

Abstract
Integrin α7 (ITGA7) is a tumor-suppressor gene that is critical for suppressing the growth of malignant tumors; however, the mechanisms allowing ITGA7 to suppress the growth of cancer cells remain unclear. Herein, we show that ITGA7 binds to tissue inhibitor of metalloproteinase 3 (TIMP3) in prostate cancer cells. The ITGA7-TIMP3 binding led to a decreased protein level of tumor necrosis factor α, cytoplasmic translocation of NF-κB, and down-regulation of cyclin D1. These changes led to an accumulation of cells in G0/G1 and a dramatic suppression of cell growth. Knocking down TIMP3 or ITGA7/TIMP3 binding interference largely abrogated the signaling changes induced by ITGA7, whereas a mutant ITGA7 lacking TIMP3 binding activity had no tumor-suppressor activity. Interestingly, knocking down ITGA7 ligand laminin β1 enhanced ITGA7-TIMP3 signaling and the downstream tumor-suppressor activity, suggesting the existence of a counterbalancing role between extracellular matrix and integrin signaling. As a result, this report demonstrates a novel and critical signaling mechanism of ITGA7, through the TIMP3/NF-κB/cyclin D1 pathway.
Integrin α7 (ITGA7) is a member of the extracellular matrix binding proteins. As a major class of cell adhesion molecules in mammalian cells, integrins are involved in many cellular processes, including development, immune responses, leukocyte trafficking, and hemostasis.1 The integrin superfamily consists of 24 members, each of which mediates a unique function in mammals. The regulation of integrin expression is critical for certain aspects of tissue differentiation and regeneration (eg, keratinocyte differentiation, hair follicle formation, and skeletal muscle development),2–4 and abnormal integrin expression is associated with several human diseases (eg, muscular dystrophy, Glanzmann’s thrombasthenia, and congenital cardiac myopathy).4–6
ITGA7 forms a heterodimer with integrin β1 in the plasma membrane and is responsible for communication between the extracellular matrix and cells.7 Itga7-deficient mice display significant hyperplasia and hypertrophy of arteries and arterioles and a malformation of skeletal muscles.4,8 Recent mutational analysis revealed ITGA7 mutations in prostate cancer, hepatocellular carcinoma, soft tissue leiomyosarcoma, and glioblastoma multiforme, with frequencies ranging from 25% to 83%.9 Many of these mutations resulted in truncation, microdeletion, or frameshift of the protein. Interestingly, patients with prostate cancer or hepatocellular carcinoma harboring ITGA7 mutations also had a higher rate of clinical relapse.
A meta-analysis of previously published microarray data10–15 indicated that ITGA7 was down-regulated in nonmetastatic prostate cancer and leiomyosarcoma, but the magnitude of the down-regulation was larger in metastatic cancers. Also, prostate cancer and soft tissue leiomyosarcoma, with focal or no ITGA7 expression, were associated with a shorter metastasis-free survival time. The forced expression of normal ITGA7 in prostate cancer and leiomyosarcoma cell lines suppressed tumor growth and cancer cell migration in vitro. A mouse model of PC3 and DU145 xenograft prostate tumors showed a dramatic reduction in tumor volume, metastatic rate, and mortality rate when ITGA7 expression was restored. However, the molecular mechanism of ITGA7-mediated tumor-suppressor activity remains unclear. Herein, we report that the tissue inhibitor of metalloproteinase 3 (TIMP3), a matrix proteinase and a tumor suppressor, interacts with the C-terminus of ITGA7. We further show that the activation of ITGA7 leads to redistribution of NF-κB to the cytoplasm, the down-regulation of cyclin D1, and cell growth arrest.
Materials and Methods
Cell Lines and Cell Culture
The prostate cancer cell lines, DU145 and PC3, and an immortalized prostate epithelial cell line, RWPE-1, were purchased from ATCC (Manassas, VA) in 2007. These cell lines underwent one cycle of growth before being stored in liquid nitrogen. Cells were used for transfections within 2 weeks of thawing. DU145 cells were cultured in modified Eagle’s medium (Invitrogen, Carlsbad, CA), and PC3 cells were cultured in F12K (Invitrogen) supplemented with 10% fetal bovine serum (Cell Gro, Manassas), both at 37°C and 5% CO2. RWPE-1 cells were cultured in keratinocyte serum-free medium supplemented with 0.05 mg/mL bovine pituitary extract and 5 ng/mL recombinant epidermal growth factor (Invitrogen).
Plasmid Construction
For the construction of pBD-ITGA7c fusion proteins, a mutagenic primer set (5′-TGTGGCCTATAATCATATGACCTTTCTGGAGGAGTACTCA-3′ and 5′-TTCATTTCAAGCAAAGTCGACGCCTGGATCTGCTCTGCGCCCCTC-3′) was designed to generate two restriction sites, NdeI and SalI, in the C-terminal end (252 amino acids) of ITGA7, so that the resulting PCR product could be ligated into a pGBKT7 vector (Clontech Laboratories Inc., Mountain View, CA). PCR was performed on the cDNA template from the donor prostate (Clontech Laboratories Inc.) using the following conditions: 94°C for 1 minute, followed by 35 cycles of 94°C for 30 seconds, 68°C for 3 minutes, and a final 3-minute extension step at 68°C. The PCR product was cut with NdeI and SalI (New England Biolabs Inc., Ipswich, MA), gel purified, and ligated into a similarly digested pGBKT7 vector. The fusion protein contained 156 amino acids from the ITGA7 C-terminus. A similar strategy was used for pBD-ITGA7n (amino acid 2 to 986) construction, except the reverse primer was replaced with 5′-GCAGAATGCGGATCCCTAGCTGTTCCAGAGACGGCC-3′. The constructs were transformed into One Shot competent cells (Invitrogen). Plasmid DNA was extracted from selected transformed cells and digested with NdeI and SalI to detect the presence of the insert. The coding frame was confirmed by automated sequencing.
For construction of pGST-ITGA7, a glutathione S-transferase (GST) fusion protein, a mutagenic primer set (5′- AGGAATTCCCGGGTCGACGCCGGGGCTCGGAGCCGCGAC-3′ and 5′-AGTCACGATGCGGCCGCTGCCCCTGAGGAAGCCGATCCT-3′) was designed to generate SalI and NotI restriction sites within the ITGA7 coding region that encompasses 1136 amino acids of ITGA7. PCR was performed using these primers under the following conditions: 94°C for 1 minute, followed by 35 cycles of 94°C for 30 seconds, 68°C for 3 minutes, and a final 10-minute extension step at 68°C. The PCR product was gel purified and ligated into a pCR2.1 TA cloning vector (Invitrogen). A similar strategy was used for constructing pGST-ITGA7c (the forward primer sequence was 5′- AGGAATTCCCGGGTCGACGGGCAGGGGCCTGGGCAGAAA-3′), in which 252 amino acids of the ITGA7 C-terminus were encoded. The plasmid DNA was transformed into Escherichia coli. The plasmid DNA from the selected transformants was digested with SalI and NotI and was in frame ligated into a similarly restricted pGEX-5X-3 vector. A series of deletions, including 5′ or 3′ deletions, of pGST-ITGA7c was then performed using the primer sets described previously.16 The procedures for generating these mutants were similar to those described for pGST-ITGA7c. The pGST-ITGA7c and its mutants were transformed into E. coli BL21 cells for recombinant protein production.
Yeast Transformation and Library Screening
The yeast AH109 competent cell preparation was described previously.17 Freshly prepared AH109 competent cells (100 μL) were mixed with 0.25 to 0.50 μg pBD-ITGA7c plasmid and 0.5 μg plasmid DNA from a prostate cDNA library (constructed in pACT2), in 0.6 mL of polyethylene glycol/lithium acetate, and then incubated at 30°C for 30 minutes. After this initial incubation with plasmid DNA, the cell solution was combined with 70 μL of dimethyl sulfoxide (Sigma, St. Louis, MO) and subjected to 15 minutes of incubation at 42°C. The cells were pelleted, resuspended in 0.5 mL yeast peptone dextrose adenine medium, and plated on synthetic defined (SD) agar plates with different stringency. The transformants were plated directly to the low- (SD-Leu/-Trp), medium- (SD-Leu/-Trp/-His), or high-stringency (SD-Ade/-His/-Leu/-Trp and X-α-Gal) plates, or the colonies were grown on the low- and medium-stringency plates and were then replicated onto high-stringency plates. The grown colonies were subjected to the colony-lift filter β-galactosidase assay, as described previously.18 pBD-53 and pAD-T antigens were cotransformed into AH109 as positive controls; pBD7-Lamin C and pAD-T antigens were cotransformed as negative controls. pCL1 was transformed into AH109, as a positive control for the β-galactosidase assay.
Plasmid DNA from positive clones was isolated from yeast using the Zymoprep Yeast Plasmid Miniprep kit (Zymo Research, Orange, CA). The plasmids were transfected into E. coli and grown on Luria-Dulbecco agar plates with 100 μg/mL ampicillin to select pAD-target fusion gene plasmid. The ampicillin-resistant colonies that harbored pAD-target fusion gene DNA were propagated in luria-bertani medium containing 100 μg/mL ampicillin. The pACT2 plasmid DNA was purified and sequenced. Insert DNA sequences were analyzed for nucleotide homology using National Center for Biotechnology Information Blast programs.
Validation of Protein Interactions in AH109
Plasmid DNA from positive clones (ie, blue colonies on high-stringency plates) were isolated from yeast, transformed into E. coli, and selected with 100 μg/mL ampicillin to obtain gene-ACT2 fusion proteins interacting with the bait-domain fusion protein. The individual purified pACT2/library plasmid DNA was then cotransformed with pBD-ITGA7c, or pGBKT7 alone, into AH109 yeast cells and grown on SD-Ade/-His/-Leu/-Trp high-stringency medium. Colony-lift filter β-galactosidase activity was assayed on those cells grown in this medium, and the positive clones were sequenced.
GST Fusion Protein Pull Down
The E. coli cells harboring GST or GST-ITGA7c were grown in 5 mL of LB with 100 μg/mL ampicillin overnight at 37°C, diluted in 20× LB, incubated with shaking until the solution reached an OD of 0.6 to 1.0, and then induced by isopropyl-β-d-thiogalactopyranoside (final concentration, 1 mmol/L) for 3 hours. The cells were then pelleted, resuspended in 1× PBS, and sonicated for 2 minutes. The proteins were solubilized in 1% Triton X-100 (Sigma-Aldrich, St. Louis, MO), centrifuged at 15,000 × g for 5 minutes, and then the supernatant was collected. The GST and GST-ITGA7c fusion proteins were purified on Glutathione Sepharose 4B columns (Amersham Pharmacia Biotech Inc., Piscataway, NJ). The E. coli protein extract was pre-incubated with the column for 15 minutes at 4°C and washed. The HisTAG-purified HisTAG-TIMP3 was then incubated with GST fusion protein–packed Glutathione Sepharose 4B at 4°C for 2 hours. The column was spun at 3000 × g for 1 minute and washed twice with PBS. The protein was eluted from the column with 40 μL of SDS-PAGE gel sample loading dye. SDS-PAGE and Western blot analysis were subsequently conducted.
Immunofluorescent Staining
RWPE1 cells were cultured on chamber slides and were washed three times with PBS. The cells were fixed with 4% paraformaldehyde for 1 hour at room temperature. After washing the slides twice again with PBS, the cells were blocked with 10% donkey serum containing 0.4% Triton X-100. The cells were then incubated with rabbit antisera against ITGA7 and goat antisera against TIMP3 (Santa Cruz Biotechnology Inc., Santa Cruz, CA) at room temperature for 1 hour. The slides were again washed twice with PBS. Secondary antibodies from donkey directed against goat (fluorescein conjugated) and against rabbit (rhodamine conjugated) were added and incubated at room temperature for 1 hour. The slides were then washed twice with PBS before the addition of DAPI. After additional washes with PBS, slides were mounted with Prolong Gold Anti-fade Reagent (Invitrogen). Immunofluorescence staining was examined with a confocal microscope.
BrdU Labeling Analysis
To perform bromodeoxyuridine (BrdU) labeling analysis, 10 μL of BrdU solution (1 mmol/L BrdU in 1× PBS) was added to 1 mL of tissue culture media. The treated cells were then incubated for 3 hours at 37°C. Cells were then resuspended with 100 μL of BD Cytofix/Cytoperm Buffer per sample (BD-Pharmagen, San Jose, CA) and incubated for an additional 30 minutes at room temperature. The cells were then pelleted and washed with 1 mL of 1× BD Perm/Wash Buffer (BD-Pharmagen). The cells were then incubated with Cytoperm Plus Buffer (BD-Pharmagen) for 10 minutes on ice. The permeation procedure was repeated twice. The cells were resuspended with 100 μL of diluted DNaseI (diluted to 300 μg/mL in Dulbecco’s PBS or 30 μg of DNaseI per tube), and incubated for 1 hour at 37°C. The cells were then washed with 1× BD Perm/Wash Buffer and incubated with 50 μL of BD Perm/Wash Buffer containing diluted fluorescent anti-BrdU antibody (1:50) and propidium iodide for 20 minutes at room temperature. The incubation was then washed with 1 mL of 1× BD Perm/Wash Buffer. The immunoreactivity of BrdU was assessed using an LSC-II flow cytometer (BD-Pharmagen).
For siRNA analysis, 125 pmol siRNA specific for TIMP3 (5′-GCAGUACAUCCAUACGGAAGCUUCC/5Phos/GGAAGCUUCCGUAUGGAUGUACUGCAC-3′), siRNA specific for β1-laminin (5′-GAAAUGAAACUCAGCCUCCAGGUCCAG/5Phos/GGACCUGGAGGCUGAGUUUCAUUTC-3′), or non-specific control (5′-UAAUGUAUUGGAACGCAUAUU/UAUGCGUUCCAAUACAUUA-3′) was transfected into PITT1 or PITT2 cells using the Lipofectamine 2000 transfection kit (Invitrogen). Immunoblots and fluorescence-activated cell sorter analyses were performed 24 hours after transfection.
Immunoblot Assays
The immunoblot procedure was previously described.9,17–25 Briefly, cells were washed with PBS and lysed by radioimmunoprecipitation assay buffer (50 mmol/L Tris-HCl at pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L phenylmethylsulfonyl fluoride, 1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 μg/mL pepstatin, and 1 mmol/L Na3VO4). The lysates were sonicated and centrifuged at 12,000 × g at 4°C for 30 minutes to remove the insoluble materials. The proteins were separated by SDS-PAGE in 8.5% polyacrylamide gels, and bands were blotted onto a polyvinylidene difluoride membrane. The membrane was blocked with 5% powdered skim milk in Tris-Tween 20 buffer (0.1 mol/L Tris-HCl and 0.1% Tween-20, pH 7.4) for 1 hour at room temperature, followed by a 2-hour incubation with primary anti-ITGA7 antibodies (1:500 dilution), anti-TIMP3 antibodies (1:500 dilution; Santa Cruz Biotechnology Inc., Dallas, TX), anti–tumor necrosis factor (TNF)-α antibodies (1:500 dilution; Santa Cruz Biotechnology Inc., Santa Cruz, CA), anti-laminin β1 antibodies (1:500 dilution; Santa Cruz Biotechnology Inc., Santa Cruz, CA), anti-p65 antibodies (1:500 dilution; Santa Cruz Biotechnology Inc., Santa Cruz, CA), or anti-cyclin D1 antibodies (1:500 dilution; Santa Cruz Biotechnology Inc., Santa Cruz, CA). The membrane was then washed three times with Tris-Tween 20 buffer and incubated with a horseradish peroxidase–conjugated secondary antibody specific for rabbit (anti-ITGA7; 1:1000 dilution), mouse (anti-TIMP3, anti-p65, and anti–TNF-α; 1:1000 dilution) or goat (anti-cyclin D1 and anti-laminin β1; 1:1000 dilution) for 1 hour at room temperature. The protein expression was detected with the electrochemiluminescence system (Amersham Life Science, Piscataway, NJ), according to the manufacturer’s protocols.
Colony Formation Assay
The colony formation assay was performed as previously described.9,16–18,20,22,23,26–29 Five thousand cells were cultured in 60-mm dishes. Triplicate experiments were performed for each cell clone. Medium containing 10% fetal bovine serum (Invitrogen) was changed every 4 days. On the seventh day, the plates were stained with 1% crystal violet and colonies with a diameter of >2 mm were counted.
Chromatin Association Assay of the p65 Subunit of NF-κB
PITT1, PITT2, C5, or C8 cells were cultured to 75% confluence, synchronized in serum-free medium for 24 hours, and then treated with tetracycline for 24 or 48 hours. The procedure is similar to those previously described.24,27 Cells were washed with PBS, trypsinized, and then resuspended in 1 mL of Buffer A (110 mmol/L KC2H3O2, 15 mmol/L NaC2H3O2, 2 mmol/L MgC2H3O2, 0.5 mmol/L EGTA, and 20 mmol/L HEPES, pH 7.3). The cell suspension was treated with a final concentration of 2 mmol/L dithiothreital and 50 μg/mL digitonin. The cells were incubated at 4°C for 10 minutes in a rotator. Nuclei were pelleted by centrifugation at 1500 × g for 10 minutes and were then resuspended in a hypotonic buffer (Buffer B: 1 mmol/L HEPES, pH 7.5, and 0.5 mmol/L EDTA supplemented with 0.5% NP-40). The nuclear suspensions were then incubated at 4°C for 15 minutes in a rotator, layered onto a 10-mL sucrose cushion (100 mmol/L sucrose and 0.5 mmol/L Tris-HCl, pH 8.5), and centrifuged at 3500 × g for 15 minutes at 4°C. The chromatin pellets were suspended in 0.25 mmol/L EDTA, pH 8.0, and sonicated for three bursts of 10 seconds each. The chromatin suspensions were then centrifuged twice at high speed for 10 minutes at 4°C, and the supernatants were retained and stored.
Results
The intracellular domain of ITGA7 is located in the C-terminal end of the protein. To investigate the intracellular proteins that interact with ITGA7, a yeast two-hybrid screening analysis was performed using the 252 C-terminal amino acids of ITGA7 on a prostate cDNA library. Thirty-seven positive clones were identified after three rounds of metabolic screening. These clones grew in high-stringency medium agar plates (ie, SD-Trp-Leu-Ade-His) and were also positive for β-galactosidase activity. One of these clones contained cDNA encoding TIMP3, a tumor suppressor that plays a critical role in inhibiting metalloproteinase and blocks invasion and metastasis of cancers.
To validate this interaction, pAD-TIMP3, a plasmid that encodes the activation domain of GAL4 fused to TIMP3, and pBD-ITGA7c, which encodes a fusion of the DNA binding domain of GAL4 and the ITGA7 C-terminus, were cotransfected into yeast AH109 cells, grown in high-stringency medium, and tested for β-galactosidase activity. As shown in Figure 1A, TIMP3 transactivated the GAL4-ITGA7 fusion protein and mediated the transcription of necessary nutrients to survive in high-stringency agar and express β-galactosidase activity. In contrast, yeast cells transformed with pAD-Lam/pBD-ITGA7 alone did not survive on the high-stringency agar plate (negative control) (Figure 1A).
Figure 1.

ITGA7 binds TIMP3. A: β-Galactosidase activity of yeast harboring pBD-ITGA7c (amino acids 982 to 1138) and pAD-TIMP3, pBD-p53, and pAD-T antigen (positive control), or pBD-ITGA7n (amino acids 2 to 986) and pAD-TIMP3 or pBD-ITGA7c and pGADT7, or pGBKT7 and pAD-TIMP3, or pGBKT7 and pGADT7, or pBD-Lamin C and pAD-T antigen. B: Co-IP of ITGA7 and TIMP3. Proteins were extracted from RWPE1 and induced PITT1 cells (pCDNA4-ITGA7/pCDNA6TO transfected PC3 clone). The lysates were IP with the indicated antibodies and detected by Western blot (WB) analysis with either ITGA7 or TIMP3 antibodies. Lanes 13 to 20: RWPE1- or tetracycline-induced PITT1 cells were transfected with pCMV-FLAG or pCMV-TBM-FLAG (TIMP3 binding motif-FLAG). Immunoprecipitations and Western blot analyses were performed using the indicated antibodies. C: ITGA7 and TIMP3 colocalized in RWPE1. Immunostaining was performed using antibodies specific for ITGA7 or TIMP3, as described in Materials and Methods. Original magnification, ×20. D: ITGA7 binds with TIMP3 in vitro. Left panel: Diagrams of GST-ITGA7c mutants. Top right panel: Binding of GST-ITGA7c fusion proteins and its mutants with purified Histag-TIMP3 from bacterial extracts. After extensive washes, the bound proteins were eluted and immunoblotted with anti-TIMP3 antibodies. Bottom right panel: Coomassie Brilliant Blue staining of fusion proteins.
To investigate the binding of ITGA7 and TIMP3 in prostate cells, co-immunoprecipitation (co-IP) assays with ITGA7 or TIMP3 antibodies were performed using cell lysates of RWPE1, an immortalized prostate epithelial cell line. Immunoblot analysis indicated that TIMP3 was present in ITGA7 co-IP complexes. Conversely, ITGA7 was detected in TIMP3 co-IP complexes (Figure 1B). Similar results were obtained in co-IP assays of PC3 clones with inducible expression of ITGA7. Immunofluorescence staining of RWPE1 cells, using anti-ITGA7 and anti-TIMP3 antibodies, also detected the colocalization of these proteins in the cytoplasm and membranous regions (Figure 1C).
We then ligated the cDNA encoding 252 amino acids at the C-terminal end of ITGA7 into pGEX-5X-3, to generate a GST-ITGA7c fusion protein. A cell-free HisTAG-TIMP3/GST-ITGA7c binding analysis was then performed. The binding assay indicated that the TIMP3 and ITGA7 C-termini bind each other in vitro (Figure 1D). Subsequently, we generated a series of N- and C-terminus deletion mutants of the GST-ITGA7c fusion protein through mutational PCR. In vitro binding assays were performed with recombinant HisTag-TIMP3 expressed from E. coli. By overlapping the results of the binding assays, a 67–amino acid (1037 to 1103) motif was identified as critical for the ITGA7/TIMP3 interaction (Figure 1D).
To investigate the functional significance of the ITGA7/TIMP3 interaction, we analyzed the effect of TIMP3 on cell growth suppression induced by ITGA7. Because PC3 cells have minimal endogenous expression of ITGA7, we generated two PC3 cell clones (PITT1 and PITT2) with inducible ITGA7 expression by transforming pCDNA4-ITGA7/pCDNA6. We then used these clones to examine the impact of ITGA7 expression on the cell cycle. As shown in Figure 2, A and B, the induction of ITGA7 expression reduced colony formation of these cells by 64% to 65% (P < 0.001). The siRNA-mediated knockdown of TIMP3 largely abrogated the colony formation suppression effect by ITGA7 (within 8% of the controls; P = 0.12 and P = 0.4, respectively).
Figure 2.

TIMP3 is essential for ITGA7-mediated suppression of cell growth. A: Immunoblot analysis of ITGA7, TIMP3, and β-actin. PITT1 or PITT2 cells (pCDNA4-ITGA7/pCDNA6TO transfected PC3 clones) were induced with or without 5 μg/mL tetracycline (Tet) and treated with siRNA specific for TIMP3 (siTim) or scrambled control (Scr) for 24 hours. The protein extracts were separated by SDS-PAGE and immunoblotted with antibodies specific to ITGA7, TIMP3, or β-actin. B: Colony formation assays of replica samples of A. Triplicate experiments were performed on each assay. ∗∗P < 0.01 (t-test). C: BrdU labeling and cell cycle analyses of replica samples of A. Triplicate experiments were performed on each assay. ∗∗∗P < 0.001 (t-test).
The critical role of TIMP3 on cell growth suppression by ITGA7 was also reflected in cell cycle analyses in which PITT1 and PITT2 cells were induced with tetracycline, treated with siRNA specific for TIMP3, and labeled with BrdU. As shown in Figure 2C, the induction of ITGA7 in PITT1 cells increased the proportion of cells in the G0/G1 phase, from 46% to 76% (P < 0.001), whereas cells in the S phase decreased, from 41% to 12% (P < 0.001). Similarly, in PITT2 cells, the proportion of cells in G0/G1 increased, from 45% to 75% (P < 0.001), and those in the S phase decreased, from 42% to 11% (P < 0.001). Treatment of these cells with siRNA specific for TIMP3 completely abolished such effects (Figure 2C). These results suggest that TIMP3 mediates the critical cell growth suppression signaling by ITGA7.
One of the critical molecules that mediate TIMP3 tumor-suppressor activity is TNF-α.30,31 To investigate the impact of ITGA7/TIMP3 interaction on the TNF-α protein level, immunoblot analyses using antibodies specific for TNF-α were performed on protein extracts from PITT1 and PITT2 cells. The results indicate significantly lower levels of TNF-α in these cells when ITGA7 expression was induced (Figure 3). TNF-α signaling induces translocation of NF-κB from the cytosol to the nucleus.32 We then investigated whether expression of ITGA7 affected the cellular distribution of NF-κB by analyzing the subcellular localization of the p65 subunit of NF-κB in PITT1 and PITT2 cells when they were induced with tetracycline. As shown in Figure 3, the induction of ITGA7 reduced the chromatin association of NF-κB, while increasing its presence in the cytosol. Consistent with the redistribution of NF-κB in these cells, one of the main targets of NF-κB transcriptional activity, cyclin D1, was also found to be down-regulated in ITGA7-induced cells (Figure 3). Knockdown of TIMP3, using TIMP3-specific siRNA, largely eliminated the redistribution effect on NF-κB induced by ITGA7 and restored the expression of cyclin D1 (Figure 4). These data suggest that TIMP3 plays a key role in inhibiting the transcriptional activity of NF-κB.
Figure 3.
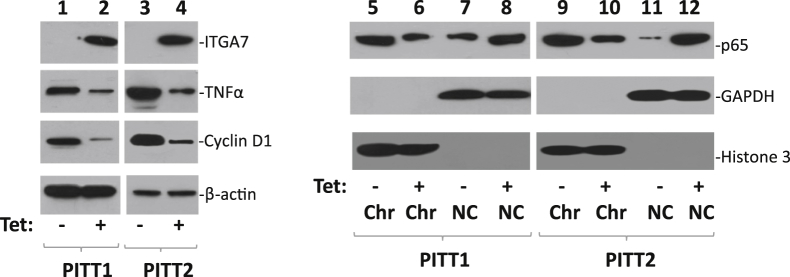
Induction of ITGA7 expression decreases TNF-α level and reverses nuclear localization p65 NF-κB subunit. Lanes 1–4: Immunoblots of ITGA7, TNF-α, cyclin D1, and β-actin from PITT1 and PITT2 cells induced with or without tetracycline (Tet). Lanes 5–12: Chromatin association of p65 NF-κB subunit. PITT1 and PITT2 cells were fractionated into chromatin (Chr) and non-chromatin (NC) fractions. Immunoblots were generated using antibodies specific for p65, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), or histone 3.
Figure 4.

TIMP3 is required for ITGA7-induced cyclin D1 down-regulation. PITT1 and PITT2 cells were induced with or without tetracycline (Tet) and treated with siRNA specific for TIMP3 (siTim) or scrambled (Scr) controls for 24 hours. Lanes 1 to 8: Immunoblots were generated from the protein extracts using antibodies specific for ITGA7, TIMP3, TNF-α, cyclin D1, or β-actin. Lanes 9 to 24: Chromatin association of p65 NF-κB subunit. PITT1 and PITT2 cells were fractionated into chromatin (Chr) and non-chromatin (NC) fractions. Immunoblots were generated using antibodies specific for p65, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), or histone 3.
To investigate whether the interaction between ITGA7 and TIMP3 is essential for suppressing the TNF-α signaling pathway, a mutant ITGA7 lacking the TIMP3 binding motif (amino acids 1037 to 1103) was cloned into pCDNA4 to generate pCDNA4-ITGA7Δ1037-1103. This ITGA7 mutant was then transfected into PC3 cells to examine the impact of such a mutation on cell growth and cell cycle progression. In comparison with cells transduced with wild-type ITGA7, pCDNA4-ITGA7Δ1037-1103–transformed PC3 clones (C5 and C8) were not growth suppressed (Figure 5) and displayed no cell cycle inhibition (pCDNA4-ITGA7C). Furthermore, the ITGA7 mutant had no impact on NF-κB distribution, and did not inhibit cyclin D1 expression. These experiments suggest that cell cycle progression and cyclin D1 expression suppression, induced by ITGA7, are dependent on TIMP3 activation. To exclude potential allosteric or non–TIMP3-binding related effect of motif deletion in ITGA7 on the function of ITGA7 molecule, a fragment of cDNA corresponding to ITGA7 amino acids 1037 to 1103 (in addition to ATG start codon and 5′ UTR of ITGA7) was constructed into pCMV-FLAG to generate a TIMP3 minimal binding FLAG (TBM-FLAG) decoy peptide to interfere with the ITGA7/TIMP3 interaction. The results showed that the expression of this peptide reduced ITGA7/TIMP3 binding (Figure 1B) and significantly reversed the cell growth inhibition and cell cycle blocking effect of ITGA7 (Figure 6). Taken together, these results indicate a critical role of the ITGA7/TIMP3 interaction for ITGA7 tumor suppression.
Figure 5.

Interaction with TIMP3 is essential for ITGA7-mediated cell growth suppression. A: Mutant ITGA7 that does not interact with TIMP3 failed to suppress TNF-α and cyclin D1 expression and had no impact on NF-κB nuclear distribution. Lanes 1–8: Immunoblots were generated from the protein extracts from PITT1, PITT2, or pCDNA4-ITGA7Δ1037-1103/pCDNA6TO transfected PC3 clones (C5 and C8), induced with or without tetracycline (Tet), using antibodies specific for ITGA7, TIMP3, TNF-α, cyclin D, or β-actin. ΔITGA7-FLAG denotes ITGA7-FLAG mutant that lacks amino acids 1037 to 1103 in ITGA7 protein. Lanes 9–24: Chromatin association of p65 NF-κB subunit. PITT1 and PITT2 cells were fractionated into chromatin (Chr) and non-chromatin (NC) fractions. Immunoblots were generated using antibodies specific for p65, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), or histone 3. B: Colony formation assays of replica samples of A. Triplicate experiments were performed on each assay. C: BrdU labeling and cell cycle analyses of replica samples of A. Triplicate experiments were performed on each assay. ∗∗P < 0.01 (t-test).
Figure 6.

TIMP3 binding motif peptide interferes with cell growth suppression by ITGA7. A: Immunoblot analysis of ITGA7, TBM-FLAG, and β-actin. PITT1 and PITT2 cells were transfected with pCMV-TBM-FLAG (TIMP3 binding motif-FLAG) or vector control (V). These cells were treated with or without tetracycline (Tet). Immunoblot assays were performed using antibodies specific for ITGA7, FLAG, or β-actin. B: Colony formation assays of duplicate experiments of A. Triplicate experiments were performed on each assay. C: BrDU labeling and cell cycle analysis of duplicate experiments of A. Triplicate experiments were performed on each assay. ∗∗P < 0.01 (t-test).
Previous studies have shown that laminin plays a protective role against the cell death induced by ITGA7.16 To investigate the impact of laminin on ITGA7-mediated cell growth suppression, PITT1 cells were treated with siRNA specific for β1-laminin to block the formation of the functional laminin α1β1γ1 complex. As shown in Figure 7, A and B, knocking down β1-laminin exacerbated the suppression of cell growth induced by ITGA7, in comparison with a scrambled control (from 53% to 82%, P < 0.001), suggesting that β1-laminin plays a role in moderating and neutralizing the tumor-suppressor activity of ITGA7. Treatment of siβ1Lam also significantly enhanced the cell cycle blockade effect induced by ITGA7, increasing the proportion of cells in G0/G1 from 76% to 89% (P < 0.001), and decreasing cells in the S phase from 11% to 5% (P < 0.001).
Figure 7.

Laminin inhibits ITGA7-TIMP3 signaling and growth suppression. A: Knocking down of β1-laminin enhanced ITGA7-TIMP3 signaling. Lanes 1 to 8: Immunoblots were generated from the protein extracts from PITT1 and C5 cells induced with or without tetracycline (Tet) and treated with siRNA specific for β1-laminin (siLam) or scrambled control (Scr), using antibodies specific for ITGA7, β1-laminin, cyclin D1, TNF-α, or β-actin. Lanes 9 to 24: Chromatin association of p65 NF-κB subunit. PITT1 and C5 cells were fractionated into chromatin (Chr) and non-chromatin (NC) fractions. Immunoblots were generated using antibodies specific for p65, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and histone 3. B: Colony formation assays of replica samples of A. C: BrdU labeling and cell cycle analyses of replica samples of A. ∗∗P < 0.001.
Although laminin also interacts with other extracellular matrix proteins, the most significant effect on cell growth from β1-laminin suppression was for cells that had concomitant ITGA7 induction (Figure 7, A–C). Conversely, the effect of β1-laminin knockdown in ITGA7-negative cells was not statistically significant. These results suggest that ITGA7 is the critical molecule regulating laminin survival signals. Subsequently, we treated pCDNA4-ITGA7Δ1037-1103/pCDNA6 cotransfected cells (C5) with siRNA specific for β1-laminin and examined what role ITGA7-TIMP3 signaling plays for extracellular matrix–mediated cell growth. As shown in Figure 7C, mutation of the TIMP3 binding site in ITGA7 completely abrogated the effect of laminin on cell growth and cell cycle blockade, compared with the wild-type ITGA7, suggesting that a counterbalancing ITGA7-TIMP3 growth-suppressing signal is the main mechanism of laminin progrowth activity.
Discussion
As one of the major extracellular matrix receptors, ITGA7 forms heterodimers using its β1 chain to transduce signals from the matrix to cells. It is well established that ITGA7 is essential for muscle cell differentiation and migration. Recent studies suggest that ITGA7 also plays a tumor-suppressor role in several human malignancies.33–39 However, the signaling mechanism of ITGA7 tumor-suppressor activity remains elusive. Our analyses in this study suggest that TIMP3, a tumor-suppressor gene, is a downstream signaling molecule of ITGA7. Several lines of evidence support the ITGA7 interaction with TIMP3. First, the C-terminus of ITGA7 binds TIMP3, both in the yeast two-hybrid system and in cell-free in vitro binding assays. The cell-free assays suggest that no bridge protein is involved in the ITGA7/TIMP3 interaction. Second, both ITGA7 and TIMP3 are readily co-immunoprecipitated from prostate epithelial cells, by antibodies against either TIMP3 or ITGA7, suggesting the ITGA7/TIMP3 interaction occurs intracellularly. Third, ITGA7 and TIMP3 are colocalized in the membranous and cytoplasmic regions of immortalized prostate epithelial cells. The TIMP3 binding motif in ITGA7-spanning portions of transmembrane and extracellular domains is intriguing. It suggests that ITGA7 may interact with TIMP3 in the context of plasma membrane or other plasma membrane–associated proteins, even though ITGA7 and TIMP3 binding is independent of other molecules. It is intriguing that the TIMP3 binding site in ITGA7 may be lost in many ITGA7 mutants because it is located in the truncated portion of the molecule.9
By identifying the ITGA7/TIMP3 interaction, this study reveals a critical role of ITGA7 as a transmitter of signals from the extracellular matrix to DNA replication. Extracellular matrix proteins, such as laminin, are some of the most critical ligands that maintain cell survival and growth. Indeed, stromal components of the prostate gland are vital to prostate gland differentiation and growth. The mechanism for extracellular matrix–mediated proproliferative activity in prostate cancer and immortalized epithelial cells has been elusive. This study, to our knowledge, is the first that identifies the laminin-ITGA7-TIMP3 axis as a critical signaling pathway for laminin-mediated cell proliferation.
TIMP3 is a natural inhibitor of the matrix metalloproteinases and inhibits degradation of the extracellular matrix. TIMP3 is also a potent angiogenesis inhibitor.40,41 Mutation of TIMP3 was found in some rare congenital vascular disorders.40,42 Unrelated to its inhibitory activity of metalloproteinases, TIMP3 is thought to inhibit vascular epithelial growth factor–mediated angiogenesis by blocking the binding of vascular epithelial growth factor to its receptor. Also, the overexpression of TIMP3 induces apoptosis in stromal cells.43–45 The finding of the TIMP3/ITGA7 interaction suggests that TIMP3 plays a role in regulating extracellular matrix signaling. Its activation by ITGA7 leads to a redistribution of NF-κB, from the nucleus to the cytoplasm, and reduces the transcriptional induction of cyclin D1. The dependency of ITGA7 on TIMP3 to down-regulate cyclin D1 and subsequently induce cell growth arrest suggests that the ITGA7-TIMP3 signaling pathway is the main mechanism of ITGA7-induced cell growth inhibition. Knocking down of TIMP3 alone significantly enhanced PC3 cell growth and colony formation activity. It suggests that TIMP3 is a likely downstream signaling molecule for ITGA7, and other signaling molecules may interact and regulate TIMP3 tumor-suppressor activity.
The current thinking is that ITGA7 is primarily involved in smooth and skeletal muscle development; however, this probably represents only one aspect of ITGA7’s function. ITGA7 is expressed widely among all tissues and organs. In a previous study, we found that ITGA7 is expressed abundantly in terminally differentiated prostate acinar cells, whereas its expression in basal cells is minimal. In terms of its putative role in dysplasia, ITGA7 mutations were found in multiple malignancies9 and large genome deletions encompassing ITGA7 were also mapped to a subset of colorectal cancers.36 In addition, the alteration of ITGA7 expression was found in a variety of human cancers.15 Herein, we corroborate these findings by demonstrating that ITGA7 suppresses cell growth. Furthermore, we show that the interaction between laminin and ITGA7 inhibits its growth suppression signaling. Previous studies suggest that there is a more significant expression of laminin 2 in acinar cell layers than in basal cells. Based on these findings, it appears that one of the physiological functions of ITGA7 in the prostate gland is to play a pivotal role in preventing hyperplasia by inhibiting acinar cell growth, especially when those cells lose contact with the extracellular matrix. This may partly explain how the decrease in ITGA7 (either by mutation or at the protein level) promotes the development of prostate cancer, hepatocellular carcinoma, glioblastoma multiformes, and soft tissue leiomyosarcoma.
Acknowledgment
We thank Baoguo Ren for technical support.
Footnotes
Supported, in part, by American Cancer Society grant RSG-08-137-01-CNE (Y.P.Y.) and National Cancer Institute grant RO1 CA098249 (J.-H.L.).
References
- 1.Hynes R.O. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Brakebusch C., Grose R., Quondamatteo F., Ramirez A., Jorcano J.L., Pirro A., Svensson M., Herken R., Sasaki T., Timpl R., Werner S., Fassler R. Skin and hair follicle integrity is crucially dependent on beta 1 integrin expression on keratinocytes. EMBO J. 2000;19:3990–4003. doi: 10.1093/emboj/19.15.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Werner A., Willem M., Jones L.L., Kreutzberg G.W., Mayer U., Raivich G. Impaired axonal regeneration in alpha7 integrin-deficient mice. J Neurosci. 2000;20:1822–1830. doi: 10.1523/JNEUROSCI.20-05-01822.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayer U., Saher G., Fassler R., Bornemann A., Echtermeyer F., von der Mark H., Miosge N., Poschl E., von der Mark K. Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat Genet. 1997;17:318–323. doi: 10.1038/ng1197-318. [DOI] [PubMed] [Google Scholar]
- 5.Basani R.B., French D.L., Vilaire G., Brown D.L., Chen F., Coller B.S., Derrick J.M., Gartner T.K., Bennett J.S., Poncz M. A naturally occurring mutation near the amino terminus of alphaIIb defines a new region involved in ligand binding to alphaIIbbeta3. Blood. 2000;95:180–188. [PubMed] [Google Scholar]
- 6.Hayashi Y.K., Chou F.L., Engvall E., Ogawa M., Matsuda C., Hirabayashi S., Yokochi K., Ziober B.L., Kramer R.H., Kaufman S.J., Ozawa E., Goto Y., Nonaka I., Tsukahara T., Wang J.Z., Hoffman E.P., Arahata K. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet. 1998;19:94–97. doi: 10.1038/ng0598-94. [DOI] [PubMed] [Google Scholar]
- 7.Echtermeyer F., Schober S., Poschl E., von der Mark H., von der Mark K. Specific induction of cell motility on laminin by alpha 7 integrin. J Biol Chem. 1996;271:2071–2075. doi: 10.1074/jbc.271.4.2071. [DOI] [PubMed] [Google Scholar]
- 8.Flintoff-Dye N.L., Welser J., Rooney J., Scowen P., Tamowski S., Hatton W., Burkin D.J. Role for the alpha7beta1 integrin in vascular development and integrity. Dev Dyn. 2005;234:11–21. doi: 10.1002/dvdy.20462. [DOI] [PubMed] [Google Scholar]
- 9.Ren B., Yu Y.P., Tseng G.C., Wu C., Chen K., Rao U.N., Nelson J., Michalopoulos G.K., Luo J.H. Analysis of integrin alpha7 mutations in prostate cancer, liver cancer, glioblastoma multiforme, and leiomyosarcoma. J Natl Cancer Inst. 2007;99:868–880. doi: 10.1093/jnci/djk199. [DOI] [PubMed] [Google Scholar]
- 10.Luo J.H. Gene expression alterations in human prostate cancer. Drugs Today (Barc) 2002;38:713–719. doi: 10.1358/dot.2002.38.10.704653. [DOI] [PubMed] [Google Scholar]
- 11.Luo J.H., Yu Y.P., Cieply K., Lin F., Deflavia P., Dhir R., Finkelstein S., Michalopoulos G., Becich M. Gene expression analysis of prostate cancers. Mol Carcinog. 2002;33:25–35. doi: 10.1002/mc.10018. [DOI] [PubMed] [Google Scholar]
- 12.Luo J.H., Yu Y.P. Genetic factors underlying prostate cancer. Expert Rev Mol Med. 2003;5:1–26. doi: 10.1017/S1462399403006057. [DOI] [PubMed] [Google Scholar]
- 13.Ren B., Yu Y.P., Jing L., Liu L., Michalopoulos G.K., Luo J.H., Rao U.N. Gene expression analysis of human soft tissue leiomyosarcomas. Hum Pathol. 2003;34:549–558. doi: 10.1016/s0046-8177(03)00014-5. [DOI] [PubMed] [Google Scholar]
- 14.Yu Y.P., Landsittel D., Jing L., Nelson J., Ren B., Liu L., McDonald C., Thomas R., Dhir R., Finkelstein S., Michalopoulos G., Becich M., Luo J.H. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 15.Tseng G.C., Cheng C., Yu Y.P., Nelson J., Michalopoulos G., Luo J.H. Investigating multi-cancer biomarkers and their cross-predictability in the expression profiles of multiple cancer types. Biomark Insights. 2009;4:57–79. doi: 10.4137/bmi.s930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Z.H., Yu Y.P., Zheng Z.L., Song Y., Xiang G.S., Nelson J., Michalopoulos G., Luo J.H. Integrin alpha 7 interacts with high temperature requirement A2 (HtrA2) to induce prostate cancer cell death. Am J Pathol. 2010;177:1176–1186. doi: 10.2353/ajpath.2010.091026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu Y.P., Luo J.H. Myopodin-mediated suppression of prostate cancer cell migration involves interaction with zyxin. Cancer Res. 2006;66:7414–7419. doi: 10.1158/0008-5472.CAN-06-0227. [DOI] [PubMed] [Google Scholar]
- 18.Yu Y.P., Tseng G.C., Luo J.H. Inactivation of myopodin expression associated with prostate cancer relapse. Urology. 2006;68:578–582. doi: 10.1016/j.urology.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 19.Lin F., Yu Y.P., Woods J., Cieply K., Gooding B., Finkelstein P., Dhir R., Krill D., Becich M.J., Michalopoulos G., Finkelstein S., Luo J.H. Myopodin, a synaptopodin homologue, is frequently deleted in invasive prostate cancers. Am J Pathol. 2001;159:1603–1612. doi: 10.1016/S0002-9440(10)63006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jing L., Liu L., Yu Y.P., Dhir R., Acquafondada M., Landsittel D., Cieply K., Wells A., Luo J.H. Expression of myopodin induces suppression of tumor growth and metastasis. Am J Pathol. 2004;164:1799–1806. doi: 10.1016/S0002-9440(10)63738-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ren B., Yu G., Tseng G.C., Cieply K., Gavel T., Nelson J., Michalopoulos G., Yu Y.P., Luo J.H. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene. 2006;25:1090–1098. doi: 10.1038/sj.onc.1209134. [DOI] [PubMed] [Google Scholar]
- 22.Yu G., Tseng G.C., Yu Y.P., Gavel T., Nelson J., Wells A., Michalopoulos G., Kokkinakis D., Luo J.H. CSR1 suppresses tumor growth and metastasis of prostate cancer. Am J Pathol. 2006;168:597–607. doi: 10.2353/ajpath.2006.050620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu Y.P., Yu G., Tseng G., Cieply K., Nelson J., Defrances M., Zarnegar R., Michalopoulos G., Luo J.H. Glutathione peroxidase 3, deleted or methylated in prostate cancer, suppresses prostate cancer growth and metastasis. Cancer Res. 2007;67:8043–8050. doi: 10.1158/0008-5472.CAN-07-0648. [DOI] [PubMed] [Google Scholar]
- 24.Shi Y.K., Yu Y.P., Zhu Z.H., Han Y.C., Ren B., Nelson J.B., Luo J.H. MCM7 interacts with androgen receptor. Am J Pathol. 2008;173:1758–1767. doi: 10.2353/ajpath.2008.080363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han Y.C., Yu Y.P., Tseng G.C., Luo J.H. Trypsin and reduction method to prepare DNA from formalin-fixed paraffin-embedded samples for methylation analysis. Histopathology. 2009;54:773–775. doi: 10.1111/j.1365-2559.2009.03286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu Z.H., Yu Y.P., Shi Y.K., Nelson J.B., Luo J.H. CSR1 induces cell death through inactivation of CPSF3. Oncogene. 2009;28:41–51. doi: 10.1038/onc.2008.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han Y.C., Yu Y.P., Nelson J., Wu C., Wang H., Michalopoulos G.K., Luo J.H. Interaction of integrin-linked kinase and miniature chromosome maintenance 7-mediating integrin {alpha}7 induced cell growth suppression. Cancer Res. 2010;70:4375–4384. doi: 10.1158/0008-5472.CAN-09-4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi Y.K., Yu Y.P., Tseng G.C., Luo J.H. Inhibition of prostate cancer growth and metastasis using small interference RNA specific for minichromosome complex maintenance component 7. Cancer Gene Ther. 2010;17:694–699. doi: 10.1038/cgt.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng Z.L., Tan L.Z., Yu Y.P., Michalopoulos G., Luo J.H. Interaction of CSR1 with XIAP reverses inhibition of caspases and accelerates cell death. Am J Pathol. 2012;181:463–471. doi: 10.1016/j.ajpath.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amour A., Slocombe P.M., Webster A., Butler M., Knight C.G., Smith B.J., Stephens P.E., Shelley C., Hutton M., Knauper V., Docherty A.J., Murphy G. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435:39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- 31.Brennan F.M., Green P., Amjadi P., Robertshaw H.J., Alvarez-Iglesias M., Takata M. Interleukin-10 regulates TNF-alpha-converting enzyme (TACE/ADAM-17) involving a TIMP-3 dependent and independent mechanism. Eur J Immunol. 2008;38:1106–1117. doi: 10.1002/eji.200737821. [DOI] [PubMed] [Google Scholar]
- 32.Joyce D., Albanese C., Steer J., Fu M., Bouzahzah B., Pestell R.G. NF-kappaB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev. 2001;12:73–90. doi: 10.1016/s1359-6101(00)00018-6. [DOI] [PubMed] [Google Scholar]
- 33.Buganim Y., Madar S., Rais Y., Pomeraniec L., Harel E., Solomon H., Kalo E., Goldstein I., Brosh R., Haimov O., Avivi C., Polak-Charcon S., Goldfinger N., Barshack I., Rotter V. Transcriptional activity of ATF3 in the stromal compartment of tumors promotes cancer progression. Carcinogenesis. 2011;32:1749–1757. doi: 10.1093/carcin/bgr203. [DOI] [PubMed] [Google Scholar]
- 34.Zhu Z.H., Yu Y.P., Zheng Z.L., Song Y., Xiang G.S., Nelson J., Michalopoulos G., Luo J.H. Integrin alpha 7 interacts with high temperature requirement A2 (HtrA2) to induce prostate cancer cell death. Am J Pathol. 2010;177:1176–1186. doi: 10.2353/ajpath.2010.091026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juengel E., Bhasin M., Libermann T., Barth S., Michaelis M., Cinatl J., Jr., Jones J., Hudak L., Jonas D., Blaheta R.A. Alterations of the gene expression profile in renal cell carcinoma after treatment with the histone deacetylase-inhibitor valproic acid and interferon-alpha. World J Urol. 2011;29:779–786. doi: 10.1007/s00345-010-0582-y. [DOI] [PubMed] [Google Scholar]
- 36.Aytekin T., Ozaslan M., Cengiz B. Deletion mapping of chromosome region 12q13-24 in colorectal cancer. Cancer Genet Cytogenet. 2010;201:32–38. doi: 10.1016/j.cancergencyto.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 37.Han Y.C., Yu Y.P., Nelson J., Wu C., Wang H., Michalopoulos G.K., Luo J.H. Interaction of integrin-linked kinase and miniature chromosome maintenance 7-mediating integrin {alpha}7 induced cell growth suppression. Cancer Res. 2010;70:4375–4384. doi: 10.1158/0008-5472.CAN-09-4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samson T., Will C., Knoblauch A., Sharek L., von der Mark K., Burridge K., Wixler V. Def-6, a guanine nucleotide exchange factor for Rac1, interacts with the skeletal muscle integrin chain alpha7A and influences myoblast differentiation. J Biol Chem. 2007;282:15730–15742. doi: 10.1074/jbc.M611197200. [DOI] [PubMed] [Google Scholar]
- 39.Ziober B.L., Chen Y.Q., Ramos D.M., Waleh N., Kramer R.H. Expression of the alpha7beta1 laminin receptor suppresses melanoma growth and metastatic potential. Cell Growth Differ. 1999;10:479–490. [PubMed] [Google Scholar]
- 40.Qi J.H., Dai G., Luthert P., Chaurasia S., Hollyfield J., Weber B.H., Stohr H., Anand-Apte B. S156C mutation in tissue inhibitor of metalloproteinases-3 induces increased angiogenesis. J Biol Chem. 2009;284:19927–19936. doi: 10.1074/jbc.M109.013763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qi J.H., Ebrahem Q., Moore N., Murphy G., Claesson-Welsh L., Bond M., Baker A., Anand-Apte B. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med. 2003;9:407–415. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- 42.Shovlin C.L., Scott J. Inherited diseases of the vasculature. Annu Rev Physiol. 1996;58:483–507. doi: 10.1146/annurev.ph.58.030196.002411. [DOI] [PubMed] [Google Scholar]
- 43.Matthews F.J., Cook S.D., Majid M.A., Dick A.D., Smith V.A. Changes in the balance of the tissue inhibitor of matrix metalloproteinases (TIMPs)-1 and -3 may promote keratocyte apoptosis in keratoconus. Exp Eye Res. 2007;84:1125–1134. doi: 10.1016/j.exer.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 44.Finan K.M., Hodge G., Reynolds A.M., Hodge S., Holmes M.D., Baker A.H., Reynolds P.N. In vitro susceptibility to the pro-apoptotic effects of TIMP-3 gene delivery translates to greater in vivo efficacy versus gene delivery for TIMPs-1 or -2. Lung Cancer. 2006;53:273–284. doi: 10.1016/j.lungcan.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 45.Kallio J.P., Hopkins-Donaldson S., Baker A.H., Kähäri V.M. TIMP-3 promotes apoptosis in nonadherent small cell lung carcinoma cells lacking functional death receptor pathway. Int J Cancer. 2011;128:991–996. doi: 10.1002/ijc.25404. [DOI] [PubMed] [Google Scholar]