

Abstract
Alpha-1-antitrypsin (a1AT) deficiency is caused by homozygosity for the a1AT mutant Z gene and occurs in 1 in 2,000 Americans. The Z mutation confers an abnormal conformation on the a1AT mutant Z protein, resulting in accumulation within the endoplasmic reticulum of hepatocytes and chronic liver injury. Autophagy is one of several proteolytic mechanisms activated to cope with this hepatocellular protein burden, and is likely important in disposal of the unique polymerized conformation of the a1AT mutant Z protein, which is thought to be especially injurious to the cell. Recent data indicates that rapamycin may more efficiently upregulate autophagy when given in weekly dose pulses, as compared to a daily regimen. Therefore, we evaluated the effect of rapamycin on PiZ mice, a well characterized model which recapitulates human a1AT liver disease. Daily dosing had no effect on autophagy, on accumulation of a1AT mutant Z protein, or on liver injury. Weekly dosing of rapamycin did increase autophagic activity, as shown by increased numbers of autophagic vacuoles. This was associated with reduction in the intrahepatic accumulation of a1AT mutant Z protein in the polymerized conformation. Markers of hepatocellular injury, including cleavage of caspase 12 and hepatic fibrosis, were also decreased. In conclusion, this is the first report of a successful in vivo method for reduction of intrahepatic a1AT mutant Z polymerized protein. Application of this finding may be therapeutic in patients with a1AT deficiency by reducing the intracellular burden of the polymerized, mutant Z protein and by reducing the progression of liver injury.
Keywords: Caspase 12, autophagy, proliferation, endoplasmic reticulum
Introduction
Alpha-1-antitrypsin (a1AT) deficiency is a genetic disease, which occurs in 1 in 2,000 Americans and is caused by homozygosity for the a1AT mutant Z gene[1, 2]. The Z mutation in the a1AT gene confers an abnormal conformation on the resultant nascent polypeptide[3]. This abnormal, mutant protein accumulates within the endoplasmic reticulum (ER) of hepatocytes, unlike the wild type protein which is efficiently glycosylated, folded and secreted. The result of the intracellular accumulation of the a1AT mutant Z protein is that homozygous, ZZ individuals have an increased risk of chronic liver disease and hepatocellular carcinoma (HCC)[4] [5, 6]. No specific treatment for a1AT associated liver disease is available, other then liver transplantation. Studies of the mutant Z protein molecule have shown that it has the tendency to form unique protein polymers within the hepatocellular ER. In some, but not all hepatocytes the accumulations of polymerized a1AT mutant Z protein become large enough to be seen under light microscopy as the cytoplasmic “globules” classically described in this disease[7]. Published data suggests that the liver injury in a1AT deficiency is directly related to the hepatic accumulation of the polymerized a1AT mutant Z protein[8–12].
Multiple, intracellular proteolytic pathways are active within the hepatocyte to degrade the accumulated a1AT mutant Z protein[13–18]. Studies in patient specimens suggest effective intracellular proteolytic degradation of the mutant Z protein is related to protection from liver injury[12, 19]. The systems involved in a1AT mutant Z intracellular proteolysis include ubiquitin dependent and ubiquitin independent proteasomal pathways and autophagy[16–18, 20]. Autophagy is a process of intracellular degradation within specialized vacuoles, called autophagosomes, used for the disposal of senescent organelles, mis-folded proteins and which is activated during starvation and at specific times during development[16, 20–22]. Autophagy is known to be activated in response to the intracellular accumulation of a1AT mutant Z protein in both human liver and in model systems[10, 16, 20, 23]. These studies have included transmission electron microscopy analysis showing that the vacuoles in question have the classical morphology of autophagic vacuoles, and that these vacuoles label with multiple markers known to label autophagic vacuoles. These marker studies included immune-EM for appropriate membrane markers and acidification, as expected for autophagic vacuoles. The autophagic vacuoles also label for, and appear to contain, a1AT mutant Z protein based on immuno-EM studies and fluorescent label light microscopy studies. Fluorescent labeling for acidification and for the autophagy marker, MDC, was also positive. In tissue culture models of a1AT mutant Z intracellular degradation, biochemical inhibition of autophagy using 3-methyladenine and wortmannin, partially inhibits intracellular degradation of a1AT mutant Z protein. Autophagy may be especially important in the disposal of the polymerized form of a1AT mutant Z protein, since aggregations of the a1AT mutant Z polymers appear to be abnormally long-lived in autophagy deficient cell lines [24]. Based on this large body of previous work, we have proposed that drugs, or other therapeutic maneuvers, which would increase autophagic activity might result in increased degradation of the accumulated a1AT mutant Z protein, and thereby reduce liver injury. Therefore, in this study we examined the effect rapamycin, an FDA approved drug used for immunosuppression, but which also is known to induce increased autophagy, on a1AT mutant Z protein accumulation in the PiZ mouse[25, 26]. The PiZ mouse is a well characterized in vivo model which recapitulates many features of the human a1AT deficiency liver disease[2, 7–11, 16, 20, 27]. These findings may be useful in the planning of future treatments for this human metabolic liver disease.
Experimental Procedures
PiZ mice were maintained on a C57Bl/6J background and C57Bl/6J mice were used as controls as previously described[2, 7–9, 11, 27]. All experiments were approved by the Animal Studies Committees of St. Louis University and Washington University, where applicable, and were conducted in accordance with the criteria outlined in the “Guide for Care and Use of Laboratory Animals.” Monomer-Polymer Assay was performed as previously described[7, 9, 11]. Briefly, lipid bilayers are disrupted in a non-denaturing buffer (0.5% Triton plus mechanical disruption) allowing an insoluble fraction of the liver homogenate to be separated by centrifugation. The a1AT polymers are large, insoluble aggregates which are only suspended in the buffered homogenate, not solubilized. Electron microscopy of the samples and immunoblot on spot membranes with polymer specific antibody confirms the presence of a1ATmutant Z polymers only in the insoluble fraction. The separated soluble and insoluble fractions are then subjected to analysis, in this case denatured SDS-PAGE. The a1AT mutant Z polymers are not linked by covalent bonds, therefore denaturation returns the polymers in the insoluble fraction to monomers allowing them to run in the gel at the monomeric molecular weight for direct, quantitative comparison to the monomer fraction. This method recovers >98% of a1AT mutant Z polymers with a high degree of reproducibility,
Histologic and immunohistochemistry analysis was performed using standard techniques, as described[7, 10, 28]. Liver were fixed in 10% formalin for 24 hours and stored in 70% EtOH until processed and embedded into paraffin blocks and sectioned. Six micron sections were rehydrated, blocked, and then incubated overnight at 4C with goat anti-human alpha-1-antitrypsin (Cappel, Cappel Products Division of Organon Teknika Corporation, Akzo Nobel, The Netherlands), then with anti-goat biotinylated (Sigma, St. Louis, MO, USA) for 1 hour at room temperature, then with DAKO (Denmark) anti-biotin/streptavidin HRP conjugated for 1 hour at room temp. Immunoblots were performed as previously described (anti-a1AT antibody from DiaSorin, Stillwater, MN, USA) in triplicate with equal protein loading and antibodies previously used in these systems, and then quantified by means of the densitometric scanning of the replicates [16, 18]. Anti-LC3 antibody (#PM036 MBL; Medical and Biological Laboratories, Nagoya, Japan) was employed as described, and included the presence of protease inhibitors during processing [29]. LC3 is a protein found in the membrane of the autophagic vacuole and increases in the lower band, LC3-II has been shown to correlate with increased numbers of autophagic vacuoles and increased autophagic activity in many systems. The positive control for LC3 was a WT mouse fasted 12h, which is known to increase autophagy and is associated with increased recovery of LC3-II on immunoblot. Equal gel loading was confirmed with either a total protein stain of the membrane or immunoblot for actin, as described previously [9, 11]. A1AT serum levels in mice were determined by immunoassay as described[30].
Morphometric quantification of autophagy, bromodeoxy uridine (BrdU) labeling, and counting were accomplished as previously described[8, 9, 16]. For morphometric quantification of autophagy, plastic embedded, thin sections were examined by transmission electron microscopy and early and late autophagic vacuoles were identified by the standard morphology of double membrane bound, early autophagic vacuoles arising near the ER containing organelles or debris, which then progress to multilamellar late autophagic vacuoles fusing with lysosomes forming electron dense residual bodies. Fifteen whole hepatocytes with nuclei were randomly selected, counting grids were overlaid, and the percent area cytoplasm occupied by autophagic vacuoles was determined by a single, blinded examiner. Confirmation that these structures represent autophagic vacuoles has been performed in ZZ human liver, PiZ mouse liver, and in tissue culture systems by immune label for proteins present within autophagic vacuoles, by immune-EM and immunohistochemistry (calnexin, degradative enzymes), and by detection of acidification by immune-EM and by fluorescent label (autophagic vacuoles acidify early in their biogenesis). In addition, the structures label with the fluorescent autophagic vacuole marker, MDC. BrdU labeling was performed by implantation in the mice of a time release BrdU osmotic pump 72 hours prior to sacrifice, which labels all nuclei which have divided in that 72 hour period. Immunohistochemical stain for BrdU then labels those nuclei which are then counted as a percent of all hepatocytes by a single, blinded examiner. All histopathologic examinations, analysis of globules, dysplasia, and counting were done by a single blinded examiner (EB or JT). Statistically analysis was accomplished with Statistical Package for the Social Sciences (SPSS, Chicago, IL).
A total of 14 PiZ mice were treated with daily dose rapamycin oral suspension in drinking water from age 3 months to 6 months, and were compared to 10 matched, litter mate PiZ mice treated with vehicle (water) alone. A total of 24 PiZ mice (12 males and 12 females) were treated with once weekly intraperitoneal injection of rapamycin from age 3 months to 6 months compared to 24 matched, litter mate PiZ mice (12 males and 12 females) treated with vehicle alone. All the analyses included samples from all the mice above, unless specifically noted. One mouse died in each of the weekly treatment and control groups of unknown causes, although there were no signs of infection on necropsy. There were no obvious infectious complications of any mouse in either group analyzed, although a few millimeters of induration and erythema were observed at some rapamycin injection sites.
Preparation of the weekly dosing agent was as follows: Both rapamycin reagent (Rapamune 1mg/ml (sirolimus) oral solution from Wyeth) and vehicle reagent (0.8% NaCl, 1.0% EtOH, 0.1% Tween-80 in water) were drawn up into a 1ml luer-lock syringe (Millipore Millex GV filter 0.22um pore PVDF membrane) then pushed slowly through syringe filter directly into the back of an insulin syringe (plunger removed). The plunger was then replaced and air bubbles were drawn out. This was all done on ice in a sterile biosafety cabinet. Dose: Rapamune 10mg/kg/once per week for 12 weeks. Time of administration was between the hours of 9 am and 10 am once per week on Wednesday.
Hepatic mRNA was prepared using Trizol (Invitrogen, Carlsbad, CA, USA) and the reverse transcribed SuperScript Choice System (Invitrogen) using methods as described. Taq DNA polymerase and SYBR from Biorad (Hercules, CA,USA) were used with the following primers: forward 5′-GGCCATACCCATGTCTATCC and reverse 5′-TTCACCACTTTTCCCATGAA. These data were standardized to beta2-microglobulin expression and specificity of the assay was verified by confirmation of the predicted product size and uniformity melt curves and by agarose electrophoresis.
Results
Weekly, pulsatile dosing of rapamycin reduces intrahepatic accumulation of a1AT mutant Z protein polymer in PiZ mice
Accumulation of a1AT mutant Z protein within hepatocytes leads to liver injury in a1AT deficiency. Intracellular proteolytic pathways, including autophagy, are involved in reducing the burden of the accumulated a1AT mutant Z protein within the hepatocyte, and reducing the risk of liver injury. We hypothesize that acceleration of intracellular proteolysis, especially increased autophagy acting to degrade the toxic, polymerized conformation of a1AT mutant Z protein will thereby, reduce liver injury. We examined this hypothesis in the PiZ mouse, a well characterized model transgenic for the human mutant Z gene, which recapitulates many aspects of the human a1AT deficiency liver injury. Rapamycin is known, among other effects, to induce increased autophagy within cells. Rapamycin has been previously shown in fly and murine models of Huntington Disease to increase autophagic activity in various daily and every other day dose regimens[31, 32]. Therefore, we examined the effect of rapamycin on PiZ mice treated with oral rapamycin daily in drinking water compared to control groups treated with vehicle alone. Dosing of approximately 10mg/g/week were chosen as previously associated with effectiveness in murine models, and blood levels were determined to be similar to the human therapeutic range (mean of 5.2ug/ml). After 3 months of treatment the PiZ livers treated with daily rapamycin were compared to daily dosed vehicle treated controls, and no difference was found in autophagic activity as assayed by morphometric counting of percent cytoplasm occupied by autophagic vacuoles by a blinded examiner under transmission electron microscopy (figure 1a). Next, lysates were made of samples of these livers and analyzed for a1AT mutant Z protein content by SDS-PAGE followed by anti-a1AT immunoblot. There was no difference in the daily treated group compared to controls in the hepatic content of a1AT mutant Z protein (either the total hepatic burden, or the separate monomer or polymer conformations), and no difference in the histopathologic appearance of the liver (figure 1b). Densitometric quantification of repeated runs confirmed no difference[7]. These results were the same when the groups of males and females were analyzed individually (not shown).
Figure 1. Lack of effect of daily dosing rapamycin on autophagy and a1AT protein in the liver in PiZ mice.

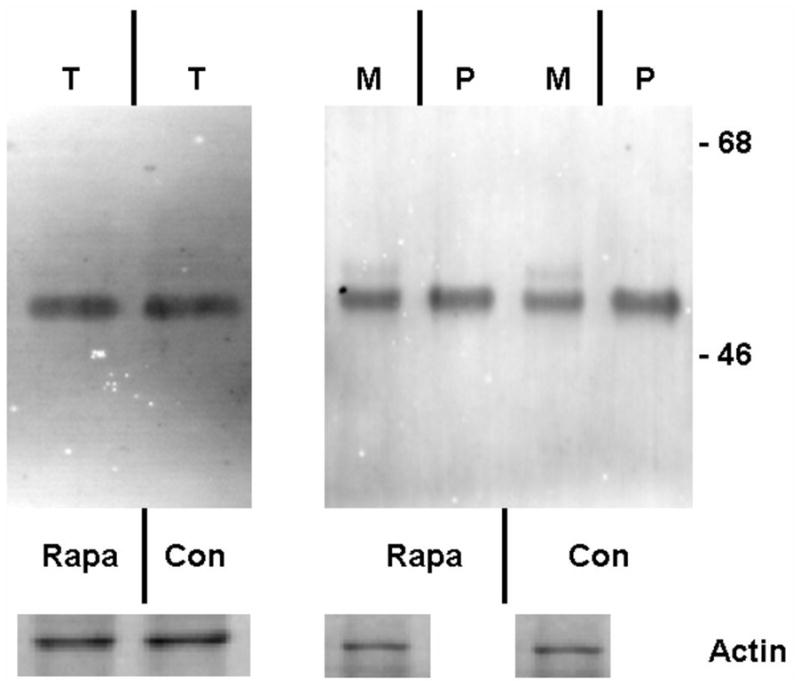
Panel a, percent area of hepatocellular cytoplasm occupied by autophagic vacuoles, determined as described by morphometric counting by a single blinded examiner, in PiZ mice treated with daily dosing rapamycin versus vehicle control PiZ mice (p>0.4 by ANOVA). Bars +/− S.D. Panel b, representative examples of SDS-PAGE followed by immunoblot for a1AT from PiZ mouse liver treated with daily dose rapamycin (rapa) or vehicle control (con) of total hepatic protein lysate (T) or after separation of a1AT monomers (M) from polymers (P). Actin loading controls below, as shown. Note, the polymers are denatured to monomers before loading and therefore run at the monomeric 52kDa of the intracellular form of a1AT protein. Also, actin is only recovered in the M fraction. Molecular weight markers, Mrx103 as shown.
We noted that some published regimens had used less frequent dosing schemes, and preliminary observations in other studies (SK, personal communications) had suggested the possibility of increased autophagic activity with weekly dosing. Specifically, an analysis of rapamycin induced autophagy associated with disease in murine retina was found to be maximally upregulated by weekly dosing (SK, personal communication). Therefore, we next examined PiZ mice treated with once weekly dosing compared to weekly dosed vehicle treated controls, but using the same total dose of 10mg/g/week. Groups of mice were again treated for 3 months and again compared to matched, vehicle treated controls. The results revealed an increase in autophagic vacuoles associated with the weekly rapamycin treatment (figure 2a). This was associated with increased detection of a LC3-II reactive band on immunoblot of liver lysates. LC3 is a protein found in the membrane of the autophagic vacuole and increases in the lower, LC3-II band correlate with increased numbers of autophagic vacuoles, and therefore suggests increased autophagic activity (figure 2b). We next determined the total hepatic content of a1AT mutant Z protein by immunoblot and found a significant reduction associated with rapamycin treatment (figure 2c) (p<0.03 by t test of gel densitometry of all replicates, figure 2d). We then examined the hepatic content of the polymerized a1AT mutant Z protein, since it is thought that autophagy is especially important in disposal of the accumulated, intracellular a1AT mutant Z protein polymer (figure 2b). Densitometric quantification of the results showed significantly less intrahepatic a1AT mutant Z protein polymer in the weekly rapamycin treated mice than in vehicle treated controls (figure 2d)(p<0.02 by t test with multiple comparisons). However, examination of the groups of treated males compared to male controls and treated females to female controls separately revealed that the effect was significantly greater in the male mice (p<0.05 by t test with multiple comparisons). Previous data has indicated that in both humans and in this mouse model that there is a sex-based difference in liver injury in a1AT deficiency. The human studies suggest that homozygous ZZ males have more severe courses and in the PiZ mouse there is increased de novo liver injury and increased liver injury after stress in male mice[8, 9].
Figure 2. Effect of weekly pulse dosing rapamycin on autophagy and a1AT protein in the liver in PiZ mice.

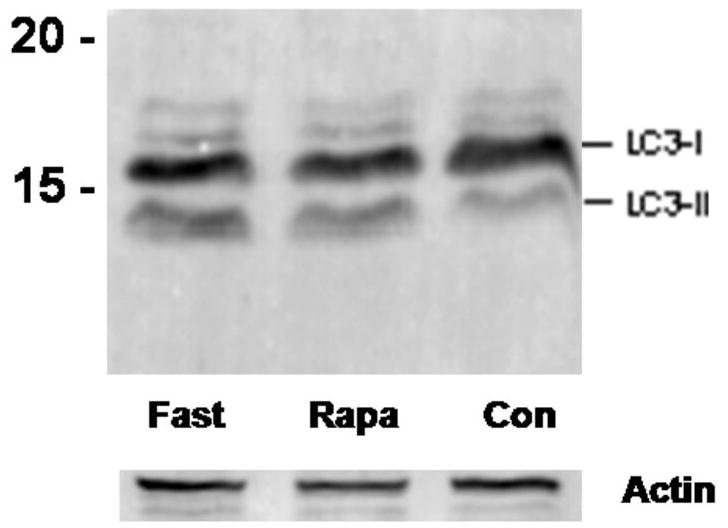
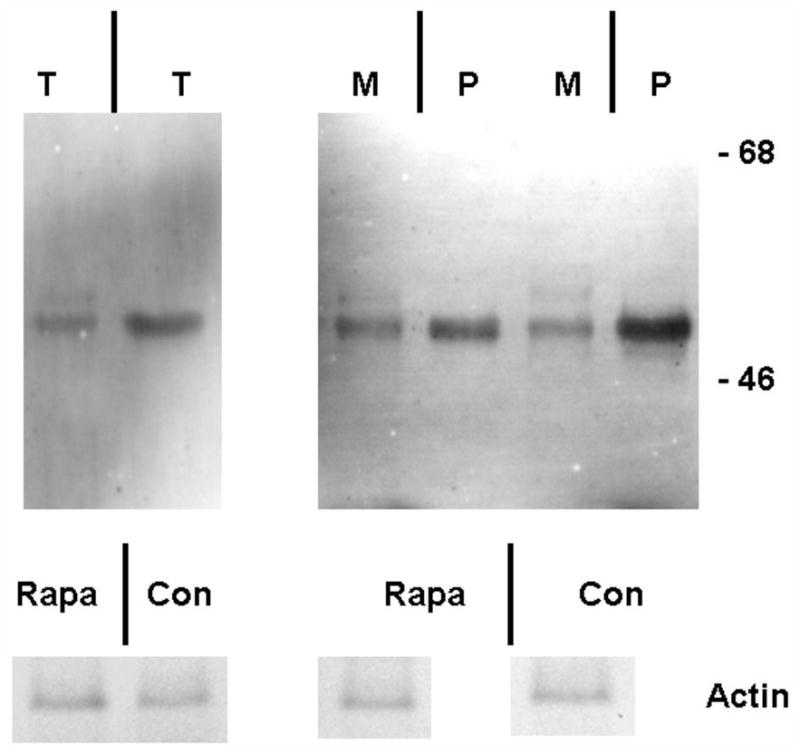
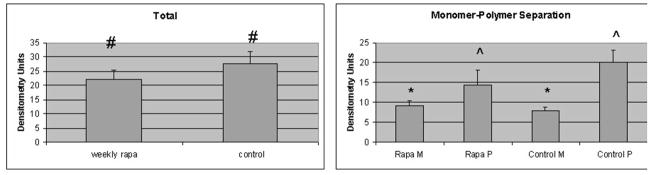
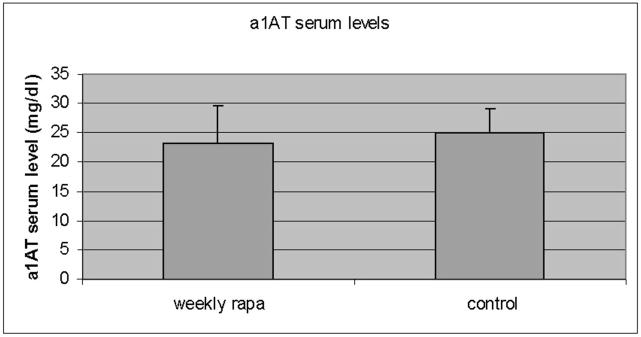
Panel a, percent area of hepatocellular cytoplasm occupied by autophagic vacuoles in PiZ mice treated with weekly dosing rapamycin versus vehicle control PiZ mice (p<0.5 by ANOVA). Bars +/− S.D. Panel b, representative example of SDS-PAGE followed by immunoblot for LC3 from a WT mouse liver fasted 12 hours (fast) as a positive control, PiZ mouse liver treated with weekly dose rapamycin (rapa) or vehicle control (con) of total hepatic protein lysate. LC3-I and LC3-II reactive bands are shown. Actin loading controls as shown, below, and molecular weight markers, Mrx103 as shown. Panel c, representative examples of SDS-PAGE followed by immunoblot for a1AT from PiZ mouse liver treated with weekly dose rapamycin (rapa) or vehicle control (con) of total hepatic protein lysate (T) or after separation of a1AT monomers (M) from polymers (P). Note, the polymers are denatured to monomers before loading and therefore run at the monomeric 52kDa of the intracellular form of a1AT protein. Molecular weight markers, Mrx103 as shown. Panel d, quantification by electronic gel scanning of the means of the weekly treated mice and controls, as from panel b. Left panel is total (Total) intrahepatic a1AT mutant Z protein, right panel is monomer (M) and polymer (P) separation. Comparisons as noted, #; p<0.03, *; p<0.02, ^; p<0.02. Panel e, mean serum a1AT protein levels at sacrifice in PiZ mice treated with weekly rapamycin compared to controls. Bars +/− S.D. Comparison p>0.4.
We next examined the monomeric a1AT mutant Z protein content of the livers (figure 2c). Surprisingly, quantification revealed that the hepatic content of monomeric a1AT mutant Z protein was modestly increased (approximately 23% of the magnitude of the decrease in the polymer content) in the rapamycin treated group compared to controls (figure 2d), but again the effect was predominant in the male mice (p<0.01 by t test with multiple comparisons). It has been shown that during synthesis of the a1AT mutant Z protein that the nascent polypeptide chain enters the ER lumen and binds with chaperone proteins which assist in folding, in order to reach the final, secretion-competent conformation. During typical synthesis of the WT a1AT protein, there is then rapid transport through the secretory pathway and secretion from the hepatocyte. However, the mutant Z protein is retained in the ER lumen for minutes to hours, after which time most of the molecules are degraded. It has been shown that a small portion of the a1AT mutant Z molecules in the ER convert to the insoluble, polymerized conformation[11, 12, 18, 33, 34]. The polymers then persist within the cell for a much longer time, possibly the life of the cell [7, 20, 24]. The ER retention of a1AT mutant Z molecules results in a much lower blood level (15% of normal), compared to the WT a1AT serum protein level. With these data in mind, we examined the serum levels of the human a1AT mutant Z protein in the PiZ mice to see if the increased intrahepatic monomer level led to increased secretion, or simply an increase in the steady state of the intrahepatic monomer which is not secreted (figure 2e). The result revealed no significant difference in a1AT mutant Z serum levels in the PiZ mice treated with weekly rapamycin compared to the weekly vehicle treated controls. This indicated that there was no increased secretion of the mutant Z protein, but raised the possibility that the increased degradation of the polymer in the hepatocyte led to further alterations in the intracellular compartmentalization of the a1AT mutant Z protein.
Rapamycin is known in some systems to alter protein synthesis. Since we had detected a modest increase in steady state a1AT mutant Z monomer associated with rapamycin noted above, we next wanted to determine if there was evidence of altered protein synthesis in the rapamycin treated PiZ mouse livers. We found no difference in total protein per gram mouse liver between rapamycin treated and controls in either males or females. We also compared serum murine albumin levels and found no difference between rapamycin treated and controls in either males or females. We next quantified steady state liver a1AT mRNA and found no significant difference between all the combined groups of rapamycin treated versus control PiZ mice. However, when the male sub group was analyzed alone there was a 55% increase in a1AT mRNA in the rapamycin treated group compared to controls which reached statistical significance (p=0.042). This was the same group of males which had a similar, small increase in the monomer a1AT mutant Z protein, suggesting a modest increase in a1AT synthesis as the mechanism. Previous studies have suggested, in both ZZ humans and in the PiZ mouse model, that there are sex hormone based alterations in a1AT mutant Z protein synthesis and accumulation, as noted above.
Weekly, pulsatile dosing of rapamycin is associated with reduced cytoplasmic a1AT mutant Z polymer accumulation
The classically described histopathologic lesion of a1AT deficiency liver disease is the appearance of “globules” of a1AT mutant Z polymerized protein within massively dilated single cisternae of ER. In human liver, and in the PiZ model, these globules appear in many, but not all hepatocytes; they grow larger as the organism grows older, and they can become as large as the hepatocyte nucleus[7, 20, 24]. However, previous studies have shown that a1AT mutant Z protein polymers are also present in non-dilated ER within the hepatocellular cytoplasm, even in hepatocytes without globules[7, 16]. We next examined the PiZ livers treated with weekly rapamycin compared to the weekly vehicle treated controls by histologic examination to determine if there were changes in globule accumulation (figure 3a). The results showed no difference in either globule number or in globule size in the rapamycin treated mice compared to controls (there was an indication of a trend of reduced globules, but it did not reach statistical significance). We then performed immunohistochemistry for a1AT, which labels both a1AT globules and also any areas of cytoplasm devoid of globules, but which contain a1AT mutant Z protein. Both the human and the PiZ mouse liver are known to be heterogeneous with respect to a1AT protein synthesis and in hepatocellular globule accumulation. In previous studies immunohistochemistry has clearly delineated hepatocytes with intracellular accumulation of a1AT mutant Z protein, even if there was no globule formation [16, 20]. The results of a1AT immunostaining in the rapamycin treated mice show that cytoplasmic a1AT staining is not present in the globule devoid areas. This is in significant contrast to the control PiZ mice which show typical dense staining, even in many areas of the hepatic lobule devoid of globules, which has been previously shown to represent a1AT mutant Z polymers not incorporated into globules (figure 3b)[7, 16]. This result indicates that reduction in a1AT mutant Z protein polymers not within globules is associated with weekly rapamycin dosing, and could suggest that autophagy is important in degrading a1AT mutant Z polymer proteins which have been recently synthesized and not yet incorporated into globules.
Figure 3. Effect of weekly pulse dosing rapamycin on the distribution of a1AT protein in the liver in PiZ mice.
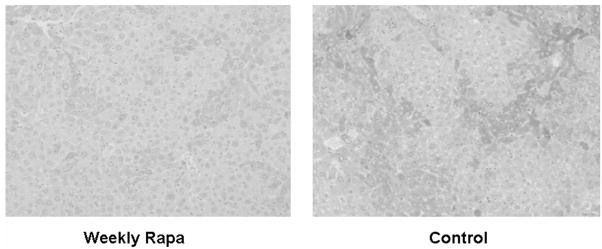
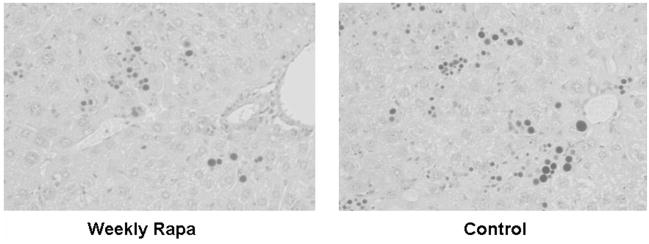
Panel a, a1AT immunohistochemical staining of globule devoid areas of PiZ mouse liver from weekly rapamycin treated (left panel) and controls (right panel). Dark areas indicate increased a1AT immunoreactivity. Panel b, photomicrograph of PiZ mouse liver treated with weekly dose rapamycin (left panel) versus control (right panel) stained with Periodic Acid-Schiff and digestion to highlight globules (dark hepatocellular inclusions shown on light background) of a1AT mutant Z protein.
Liver injury is reduced in PiZ mice treated with weekly, pulsatile dosing of rapamycin
There is a complex injury cascade in human liver, and in PiZ mouse model liver resulting from the intracellular accumulation of a1AT mutant Z protein, which is likely maximally stimulated by accumulation of the mutant Z protein specifically in the polymerized conformation [2, 11, 35, 36]. This includes ER stress with cleavage of the murine ER stress-related protein, caspase 12, increased apoptosis of globule containing cells, a compensatory hepatocellular proliferative response, and dysplasia. Since rapamycin might have had effects on the inflammatory response through its immunosuppressive activity, we first examined scoring of inflammation under histopathologic examination by a single, blinded examiner (EB) (figure 4a). The standard analysis in clinical hepatology studies uses scoring range of 1–4 (1 = mild expansion of portal tract with leukocytes to 4 = sheets of infiltrating leukocytes in the lobule and portal areas deforming the architecture). The results showed no reduction in the modest lobular, lymphocytic infiltrate present (mean 0.8 and 0.9 rapamycin and control PiZ livers respectively) in the liver of the PiZ mice treated with weekly rapamycin compared to controls. This is not unexpected since attack by leukocytes is not thought to be a large part of the pathophysiological injury cascade in this disease. Previous publications have also shown that TUNEL stain is not informative in quiescent liver in this model [11]. However, several investigators have shown that apoptotic signaling, including caspase 12 cleavage, which is thought to be specifically related to ER stress apoptosis, is associated with a1AT mutant Z polymer protein cell injury[8, 9, 11, 28, 37–39]. Therefore, we next examined the cleavage of caspase 12 by immunoblot of the homogenized livers. The results showed significantly decreased cleaved caspase 12 in the weekly rapamycin treated PiZ mice compared to controls (figure 4b). Next, we examined hepatocellular proliferation by bromodeoxy uridine incorporation into dividing hepatocellular nuclei to assay the hepatic proliferative response which compensates for increase hepatocellular death [8, 9]. Previous studies in the PiZ mouse have shown that increased injury results in increased hepatocellular death and a compensatory increase in the proliferation of the remaining hepatocytes to preserve hepatocellular mass. Previously published studies demonstrated that increased a1AT mutant Z protein polymer in the liver was associated with increased compensatory hepatocellular proliferation. Our results here show that weekly rapamycin treatment is associated with a significantly decreased hepatocellular proliferation, which suggests a decrease in hepatocyte-injury related reactive proliferation (figure 4c). Finally, we examined the effect of rapamycin on hepatic fibrosis using Sirius red staining which labels as red fibrous tissue in the hepatic parenchyma. Previous studies of the PiZ model have shown increased susceptibility to hepatic fibrosis compared to wild type mice ([28] and manuscript under review). Our results revealed a significant reduction in hepatic fibrosis associated with rapamycin treatment. Scoring by single, blinded examiner (JT) using the Ishak System commonly used in clinical hepatology studies to evaluate progression of fibrotic liver injury (Ishak system 1–6, 1 = normal, 2 = portal tract expanded by fibrosis, 3 = fibrosis bridging between portal tracts, 4 = complete bridging forming nodules, 5 = regenerative nodules with loss of central vein and other normal structures, 6 = severe cirrhotic regenerative nodules) resulted in mean scores of WT = 1.0, PiZ rapa = 1.7, PiZ control = 2.6 (p<0.05 PiZ rapa compared to PiZ control).
Figure 4. Effect of weekly pulse dosing rapamycin on liver injury in PiZ mice.
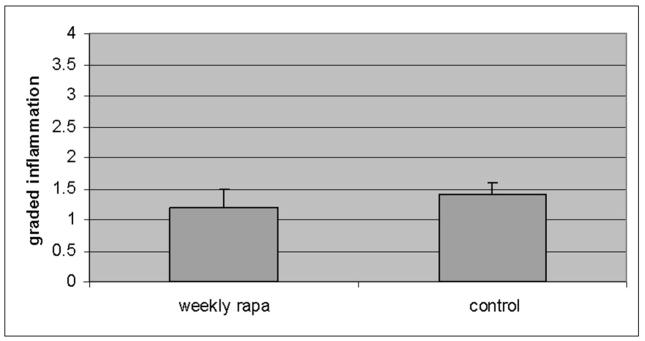
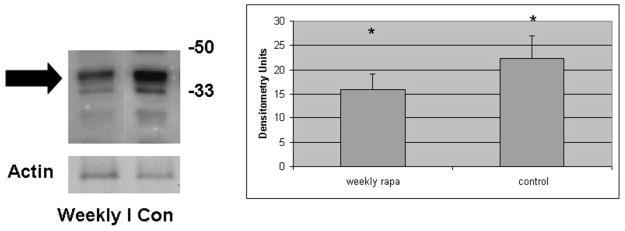
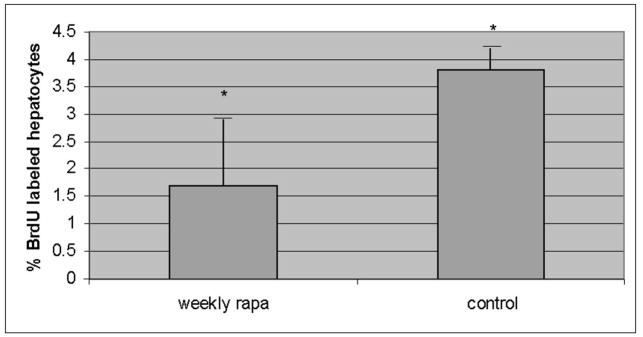
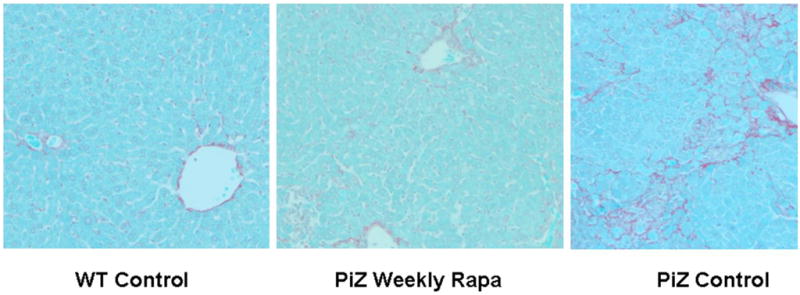
Panel a, inflammation graded 1+ to 4+ by blinded examiner and shown as means in weekly rapamycin treated (left panel) versus controls (right panel). Bars +/− S.D. Comparison p>0.3. Panel b, example of SDS-PAGE followed by immunoblot for caspase 12 (left panel) and means of densitometric quantification of replicates (right panel) of PiZ mice treated with weekly rapamycin versus controls. Arrow indicates 37–39kDa cleaved (activated) caspase 12. Bars +/− S.D. Molecular weight markers, Mrx103 as shown. Comparison * p<0.04. Panel c, quantification of mean percent BrdU labeled hepatocellular nuclei from male PiZ mice treated with weekly rapamycin compared to controls. Bars +/− S.D. Comparison * p<0.03. Panel d, hepatic fibrosis as shown by red fibers in representative photomicrographs stained by Sirius red in WT mice (left panel), weekly PiZ rapa treated (middle panel) compared to control (right panel) PiZ mice.
Discussion
These data suggest the possibility of a new approach to a1AT deficiency, a metabolic liver disease which at present has no specific treatment other than liver transplantation. A large number of studies over the last decade have confirmed the hypothesis that the intrahepatic accumulation of the a1AT mutant Z protein, especially the pool in the polymerized conformation, is the cause of the liver injury in a1AT deficiency[2, 11, 35, 36]. Here for the first time, we present data on an in vivo method associated with reduced accumulation of a1AT mutant Z protein polymers. However, additional future work will be required as many questions remain.
First, what aspects of the regulation of autophagy respond more strongly to weekly, pulsatile dosing rather than daily dosing of rapamycin. We are presently examining this question. Detailed analysis of this, and other pharmacokinetic issues will be needed in order to plan a possible evaluation of this treatment in human patients. Second, why did the effect of increased proteolysis appear to be greatest on the a1AT mutant Z polymers not incorporated into globules? The role of globule formation in this disease is still poorly understood, although our previous work has shown that the globules are pure a1AT mutant Z protein in the polymerized conformation and that globule containing cells are more susceptible to apoptotic death [11]. However, it is not yet clear if the globule is an indicator of injury to an individual hepatocyte, or if globule formation is a method of sequestration of the cytotoxic polymer as a means of protecting an already injured cell from further damage[40, 41]. Previous data show that globule containing cells are more likely to undergo apoptosis than globule devoid cells, and that globule devoid cells tend to be dividing more frequently to replace dying, globule containing hepatocytes[11]. Therefore, we would propose that the hepatocytes form globules only after the active proteolytic mechanisms have been unable to keep the steady state level of intrahepatic a1AT mutant Z protein polymer accumulation below some threshold level. Then the globule forms, which could be a marker for a cell spiraling down the injury cascade to apoptosis. If this hypothesis is correct, then the effect of rapamycin on reducing a1AT mutant Z protein polymer in cells which have not yet “lost the battle” in resisting globule formation, might be especially beneficial to the liver in reducing cell death and liver injury.
Our data further support this view by showing that the compensatory hepatocellular proliferation, which has been well documented to be related to liver injury in this, and in other disease models, was reduced by the rapamycin weekly treatment. It is possible that we did not see a larger effect on globule accumulation because we started treating mice at full maturity. We chose this age, 3 months, because past work in this model had suggested that at that age there is significant ongoing injury in the liver. Further, we felt that initiating a potentially beneficial treatment at this age of high injury would experimentally increase our chances of observing reduced injury, such as the reduced caspase 12 cleavage we documented. Our past work has shown that globules are already well formed, and grow only slowly in mice older than 3 months.
The cautionary aspect of this work is the detection of the small increase in a1AT mutant Z protein monomers in the male mice, and which appeared to be associated with a small, but statistically significant increase in a1AT mRNA in those same males. Although the magnitude of a 55% increase is fairly modest when considering the physiological significance of mRNA measurements, it was at a statistically significant P value of P = 0.042. However, the overall hepatic burden of a1AT protein in those livers was decreased due to a much greater magnitude in the reduction of the polymerized mutant Z protein. There was no change in the serum levels associated with rapamycin treatment, which suggested that this small increase in monomer protein was disposed of by intracellular proteolysis. These issues will require careful study in future efforts to bring pro-autophagic treatments to humans with a1AT deficiency.
In conclusion, we have shown that rapamycin is associated with increased autophagy and reduced injury in this in vivo model of a1AT deficiency. Additional biochemical and cell biological analysis will be needed to understand the exact mechanism of this drug in mitigating liver injury in the PiZ mice so that it can be developed into a viable treatment for human disease.
Acknowledgments
Financial Support: NIH DK-067489 (JT), The American Liver Foundation, Alpha-1 Foundation, Cardinal Glennon Children’s Medical Center Pediatric Research Institute, and Washington University Digestive Disease Research Core Center NIH PO3 DK52574. We appreciate the invaluable mentoring and leadership of Dr. David Perlmutter.
Abbreviations
- a1AT
alpha-1-antitrypsin
- ER
Endoplasmic reticulum
- HCC
Hepatocellular carcinoma
- BrdU
Bromodeoxy Uridine
- Rapa
Rapamycin
Footnotes
Author contributions: SK, JHT, MA: concept and design; JHT, KB: acquisition of data; JHT, EMB, DH, KB: analysis and interpretation; JHT: drafts and revisions.
References
- 1.Greene CM, Miller SD, Carroll T, McLean C, O’Mahony M, Lawless MW, O’Neill SJ, Taggart CC, McElvaney NG. Alpha-1 antitrypsin deficiency: a conformational disease associated with lung and liver manifestations. J Inherit Metab Dis. 2008;31(1):21–34. doi: 10.1007/s10545-007-0748-y. [DOI] [PubMed] [Google Scholar]
- 2.Teckman JH. Alpha1-antitrypsin deficiency in childhood. Semin Liver Dis. 2007;27(3):274–81. doi: 10.1055/s-2007-985072. [DOI] [PubMed] [Google Scholar]
- 3.Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992;357(6379):605–7. doi: 10.1038/357605a0. [DOI] [PubMed] [Google Scholar]
- 4.Perlmutter DH, Brodsky JL, Balistreri WF, Trapnell BC. Molecular pathogenesis of alpha-1-antitrypsin deficiency-associated liver disease: a meeting review. Hepatology. 2007;45(5):1313–23. doi: 10.1002/hep.21628. [DOI] [PubMed] [Google Scholar]
- 5.Teckman JH, Lindblad D. Alpha-1-antitrypsin deficiency: diagnosis, pathophysiology, and management. Curr Gastroenterol Rep. 2006;8(1):14–20. doi: 10.1007/s11894-006-0059-8. [DOI] [PubMed] [Google Scholar]
- 6.American Thoracic Society/European Respiratory Society Statement: Standards for the Diagnosis and Management of Individuals with Alpha-1 Antitrypsin Deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900. doi: 10.1164/rccm.168.7.818. [DOI] [PubMed] [Google Scholar]
- 7.An JK, Blomenkamp K, Lindblad D, Teckman JH. Quantitative isolation of alphalAT mutant Z protein polymers from human and mouse livers and the effect of heat. Hepatology. 2005;41(1):160–7. doi: 10.1002/hep.20508. [DOI] [PubMed] [Google Scholar]
- 8.Rudnick DA, Liao Y, An JK, Muglia LJ, Perlmutter DH, Teckman JH. Analyses of hepatocellular proliferation in a mouse model of alpha-1-antitrypsin deficiency. Hepatology. 2004;39(4):1048–55. doi: 10.1002/hep.20118. [DOI] [PubMed] [Google Scholar]
- 9.Rudnick DA, Shikapwashya O, Blomenkamp K, Teckman JH. Indomethacin increases liver damage in a murine model of liver injury from alpha-1-antitrypsin deficiency. Hepatology. 2006;44(4):976–82. doi: 10.1002/hep.21326. [DOI] [PubMed] [Google Scholar]
- 10.Teckman JH, An JK, Blomenkamp K, Schmidt B, Perlmutter D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am J Physiol Gastrointest Liver Physiol. 2004;286(5):G851–62. doi: 10.1152/ajpgi.00175.2003. [DOI] [PubMed] [Google Scholar]
- 11.Lindblad D, Blomenkamp K, Teckman J. Alpha-1-antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology. 2007;46(4):1228–35. doi: 10.1002/hep.21822. [DOI] [PubMed] [Google Scholar]
- 12.Wu Y, Whitman I, Molmenti E, Moore K, Hippenmeyer P, Perlmutter DH. A lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc Natl Acad Sci U S A. 1994;91(19):9014–8. doi: 10.1073/pnas.91.19.9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brodsky JL, Scott CM. Tipping the delicate balance: defining how proteasome maturation affects the degradation of a substrate for autophagy and endoplasmic reticulum associated degradation (ERAD) Autophagy. 2007;3(6):623–5. doi: 10.4161/auto.4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Y, Swulius MT, Moremen KW, Sifers RN. Elucidation of the molecular logic by which misfolded alpha 1-antitrypsin is preferentially selected for degradation. Proc Natl Acad Sci U S A. 2003;100(14):8229–34. doi: 10.1073/pnas.1430537100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sifers RN. Cell biology. Protein degradation unlocked. Science. 2003;299(5611):1330–1. doi: 10.1126/science.1082718. [DOI] [PubMed] [Google Scholar]
- 16.Teckman JH, Perlmutter DH. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol. 2000;279(5):G961–74. doi: 10.1152/ajpgi.2000.279.5.G961. [DOI] [PubMed] [Google Scholar]
- 17.Teckman JH, Burrows J, Hidvegi T, Schmidt B, Hale PD, Perlmutter DH. The proteasome participates in degradation of mutant alpha 1-antitrypsin Z in the endoplasmic reticulum of hepatoma-derived hepatocytes. J Biol Chem. 2001;276(48):44865–72. doi: 10.1074/jbc.M103703200. [DOI] [PubMed] [Google Scholar]
- 18.Teckman JH, Gilmore R, Perlmutter DH. Role of ubiquitin in proteasomal degradation of mutant alpha(1)-antitrypsin Z in the endoplasmic reticulum. Am J Physiol Gastrointest Liver Physiol. 2000;278(1):G39–48. doi: 10.1152/ajpgi.2000.278.1.G39. [DOI] [PubMed] [Google Scholar]
- 19.Teckman JH, Perlmutter DH. The endoplasmic reticulum degradation pathway for mutant secretory proteins alpha1-antitrypsin Z and S is distinct from that for an unassembled membrane protein. J Biol Chem. 1996;271(22):13215–20. doi: 10.1074/jbc.271.22.13215. [DOI] [PubMed] [Google Scholar]
- 20.Teckman JH, An JK, Loethen S, Perlmutter DH. Fasting in alpha1-antitrypsin deficient liver: constitutive [correction of consultative] activation of autophagy. Am J Physiol Gastrointest Liver Physiol. 2002;283(5):G1156–65. doi: 10.1152/ajpgi.00041.2002. [DOI] [PubMed] [Google Scholar]
- 21.Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci. 2005;118(Pt 1):7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fortun J, Dunn WA, Jr, Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23(33):10672–80. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perlmutter DH. Alpha1-antitrypsin deficiency: liver disease associated with retention of a mutant secretory glycoprotein in the endoplasmic reticulum. Methods Mol Biol. 2003;232:39–56. doi: 10.1385/1-59259-394-1:39. [DOI] [PubMed] [Google Scholar]
- 24.Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, Yoshimori T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem. 2006;281(7):4467–76. doi: 10.1074/jbc.M509409200. [DOI] [PubMed] [Google Scholar]
- 25.Paglin S, Lee NY, Nakar C, Fitzgerald M, Plotkin J, Deuel B, Hackett N, McMahill M, Sphicas E, Lampen N, Yahalom J. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 2005;65(23):11061–70. doi: 10.1158/0008-5472.CAN-05-1083. [DOI] [PubMed] [Google Scholar]
- 26.Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell. 2005;19(1):15–26. doi: 10.1016/j.molcel.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 27.Cruz PE, Mueller C, Cossette TL, Golant A, Tang Q, Beattie SG, Brantly M, Campbell-Thompson M, Blomenkamp KS, Teckman JH, Flotte TR. In vivo post-transcriptional gene silencing of alpha-1 antitrypsin by adeno-associated virus vectors expressing siRNA. Lab Invest. 2007;87(9):893–902. doi: 10.1038/labinvest.3700629. [DOI] [PubMed] [Google Scholar]
- 28.Mencin A, Seki E, Osawa Y, Kodama Y, De Minicis S, Knowles M, Brenner DA. Alpha-1 antitrypsin Z protein (PiZ) increases hepatic fibrosis in a murine model of cholestasis. Hepatology. 2007;46(5):1443–52. doi: 10.1002/hep.21832. [DOI] [PubMed] [Google Scholar]
- 29.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA, Jr, Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovacs AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, Lopez-Otin C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Melendez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl T, Munz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nurnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri I, Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov O, Settleman J, Shacka JJ, Shapiro IM, Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Talloczy Z, Tanaka K, Tanida I, Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcategui NL, van der Klei I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z, Yuzaki M, Zabirnyk O, Zheng X, Zhu X, Deter RL. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A. 2000;97(4):1796–801. doi: 10.1073/pnas.97.4.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 32.Sarkar S, Rubinsztein DC. Huntington’s disease: degradation of mutant huntingtin by autophagy. Febs J. 2008;275(17):4263–70. doi: 10.1111/j.1742-4658.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- 33.Qu D, Teckman JH, Omura S, Perlmutter DH. Degradation of a mutant secretory protein, alpha1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J Biol Chem. 1996;271(37):22791–5. doi: 10.1074/jbc.271.37.22791. [DOI] [PubMed] [Google Scholar]
- 34.Lomas DA, Mahadeva R. Alpha1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest. 2002;110(11):1585–90. doi: 10.1172/JCI16782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perlmutter DH. The cellular response to aggregated proteins associated with human disease. J Clin Invest. 2002;110(9):1219–20. doi: 10.1172/JCI16780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawless MW, Greene CM, Mulgrew A, Taggart CC, O’Neill SJ, McElvaney NG. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J Immunol. 2004;172(9):5722–6. doi: 10.4049/jimmunol.172.9.5722. [DOI] [PubMed] [Google Scholar]
- 37.Miller SD, Greene CM, McLean C, Lawless MW, Taggart CC, O’Neill SJ, McElvaney NG. Tauroursodeoxycholic acid inhibits apoptosis induced by Z alpha-1 antitrypsin via inhibition of Bad. Hepatology. 2007;46(2):496–503. doi: 10.1002/hep.21689. [DOI] [PubMed] [Google Scholar]
- 38.Rudnick DA, Perlmutter DH. Alpha-1-antitrypsin deficiency: a new paradigm for hepatocellular carcinoma in genetic liver disease. Hepatology. 2005;42(3):514–21. doi: 10.1002/hep.20815. [DOI] [PubMed] [Google Scholar]
- 39.Hidvegi T, Schmidt BZ, Hale P, Perlmutter DH. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J Biol Chem. 2005;280(47):39002–15. doi: 10.1074/jbc.M508652200. [DOI] [PubMed] [Google Scholar]
- 40.Granell S, Baldini G. Inclusion bodies and autophagosomes: are ER-derived protective organelles different than classical autophagosomes? Autophagy. 2008;4(3):375–7. doi: 10.4161/auto.5605. [DOI] [PubMed] [Google Scholar]
- 41.Granell S, Baldini G, Mohammad S, Nicolin V, Narducci P, Storrie B. Sequestration of mutated alpha1-antitrypsin into inclusion bodies is a cell-protective mechanism to maintain endoplasmic reticulum function. Mol Biol Cell. 2008;19(2):572–86. doi: 10.1091/mbc.E07-06-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]