

Abstract
Antidepressant usage in prodromal Huntington Disease (HD) remains uncharacterized, despite its relevance in designing experiments, studying outcomes of HD, and evaluating the efficacy of therapeutic interventions. We searched baseline medication logs of 787 prodromal HD and 215 healthy comparison (HC) participants for antidepressant use. Descriptive and mixed-effects logistic regression modeling characterized usage across participants. At baseline, approximately one in five prodromal HD participants took antidepressants. Of those, the vast majority took serotonergic antidepressants (selective serotonin reuptake inhibitor (SSRI) or serotonin/norepinephrine reuptake inhibitor (SNRI)). Significantly more prodromal HD participants used serotonergic antidepressants than their HC counterparts. Because of the prevalence of these medications, further analyses focused on this group alone. Mixed-effects logistic regression modeling revealed significant relationships of both closer proximity to diagnosis and female sex with greater likelihood to be prescribed a serotonergic antidepressant. More prodromal HD participants took antidepressants in general and specifically the subclass of serotonergic antidepressants than their at-risk counterparts, particularly when they were closer to predicted time of conversion to manifest HD. These propensities must be considered in studies of prodromal HD participants.
Keywords: Psychiatric, Antidepressant, Neuroprotection, Clinical trials, SSRI
1. Introduction
Huntington disease (HD) is a progressive neurodegenerative disease with an autosomal dominant inheritance pattern, which typically leads to symptoms in three domains: cognition, behavior, and movement. HD is caused by a CAG (glutamine) repeat expansion (≥36) in the IT15/HTT gene which encodes the protein huntingtin. Longer repeat expansions are associated with earlier onset of manifest HD symptoms. Manifest HD is diagnosed on the basis of unequivocal evidence of an extrapyramidal movement disorder (dystonia, dysarthria, chorea, gait disturbance, postural instability, oculomotor dysfunction) (Huntington Study Group, 1996), though cognitive and behavioral symptoms are often observed years before motor signs develop (Paulsen et al., 2008).
Prodromal HD is defined by the presence of the CAG repeat expansion (as determined by genetic testing) in the absence of signs of a diagnosable movement disorder. Studies of prodromal HD allow investigations into the earliest stages of cognitive and motor decline (Paulsen et al., 2006), as well as psychiatric changes including irritability, depression, apathy, anxiety, obsessive–compulsive behaviors (OCB), and psychosis (Cummings, 1995; Naarding et al., 2001; Paulsen et al., 2001; Duff et al., 2007; van Duijn et al., 2007; Beglinger et al., 2008), which can be amenable to pharmacological intervention.
Leroi et al. (2002) reported that over the course of HD, major depression affects 28.6% of patients, while an additional 14.3% are affected by “non-major” depression according to formal Diagnostic and Statistical Manual - IV (DSM-IV) criteria. By comparison, Hasin et al. (2005) reported that the prevalence of 12-month and lifetime diagnosis of Major Depressive Disorder was 5.28% and 13.23%, respectively, in a sample of more than 43,000 healthy adults aged 18 and older residing in households and group quarters in the United States. Sixty-four percent of HD patients show a history of irritability and agitation (Pflanz et al., 1991), whereas 15–50% demonstrate obsessive and compulsive symptoms (Beglinger et al., 2007). However, the prevalence and incidence of psychiatric symptoms in prodromal HD remain unclear as they vary widely between studies (Naarding et al., 2001; Duff et al., 2007; van Duijn et al., 2007). The extreme variation in reported prevalence and incidence of these symptoms makes it difficult to determine whether these symptoms are being adequately managed and to ascertain whether these symptoms are affecting the overall progression of the disease.
Few randomized, placebo-controlled trials have been conducted to examine the effectiveness of symptomatic treatments in HD. Case studies of HD patients show that depression is improved by treatment with atypical antipsychotics, selective serotonin reuptake inhibitors (SSRIs), monoamine oxidase inhibitors (MAOIs), and tetracyclic, and tricyclic antidepressants (Patel et al., 1996; Squitieri et al., 2001; Bonelli et al., 2003; Ciammola et al., 2009). SSRIs can also decrease irritability, agitation, and obsessive–compulsive behaviors in individuals with HD (Ranen et al., 1996; Como et al., 1997; Patzold and Brune, 2002; Royuela Rico et al., 2003). While anecdotal evidence supports the use of newer-generation antidepressants, controlled clinical trials are needed to objectively measure their efficacy in HD (Adam and Jankovic, 2008).
Antidepressant usage may explain some of the difficulty in assessing the natural progression of psychiatric symptoms in prodromal HD. Studies have failed to reveal consistent patterns of significant psychiatric symptoms in prodromal HD (Soliveri et al., 2002; Julien et al., 2007; Witjes-Aneetal., 2007), which may be related to the efficacy of medications, nonadherence to treatment regimens, or under-diagnosis of emotional disturbance. Furthermore, observational studies of psychiatric symptoms in HD are not randomized, rendering findings difficult to interpret.
Antidepressant usage remains unreported for prodromal HD participants, despite burgeoning interest in the efficacy of these compounds for various therapeutic interventions. Patterns of antidepressant use are important to consider when designing clinical trials in HD. First, the base rate of antidepressant use is necessary to inform recruitment strategies in an already rare participant population. Second, antidepressant usage will likely need to be controlled in clinical trials because of possible interactions with drugs of interest. Third, antidepressant medications may have interactions with the natural course of HD progression, which needs to be controlled in the experimental setting.
1.1. Purpose
The purpose of the current study was to provide descriptive information about the patterns of antidepressant use in a large cohort of prodromal Huntington disease participants and a healthy at-risk comparison group. We also examined demographic and clinical predictors of serotonergic antidepressant use and asked whether this use was related to the extent to which individuals exhibited motor symptoms on the Unified Huntington Disease Rating Scale (UHDRS).
2. Methods
2.1. Participants
Participants were recruited to the PREDICT-HD study, an ongoing multi-site longitudinal investigation of the biological and neurobehavioral predictors of early disease in people with the gene expansion for HD and gene non-expanded comparison participants from HD-affected families (Paulsen et al., 2006; Paulsen et al., 2008). PREDICTHD study participants complete biennial magnetic resonance imaging (MRI) and annual visits that include a detailed motor examination, cognitive testing, psychiatric rating scales, and medication usage reporting. Exclusion criteria include a history of other central nervous system disease or events (seizures, trauma), developmental cognitive disorders, pacemakers, metallic implants, prescribed antipsychotic or phenothiazine derivative medications in the past 6 months, or clinical evidence of unstable medical or psychiatric illness. There are no restrictions for over-the-counter and natural remedies. Participants are included if they have a family history of HD, are 18 years or older, and have completed voluntary and independent genetic testing prior to enrollment in the study. As a result of their genetic testing, all participants are aware of their CAG repeat status prior to enrollment in the PREDICT-HD study. The study was approved by institutional review boards at all study and data-processing sites. Participants provided informed consent for participation, and all aspects of the study are in compliance with national legislation and the Declaration of Helsinki.
The study collects data from two groups: (1) people with the pathologic gene expansion (CAG≥36) but without motor signs sufficient for clinical HD diagnosis at the time of enrollment (prodromal HD), and (2) those at risk due to having a parent with HD, but without the pathologic gene expansion (CAG<30; healthy comparison (HC)). Confirmation of polyglutamine (CAG) repeat length is determined from baseline blood draws. Data from 1002 participants (787 prodromal HD, 215 HC) enrolled between September 2002 and November 2008 were utilized for the current study. The total participant pool was 97.80% Caucasian, 88.42% right-handed, 66.83% married, and 63.67% female. See Table 1 for additional demographic information. Participants who show diagnosable motor signs at baseline are excluded from the study. However, a proportion of the participants who enrolled without diagnosable motor symptoms and developed them over the course of the study were considered to have “converted” to manifest HD. These participants continued to take part in the study and were included in the current report as another comparison group. The overall number of “converted” participants was 108 (36 men, 72 women).
Table 1.
Demographics.
| prHD | HC | Statistic | P-value | |
|---|---|---|---|---|
| Age | 40.82 (9.76)a | 43.91 (11.59)a | t= −3.58 | <0.001 |
| Education | 14.32 (2.68) | 14.67 (2.69) | t= −1.74 | 0.08 |
| Sex | 63.41% F (499F:288M) | 64.65% F (139F:76M) | χ2 = 0.11 | 0.74 |
Abbreviations: prHD=prodromal Huntington disease, HC=healthy comparison. Study evaluates 787 prHD and 215 HC participants.
Significant difference in age between groups, t-test used Satterthwaite adjustment for unequal variances.
2.2. UHDRS motor assessment
At each visit to a PREDICT-HD study site, participants are examined on motor, cognitive, behavioral, and physical measures. The motor assessment measures the following motor dimensions of HD: ocular pursuit, saccade initiation, saccade velocity, dysarthria, tongue protrusion, finger tapping speed, pronation–supination of hands, Luria test of motor sequencing, arm rigidity, bradykinesia, maximal dystonia and chorea, gait, tandem walking, and the retropulsion pull test (Huntington Study Group, 1996). In the current analysis we use the total motor score summed across all these dimensions as the outcome variable for the longitudinal naturalistic study of serotonergic antidepressant users' motor sign progression compared to the progression of non-users. A higher score on the motor assessment is indicative of more impaired performance.
2.3. Diagnostic confidence level (DCL)
The diagnostic confidence level refers to the level of certainty a motor-rater has about assigning a clinical diagnosis of HD based on the presence of overt motor abnormalities observed during the motor assessment of the UHDRS. DCL is rated on a 5-point scale that ranges from 0 (normal) to 4 (99% confidence that the patient has manifest HD) (Huntington Study Group, 1996). This measure is subjective and thus highly variable, but is a useful proxy for the examiner's impression of the participant's stage in prodromal HD progression. This measure has been widely used clinically as a scale of disease progression. At baseline, participants with a DCL of 4 were excluded because they did not meet criteria to participate in a prodromal study. However, participants who enrolled with a DCL<4 and received a rating of 4 later in the study were retained and used as a diagnosed comparison group.
2.4. Estimated time to diagnosis
For our logistic regression and naturalistic analyses, we considered antidepressant usage rates across groups defined by estimated time to diagnosis, wherein participants were classified as far from diagnosis (≥15 years), midway to diagnosis (9–15 years), and near to diagnosis (<9 years). Estimated time to diagnosis was based on the Langbehn et al. formula which uses CAG repeat expansion length and current age at baseline visit to model estimated years to diagnosis (Langbehn et al., 2004).
2.5. Medication logs
Participants' medication logs were examined for the generic and trade names of all antidepressants approved for use in countries with at least one study site. Medications were analyzed based on groups as defined in Appendix A. The serotonergic antidepressant group (presented in Fig. 2) included both the SSRI group and the less selective group of serotonin/norepinephrine reuptake inhibitors (SNRIs). The medication classification scheme we used involved separating pure SSRIs from the less specific SNRIs because these medications have different mechanisms of action and may be of research interest separately. It should be further noted that because of its novel mechanism and frequency of usage, bupropion was considered to be in a class of its own. The frequency of antidepressant usage in the sample was then determined using descriptive analyses. For the purpose of this analysis, participants were considered antidepressant users if they reported having a prescription for any antidepressant at any dose. Dosage level, reason for treatment, and adherence to prescription were not analyzed in the current study because the data were self-reported with unknown reliability. In addition, it should be noted that bupropion is the second most commonly used antidepressant in our analysis and it is often prescribed as a smoking cessation aid, independent of psychiatric symptom manifestation. This type of usage may further complicate the picture of psychiatric symptoms during prodromal HD.
Fig. 2.
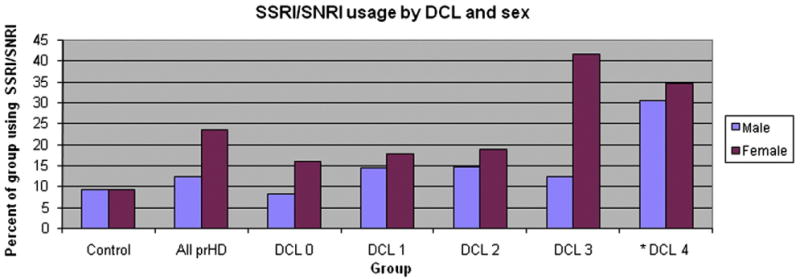
Comparison of serotonergic antidepressant usage across diagnostic confidence levels by sex. Note: serotonergic antidepressant usage is expressed as the percent of the specified sex and diagnostic group that identifies a current SSRI or SNRI prescription at baseline visit (e.g., 16.04% of female participants who have a DCL of 0 took an SSRI or SNRI at baseline). *See note for Fig. 1.
2.6. Analysis
Descriptive analyses examined the rates of usage of all classes of antidepressants at the baseline visit of all participants. In this analysis, we used diagnostic confidence level as the measure of disease progression because it is a useful proxy for the examiner's impression of the participant's symptoms, which we hypothesized would be related to the likelihood of receiving serotonergic antidepressant treatment. Antidepressant usage rates in diagnosed participants were based on usage at the first visit at which a particular participant was considered diagnosed (DCL 4 or “diagnosed” group).
Next, because of the very high frequency of SSRI and SNRI usage and the hypothesized effects of these compounds on prodromal HD progression, mixed-effects logistic regression examined the use of these medications in relation to gene status (prodromal HD or HC), estimated time to diagnosis, research visit number, and research visit year, controlling for age, sex, research site, and repeated visits. Research visit number was included in the model because of preliminary analyses suggesting increased likelihood to be prescribed an SSRI or SNRI with increasing visit number. We added visit year as a covariate to ascertain whether there were temporal trends of increased or decreased prescription rates based on the calendar year. Baseline descriptive analyses are presented in terms of the participants' proximity to diagnosis and sex because these were significant predictors of antidepressant usage in the logistic regression.
2.7. Longitudinal naturalistic study of SSRI/SNRI treatment in prodromal HD participants
We compared the motor performance of prodromal HD participants using SSRIs or SNRIs to the performance of prodromal HD participants not using these compounds longitudinally to test whether such drugs had a demonstrable association with longterm prodromal HD progression. We examined this effect by fitting a mixed model (Brown and Prescott, 1999) with total motor score on the UHDRS as the outcome and subject as a random effect. SSRI/SNRI use was a time-dependent variable, as was prodromal HD group (far, mid, near, diagnosed), since conversion to diagnosed status occurred longitudinally. We fit interactions of group and visit number as an indication of mean motor score per visit within each group. An additional interaction between group and SSRI/SNRI use in the interval prior to the current visit was also fit, allowing group-specific examination of differences in motor score between subjects on and off SSRI/SNRIs. We used group-specific first order autoregressive moving-average autocorrelation matrices, supported by the Akaike Information Criterion (AIC) comparisons to alternative covariance structures (Brown and Prescott, 1999), to account for within-subject correlation and notable residual heterogeneity among groups in motor score variance. We fit additional similar mixed models that incorporated higher order interactions of SSRI/SNRI use, visit, and group in order to test whether SSRI/SNRI use had a significant effect on rates of motor score change. For these analyses, an SSRI/SNRI user was defined as someone who was on an SSRI/SNRI in the interval prior to the current visit or at the time of the current visit. A non-user was someone who did not use an SSRI/SNRI in the interval prior to the current visit or at the time of the current visit. We did not control for lifetime history of antidepressant usage prior to study enrollment. Data from the 108 participants who enrolled without diagnosable motor symptoms but who converted to “manifest HD” during the course of the study were included in this analysis. For these participants, all visits at which they were considered prodromal were included in the prodromal categories and then their first visit as a manifest HD participant was included in the “DCL 4” group.
3. Results
Demographic comparison of prodromal HD and HC groups revealed no statistically significant differences in education or sex, but it did show that the HC participant group was significantly older than the prodromal HD group (see Table 1).
See Fig. 1 for a summary of results. At baseline, 21.86% of prodromal HD participants took antidepressants, while only 13.02% of HC participants took antidepressants. Of the prodromal HD participants taking antidepressants, 72.67% took SSRIs or SNRIs. Slightly more prodromal HD participants used SSRIs or SNRIs than their HC counterparts (15.98% vs. 9.30%; χ2=6.06, p=0.01; see Fig. 1). Because of the relative prevalence of these compounds and their hypothesized neuroprotective effects in prodromal HD, further analyses focused on the serotonergic antidepressant group (SSRI or SNRI).
Fig. 1.
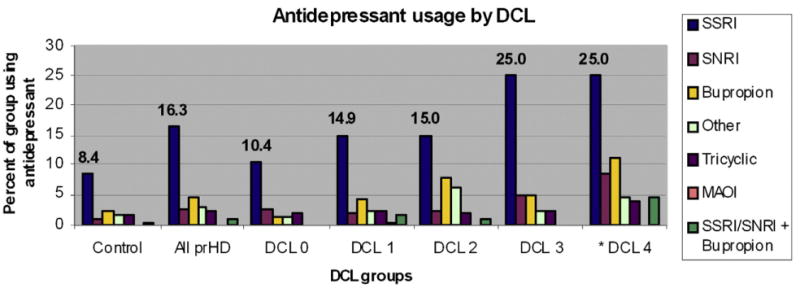
Use of different classes of antidepressant therapy across diagnostic confidence levels in prodromal HD and HC participants of the PREDICT-HD study. Note: diagnostic confidence level (DCL) refers to the motor rater's confidence that the participant is showing overt motor signs of Huntington disease. DCL 0 = normal, 1 ≤50% confidence, 2 = 50–89% confidence, 3 =90-98% confidence, 4≥ 99% confidence. Participant sex breakdown by group is as follows: comparison (76 men, 139 women), DCL 0 (120 men, 188 women), DCL 1 (111 men, 224 women), DCL 2 (41 men, 60 women), DCL 3 (16 men, 26 women), DCL 4 (36 men, 72 women). *Data for all groups except DCL 4 is taken from baseline visit. DCL 4 group is composed of the 108 participants who were considered prodromal HD at baseline but who converted to “manifest HD” during the course of the study. Medication usage for this group is taken from the participant's first visit after converting to manifest HD.
Mixed-effects logistic regression modeling showed a significant, positive relationship between elevated serotonergic antidepressant use and nearer estimated time to diagnosis ([F3,2460]=6.83, p=0.0001). Additionally, there was a significant, positive relationship of increased serotonergic antidepressant use and higher DCL ([F4,2230]=4.21, p=0.0021) (Fig. 1). Mixed-effects logistic regression modeling revealed that, among demographic variables, there was a statistically significant relationship between female sex and serotonergic antidepressant usage ([F1, 763.5]=11.61, p=0.0007) (Fig. 2). Trends relating age, prescription year, and visit number were non-significant (p>0.05) in the logistic regression model.
Longitudinal linear modeling of the association between serotonergic antidepressant treatment and motor signs revealed that diagnosed participants using these medications had higher total motor scores than those who weren't (see Table 2). In contrast, there were no significant motor differences in any of the prodromal HD groups or in comparison participants. The longitudinal model showed no significant evidence of different rates of motor score change between participants on and off serotonergic antidepressants ([F1,1134]=0.73, p=0.39). Finally, the potential three-way interaction between serotonergic antidepressant use, study visit, and group failed to show evidence of an association between rate of motor score change and usage of these medications ([F5,602]=1.54, p=0.18).
Table 2.
Naturalistic examination of total motor scores as a function of serotonergic antidepressant usage across groups.
| Group | Motor score: users | Motor score: non-users | Users–non-users | |
|---|---|---|---|---|
|
|
|
|
||
| Mean (95% CI) | Mean (95% CI) | Mean (95% CI) | P-value | |
| HC | 2.60 (1.65, 3.54) | 2.82 (2.41, 3.22) | −0.22 (−1.18, 0.73) | 0.65 |
| Far | 3.69 (2.87, 4.51) | 3.51 (3.08, 3.94) | 0.18 (−0.65, 1.01) | 0.67 |
| Mid | 5.29 (4.47, 6.12) | 5.07 (4.55, 5.58) | 0.22 (−0.62, 1.08) | 0.60 |
| Near | 8.28 (6.90, 9.66) | 8.32 (7.52, 9.11) | −0.04 (−1.46, 1.38) | 0.96 |
| DCL 4 | 23.94 (21.15, 26.73) | 19.52 (17.19, 21.84) | 4.42 (1.86, 6.99) | 0.0008 |
Note: participant groups are defined by estimated time to diagnosis: far from diagnosis (≥15 years), midway to diagnosis (9–15 years), and those near to diagnosis (<9 years). Estimated time to diagnosis considers CAG repeat expansion length and current age at baseline visit to model estimated years to diagnosis. Means reflect total motor score generated from the Unified Huntington's Disease Rating Scale (UHDRS). A serotonergic antidepressant user is defined as someone who was on an SSRI or SNRI in the interval prior to the current visit or at the time of the current visit A non-user was someone who did not use an SSRI or SNRI in the interval prior to the current visit or at the time of the current visit. The DCL 4 group is composed of the 108 participants who were considered prodromal at baseline but who converted to “manifest HD” during the course of the study. Healthy comparison participants were followed for an average of 2.86 years (range 1–6 years) and prodromal HD participants were followed for an average of 3.65 years (range 1–6 years). Abbreviations: HC = healthy comparison, DCL = diagnostic confidence level.
4. Discussion
Our findings indicate that significantly more prodromal HD participants take antidepressants than their HC counterparts (prodromal HD: 21.86%; HC: 13.02%). Of those, the most common class of drug is the serotonergic antidepressant (SSRI/SNRI) (prodromal HD: 15.98%; HC: 9.30%). Serotonergic antidepressant use was more frequent for individuals closer to diagnosis and for women. Of note, potential participants were excluded from the study if they use antipsychotic medications or have clinical evidence of unstable psychiatric illness. These exclusion criteria may cause under-reporting of antidepressant usage levels compared to the general prodromal HD population, suggesting that our findings may be a conservative underestimate of the real usage of these medications.
Stepwise increases in serotonergic antidepressant usage as individuals approached diagnosis were punctuated by a notable jump in usage at DCL 3 (see Fig. 1), which may reflect many factors. It is possible that as participants proceed through the prodromal phase of HD they develop more mood, obsessive, and/or anxious symptoms as a natural outcome of neurodegeneration. This degeneration may accelerate more rapidly as participants approach diagnosis. This hypothesis is supported by the significant overlap of brain regions affected by HD neuropathology with those implicated in Major Depressive Disorder. In particular, prodromal HD has been associated with decreased caudate nucleus, putamen, and global white matter volumes (Beglinger et al., 2005). These structural changes could be related to depressive symptom manifestation, as demonstrated by the work of Mayberg (1997), Drevets et al. (1992), Alexopoulos et al. (1997), Krishnan et al. (1997). Alternatively, the familial and environmental pressures of living with HD may increase notably when participants are faced with imminent development of manifest motor symptoms, causing an increase in depressive symptoms which may lead to antidepressant treatment in some cases. It is also possible that as individuals show more overt signs of HD development, the prescribing physician is more likely to recommend the use of an antidepressant.
Women were observed to be more likely to take an antidepressant than men at all stages of prodromal HD and during manifest HD (Fig. 2). The finding of increased antidepressant usage in prodromal HD women is consistent with the vast majority of depression studies in the non-HD segment of the population. For example, Weissman and Klerman (1977) found a 2:1 female to male ratio among depressed individuals to be consistent across 12 studies conducted in the United States from 1936 to 1973. Hasin et al. (2005) later found female sex to be associated with increased risk for developing depressive symptoms and receiving pharmacological treatment. Mojtabai (2008) further showed that women were twice as likely to have a current prescription for an antidepressant as men between 2001 and 2003.
For a comparison to our study, Mojtabai (2008) found that 10.1% of respondents in a nationally representative cross-sectional survey of households in the U.S. between 2001 and 2003 had received prescriptions for antidepressants in the past year. Our findings suggest that the rate of antidepressant use in prodromal HD participants is more than double the usage of the general population. The rate among healthy comparison participants is likewise elevated, though not to as great an extent, which may reflect the pressures and stresses of living in a family affected by HD. Some of this elevation may reflect effects of sex on antidepressant use: participants in the Mojtabai study were 57% female, while those in our study were 63% female. However, it should be noted that our finding of increased antidepressant use in women is in line with a recently published study by Pang et al. (2009) which showed a female-specific depression-associated behavioral phenotype in transgenic HD mice. The finding of elevated antidepressant usage in the men with prodromal HD in the current study was inconsistent with the findings by Pang et al. (2009), and there are several possible explanations for the discrepancy. First, there are substantive differences in the manifestation of HD and depression between human participants and animal models of disease, not least of which in this case is the extent of CAG repeat expansion. The average CAG repeat expansion in the human participants in the current study was 42.48 (standard deviation (S.D.)=2.55), while the R6/1 transgenic line used by Pang and colleagues contained 116 CAG repeats. That difference, in addition to the greater genetic homogeneity in the mouse models due to the use of inbred strains, could result in any number of inconsistencies between studies in humans and mouse models. In addition, there may be other uniquely human factors playing into the relationship, such as psychosocial pressures and treatment with SSRIs for indications other than depression (for example, using bupropion as a smoking cessation aid), which would not necessarily be detectable in the mouse model. One final difference could have led to the observed findings: the sample included in the current study was 25 times larger than the sample included in the Pang study, which could have resulted in the detection of variability in the current sample that was undetectable in a smaller group of animals.
These findings have implications for HD research and experimental design. Any studies requiring drug-free participants will need to consider how the prevalence of antidepressant usage affects the number of eligible participants and whether drug-free participants would be representative of the true target population. On the other hand, inclusion of such participants may lead to drug interactions as well as antidepressant effects on other experimental psychotherapeutic interventions. Therefore, it may be prudent to exclude those taking an antidepressant, although this could lead to problems of sample size and limited statistical power.
Moreover, existing studies of psychiatric symptoms in prodromal HD may have been confounded with usage patterns and efficacy of antidepressant treatments if these variables were not assessed and controlled. These studies have typically produced mixed and inconclusive patterns, which may be due to differential medication effects (van Duijn et al., 2007). In future studies, it will be especially important to consider medication effects as they pertain to the prevalence of DSM-defined major depression or other categorical mood disorders in prodromal HD.
Research in humans with HD suggests that serotonergic antidepressants may positively affect the course of prodromal HD in a variety of capacities including motor, behavioral, and cognitive symptom manifestation, though not consistently (Como et al., 1997). De Marchi et al. (2001) found that treatment with fluoxetine improved motor and behavioral symptoms in two HD participants with significant obsessive and compulsive behaviors. Additionally, one of the participants showed improved cognitive function. These benefits were maintained for 2–6 years, despite the progressive nature of HD. More recent experiments with HD mouse models have revealed that treatment with common SSRIs (paroxetine, fluoxetine, and sertraline) increased survival time, improved motor and behavioral function, decreased weight loss, improved glucose metabolism, suppressed neurodegeneration in the striatum, promoted neurogenesis in the hippocampus, improved cognitive performance on a T-maze paradigm, increased brain-derived neurotrophic factor (BDNF) levels, and helped maintain pro-apoptotic proteins at appropriate levels in the brain. (Duan et al., 2004; Duan et al., 2008; Peng et al., 2008; Mostert et al., 2008; Grote et al., 2005). With a growing body of animal model research supporting neuroprotective effects of SSRIs, studies in prodromal HD human participants are becoming critical. There has been recent interest in conducting naturalistic studies of the proposed neuroprotective effects in prodromal HD human participants who use antidepressants. Our preliminary naturalistic analysis revealed a finding that runs counter to the hypothesis of SSRIs as neuroprotective agents in prodromal HD (Duan et al., 2004; Grote et al., 2005; Duan et al., 2008; Mostert et al., 2008; Peng et al., 2008). We found that diagnosed participants who had prescriptions for SSRIs or SNRIs showed more motor signs of HD, as compared with the participants who did not take these medications (Table 2). However, participants were not randomly assigned to treatment groups, leaving the potential for confounding by indication. This obviously limits the interpretability of such naturalistic observations. In addition to the effects of sex and disease progression, we considered the effects of contact with the research environment and prescribing trends by year because of their potential for confounding the current naturalistic analysis. In consideration of the prescribing trends, we found that there was no significant temporal trend in serotonergic antidepressant prescription rates by calendar year (p = 0.3257) in the current sample. However, we did find that there was a non-significant trend toward increased likelihood to be prescribed an SSRI with increased contact with the research team and/or clinicians (p = 0.0943). Importantly, when controlling for the effects of calendar year and number of research visits, the relationship of DCL with serotonergic antidepressant use remains significant (p = 0.0013). Additionally, there appear to be effects of any number of traits that are specific to participants who choose to volunteer for prodromal HD research which limit the interpretability of the current analyses. These trends underscore the necessity of randomized controlled clinical trials of serotonergic antidepressant use in prodromal HD in order to appropriately answer the question of their potential neuroprotective effects.
There are several avenues for future investigation of the effects of serotonergic antidepressants in prodromal HD. The activity of motor nuclei heavily innervated by serotonergic afferents could be studied in animal models. The impact of serotonergic antidepressant treatment on structural brain measures such as striatal volume in prodromal HD participants could be analyzed longitudinally. This method may prove more sensitive to serotonin-mediated changes in prodromal HD participants than total motor score on the UHDRS, though analysis of such observational data could be fraught with many of the same confounding factors, requiring careful matching by propensity to receive antidepressants. Randomization of prodromal HD participants to serotonergic antidepressants could control for many of the factors that we found to be inextricably related to usage in our analyses.
Supplementary Material
Acknowledgments
This research is supported by the National Institutes for Health, National Institute of Neurological Disorders and Stroke (NS40068) and CHDI Foundation, Inc.
The authors would like to acknowledge Jessica Wood, for pharmacological input, Justin O'Rourke, Christine Werling, Sean Thompson, and Kristine Bjork, for editorial assistance, all at The University of Iowa. We would also like to thank the participants in the PREDICT-HD study for giving their time in support of this study.
Footnotes
Data previously presented at James F. Jakobsen Graduate Conference, The University of Iowa, March 27, 2010.
Appendix A. Supplementary data:Supplementary data to this article can be found online at doi:10.1016/j.psychres.2011.09.005.
References
- Adam OR, Jankovic J. Symptomatic treatment of Huntington disease. Neurotherapeutics. 2008;5:181–197. doi: 10.1016/j.nurt.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulos GS, Meyers BS, Young RC, Campbell S, Silbersweig D, Charlson M. ‘Vascular depression’ hypothesis. Archives of General Psychiatry. 1997;54:915–922. doi: 10.1001/archpsyc.1997.01830220033006. [DOI] [PubMed] [Google Scholar]
- Beglinger LJ, Nopoulos PC, Jorge RE, Langbehn DR, Mikos AE, Moser DJ, Duff K, Robinson RG, Paulsen JS. Cognitive and Behavioral Neurology. 2005;18:102–107. doi: 10.1097/01.wnn.0000152205.79033.73. [DOI] [PubMed] [Google Scholar]
- Beglinger LJ, Langbehn DR, Duff K, Stierman L, Black DW, Nehl C, Anderson K, Penziner E, Paulsen JS, Huntington Study Group Investigators Probability of obsessive and compulsive symptoms in Huntington's disease. Biological Psychiatry. 2007;61:415–418. doi: 10.1016/j.biopsych.2006.04.034. [DOI] [PubMed] [Google Scholar]
- Beglinger LJ, Paulsen JS, Watson DB, Wang C, Duff K, Langbehn DR, Moser DJ, Paulson HL, Aylward EH, Carlozzi NE, Queller S, Stout JC. Obsessive and compulsive symptoms in prediagnosed Huntington's disease. The Journal of Clinical Psychiatry. 2008;69:1758–1765. doi: 10.4088/jcp.v69n1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonelli RM, Mayr BM, Niederwieser G, Reisecker F, Kapfhammer HP. Ziprasidone in Huntington's disease: the first case reports. Journal of Psychopharmacology. 2003;17:459–460. doi: 10.1177/0269881103174009. [DOI] [PubMed] [Google Scholar]
- Brown H, Prescott R. Applied Mixed Models in Medicine. Wiley; New York: 1999. [Google Scholar]
- Ciammola A, Sassone J, Colciago C, Mencacci NE, Poletti B, Ciarmiello A, Squitieri F, Silani V. Aripiprazole in the treatment of Huntington's disease: a case series. Neuropsychiatric Disease and Treatment. 2009;5:1–4. [PMC free article] [PubMed] [Google Scholar]
- Como PG, Rubin AJ, O'Brien CF, Lawler K, Hickey C, Rubin AE, Henderson R, McDermott MP, McDermott M, Steinberg K, Shoulson I. A controlled trial of fluoxetine in nondepressed patients with Huntington's disease. Movement Disorders. 1997;12:397–401. doi: 10.1002/mds.870120319. [DOI] [PubMed] [Google Scholar]
- Cummings JL. Behavioral and psychiatric symptoms associated with Huntington's disease. Advances in Neurology. 1995;65:179–186. [PubMed] [Google Scholar]
- De Marchi N, Daniele F, Ragone MA. Fluoxetine in the treatment of Huntington's disease. Psychopharmacology. 2001;153:264–266. doi: 10.1007/s002130000575. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Videen TO, Price JL, Preskorn SH, Carmichael ST, Raichle ME. A functional anatomical study of unipolar depression. The Journal of Neuroscience. 1992;12:3628–3641. doi: 10.1523/JNEUROSCI.12-09-03628.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ladenheim B, Xu X, Cadet JL, Mattson MP. Paroxetine retards disease onset and progression in Huntingtin mutant mice. Annals of Neurology. 2004;55:590–594. doi: 10.1002/ana.20075. [DOI] [PubMed] [Google Scholar]
- Duan W, Peng Q, Masuda N, Ford E, Tryggestad E, Ladenheim B, Zhao M, Cadet JL, Wong J, Ross CA. Sertraline slows disease progression and increases neurogenesis in N171-82Q mouse model of Huntington's disease. Neurobiology of Disease. 2008;30:312–322. doi: 10.1016/j.nbd.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Paulsen JS, Beglinger LJ, Langbehn DR, Stout JC, Predict-HD Investigators of the Huntington Study Group Psychiatric symptoms in Huntington's disease before diagnosis: the predict-HD study. Biological Psychiatry. 2007;62:1341–1346. doi: 10.1016/j.biopsych.2006.11.034. [DOI] [PubMed] [Google Scholar]
- Grote HE, Bull ND, Howard ML, van Dellen A, Blakemore C, Bartlett PF, Hannan AJ. Cognitive disorders and neurogenesis deficits in Huntington's disease mice are rescued by fluoxetine. The European Journal of Neuroscience. 2005;22:2081–2088. doi: 10.1111/j.1460-9568.2005.04365.x. [DOI] [PubMed] [Google Scholar]
- Hasin DS, Goodwin RD, Stinson FS, Grant BF. Epidemiology of major depressive disorder: results from the national epidemiologic survey on alcoholism and related conditions. Archives of General Psychiatry. 2005;62:1097–1106. doi: 10.1001/archpsyc.62.10.1097. [DOI] [PubMed] [Google Scholar]
- Huntington Study Group. Unified Huntington's disease rating scale: reliability and consistency. Movement Disorders. 1996;11:136–142. doi: 10.1002/mds.870110204. [DOI] [PubMed] [Google Scholar]
- Julien CL, Thompson JC, Wild S, Yardumian P, Snowden JS, Turner G, Craufurd D. Psychiatric disorders in preclinical Huntington's disease. Journal of Neurology, Neurosurgery, and Psychiatry. 2007;78:939–943. doi: 10.1136/jnnp.2006.103309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan KRR, Hays JC, Blazer DG. MRI-defined vascular depression. The American Journal of Psychiatry. 1997;154:497–501. doi: 10.1176/ajp.154.4.497. [DOI] [PubMed] [Google Scholar]
- Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR, Group International Huntington's Disease Collaborative A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clinical Genetics. 2004;65(267):268–277. doi: 10.1111/j.1399-0004.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- Leroi I, O'Hearn E, Marsh L, Lyketsos CG, Rosenblatt A, Ross CA, Brandt J, Margolis RL. Psychopathology in patients with degenerative cerebellar diseases: a comparison to Huntington's disease. The American Journal of Psychiatry. 2002;159:1306–1314. doi: 10.1176/appi.ajp.159.8.1306. [DOI] [PubMed] [Google Scholar]
- Mayberg HS. Limbic-cortical dysregulation: a proposed model of depression. Journal of Neuropsychiatry. 1997;9:471–481. doi: 10.1176/jnp.9.3.471. [DOI] [PubMed] [Google Scholar]
- Mojtabai R. Increase in antidepressant medication in the US adult population between 1990 and 2003. Psychotherapy and Psychosomatics. 2008;77:83–92. doi: 10.1159/000112885. [DOI] [PubMed] [Google Scholar]
- Mostert JP, Koch MW, Heerings M, Heersema DJ, De Keyser J. Therapeutic potential of fluoxetine in neurological disorders. CNS Neuroscience & Therapeutics. 2008;14:153–164. doi: 10.1111/j.1527-3458.2008.00040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naarding P, Kremer HP, Zitman FG. Huntington's disease: a review of the literature on prevalence and treatment of neuropsychiatric phenomena. European Psychiatry. 2001;16:439–445. doi: 10.1016/s0924-9338(01)00604-6. [DOI] [PubMed] [Google Scholar]
- Pang TY, Du X, Zajac MS, Howard ML, Hannan AJ. Altered serotonin receptor expression is associated with depression-related behavior in the R6/1 transgenic mouse model of Huntington's disease. Human Molecular Genetics. 2009;18:753–766. doi: 10.1093/hmg/ddn385. [DOI] [PubMed] [Google Scholar]
- Patel SV, Tariot PN, Asnis J. l-deprenyl augmentation of fluoxetine in a patient with Huntington's disease. Annals of Clinical Psychiatry. 1996;8:23–26. doi: 10.3109/10401239609149087. [DOI] [PubMed] [Google Scholar]
- Patzold T, Brune M. Obsessive compulsive disorder in Huntington disease: a case of isolated obsessions successfully treated with sertraline. Neuropsychiatry, Neuropsychology, and Behavioral Neurology. 2002;15:216–219. [PubMed] [Google Scholar]
- Paulsen JS, Hayden M, Stout JC, Langbehn DR, Aylward E, Ross CA, Guttman M, Nance M, Kieburtz K, Oakes D, Shoulson I, Kayson E, Johnson S, Penziner E, Predict-HD Investigators of the Huntington Study Group Preparing for preventive clinical trials: the Predict-HD study. Archives of Neurology. 2006;63:883–890. doi: 10.1001/archneur.63.6.883. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, Duff K, Kayson E, Biglan K, Shoulson I, Oakes D, Hayden M, Predict-HD Investigators Coordinators of the Huntington Study Group Detection of Huntington's disease decades before diagnosis: the Predict-HD study. Journal of Neurology, Neurosurgery, and Psychiatry. 2008;79:874–880. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Ready RE, Hamilton JM, Mega MS, Cummings JL. Neuropsychiatric aspects of Huntington's disease. Journal of Neurology, Neurosurgery, and Psychiatry. 2001;71:310–314. doi: 10.1136/jnnp.71.3.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, Masuda N, Jiang M, Li Q, Zhao M, Ross CA, Duan W. The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington's disease mouse model. Experimental Neurology. 2008;210:154–163. doi: 10.1016/j.expneurol.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflanz S, Besson JA, Ebmeier KP, Simpson S. The clinical manifestation of mental disorder in Huntington's disease: a retrospective case record study of disease progression. Acta Psychiatrica Scandinavica. 1991;83:53–60. doi: 10.1111/j.1600-0447.1991.tb05511.x. [DOI] [PubMed] [Google Scholar]
- Ranen NG, Lipsey JR, Treisman G, Ross CA. Sertraline in the treatment of severe aggressiveness in Huntington's disease. The Journal of Neuropsychiatry and Clinical Neurosciences. 1996;8:338–340. doi: 10.1176/jnp.8.3.338. [DOI] [PubMed] [Google Scholar]
- Royuela Rico A, Gil-Verona JA, Macias Fernandez JA. A case of obsessive symptoms in Huntington's disease. Actas Españolas de Psiquiatría. 2003;31:367–370. [PubMed] [Google Scholar]
- Soliveri P, Monza D, Piacentini S, Paridi D, Nespolo C, Gellera C, Mariotti C, Albanese A, Girotti F. Cognitive and psychiatric characterization of patients with Huntington's disease and their at-risk relatives. Neurological Sciences. 2002;23(Suppl 2):S105–S106. doi: 10.1007/s100720200091. [DOI] [PubMed] [Google Scholar]
- Squitieri F, Cannella M, Piorcellini A, Brusa L, Simonelli M, Ruggieri S. Short-term effects of olanzapine in Huntington disease. Neuropsychiatry, Neuropsychology, and Behavioral Neurology. 2001;14:69–72. [PubMed] [Google Scholar]
- van Duijn E, Kingma EM, van der Mast RC. Psychopathology in verified Huntington's disease gene carriers. The Journal of Neuropsychiatry and Clinical Neurosciences. 2007;19:441–448. doi: 10.1176/jnp.2007.19.4.441. [DOI] [PubMed] [Google Scholar]
- Weissman MM, Klerman GL. Sex differences and the epidemiology of depression. Archives of General Psychiatry. 1977;34:98–111. doi: 10.1001/archpsyc.1977.01770130100011. [DOI] [PubMed] [Google Scholar]
- Witjes-Ane MN, Mertens B, van Vugt JP, Bachoud-Lévi AC, van Ommen GJ, Roos RA. Longitudinal evaluation of “presymptomatic” carriers of Huntington's disease. The Journal of Neuropsychiatry and Clinical Neurosciences. 2007;19:310–317. doi: 10.1176/jnp.2007.19.3.310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.