was stable and characteristic of a homogenous dimeric sample, for NprR(ΔHTH) as well as for the full-length protein (Figure 1A and B). The effect of signaling peptide binding on the oligomeric state of NprR was then analyzed. Electrophoretic analysis of the purified proteins in non-denaturing conditions demonstrated that addition of the heptapeptide NprX7 reduced the mobility of NprR (Figure 1C). DLS analysis indicated that the hydrodynamic diameter of the apo protein and of the protein–peptide complex corresponded to a dimer and a tetramer, respectively (Table 1). The same results were obtained with NprR(ΔHTH) as with the full-length NprR, demonstrating that the absence of the HTH domain did not alter the conformational switch induced by peptide binding. Together with the functional experiments showing that NprR requires the presence of the NprX peptide to induce the transcription of the nprA gene (26), these results suggest that NprX binding induces a structural change of the NprR transcriptional activator from an inactive dimer to an active tetramer. DSC measurements showed that addition of peptide increased the Tm of NprR from 45.2°C to 50.3°C, suggesting that the tetrameric complex is more stable than the dimeric apo form.
Figure 1.

Oligomerization state analysis. (A) SEC-MALS analysis of full length NprR. The retention volume of 13.2 ml and the log(Mw) under the peak correspond to a homogenous sample of 115 kDa, consistent with a dimer of two subunits of Mw = 51 kDa. The elution profiles are represented according to retention volume (in ml) with the refractive index (in mV) indicated on the left axis and the logarithm of molecular weight (Mw) on the right axis. (B) SEC-MALS analysis of NprR(ΔHTH). The peak retention volume of 13.8 ml and the log(Mw) value under the peak correspond to a molecular weight of 97 kDa, consistent with a dimer of two subunits of Mw = 44,5 kDa. (C) Effect of peptide binding on the NprR electrophoretic mobility. Native gel analysis of 30 µM NprR and NprR (ΔHTH) samples in the presence (+) and in the absence (−) of 1 mM NprX7. Full-length NprR was loaded in lanes 1 and 2 and NprR(ΔHTH) in lanes 4 and 5 from the same gel. The gels were stained using Coomassie blue. See also Supplementary Figure S1. (D) NprR activation by NprX7 and NprX8n. Complementation of the ΔRX nprA’Z [pHT304-R] mutant strain with 5 µM of synthetic peptides. The bacteria were grown at 37°C in HCT medium, and each peptide were added independently 1 h after the onset of the stationary phase. β-Galactosidase activity of the ΔRX nprA’Z [pHT304-R] mutant strain was measured 3 h later. The control corresponds to the β-galactosidase activity of the ΔRX nprA’Z [pHT304-R] mutant strain without addition of peptide. Data are averages of three independent experiments (vertical bars are SEM from mean values).
Table 1.
DLS analysis of NprR and NprR(ΔHTH) in the presence and absence of peptide
| Samples | Rha (nm) | Pdb (%) | MWc (kDa) | Oligomeric state |
|---|---|---|---|---|
| NprR (51 kDa) | 4.4 | 4.5 | 109 | Dimeric |
| NprR/NprX7 | 5.8 | 9.9 | 208 | Tetrameric |
| NprRΔHTH (45 kDa) | 4.2 | 4.2 | 99 | Dimeric |
| NprRΔHTH/NprX7 | 5.7 | 11.8 | 198 | Tetrameric |
aRh, hydrodynamic radius.
bPd, polydispersity.
cMW, molecular weight.
The minimal active form of NprX has been shown to be the heptapeptide SKPDIVG (26). ITC experiments (Supplementary Figure S1) allowed us to demonstrate that the conserved motif SKPDI is not recognized by the NprR protein, explaining why it was found to be inactive. Addition of a serine residue at the N-terminal extremity of the heptapeptide to form the NprX8n SSKPDIVG octapeptide increased its affinity for the NprR(ΔHTH) protein (Kd decreasing from 0.10 ± 0.02 µM to 0.03 ± 0.01 µM). However, addition of a second residue to form NprX9n YSSKPDIVG had the opposite effect (Kd increasing to 0.22 ± 0.03 µM). C-terminal elongation of the peptide decreased its affinity for NprR to a Kd of ∼1 µM observed with NprX8c SKPDIVGQ. Functional analysis using β-galactosidase assays (Figure 1D) confirmed that NprX8n efficiently activates NprR. Based on these results, we used both NprX7 and NprX8n peptide forms in the co-crystallization assays.
The NprR/NprX tetramer is a dimer of dimers
To determine the structural basis of the activation mechanism of NprR, we tried to crystallize NprR alone and in complex with NprX. The apo form of the protein did not crystallize, but we obtained crystals of the truncated form NprR(ΔHTH) in complex with the heptapeptide NprX7. The crystals diffracted up to 3.5 Å resolution. They belong to space group P1 and contained 12 molecules per asymmetric unit. Initial crystallographic phases were determined by the SAD method using a selenomethionine labeled form of NprR(ΔHTH) co-crystallized with NprX7. The positions of 120 selenium atoms corresponding to the 11 methionines contained in each NprR(ΔHTH) chain, except the N-terminal residue, were determined. The anomalous signal of Met417 at the C-terminal extremity of the protein was weak but visible. This helped us assign the complete sequence of the protein, despite the low resolution of the diffraction data set. A native data set obtained with NprR(ΔHTH) in complex with the octapeptide NprX8n diffracted up to 3.2 Å resolution. It was used for final refinement of the model. Data processing and refinement statistics are given in Table 2.
Pairwise superposition of the 12 NprR(ΔHTH) molecules of the asymmetric unit results in an rms distance below 1 Å for ∼330 aligned Cα atoms. Each monomer is composed of 18 α−helices forming nine TPR motifs arranged in a two-layered right-handed superhelix (Figure 2A). In most chains, there was no visible electron density for the surface loop connecting helices α15 and α16 from TPR 8. The C-terminal His-tag was not visible, and five to ten residues were also disordered at both extremities of each chain. The electron density map of the last helix α18 was of poorly defined, most probably owing to the proximity of the flexible His-tag. The 12 molecules form three tetramers displaying a maximal rms distance of ∼1 Å for 1320 aligned Cα atoms. Each NprR(ΔHTH) tetramer (Figure 2B) is characterized by an overall interface area of ∼3800 Å2 and a dissociation energy ΔG of ∼11 kcal/mol.
Figure 2.

Crystal structure of the NprR(ΔHTH)/NprX complex. (A) Subunit fold. NprR(ΔHTH) is shown as cartoon colored by TPR from blue (TPR1) to purple (TPR9). The numbering of the helices and of the TPR motifs is indicated. The bound peptide is highlighted as spheres colored by spectrum from blue to red. (B) Quaternary structure. The NprR(ΔHTH) tetramer is shown as cartoon and colored by chain. The bound peptides are represented as in panel A. (C) Type I dimer. The two subunits colored by chain as in panel B are shown as cartoon and surface. The contact region is highlighted and a close view of the interface residues Y223 and F225 is shown. (D) Type II dimer. The two subunits colored by chain as in panel B are shown as cartoon and surface. The contact region is highlighted and a close view of the interface residues N407 and Y410 is shown. See also Supplementary Figure S2.
Analysis of subunit interactions in the tetramer showed that it contained two types of dimers, with equivalent interface areas of ∼950 Å2 and a solvation energy ΔiG of ∼−10 kcal/mol. The dimer formed by two antiparallel chains (Figure 2C) has been called type I dimer. It is mainly stabilized by van der Waals contacts between symmetry related residues from loops α8-α9 and α10-α11. In particular, a strong stacking interaction is observed between residues Y223 and F225 of loop α8-α9 from each subunit. These residues belong to a highly conserved motif observed in all NprR pherogroups (Supplementary Figure S2). In the so-called type II dimer (Figure 2D), two chains interact mainly through hydrophobic contacts between symmetry related residues from helices α9 and α18. In particular, residues Y410, conserved among all pherogroups of NprR (Supplementary Figure S2), and N407 participate to this type II interface.
The interface of the crystallographic type II dimer is conserved in the apo NprR dimer
Mutations affecting the subunit contacts observed in the NprR(ΔHTH)/NprX crystal structure were engineered to assess the conformation of the apo dimer. The Y223A/F225A and N407A/Y410A mutations were introduced in NprR(ΔHTH) to disrupt the type I and type II interfaces, respectively. Analytical size exclusion chromatography was used to assess the oligomerization state of the interface mutant proteins. The Y223A/F225A mutant protein displayed the same elution volume than the dimeric NprR(ΔHTH) sample, whereas the N407A/Y410A mutant protein was eluted as a monomer according to the calibration of the column (Figure 3A). These results suggest that the apo dimer is stabilized by the interactions observed in the crystallographic dimer II. Native gel electrophoresis (Figure 3B) showed that NprR(ΔHTH)(N407A/Y410A) migrated faster than the Y223A/F225A mutant protein and that addition of peptide did not affect the mobility of the two mutant proteins. This result suggested that none of the two mutant proteins could form the active tetramer. However, ITC experiments demonstrated that the mutations did not induce a loss of affinity for the peptide: both mutant proteins displayed a Kd value of ∼0.1 µM similar to that of the wild-type protein (Figure 3C). Taken together, these results indicate that NprX binding cannot promote the formation of the active tetramer neither by association of apo dimers with modified type I interface residues (Y223A/F225A mutant) nor from NprR monomers with modified type II interface residues (N407A/Y410A). Transcription assays performed with the two NprR mutant proteins confirmed that no expression of the nprA’lacZ reporter gene could be observed on addition of NprX on the NprR(Y223A/F225A) and NprR(N407A/Y410A) mutant proteins (Figure 3D). These results suggest that the role of NprX binding consists in promoting the association of two apo dimers stabilized by type II interactions, resulting in the formation of an active tetramer stabilized by type I interactions.
Figure 3.

Characterization of the NprR(ΔHTH) interface mutant proteins. (A) Size exclusion chromatography analysis. In all, 100 µl of protein samples at 5 mg/ml have been loaded in the absence of peptide on an analytical SuperdexTM 200 10/30 column (GE Healthcare) pre-equilibrated in 20 mM Tris–HCl (pH 8) 100 mM NaCl. The elution profiles of NprR(ΔHTH) in pink, the type I dimer interface mutant NprR(ΔHTH)(Y223A/F225A) in cyan and the type II dimer interface mutant NprR(ΔHTH)(N407A/Y410A) in green have been superimposed. The calibration of the column is shown at the top of the figure. (B) Native gel analysis. The electrophoretic mobility of the interface mutant proteins is shown in the presence (+) and in the absence (−) of the peptide NprX7. (C) ITC characterization of the NprR interface mutants (Y223A/F225A) and (N407A/Y410A). The isothermal titration curves between both NprR interface mutants and NprX7 are shown with their respective calculated Kd values. (D) Transcription assays with the NprR interface mutants. The specific activity of the Bacillus ΔRX nprA′Z mutant strain carrying the plasmids pHT-R[Y223A-F225A]X or pHT-R[N407A-Y410A]X (Supplementary Table S2) is expressed in units of ß-galactosidase per milligram of protein (Miller units). Time zero was defined as the onset of the stationary phase.
NprX activation mechanism involves the NprR conserved residue Arg126
Crystal structure analysis of the NprR(ΔHTH)/NprX7 and NprR(ΔHTH)/NprX8n complexes showed that the peptide binds in a deep cleft in the middle of the TPR domain of the protein. Clear electron density corresponding to the bound peptide was observed in the TPR domain of both structures (Figure 4A). The interface area covers ∼725 Å2 in the complex with NprX7 and 780 Å2 with NprX8n, explaining the 4-fold increase of the affinity observed by ITC. The complex is mainly stabilized by van der Waals interactions and H-bonds between main chain atoms from the peptide and protein side chains from the TPR motifs 1 to 7 (Figure 4B). The side chain of the NprX D residue corresponding to the single conserved position among the seven phylogenetic groups of NprX peptides (26) is the only one to specifically interact with the protein. It forms H-bonds with the side chain of the conserved residue Arg126 from loop α3-α4 in the TPR2 motif of NprR (Supplementary Figure S2). Arg126 also interacts with the C-terminal carboxylate of the peptide. ITC experiments demonstrated that the loss of the D-Arg126 interaction increased the Kd value from 0.10 µM to 0.64 µM while substitution of the COO− by a CO-NH group had an even more drastic effect with a 50-fold increase of the Kd that reached the value of 5.5 µM (Figure 4C). Interestingly, besides the fact that the loss of the D-Arg126 interaction reduced the affinity of the protein for the peptide, it also abolished the band shift characteristic of tetramer formation observed on native gel on addition of peptide (Supplementary Figure S3). This result was confirmed by transcription assays showing that NprX did not activate the NprR(R126A) mutant protein (Figure 4D). Taken together, these results strongly suggest that the D-Arg126 interaction plays an essential role in the activation mechanism of NprR by NprX.
Figure 4.

Analysis of the NprR/NprX interaction. (A) Electron density map of the peptide-binding site. The (2Fo-Fc) electron density map is represented in gray mesh at a contour level of 1σ around the bound NprX8n (SSKPDIVG) shown in sticks and colored by atoms. The side chains of the three protein residues implicated in the activation mechanism (Arg126, Trp167 and His201) are shown as sticks in their electron density and labeled. The protein is displayed as cartoon colored by TPR according to Figure 2A. (B) Peptide binding mode. A close view of the peptide binding site is shown with NprR(ΔHTH) displayed as cartoon and colored by TPR motifs according to Figure 2A. The protein residues directly interacting with the peptide are highlighted in sticks and labeled (Tyr193 and Asn194 from helix α7, Glu241 from α9, Ser274 and Asn275 from α11 and Asn306 and Asp309 from α13). The H-bonds are shown as yellow dashed lines. The bound NprX8n peptide (SSKPDIVG) is shown in sticks colored by atom type. See also Supplementary Figure S2. (C) ITC characterization of NprR and NprX mutants affecting the Arg126-D interaction. The isothermal titration curve of the NprR(ΔHTH)(R126A)/NprX7 interaction is compared with the results obtained with two variant forms of the NprX7 peptide, SKPAIVG and SKPDIVG-CO-NH. The calculated Kd values are indicated. (D) Transcription assays with NprR(R126A). The specific activity of the Bacillus ΔRX nprA′Z mutant strain carrying the plasmid pHT-R[R126A]X (Supplementary Table S2) is expressed in units of ß-galactosidase per milligram of protein (Miller units). Time zero was defined as the onset of the stationary phase. See also Supplementary Figure S3.
Peptide recognition specificity
Previous functional experiments demonstrated that NprR from group I was activated by both NprX7 from groups I and II, but not from the five other identified phylogenetic groups (26). We used ITC experiments to test if this result could reflect the affinity of the protein for different NprX7 sequences (Figure 5). The active peptides NprX7-I (SKPDIVG) and NprX7-II (SKPDTYG) displayed about the same affinity for NprR-I, with respective Kd values of ∼0.1 µM and 0.4 µM. Modeling of these substitutions did not induce any clashes with the surrounding protein residues. This suggests that the substituted positions 5 and 6 are not essential for binding or for activation. The inactive peptide (WKPDTLG) characteristic of groups V to VII and displaying an additional substitution in position 1 only poorly interacted with NprR-I, with Kd≈3 µM. Modeling analysis indicated that this could be explained by sterical hindrance introduced by the W substitution. A strong decrease of the affinity was also observed (Kd≈6 µM) with the inactive peptide NprX7-IV (SRPDVLT) presenting substitutions in positions 2, 5, 6 and 7. Together with the reduced affinity observed when substituting the C-terminal CO-OH group by CO-NH in NprX7-I, this result suggests that the G residue at the C-terminus of the peptide is important for proper positioning of the carboxylate interacting with the essential Arg126. More surprisingly, the inactive peptide NprX7-III (SNPDIYG) displayed a strong affinity for the protein, with a Kd of ∼0.08 µM. As the active peptide NprX7-II also contains the V to Y substitution in position 6, the inactivity of NprX7-III could be mainly induced by the K to N substitution in position 2, suggesting that the K residue also plays a role in the formation of the active tetramer.
Figure 5.

Peptide specificity analysis. The isothermal titration curves of NprR(ΔHTH) with NprX7 peptides from the different pherogroups are shown with their corresponding Kd values. The sequence of each peptide is indicated with the modified position compared with NprX7-I highlighted in bold.
DISCUSSION
Our study demonstrates that signaling peptide binding shifts the inactive apo dimer of NprR toward an active tetrameric NprR/NprX complex. In the tetrameric conformation, the N-terminal extremities of two adjacent NprR(ΔHTH) subunits are distant of ∼34 Å. This corresponds to the distance between two half sites of a B-type DNA target. In the absence of an NprR/NprX/DNA crystal structure, a hypothetical model was built by juxtaposing to the NprR(ΔHTH)/NprX complex two DNA-bound HTH dimers from the recently solved structure of the PlcR/PapR/DNA complex (PDB ID 3U3W) (43) (Figure 6). In this model, the two DNA-binding domains of the protein are positioned at opposite ends of the tetramer, in a manner similar to the one shown for the PrgX/pheromone complex (20). The two DNA target sites of PrgX have been identified, and an allosteric regulation mechanism has been proposed (44). The tetrameric conformation of the NprR/NprX complex thus suggests a similar mechanism. However, the two DNA target sites of the NprR/NprX tetramer remain to be identified to corroborate this hypothesis.
Figure 6.
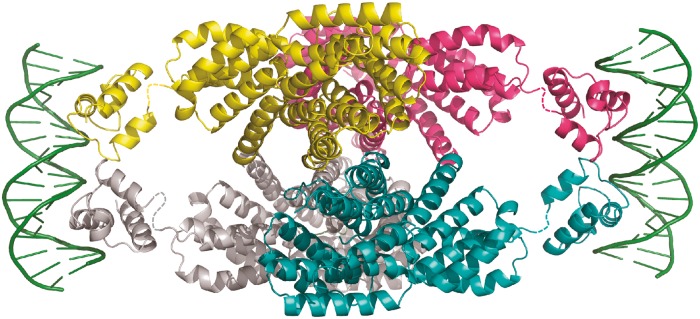
Model of full-length NprR in complex with DNA. The NprR(ΔHTH)/NprX tetramer is displayed in cartoon and colored by chain. The HTH domains from the recently solved structure of the PlcR/PapR/DNA complex (PDB ID 3U3W) are displayed in cartoon and colored according to the adjacent NprR(ΔHTH) subunit. The flexible linkers are shown as dashed lines. The DNA molecule is shown as green cartoon.
The peptide-binding mode is conserved among all members of the RNPP family. Like already reported (15), the peptide side chains are mostly involved in non-bonded hydrophobic interactions, whereas the protein mainly H-bonds with the peptide backbone via asparagines (Figure 4B). In particular, the NprR/NprX complex displays an interaction between the peptide and NprR residue Asn275 that is conserved in all RNPP proteins (15,21,45). However, the peptide-induced conformational changes are specific for each system. NprX binding triggers tetramerization in NprR, whereas the PrgX tetramer, which is different from the NprR tetramer, is alternatively destabilized by cCF10 and stabilized by iCF10 binding (46).
In the molecular mechanism that has been proposed for the activation mode of the RNPP regulator PlcR, peptide binding destabilizes the N-terminus of the PlcR dimer, allowing subsequent conformational changes on DNA binding (43). Recently, two structures of peptide-bound Rap proteins have been published (47,48), showing that peptide binding induces a drastic conformational change at the N-terminus of the protein. The 3-helix bundle and the linker helix observed in the RapH/Spo0F (22) and RapF/ComA (23) complexes adopt a classical TPR conformation in the RapJ/PhrC (47) and RapF/PhrF (48) complexes. In this conformation (PDB ID 4GYO), the RapJ fold is similar to that observed in the NprR/NprX complex. Both proteins display the same number of TPRs and an rms distance of ∼2.5 Å over 294 aligned Cα atoms.
Similarly, we hypothesized that in the absence of bound peptide, loop α3-α4 of the NprR TPR2 motif could undergo a large conformational change resulting in reorientation of the first TPR motif and of the N-terminal HTH DNA-binding domain. As a consequence, the tetramer would dissociate into two apo dimers displaying altered TPR domains. Our mutagenesis analysis further suggested that the type II interface observed in the tetramer would be conserved in this apo dimer. This hypothesis is supported by SAXS measurements performed with apo NprR(ΔHTH). We compared the experimental data with simulated curves calculated from different models based on the type II dimer of the NprR(ΔHTH)/NprX complex. After addition of the flexible N- and C-terminal extremities missing in the crystal structure, a good fit (Figure 7A) could be obtained by introducing a kink and flexibility in loop α3-α4. The resulting model of the apo NprR(ΔHTH) dimer (Figure 7B) mimicks the Rap conformational change, supporting the hypothesis that NprR might be an evolutionary intermediate between the Rap proteins and the other subgroups of the RNPP family. It supports the essential role of Arg126, which is located in this hinge region and is most probably directly involved in the peptide-induced conformational change. It also shows that the two N-terminal extremity of the NprR(ΔHTH) apo dimer are too distant (∼110 Å) to allow proper positioning of the HTH domains in the two half-sites of the DNA target as shown in Figure 5. In the apo dimer of full-length NprR, each missing HTH domain could also be sequestered in an interaction with the adjacent TPR domain, in a similar manner as observed in the complex between RapF and the HTH domain of ComA (23). The conformational change induced by peptide binding would thus have two consequences: (i) release the HTH domains and (ii) favor the tetramerization allowing HTH dimerization and DNA binding.
Figure 7.

Solution analysis of the apo NprR dimer. (A) SAXS analysis. Fit of the experimental SAXS curve of apo NprR(ΔHTH) (in blue) to the calculated curve obtained with the best model produced by DaDiModO (in red). (B) Model of apo NprR(ΔHTH). The best model of apo NprR(ΔHTH) produced by DaDiModO (see ‘Materials and Methods’ section) is shown in ribbon colored by chain (yellow and cyan) with TPR1 and TPR2 of both chains in blue. The crystal dimer II used as initial model is superimposed in cartoon with the same colors.
This activation mechanism based on a peptide-induced switch from an inactive apo dimer to an active tetrameric complex seems to be common to all NprR/NprX pairs. Indeed, multiple sequence alignments of NprR homologues including members from the seven phylogenetic subfamilies previously identified (26) demonstrated that most contact residues from both dimer I and II interfaces are conserved in the seven pherogroups (Supplementary Figure S2).
We showed that residues D and K of NprX7-I as well as its C-terminal carboxylate are essential to the activation process. The D residue, conserved among all pherogroups of NprX7, and the COO- group interact with Arg126, which belongs to a sequence motif (KRD) strictly conserved in all NprR proteins (Supplementary Figure S2). We hypothesized that the loss of the Arg126-peptide interaction could induce the conformational change proposed to occur in the TPR2 motif. Our results also suggested that the residue K of NprX7-I could play a role in the activation of NprR. Its side chain is involved in a hydrophobic stacking interaction with His201 that further interacts with Trp167 from the TPR3 motif (Figure 4). The latter is adjacent to the TPR4 motif carrying residues Y223 and F225 involved in the formation of the tetramer. We thus suggest that the K residue of the peptide could be involved in a rearrangement of the superhelix formed by the TPR domain of the apo dimer, allowing the formation of the dimer I interface observed in the complex.
Finally, our study suggests that a competition between NprX peptides from different pherogroups can occur. The characterization of the non-productive binding of peptide will provide necessary insights for the rational design of quorum-quenching molecules useful as new antimicrobial agents. As the characterization of the RNPP family, a rapid increase of the number of sensor regulators uncovered within the low GC% Gram-positive bacteria was observed, pointing out their important role in adaptative and virulence processes (26,49–51). This clearly identifies these regulators as major targets for the search of new molecules with applications in medical, food and biotechnology areas.
ACCESSION NUMBERS
PDB accession number: 4GPK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–2 and Supplementary Figures 1–3.
FUNDING
Centre National de la Recherche Scientifique (CNRS); Institut National de la Recherche Agronomique (INRA); french Agence Nationale de la Recherche [Cell.com project, N° ANR-09-BLAN-0253]. Funding for open access charge: Centre National de la Recherche Scientifique.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank staffs of beamlines Proxima-1 and Swing at the french synchrotron SOLEIL and ID14, ID23 and ID29 at the European Synchrotron Radiation Facility. They acknowledge the use of the IMAGIF crystallization platform and thank N. Lazar and B. Collinet for SEC-MALS analysis.
REFERENCES
- 1.Miller MB, Bassler BL. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001;55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- 2.Williams P. Quorum sensing, communication and cross-kingdom signalling in the bacterial world. Microbiology. 2007;153:3923–3938. doi: 10.1099/mic.0.2007/012856-0. [DOI] [PubMed] [Google Scholar]
- 3.Lyon GJ, Novick RP. Peptide signaling in Staphylococcus aureus and other Gram-positive bacteria. Peptides. 2004;25:1389–1403. doi: 10.1016/j.peptides.2003.11.026. [DOI] [PubMed] [Google Scholar]
- 4.Lazazzera BA. The intracellular function of extracellular signaling peptides. Peptides. 2001;22:1519–1527. doi: 10.1016/s0196-9781(01)00488-0. [DOI] [PubMed] [Google Scholar]
- 5.Slamti L, Lereclus D. A cell-cell signaling peptide activates the PlcR virulence regulon in bacteria of the Bacillus cereus group. EMBO J. 2002;21:4550–4559. doi: 10.1093/emboj/cdf450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pottathil M, Lazazzera BA. The extracellular Phr peptide-Rap phosphatase signaling circuit of Bacillus subtilis. Front. Biosci. 2003;8:d32–45. doi: 10.2741/913. [DOI] [PubMed] [Google Scholar]
- 7.Lanigan-Gerdes S, Dooley AN, Faull KF, Lazazzera BA. Identification of subtilisin, Epr and Vpr as enzymes that produce CSF, an extracellular signalling peptide of Bacillus subtilis. Mol. Microbiol. 2007;65:1321–1333. doi: 10.1111/j.1365-2958.2007.05869.x. [DOI] [PubMed] [Google Scholar]
- 8.Pomerantsev AP, Pomerantseva OM, Camp AS, Mukkamala R, Goldman S, Leppla SH. PapR peptide maturation: role of the NprB protease in Bacillus cereus 569 PlcR/PapR global gene regulation. FEMS Immunol. Med. Microbiol. 2009;55:361–377. doi: 10.1111/j.1574-695X.2008.00521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ji G, Beavis RC, Novick RP. Cell density control of staphylococcal virulence mediated by an octapeptide pheromone. Proc. Natl Acad. Sci. USA. 1995;92:12055–12059. doi: 10.1073/pnas.92.26.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solomon JM, Lazazzera BA, Grossman AD. Purification and characterization of an extracellular peptide factor that affects two different developmental pathways in Bacillus subtilis. Genes Dev. 1996;10:2014–2024. doi: 10.1101/gad.10.16.2014. [DOI] [PubMed] [Google Scholar]
- 11.Bouillaut L, Perchat S, Arold S, Zorrilla S, Slamti L, Henry C, Gohar M, Declerck N, Lereclus D. Molecular basis for group-specific activation of the virulence regulator PlcR by PapR heptapeptides. Nucleic Acids Res. 2008;36:3791–3801. doi: 10.1093/nar/gkn149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gominet M, Slamti L, Gilois N, Rose M, Lereclus D. Oligopeptide permease is required for expression of the Bacillus thuringiensis plcR regulon and for virulence. Mol. Microbiol. 2001;40:963–975. doi: 10.1046/j.1365-2958.2001.02440.x. [DOI] [PubMed] [Google Scholar]
- 13.Perego M, Hoch JA. Cell-cell communication regulates the effects of protein aspartate phosphatases on the phosphorelay controlling development in Bacillus subtilis. Proc. Natl Acad. Sci. USA. 1996;93:1549–1553. doi: 10.1073/pnas.93.4.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonard BA, Podbielski A, Hedberg PJ, Dunny GM. Enterococcus faecalis pheromone binding protein, PrgZ, recruits a chromosomal oligopeptide permease system to import sex pheromone cCF10 for induction of conjugation. Proc. Natl Acad. Sci. USA. 1996;93:260–264. doi: 10.1073/pnas.93.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Declerck N, Bouillaut L, Chaix D, Rugani N, Slamti L, Hoh F, Lereclus D, Arold ST. Structure of PlcR: Insights into virulence regulation and evolution of quorum sensing in Gram-positive bacteria. Proc. Natl Acad. Sci. USA. 2007;104:18490–18495. doi: 10.1073/pnas.0704501104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 17.D'Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem. Sci. 2003;28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Wintjens R, Rooman M. Structural classification of HTH DNA-binding domains and protein-DNA interaction modes. J. Mol. Biol. 1996;262:294–313. doi: 10.1006/jmbi.1996.0514. [DOI] [PubMed] [Google Scholar]
- 19.Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol. Rev. 2005;29:231–262. doi: 10.1016/j.femsre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 20.Shi K, Brown CK, Gu ZY, Kozlowicz BK, Dunny GM, Ohlendorf DH, Earhart CA. Structure of peptide sex pheromone receptor PrgX and PrgX/pheromone complexes and regulation of conjugation in Enterococcus faecalis. Proc. Natl Acad. Sci. USA. 2005;102:18596–18601. doi: 10.1073/pnas.0506163102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozlowicz BK, Shi K, Gu ZY, Ohlendorf DH, Earhart CA, Dunny GM. Molecular basis for control of conjugation by bacterial pheromone and inhibitor peptides. Mol. Microbiol. 2006;62:958–969. doi: 10.1111/j.1365-2958.2006.05434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parashar V, Mirouze N, Dubnau DA, Neiditch MB. Structural basis of response regulator dephosphorylation by Rap phosphatases. PLoS Biol. 2011;9:e1000589. doi: 10.1371/journal.pbio.1000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker MD, Neiditch MB. Structural basis of response regulator inhibition by a bacterial anti-activator protein. PLoS Biol. 2011;9:e1001226. doi: 10.1371/journal.pbio.1001226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishikawa S, Core L, Perego M. Biochemical characterization of aspartyl phosphate phosphatase interaction with a phosphorylated response regulator and its inhibition by a pentapeptide. J. Biol. Chem. 2002;277:20483–20489. doi: 10.1074/jbc.M201086200. [DOI] [PubMed] [Google Scholar]
- 25.Bongiorni C, Stoessel R, Shoemaker D, Perego M. Rap phosphatase of virulence plasmid pXO1 inhibits Bacillus anthracis sporulation. J. Bacteriol. 2006;188:487–498. doi: 10.1128/JB.188.2.487-498.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perchat S, Dubois T, Zouhir S, Gominet M, Poncet S, Lemy C, Aumont-Nicaise M, Deutscher J, Gohar M, Nessler S, et al. A cell-cell communication system regulates protease production during sporulation in bacteria of the Bacillus cereus group. Mol. Microbiol. 2011;82:619–633. doi: 10.1111/j.1365-2958.2011.07839.x. [DOI] [PubMed] [Google Scholar]
- 27.Dubois T, Faegri K, Perchat S, Lemy C, Buisson C, Nielsen-LeRoux C, Gohar M, Jacques P, Ramarao N, Kolsto AB, et al. Necrotrophism is a quorum-sensing-regulated lifestyle in Bacillus thuringiensis. PLoS Pathog. 2012;8:e1002629. doi: 10.1371/journal.ppat.1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993;26:795–800. [Google Scholar]
- 29.Sheldrick GM. A short history of SHELX. Acta Crystallogr. A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 30.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terwilliger T. SOLVE and RESOLVE: automated structure solution, density modification and model building. J. Synchrotron. Radiat. 2004;11:49–52. doi: 10.1107/s0909049503023938. [DOI] [PubMed] [Google Scholar]
- 32.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D. Biol. Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 33.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta. Crystallogr. D. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 34.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Smart OS, Vonrhein C, Womack TO. BUSTER. version 2.8.0. Cambridge, United Kingdom: Global Phasing Ltd; 2009. [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 38.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 39.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galtier N, Gouy M, Gautier C. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput. Appl. Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- 41.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 42.Evrard G, Mareuil F, Bontems F, Sizun C, Perez J. DADIMODO: a program for refining the structure of multidomain proteins and complexes against small-angle scattering data and NMR-derived restraints. J. Appl. Cryst. 2011;44:1264–1271. [Google Scholar]
- 43.Grenha R, Slamti L, Nicaise M, Refes Y, Lereclus D, Nessler S. Structural basis for the activation mechanism of the PlcR virulence regulator by the quorum-sensing signal peptide PapR. Proc. Natl Acad. Sci. USA. 2013;110:1047–1052. doi: 10.1073/pnas.1213770110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bae T, Kozlowicz B, Dunny GM. Two targets in pCF10 DNA for PrgX binding: their role in production of Qa and prgX mRNA and in regulation of pheromone-inducible conjugation. J. Mol. Biol. 2002;315:995–1007. doi: 10.1006/jmbi.2001.5294. [DOI] [PubMed] [Google Scholar]
- 45.Diaz AR, Core LJ, Jiang M, Morelli M, Chiang CH, Szurmant H, Perego M. Bacillus subtilis RapA phosphatase domain interaction with its substrate, phosphorylated Spo0F, and its inhibitor, the PhrA peptide. J. Bacteriol. 2012;194:1378–1388. doi: 10.1128/JB.06747-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fixen KR, Chandler JR, Le T, Kozlowicz BK, Manias DA, Dunny GM. Analysis of the amino acid sequence specificity determinants of the enterococcal cCF10 sex pheromone in interactions with the pheromone-sensing machinery. J. Bacteriol. 2007;189:1399–1406. doi: 10.1128/JB.01226-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parashar V, Jeffrey PD, Neiditch MB. Conformational change-induced repeat domain expansion regulates rap phosphatase quorum-sensing signal receptors. PLoS Biol. 2013;11:e1001512. doi: 10.1371/journal.pbio.1001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gallego Del Sol F, Marina A. Structural basis of rap phosphatase inhibition by phr peptides. PLoS Biol. 2013;11:e1001511. doi: 10.1371/journal.pbio.1001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng F, Ji H, Cao M, Wang C, Feng Y, Li M, Pan X, Wang J, Qin Y, Hu F, et al. Contribution of the Rgg transcription regulator to metabolism and virulence of Streptococcus suis serotype 2. Infect. Immun. 2011;79:1319–1328. doi: 10.1128/IAI.00193-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fleuchot B, Gitton C, Guillot A, Vidic J, Nicolas P, Besset C, Fontaine L, Hols P, Leblond-Bourget N, Monnet V, et al. Rgg proteins associated with internalized small hydrophobic peptides: a new quorum-sensing mechanism in streptococci. Mol. Microbiol. 2011;80:1102–1119. doi: 10.1111/j.1365-2958.2011.07633.x. [DOI] [PubMed] [Google Scholar]
- 51.Chang JC, LaSarre B, Jimenez JC, Aggarwal C, Federle MJ. Two group A streptococcal peptide pheromones act through opposing Rgg regulators to control biofilm development. PLoS Pathog. 2011;7:e1002190. doi: 10.1371/journal.ppat.1002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.