

INTRODUCTION
The role of inflammatory mediators in the pathogenesis of exercise-induced bronchoconstriction (EIB) has become increasingly clear from studies conducted in human subjects with asthma over the last 15 years. These results indicate very clearly that mediators from mast cells and other airway leukocytes are released into the airways following exercise challenge in individuals who are susceptible to EIB. The evidence that the release of such mediators plays a causal role in the pathogenesis of EIB is strongest for the cysteinyl leukotrienes (CysLTs) C4, D4, and E4, in part because of the availability of receptor antagonists and synthesis inhibitors that alter the leukotriene (LT) pathway. It is also apparent that several other mediators that may play important roles in the pathogenesis of EIB are released into the airways, but the precise roles of these mediators are inferred from animal studies and from the basic biological function of the mediators. Many unanswered questions remain, including why leukocytes become activated in the airways, how either evaporative water loss or the addition of a hyperosmolar solution to the airways initiates downstream cellular effects, and what the connection is between the epithelium and these events that leads to leukocyte activation.
ALTERATIONS IN THE AIRWAYS THAT LEAD TO EIB
As a prototypical feature of indirect airway hyperresponsiveness (AHR), EIB shares common features with other stimuli such as hypertonic aerosols and adenosine, which cause bronchoconstriction through the release of mediators.1 EIB is only weakly related to structural alterations of the lung2–4 and airway smooth-muscle hyperresponsiveness measured by direct-acting agonists of smooth-muscle contraction such as methacholine.5 In one study of 27 asthmatic children, there was no relationship between the methacholine PC20 and maximum decrease in forced expiratory volume in 1 second (FEV1) after exercise (r = −0.2, P = .40). Another study of elite athletes found that 9 of 25 elite athletes with a positive eucapnic voluntary hyperpnea (EVH) challenge, a surrogate for exercise challenge, had a positive methacholine challenge, demonstrating the discordance between these 2 different features of AHR.6 Collectively, these studies indicate that EIB is pathophysiologically distinct from other features of asthma, but shares common features with other forms of indirect AHR. A clear understanding of the pathophysiology of EIB is important, as EIB at an early age is associated with the persistence of asthma later in life.7–9 There is also evidence that chronic lung disease early in life is a risk factor for the development or persistence of EIB later in life.10
As subjects with asthma can be characterized based on the presence or absence of EIB using a dry air exercise challenge test, several studies have made comparisons between asthmatics with and without EIB to better understand the basis for EIB. An inflammatory basis of EIB is suggested by an increase in the fraction of exhaled nitric oxide (FENO) among asthmatics who are susceptible to EIB,11 especially in subjects with atopy.12 Although differences in the concentration of inflammatory lipid mediators have not been identified in studies evaluating metabolites in the urine,13 the concentrations of inflammatory lipid mediators are increased in the airways of individuals with EIB.2,4,14 In particular, the concentration of CysLTs is increased in induced sputum of adults with EIB,4 and in exhaled breath condensate (EBC) of children with EIB.2 In addition, the levels of 8-isoprostanes, nonenzymatic products of phospholipid oxidation, are increased in EBC of asthmatics with EIB, and correlate with the severity of EIB.14 There is also evidence of a reduction in the formation of protective lipid mediators in the airways such as lipoxin A4 in patients with EIB.15 Prostaglandin E2 (PGE2) is a key regulatory eicosanoid that inhibits EIB when administered by inhalation.16 The production of PGE2 relative to CysLTs is reduced in patients with EIB.4 As the epithelium is a major source of PGE2, it is notable that the number of epithelial cells shed into the induced sputum is greater in patients with EIB.4
The intensity of cellular inflammation in the airways may be an important factor in the susceptibility to EIB, as the formation of inflammatory eicosanoids such as CysLTs and PGD2 is largely restricted to myeloid cells, especially mast cells that contain leukotriene C4 (LTC4) and prostaglandin D2 (PGD2) synthases and eosinophils that also contain LTC4 synthase.17 Several studies have associated the degree of sputum eosinophilia with the severity of EIB,4 although sputum eosinophilia is not present in all patients with EIB.4 A recent study of the effect of inhaled corticosteroid (ICS) ciclesonide in steroid-naïve asthmatic patients with EIB found that the magnitude and onset of the suppression of EIB in response to high-dose, but not low-dose, ICS therapy was associated with the degree of sputum eosinophilia.18
In a genome-wide expression study of airway cells, the authors identified a unique molecular phenotype of EIB-positive asthmatics relative to asthmatic individuals without EIB, notable for the increased expression of mast cell, mucus, and epithelial repair genes.19 These results are consistent with other studies that found mast-cell involvement in EIB.20 The authors found that the mast-cell genes tryptase and carboxypeptidase A3 (CPA3) were significantly increased in EIB-positive asthmatics, but the expression of chymase was unaltered19; these findings are particularly note-worthy, because the appearance of secretory granule proteases is regulated by the peripheral tissue, and most prior studies have found that the mucosal mast cells express tryptase, but not CPA3 or chymase.21 This intraepithelial mast-cell phenotype with high expression of tryptase and CPA3, but low expression of chymase, was recently described in the T-helper type 2 (Th2) high phenotype of asthma.22,23 Because this Th2 high phenotype is driven by interleukin (IL)-13,24 it is interesting that a genetic study found an association between IL-13 gene polymorphisms and the severity of EIB, and with the treatment response to a CysLT1 receptor antagonist among these subjects with EIB.25 In making comparisons between asthmatics with and without EIB, the authors also identified a selective increase in the levels of the complement component C3a in individuals with EIB.26 Thus, patients who are susceptible to EIB have epithelial shedding, overproduction of inflammatory mediators such as CysLTs, relative underproduction of protective lipid mediators, and infiltration of the airways with eosinophils and mast cells (Fig. 1).
Fig. 1.
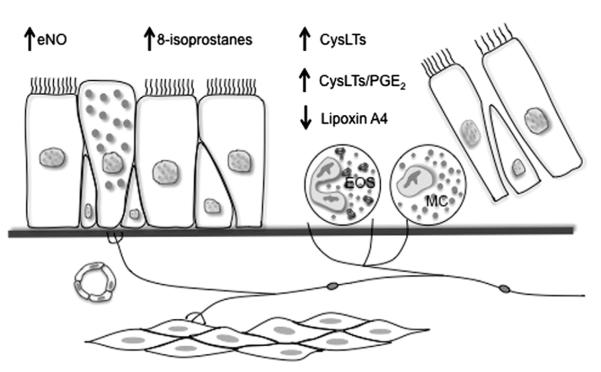
Disease model of exercise-induced bronchoconstriction (EIB) pathogenesis. Studies examining differences between asthmatics with EIB to those without EIB have identified an increased concentration of shed epithelial cells, and infiltration of the airways with eosinophils and mast cells. There is an alteration in the balance of lipid mediators notable for an increase in cysteinyl leukotrienes (CysLTs), CysLT/prostaglandin E2 (PGE2) ratio, and 8-isoprostanes, and a reduction in lipoxin A4. eNO, exhaled nitric oxide. (Adapted from Hallstrand TS, Henderson WR Jr. Role of leukotrienes in exercise-induced bronchoconstriction. Curr Allergy Asthma Rep 2009;9:18–25.)
INFLAMMATORY MEDIATOR RELEASE DURING EIB
During periods of high ventilation, large volumes of air are equilibrated to the humidified body conditions of the lower airways over a short period of time, leading to the transfer of water, with resulting stress to the epithelium and cooling of the airways, largely as a result of water transfer.27–31 During tidal breathing at rest, little if any of the conditioning of the inspired air takes place beyond the upper airway and proximal trachea; however, conditioning of the inspired air takes place farther along the tracheobronchial tree as ventilation increases, forcing incompletely conditioned air to move deeply into the distal airways before it is brought to body conditions.32 This movement of water out of the airways triggers the release of mediators predominantly from airway leukocytes. The cellular mechanism leading to the activation of leukocytes, either directly through the movement of water or via a signal from the epithelium in response to this water movement, is not known in detail. There is strong evidence that leukocyte-derived eicosanoids including CysLTs and PGD2 are released into the airways following an exercise challenge as assessed by induced sputum analysis.20,33 Similarly, in an analysis of EBC, the levels of CysLTs increased after exercise challenge, most notably in the EIB-positive group; furthermore, the change in CysLTs in EBC following challenge was correlated with the severity of EIB.34 In nonasthmatic subjects, a gene-expression profiling study of peripheral blood following exercise challenge found increases in the expression of 5-lipoxygenase (5-LO) and 5-LO–activating protein (FLAP) in response to exercise challenge, as well as increased levels of LTs in the peripheral blood.35
The formation of LTs and other eicosanoids is initiated by the release of unesterified arachidonic acid by the hydrolysis at the sn-2 position of membrane phospholipids by phospholipase A2 (PLA2) (Fig. 2). Arachidonic acid is transferred by FLAP to 5-LO, initiating the oxygenation of arachidonic acid to 5(S)-hydroperoxyeicosatetraenoic acid (5S-HpETE), followed by dehydration to the unstable epoxide leukotriene A4 (LTA4).36 The critical enzyme in the formation of CysLTs from LTA4 is the enzyme LTC4 synthase (LTC4S), part of a family of membrane-bound proteins involved in eicosanoid and glutathione metabolism including FLAP, microsomal glutathione S-transferases (MGSTs), and microsomal prostaglandin E synthase 1. LTC4S conjugates glutathione with a high degree of substrate selectivity for LTA4,37,38 leading to the generation of LTC4 that is exported from the cell and rapidly metabolized to LTD4 and then LTE4.39 The effects of LTD4, including bronchoconstriction and airway inflammation, are mediated through the CysLT1 receptor; however, the vast majority of CysLTs exist in the stable form of LTE4 that does not interact with the CysLT1 receptor.39 Recent landmark studies have identified 2 different receptors for LTE4 that participate in the development of allergic inflammation in animal models.40 The incomplete effectiveness of CysLT1 receptor antagonists may be explained in part by the presence of these additional receptors for LTE4.
Fig. 2.

Eicosanoid formation from arachidonic acid via the 5- and 15-lipoxygenase and cyclooxygenase pathways. COX, cyclooxygenase; CysLT, cysteinyl leukotriene; FLAP, 5-lipoxygenase–activating protein; HETE, hydroxyeicosatetraenoic acid; LO, lipoxygenase; PG, prostaglandin; PLA2, phospholipase A2; TX, thromboxane.
Because the expression of 5-LO is largely restricted to myeloid cells, the majority of LT synthesis occurs in leukocytes; however, arachidonic acid and intermediates such as LTA4 are permeable across cell membranes, allowing for the transcellular metabolism of eicosanoids. Several studies have demonstrated that eicosanoid production in leukocytes is increased when the leukocyte is cocultured with a structural cell such as an epithelial cell.41 An important recent study demonstrated that 5-LO–deficient mice transplanted with immune cells deficient in LTC4S were able to make normal quantities of CysLTs, demonstrating that 5-LO–containing immune cells transfer intermediates that restore LT synthetic capacity by transcellular metabolism.42
The participation of epithelial cells in the release of mediators is suggested by several observations. First, the level of the epithelial-derived 15-lipoxygenase product 15S-hydroxyeicosatetranoic acid (15S-HETE) is increased after exercise challenge, and is elevated in asthmatics compared with controls after exercise challenge.43 The other mechanism involves the relative underproduction of epithelial-derived PGE2 in patients with EIB. Following exercise challenge the level of PGE2 declines in the airways of asthmatics with EIB,20 whereas PGE2 tends to increase in normal subjects.43 This relative imbalance in epithelial-derived PGE2 when compared with leukocyte-derived LTs supports the hypothesis that transcellular transport of epithelial-derived arachidonic acid or a regulator of arachidonic acid release contributes to LT synthesis in leukocytes, favoring bronchoconstriction in the period following exercise challenge (Fig. 3).43 One explanation for these findings is that the epithelium serves to activate the production of inflammatory eicosanoids by leukocytes that are in close contact, and that there is shunting of epithelial-derived arachidonic acid away from the epithelial-derived PGE2. In cell culture, inflammatory cells cocultured with epithelial cells have increased synthesis of leukocyte-derived eicosanoids.44 Under the influence of IL-13, epithelial cells have reduced capacity for PGE2 synthesis through a reduction in the synthetic enzymes cyclooxygenase-2 (COX-2) and PGE synthase 1.45 The underproduction of PGE2 could also directly alter the formation of CysLTs and PGD2, as inhaled PGE2 regulates the levels of these eicosanoids after segmental allergen challenge46 and PGE2 directly alters CysLT formation in cultured mast cells.47
Fig. 3.
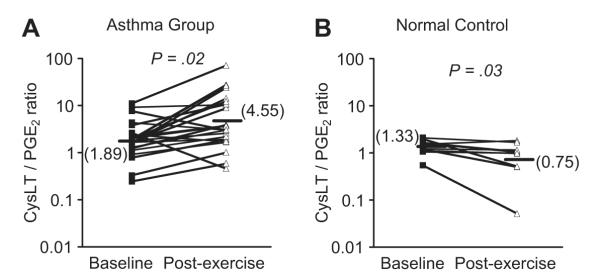
Ratio of CysLTs to PGE2 in induced sputum following exercise challenge. The levels of CysLTs and PGE2 were measured in induced sputum in a group of asthmatics with EIB (A) and in a nonasthmatic control group (B) at baseline and after exercise challenge. The results demonstrate opposing effects of an increase in the CysLT/PGE2 ratio after challenge in the asthma group, whereas the ratio decreased after challenge in the control group. (Data from Hallstrand TS, Chi EY, Singer AG, et al. Secreted phospholipase A2 group X over-expression in asthma and bronchial hyperresponsiveness. Am J Respir Crit Care Med 2007;176:1072–8.)
There is strong evidence that CysLTs play a causative role in the development of bronchoconstriction following exercise, through early proof-of-concept pharmacologic studies with CysLT1 antagonists and 5-LO inhibitors. The CysLT1 antagonist zafirlukast, given by inhalation to 9 patients with EIB 30 minutes before an exercise challenge, reduced the mean maximal percent decrease in FEV1 to 14.5%, compared with 30.2% during placebo administration.48 Oral zafirlukast was also shown to effectively inhibit EIB 2 hours after a single dose in a crossover study of 8 patients, resulting in a reduction in the mean maximal percent decrease in FEV1 after challenge from 36.0% on placebo to 21.6% on zafirlukast.49 A larger recent study showed that montelukast significantly reduced the severity of EIB at 2, 12, and 24 hours after a single dose, based on the maximum decrease in FEV1 (10.8% montelukast vs 22.3% placebo at 2 hours, P≤.001) and area under the curve for the percentage decrease in FEV1.50 A second similarly designed study of 62 patients with EIB also showed similar efficacy at 2 hours after a single dose of montelukast compared with placebo (11.7% montelukast vs 17.5% placebo, P≤.001).51 In a similar manner, the 5-LO inhibitor zileuton administered 4 times daily for 2 days reduced the decrease in FEV1 after challenge, from 28.1% on placebo to 15.6% on zileuton.52 These results clearly demonstrate a role for CysLTs in the pathogenesis of EIB but also indicate that the protection from EIB is incomplete, suggesting that mediators other than the CysLTs may play a significant role and that the loss of bronchoprotective mediators may be important. A recent study found that an ICS added to a CysLT1 antagonist had improved efficacy over treatment with either an ICS or CysLT1 antagonist alone.53
CELLULAR ACTIVATION DURING EIB
Mast cells and eosinophils are strongly implicated as the cellular sources of CysLTs and other eicosanoids such as PGD2 in EIB. The eosinophil product eosinophilic cationic protein (ECP) is released into the airways following challenge, and the amount of ECP release varies with the severity of the EIB under different experimental conditions.33 Following exercise challenge, histamine and the mast-cell protease tryptase are released into the airways, demonstrating mast-cell degranulation (Fig. 4).20 Using the urinary levels of 9α,11β-PGF2 as a marker of PGD2 metabolism and mast-cell activation in response to EVH, the release of PGD2 can be inhibited by pretreatment with a cromone or a high dose of inhaled steroid, suggesting that PGD2 release by mast cells may play an important role in the development of bronchoconstriction after exercise challenge.13,54 Using mannitol challenge as a surrogate for exercise, pharmacologic inhibitors indicate that histamine is responsible for bronchoconstriction early after challenge while the release of CysLTs is responsible for sustained bronchoconstriction.55
Fig. 4.
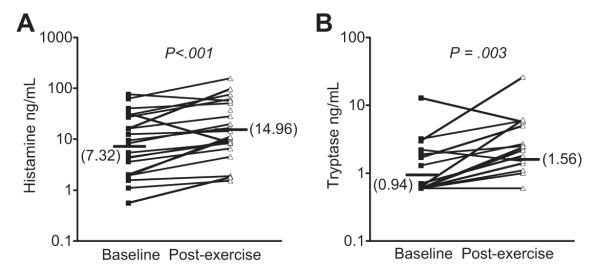
Mast-cell degranulation following exercise challenge in asthmatics with EIB. The levels of histamine (A) and tryptase (B) were measured in induced sputum supernatant at baseline and on a separate day 30 minutes after exercise challenge. Significant increases in both histamine and tryptase were observed following exercise challenge in subjects with EIB. (Data from Hallstrand TS, Moody MW, Wurfel MM, et al. Inflammatory basis of exercise-induced bronchoconstriction. Am J Respir Crit Care Med 2005;172:679–86.)
CONTRIBUTION OF SENSORY NERVES
Sensory nerves release neurokinins when activated through a process called retrograde axonal transmission, leading to bronchoconstriction and mucus release. There is evidence in animal models that the production of eicosanoids such as CysLTs mediate bronchoconstriction, at least in part through the activation of airway sensory nerves. Although sensory nerves may be activated directly by osmotic stimuli, several eicosanoids can either directly activate or alter the activation threshold of sensory nerves.56 In a dog model, a combination neurokinin-1 and neurokinin-2 receptor antagonist inhibited hyperpnea-induced bronchoconstriction (HIB) and the generation of LTs that are known in this model to cause HIB.57 In a guinea pig model of HIB, either a 5-LO inhibitor or a CysLT1 antagonist inhibited HIB and the release of neurokinins, whereas a neurokinin-2 receptor antagonist inhibited HIB but not the release of leukotrienes, suggesting that leukotrienes cause bronchoconstriction via sensory nerves during HIB.58 In humans, the effects of neurokinin-1 antagonists have been modest59,60; however, this lack of efficacy may be due to the predominance of neurokinin A, which binds predominantly to the neurokinin-2 receptor.61 As goblet-cell mucin release is initiated via neurokinin release, it is notable that mucin 5AC (MUC5AC), the predominant gel-forming mucin of goblet cells, is released into the airways during EIB in humans (Fig. 5) and is associated with the levels of CysLTs in the airways.62 Furthermore, the levels of neurokinin A and CysLTs in these individuals after exercise challenge are correlated, suggesting that CysLTs mediate the activation of sensory nerves and mucus release during EIB in humans.62
Fig. 5.
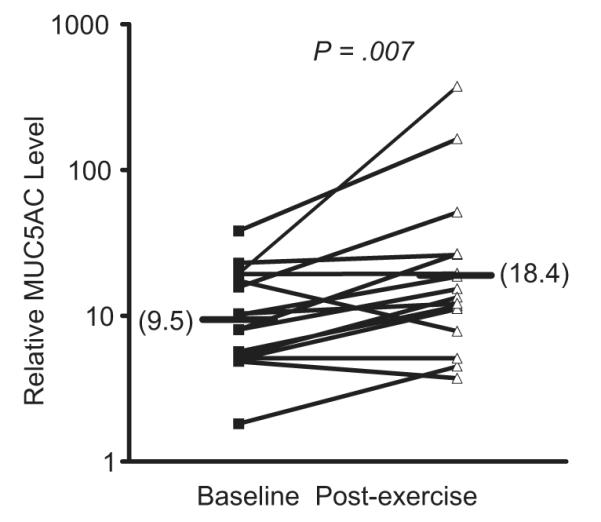
Goblet cell mucin release following exercise challenge in asthmatics with EIB. The levels of the gel-forming mucin MUC5AC were measured before and after exercise challenge in induced sputum supernatant in subjects with EIB. The induced sputum samples were dialyzed to remove the dithiothreitol (DTT) before analysis. A significant increase in MUC5AC was observed following exercise challenge. (Data from Hallstrand TS, Debley JS, Farin FM, et al. Role of MUC5AC in the pathogenesis of exercise-induced bronchoconstriction. J Allergy Clin Immunol 2007;119:1092–8.)
LATE PHASE RESPONSE
Despite the very compelling evidence that exercise challenge acutely causes the release of inflammatory mediators into the airways, several studies failed to demonstrate a cellular influx into the airways or an increase in direct AHR following an exercise challenge.63–66 A physiologic late-phase response has also been difficult to demonstrate in many studies, or has been attributed to factors such as the inevitable fluctuation in airway tone among patients with asthma.67–69 Despite this controversy, some studies indicate that a portion of patients have a second wave of airflow obstruction consistent with a late-phase response to exercise challenge,70,71 and there is clearly a late-phase response in a dog model of HIB.72 The late-phase response to exercise challenge may also be inhibited by treatment with the CysLT1 receptor antagonist montelukast.70 Recent provocative results using EBC demonstrate an increase in high-sensitivity C-reactive protein only in asthmatics with EIB following exercise challenge.73 In addition, the FENO, serum ECP, and AHR to inhaled histamine were all increased following exercise challenge in asthmatics with EIB.73 Further, RANTES and eotaxin in EBC were increased in asthmatics relative to controls, and the levels of RANTES and eotaxin were increased in EBC after exercise challenge in the group with EIB but not in asthmatics without EIB.74,75 These data indicate that inflammatory mediator release with exercise challenge may recruit leukocytes to the airways, but the magnitude of such leukocyte recruitment is small or counterregulated by anti-inflammatory pathways.
REGULATION OF EICOSANOID PRODUCTION AND LEUKOCYTE ACTIVATION BY THE EPITHELIUM
A unifying concept regarding the pathogenesis of EIB is that the epithelium may regulate the response to water loss experienced during exercise challenge. One possibility is that a regulator of eicosanoid formation is released by the airway epithelium in response to water loss. Water is transferred out of the airways during exercise, leading to the movement of water by the semipermeable and osmotically sensitive epithelium that rapidly corrects alterations in airway surface liquid (ASL) osmolarity (Fig. 6).76 A similar epithelial response with the transfer of water should occur in response to exogenous osmolar stimuli such as mannitol and hypertonic saline. In response to water loss in the luminal surface, epithelial cell height reduces as the cells transfer water to the surface.77,78 Because of the ability of the epithelium to respond to movement of water, there is little evidence that any osmotic gradient exists more than transiently in the ASL.79 However, evaporative water loss from epithelial cells initiates the release of cellular adenosine triphosphate and adenosine, leading to the activation of chloride channels and increasing intracellular calcium.76 This stress to the epithelium during exercise may explain the increased levels of adenosine in EBC following exercise challenge that correlate with the severity of EIB.80 It is also known that osmotic stimuli can directly activate inflammatory cells such as mast cells81; however, hyperosmolarity per se exists only transiently in the airways, and likely serves as a stimulus primarily to the epithelium. The connection between epithelial stress induced by water loss and the activation of leukocytes in the airways needs to be understood more completely.
Fig. 6.
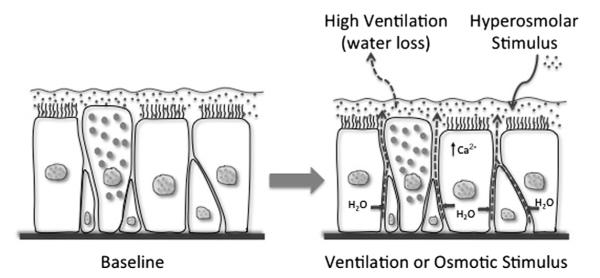
Basic overview of the epithelial response to water loss from the airway surface liquid. As ventilation increases, water is lost from the airway surface liquid, resulting in transient hyperosmolarity and the passive movement of water from airway epithelial cells (arrows) to restore the osmolarity of the airway surface liquid. The movement of water causes epithelial cells to shrink and initiates cellular signaling events, including an increase in intracellular calcium (Ca2+). The addition of osmotically active substances such as mannitol and hypertonic saline similarly cause water movement to restore the surface osmolarity.
The first rate-limiting step in eicosanoid formation is the release of arachidonic acid mediated by PLA2. It is well known that cytosolic PLA2α (cPLA2α) functions as a major regulator of efficient eicosanoid synthesis.82 More recently, a family of secreted PLA2s (sPLA2) have been described that can also serve as regulators of eicosanoid synthesis and may preferentially direct eicosanoid production toward LT synthesis.39 It has been known for some time that sPLA2 activity in the airways increases following allergen challenge in the upper and lower airways.83–85 Of the mammalian sPLA2s, groups V and X have generated the most interest because of their capacity to initiate cellular eicosanoid synthesis,86 particularly sPLA2 group X (sPLA2-X), because it is the most potent of the sPLA2s at releasing arachidonic acid from membrane phospholipids. In murine models of asthma, genetic deficiency of either sPLA2-V or sPLA2-X attenuates the development of allergen-induced inflammation, mucus release, and AHR,87,88 as does the pharmacologic inhibition of human sPLA2-X expressed in a transgenic mouse model.89 The authors have found that sPLA2-X levels are increased in the bronchoalveolar lavage fluid of patients with asthma and that sPLA2-X is predominantly expressed in the airway epithelium.90 In addition, using semiquantitative techniques they have found that sPLA2-X protein in induced sputum supernatant and epithelial cells immunostaining for sPLA2-X in induced sputum increases following exercise challenge, suggesting that activation or release of sPLA2-X may be involved in the generation of eicosanoids following exercise challenge.43
The release of sPLA2-X or other similar enzymes into the airways after exercise challenge could play an important role in the pathogenesis of EIB through the initiation of mediator formation in leukocytes, the degradation of surfactant phospholipids, and the generation of lysophospholipids. The authors examined the ability of sPLA2-X to efficiently activate CysLT formation by human eosinophils.91 Recombinant human sPLA2-X initiates arachidonic acid release and CysLT synthesis through a mechanism that is dependent on the enzymatic activity of sPLA2-X, but could also be replicated by lysophospholipids released by eosinophils in response to sPLA2-X. Of interest, the full mechanism of sPLA2-X–mediated activation of eosinophils involves the activation of the mitogen-activated kinase cascade and the activation of cPLA2α; however, CysLT formation is amplified by sPLA2-X, probably as an additional source of arachidonic acid.
Another important finding is the increased expression of transglutaminase 2 (TGM2) in the airways of patients with EIB relative to asthmatics without EIB.19 It is notable that the TGM2 gene is located on chromosome 20q11.2-12 near a cluster of genes related to epithelial barrier function, in close proximity to a region linked to both atopic dermatitis and asthma.92 Using an in vitro assay of sPLA2 activity, the authors found that recombinant human TGM2 enzymatically modifies sPLA2-X, leading to a substantial increase in the sPLA2 activity of the enzyme, suggesting that one of the mechanisms of TGM2 action in asthma is regulation of eicosanoid and lysophospholipid synthesis. The potential importance of this finding was highlighted in mouse models showing that TGM2 is induced in the airways of mice sensitized and challenged with ovalbumin in the presence of adjuvant,93 as well as in mouse models of PMA-induced atopic dermatitis and immunoglobulin E (IgE)-dependent passive cutaneous anaphylaxis.94 In one study a peptide that inhibits both TGM2 and PLA2 reduced allergen-induced airway inflammation and eicosanoid formation, but the specific role of TGM2 remains to be fully elucidated.93 A chemical inhibitor of TGM2 partially inhibited PMA-induced dermatitis and IgE-dependent cutaneous anaphylaxis.94 Collectively these studies suggest that the epithelium can serve as an important regulator of eicosanoid formation, and that the release of sPLA2-X from the airway epithelium could regulate mediator formation in EIB.
SUMMARY
Exercise challenge leads to a distinct syndrome of bronchoconstriction that occurs in a susceptible group of subjects who have epithelial shedding, overproduction of inflammatory eicosanoids, and mast-cell and eosinophil infiltration of the airways. Exercise challenge causes water loss from the airways, leading to the movement of water from the epithelium to correct the resultant transient shift in osmolarity. Although the precise mechanism by which water loss leads to the activation of leukocytes is not known, it is clear that exercise challenge initiates the release of inflammatory mediators from leukocytes such as mast cells and eosinophils. In the case of the release of CysLTs, it is clear that the release of CysLTs acts via the CysLT1 receptor to initiate bronchoconstriction, based on studies in humans using pharmacologic inhibitors. Other mediators such as 15S-HETE and PGD2 are released from the airway cells, but the specific role of these mediators in the development of bronchoconstriction is less certain. Based predominantly on animal and in vitro studies, sensory nerves seem to play an important role in either transmitting the effects of eicosanoids that are released in the airways or as direct sensors of changes in airway osmolarity, a process that is potentiated by elevated levels of eicosanoids. A possible explanation for the cellular events in the airways is that the airway epithelium, acting as the primary sensor of water loss, releases a product that serves to activate leukocytes that are in close contact with the epithelium, leading to the release of leukocyte-derived eicosanoids and activation of sensory nerves. One such product is sPLA2-X, which is avidly expressed in the airway epithelium, increased in the airway fluid of asthmatics, and serves to activate airway cells such as eosinophils to generate CysLTs. The regulation of these cellular events requires further study so that targeted therapies may be developed to modulate this important aspect of asthma.
KEY POINTS.
Patients who are susceptible to exercise-induced bronchoconstriction have epithelial shedding, infiltration of the airways with mast cells and eosinophils, and increased production of inflammatory mediators such as leukotrienes.
During exercise and hyperpnea the inspired air is equilibrated to the conditions of the lower airways, resulting in the transfer of water out of the airways.
Following exercise challenge, mediators such as cysteinyl leukotrienes and prostaglandin D2 are released into the airways from mast cells and eosinophils.
Sensory nerves may mediate the effects of cysteinyl leukotrienes and other lipid mediators, leading to smooth-muscle contraction and mucus release.
The epithelium may serve as a regulator of leukocyte activation in response to water loss or osmotic stress, but the mechanism remains incompletely understood.
Acknowledgments
Funding: Supported by National Institutes of Health grant HL089215.
T.S. Hallstrand has received research grants from the NIH, American Lung Association, has served as a consultant for Amgen and TEVA pharmaceuticals, and has received lecture fees from Merck & Co. W.R. Henderson has served on advisory boards for Gilead Sciences, has received lecture fees from Merck & Co., and has received industry-sponsored grants from Genentech and Gilead Sciences.
Footnotes
Disclosures: W.A. Altemeier and M.L. Aitken have no conflicts of interest.
REFERENCES
- 1.Joos GF, O’Connor B, Anderson SD, et al. Indirect airway challenges. Eur Respir J. 2003;21:1050–68. doi: 10.1183/09031936.03.00008403. [DOI] [PubMed] [Google Scholar]
- 2.Carraro S, Corradi M, Zanconato S, et al. Exhaled breath condensate cysteinyl leukotrienes are increased in children with exercise-induced bronchoconstriction. J Allergy Clin Immunol. 2005;115:764–70. doi: 10.1016/j.jaci.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 3.Cabral AL, Conceicao GM, Fonseca-Guedes CH, et al. Exercise-induced bronchospasm in children: effects of asthma severity. Am J Respir Crit Care Med. 1999;159:1819–23. doi: 10.1164/ajrccm.159.6.9805093. [DOI] [PubMed] [Google Scholar]
- 4.Hallstrand TS, Moody MW, Aitken ML, et al. Airway immunopathology of asthma with exercise-induced bronchoconstriction. J Allergy Clin Immunol. 2005;116:586–93. doi: 10.1016/j.jaci.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freezer NJ, Croasdell H, Doull IJ, et al. Effect of regular inhaled beclomethasone on exercise and methacholine airway responses in school children with recurrent wheeze. Eur Respir J. 1995;8:1488–93. [PubMed] [Google Scholar]
- 6.Holzer K, Anderson SD, Douglass J. Exercise in elite summer athletes: challenges for diagnosis. J Allergy Clin Immunol. 2002;110:374–80. doi: 10.1067/mai.2002.127784. [DOI] [PubMed] [Google Scholar]
- 7.Frank PI, Morris JA, Hazell ML, et al. Long term prognosis in preschool children with wheeze: longitudinal postal questionnaire study 1993-2004. BMJ. 2008;336:1423–6. doi: 10.1136/bmj.39568.623750.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riiser A, Hovland V, Carlsen KH, et al. Does bronchial hyperresponsiveness in childhood predict active asthma in adolescence? Am J Respir Crit Care Med. 2012;186:493–500. doi: 10.1164/rccm.201112-2235OC. [DOI] [PubMed] [Google Scholar]
- 9.Stern DA, Morgan WJ, Halonen M, et al. Wheezing and bronchial hyperresponsiveness in early childhood as predictors of newly diagnosed asthma in early adulthood: a longitudinal birth-cohort study. Lancet. 2008;372:1058–64. doi: 10.1016/S0140-6736(08)61447-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joshi S, Powell T, Watkins WJ, et al. Exercise-induced bronchoconstriction in school-aged children who had chronic lung disease in infancy. J Pediatr. 2012 doi: 10.1016/j.jpeds.2012.09.040. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 11.Scollo M, Zanconato S, Ongaro R, et al. Exhaled nitric oxide and exercise-induced bronchoconstriction in asthmatic children. Am J Respir Crit Care Med. 2000;161:1047–50. doi: 10.1164/ajrccm.161.3.9905043. [DOI] [PubMed] [Google Scholar]
- 12.Malmberg LP, Pelkonen AS, Mattila PS, et al. Exhaled nitric oxide and exercise-induced bronchoconstriction in young wheezy children—interactions with atopy. Pediatr Allergy Immunol. 2009;20:673–8. doi: 10.1111/j.1399-3038.2009.00858.x. [DOI] [PubMed] [Google Scholar]
- 13.Kippelen P, Larsson J, Anderson SD, et al. Effect of sodium cromoglycate on mast cell mediators during hyperpnea in athletes. Med Sci Sports Exerc. 2010;42:1853–60. doi: 10.1249/MSS.0b013e3181da4f7d. [DOI] [PubMed] [Google Scholar]
- 14.Barreto M, Villa MP, Olita C, et al. 8-Isoprostane in exhaled breath condensate and exercise-induced bronchoconstriction in asthmatic children and adolescents. Chest. 2009;135:66–73. doi: 10.1378/chest.08-0722. [DOI] [PubMed] [Google Scholar]
- 15.Tahan F, Saraymen R, Gumus H. The role of lipoxin A4 in exercise-induced bronchoconstriction in asthma. J Asthma. 2008;45:161–4. doi: 10.1080/02770900701847068. [DOI] [PubMed] [Google Scholar]
- 16.Melillo E, Woolley KL, Manning PJ, et al. Effect of inhaled PGE2 on exercise-induced bronchoconstriction in asthmatic subjects. Am J Respir Crit Care Med. 1994;149:1138–41. doi: 10.1164/ajrccm.149.5.8173753. [DOI] [PubMed] [Google Scholar]
- 17.Cai Y, Bjermer L, Halstensen TS. Bronchial mast cells are the dominating LTC4S-expressing cells in aspirin-tolerant asthma. Am J Respir Cell Mol Biol. 2003;29:683–93. doi: 10.1165/rcmb.2002-0174OC. [DOI] [PubMed] [Google Scholar]
- 18.Duong M, Subbarao P, Adelroth E, et al. Sputum eosinophils and the response of exercise-induced bronchoconstriction to corticosteroid in asthma. Chest. 2008;133:404–11. doi: 10.1378/chest.07-2048. [DOI] [PubMed] [Google Scholar]
- 19.Hallstrand TS, Wurfel MM, Lai Y, et al. Transglutaminase 2, a novel regulator of eicosanoid production in asthma revealed by genome-wide expression profiling of distinct asthma phenotypes. PLoS One. 2010;5:e8583. doi: 10.1371/journal.pone.0008583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallstrand TS, Moody MW, Wurfel MM, et al. Inflammatory basis of exercise-induced bronchoconstriction. Am J Respir Crit Care Med. 2005;172:679–86. doi: 10.1164/rccm.200412-1667OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gurish MF, Austen KF. Developmental origin and functional specialization of mast cell subsets. Immunity. 2012;37:25–33. doi: 10.1016/j.immuni.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Dougherty RH, Sidhu SS, Raman K, et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(h)2-high asthma. J Allergy Clin Immunol. 2010;125:1046–1053.e8. doi: 10.1016/j.jaci.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodruff PG, Boushey HA, Dolganov GM, et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104:15858–63. doi: 10.1073/pnas.0707413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woodruff PG, Modrek B, Choy DF, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–95. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang MJ, Lee SY, Kim HB, et al. Association of IL-13 polymorphisms with leukotriene receptor antagonist drug responsiveness in Korean children with exercise-induced bronchoconstriction. Pharmacogenet Genomics. 2008;18:551–8. doi: 10.1097/FPC.0b013e3282fe94c5. [DOI] [PubMed] [Google Scholar]
- 26.Gharib SA, Nguyen EV, Lai Y, et al. Induced sputum proteome in healthy subjects and asthmatic patients. J Allergy Clin Immunol. 2011;128:1176–1184.e6. doi: 10.1016/j.jaci.2011.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson SD, Schoeffel RE. Respiratory heat and water loss during exercise in patients with asthma. Effect of repeated exercise challenge. Eur J Respir Dis. 1982;63:472–80. [PubMed] [Google Scholar]
- 28.Anderson SD, Schoeffel RE, Black JL, et al. Airway cooling as the stimulus to exercise-induced asthma—a re-evaluation. Eur J Respir Dis. 1985;67:20–30. [PubMed] [Google Scholar]
- 29.Gilbert IA, McFadden ER., Jr Airway cooling and rewarming. The second reaction sequence in exercise-induced asthma. J Clin Invest. 1992;90:699–704. doi: 10.1172/JCI115940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strauss RH, McFadden ER, Jr, Ingram RH, Jr, et al. Influence of heat and humidity on the airway obstruction induced by exercise in asthma. J Clin Invest. 1978;61:433–40. doi: 10.1172/JCI108954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deal EC, Jr, McFadden ER, Jr, Ingram RH, Jr, et al. Role of respiratory heat exchange in production of exercise-induced asthma. J Appl Physiol. 1979;46:467–75. doi: 10.1152/jappl.1979.46.3.467. [DOI] [PubMed] [Google Scholar]
- 32.Gilbert IA, Fouke JM, McFadden ER., Jr Heat and water flux in the intrathoracic airways and exercise-induced asthma. J Appl Physiol. 1987;63:1681–91. doi: 10.1152/jappl.1987.63.4.1681. [DOI] [PubMed] [Google Scholar]
- 33.Mickleborough TD, Lindley MR, Ray S. Dietary salt, airway inflammation, and diffusion capacity in exercise-induced asthma. Med Sci Sports Exerc. 2005;37:904–14. [PubMed] [Google Scholar]
- 34.Bikov A, Gajdocsi R, Huszar E, et al. Exercise increases exhaled breath condensate cysteinyl leukotriene concentration in asthmatic patients. J Asthma. 2010;47:1057–62. doi: 10.1080/02770903.2010.512690. [DOI] [PubMed] [Google Scholar]
- 35.Hilberg T, Deigner HP, Moller E, et al. Transcription in response to physical stress—clues to the molecular mechanisms of exercise-induced asthma. FASEB J. 2005;19:1492–4. doi: 10.1096/fj.04-3063fje. [DOI] [PubMed] [Google Scholar]
- 36.Mandal AK, Jones PB, Bair AM, et al. The nuclear membrane organization of leukotriene synthesis. Proc Natl Acad Sci U S A. 2008;105:20434–9. doi: 10.1073/pnas.0808211106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ago H, Kanaoka Y, Irikura D, et al. Crystal structure of a human membrane protein involved in cysteinyl leukotriene biosynthesis. Nature. 2007;448:609–12. doi: 10.1038/nature05936. [DOI] [PubMed] [Google Scholar]
- 38.Martinez Molina D, Wetterholm A, Kohl A, et al. Structural basis for synthesis of inflammatory mediators by human leukotriene c4 synthase. Nature. 2007;448:613–6. doi: 10.1038/nature06009. [DOI] [PubMed] [Google Scholar]
- 39.Hallstrand TS, Henderson WR., Jr An update on the role of leukotrienes in asthma. Curr Opin Allergy Clin Immunol. 2010;10:60–6. doi: 10.1097/ACI.0b013e32833489c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Austen KF, Maekawa A, Kanaoka Y, et al. The leukotriene E4 puzzle: finding the missing pieces and revealing the pathobiologic implications. J Allergy Clin Immunol. 2009;124:406–14. doi: 10.1016/j.jaci.2009.05.046. [quiz: 415–6] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holgate ST, Peters-Golden M, Panettieri RA, et al. Roles of cysteinyl leukotrienes in airway inflammation, smooth muscle function, and remodeling. J Allergy Clin Immunol. 2003;111:S18–34. doi: 10.1067/mai.2003.25. [DOI] [PubMed] [Google Scholar]
- 42.Zarini S, Gijon MA, Ransome AE, et al. Transcellular biosynthesis of cysteinyl leukotrienes in vivo during mouse peritoneal inflammation. Proc Natl Acad Sci U S A. 2009;106:8296–301. doi: 10.1073/pnas.0903851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hallstrand TS, Chi EY, Singer AG, et al. Secreted phospholipase A2 group X over-expression in asthma and bronchial hyperresponsiveness. Am J Respir Crit Care Med. 2007;176:1072–8. doi: 10.1164/rccm.200707-1088OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wijewickrama GT, Kim JH, Kim YJ, et al. Systematic evaluation of transcellular activities of secretory phospholipases A2. High activity of group V phospholipases A2 to induce eicosanoid biosynthesis in neighboring inflammatory cells. J Biol Chem. 2006;281:10935–44. doi: 10.1074/jbc.M512657200. [DOI] [PubMed] [Google Scholar]
- 45.Trudeau J, Hu H, Chibana K, et al. Selective downregulation of prostaglandin E2-related pathways by the Th2 cytokine IL-13. J Allergy Clin Immunol. 2006;117:1446–54. doi: 10.1016/j.jaci.2006.01.049. [DOI] [PubMed] [Google Scholar]
- 46.Hartert TV, Dworski RT, Mellen BG, et al. Prostaglandin E2 decreases allergenstimulated release of prostaglandin D2 in airways of subjects with asthma. Am J Respir Crit Care Med. 2000;162:637–40. doi: 10.1164/ajrccm.162.2.9904038. [DOI] [PubMed] [Google Scholar]
- 47.Feng C, Beller EM, Bagga S, et al. Human mast cells express multiple EP receptors for prostaglandin E that differentially modulate activation responses. Blood. 2006;107:3243–50. doi: 10.1182/blood-2005-07-2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makker HK, Lau LC, Thomson HW, et al. The protective effect of inhaled leukotriene D4 receptor antagonist ICI 204,219 against exercise-induced asthma. Am Rev Respir Dis. 1993;147:1413–8. doi: 10.1164/ajrccm/147.6_Pt_1.1413. [DOI] [PubMed] [Google Scholar]
- 49.Finnerty JP, Wood-Baker R, Thomson H, et al. Role of leukotrienes in exercise-induced asthma. Inhibitory effect of ICI 204219, a potent leukotriene D4 receptor antagonist. Am Rev Respir Dis. 1992;145:746–9. doi: 10.1164/ajrccm/145.4_Pt_1.746. [DOI] [PubMed] [Google Scholar]
- 50.Pearlman DS, van Adelsberg J, Philip G, et al. Onset and duration of protection against exercise-induced bronchoconstriction by a single oral dose of montelukast. Ann Allergy Asthma Immunol. 2006;97:98–104. doi: 10.1016/S1081-1206(10)61377-4. [DOI] [PubMed] [Google Scholar]
- 51.Philip G, Villaran C, Pearlman DS, et al. Protection against exercise-induced bronchoconstriction two hours after a single oral dose of montelukast. J Asthma. 2007;44:213–7. doi: 10.1080/02770900701209806. [DOI] [PubMed] [Google Scholar]
- 52.Meltzer SS, Hasday JD, Cohn J, et al. Inhibition of exercise-induced bronchospasm by zileuton: a 5-lipoxygenase inhibitor. Am J Respir Crit Care Med. 1996;153:931–5. doi: 10.1164/ajrccm.153.3.8630575. [DOI] [PubMed] [Google Scholar]
- 53.Duong M, Amin R, Baatjes AJ, et al. The effect of montelukast, budesonide alone, and in combination on exercise-induced bronchoconstriction. J Allergy Clin Immunol. 2012;130:535–539.e3. doi: 10.1016/j.jaci.2012.02.051. [DOI] [PubMed] [Google Scholar]
- 54.Kippelen P, Larsson J, Anderson SD, et al. Acute effects of beclomethasone on hyperpnea-induced bronchoconstriction. Med Sci Sports Exerc. 2010;42:273–80. doi: 10.1249/MSS.0b013e3181b541b1. [DOI] [PubMed] [Google Scholar]
- 55.Currie GP, Haggart K, Lee DK, et al. Effects of mediator antagonism on mannitol and adenosine monophosphate challenges. Clin Exp Allergy. 2003;33:783–8. doi: 10.1046/j.1365-2222.2003.01688.x. [DOI] [PubMed] [Google Scholar]
- 56.Taylor-Clark TE, Nassenstein C, Undem BJ. Leukotriene D increases the excitability of capsaicin-sensitive nasal sensory nerves to electrical and chemical stimuli. Br J Pharmacol. 2008;154:1359–68. doi: 10.1038/bjp.2008.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Freed AN, McCulloch S, Meyers T, et al. Neurokinins modulate hyperventilation-induced bronchoconstriction in canine peripheral airways. Am J Respir Crit Care Med. 2003;167:1102–8. doi: 10.1164/rccm.200201-055OC. [DOI] [PubMed] [Google Scholar]
- 58.Lai YL, Lee SP. Mediators in hyperpnea-induced bronchoconstriction of guinea pigs. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:597–602. doi: 10.1007/s002109900090. [DOI] [PubMed] [Google Scholar]
- 59.Fahy JV, Wong HH, Geppetti P, et al. Effect of an NK1 receptor antagonist (CP-99,994) on hypertonic saline-induced bronchoconstriction and cough in male asthmatic subjects. Am J Respir Crit Care Med. 1995;152:879–84. doi: 10.1164/ajrccm.152.3.7663799. [DOI] [PubMed] [Google Scholar]
- 60.Ichinose M, Miura M, Yamauchi H, et al. A neurokinin 1-receptor antagonist improves exercise-induced airway narrowing in asthmatic patients. Am J Respir Crit Care Med. 1996;153:936–41. doi: 10.1164/ajrccm.153.3.8630576. [DOI] [PubMed] [Google Scholar]
- 61.Barnes PJ. Neurogenic inflammation in the airways. Respir Physiol. 2001;125:145–54. doi: 10.1016/s0034-5687(00)00210-3. [DOI] [PubMed] [Google Scholar]
- 62.Hallstrand TS, Debley JS, Farin FM, et al. Role of MUC5AC in the pathogenesis of exercise-induced bronchoconstriction. J Allergy Clin Immunol. 2007;119:1092–8. doi: 10.1016/j.jaci.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Foresi A, Mattoli S, Corbo GM, et al. Late bronchial response and increase in methacholine hyperresponsiveness after exercise and distilled water challenge in atopic subjects with asthma with dual asthmatic response to allergen inhalation. J Allergy Clin Immunol. 1986;78:1130–9. doi: 10.1016/0091-6749(86)90262-9. [DOI] [PubMed] [Google Scholar]
- 64.Gauvreau GM, Ronnen GM, Watson RM, et al. Exercise-induced bronchoconstriction does not cause eosinophilic airway inflammation or airway hyperresponsiveness in subjects with asthma. Am J Respir Crit Care Med. 2000;162:1302–7. doi: 10.1164/ajrccm.162.4.2001054. [DOI] [PubMed] [Google Scholar]
- 65.Jarjour NN, Calhoun WJ. Exercise-induced asthma is not associated with mast cell activation or airway inflammation. J Allergy Clin Immunol. 1992;89:60–8. doi: 10.1016/s0091-6749(05)80041-7. [DOI] [PubMed] [Google Scholar]
- 66.Zawadski DK, Lenner KA, McFadden ER., Jr Effect of exercise on nonspecific airway reactivity in asthmatics. J Appl Physiol. 1988;64:812–6. doi: 10.1152/jappl.1988.64.2.812. [DOI] [PubMed] [Google Scholar]
- 67.Boner AL, Vallone G, Chiesa M, et al. Reproducibility of late phase pulmonary response to exercise and its relationship to bronchial hyperreactivity in children with chronic asthma. Pediatr Pulmonol. 1992;14:156–9. doi: 10.1002/ppul.1950140304. [DOI] [PubMed] [Google Scholar]
- 68.Iikura Y, Inui H, Obata T, et al. Drug effects on exercise-induced late asthmatic responses. N Engl Reg Allergy Proc. 1988;9:203–7. doi: 10.2500/108854188779023423. [DOI] [PubMed] [Google Scholar]
- 69.Zawadski DK, Lenner KA, McFadden ER., Jr Re-examination of the late asthmatic response to exercise. Am Rev Respir Dis. 1988;137:837–41. doi: 10.1164/ajrccm/137.4.837. [DOI] [PubMed] [Google Scholar]
- 70.Melo RE, Sole D, Naspitz CK. Exercise-induced bronchoconstriction in children: montelukast attenuates the immediate-phase and late-phase responses. J Allergy Clin Immunol. 2003;111:301–7. doi: 10.1067/mai.2003.66. [DOI] [PubMed] [Google Scholar]
- 71.Crimi E, Balbo A, Milanese M, et al. Airway inflammation and occurrence of delayed bronchoconstriction in exercise-induced asthma. Am Rev Respir Dis. 1992;146:507–12. doi: 10.1164/ajrccm/146.2.507. [DOI] [PubMed] [Google Scholar]
- 72.Freed AN, Bromberger-Barnea B, Menkes HA. Dry air-induced constriction in lung periphery: a canine model of exercise-induced asthma. J Appl Physiol. 1985;59:1986–90. doi: 10.1152/jappl.1985.59.6.1986. [DOI] [PubMed] [Google Scholar]
- 73.Zietkowski Z, Skiepko R, Tomasiak-Lozowska MM, et al. Changes in high-sensitivity C-reactive protein in serum and exhaled breath condensate after intensive exercise in patients with allergic asthma. Int Arch Allergy Immunol. 2010;153:75–85. doi: 10.1159/000301582. [DOI] [PubMed] [Google Scholar]
- 74.Zietkowski Z, Skiepko R, Tomasiak-Lozowska MM, et al. RANTES in exhaled breath condensate of allergic asthma patients with exercise-induced bronchoconstriction. Respiration. 2010;80:463–71. doi: 10.1159/000264923. [DOI] [PubMed] [Google Scholar]
- 75.Zietkowski Z, Skiepko R, Tomasiak-Lozowska MM, et al. Eotaxin in exhaled breath condensate of allergic asthma patients with exercise-induced bronchoconstriction. Respiration. 2011;82:169–76. doi: 10.1159/000323180. [DOI] [PubMed] [Google Scholar]
- 76.Tarran R. Regulation of airway surface liquid volume and mucus transport by active ion transport. Proc Am Thorac Soc. 2004;1:42–6. doi: 10.1513/pats.2306014. [DOI] [PubMed] [Google Scholar]
- 77.Matsui H, Davis CW, Tarran R, et al. Osmotic water permeabilities of cultured, well-differentiated normal and cystic fibrosis airway epithelia. J Clin Invest. 2000;105:1419–27. doi: 10.1172/JCI4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Willumsen NJ, Davis CW, Boucher RC. Selective response of human airway epithelia to luminal but not serosal solution hypertonicity. Possible role for proximal airway epithelia as an osmolality transducer. J Clin Invest. 1994;94:779–87. doi: 10.1172/JCI117397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kotaru C, Hejal RB, Finigan JH, et al. Desiccation and hypertonicity of the airway surface fluid and thermally induced asthma. J Appl Physiol. 2003;94:227–33. doi: 10.1152/japplphysiol.00551.2002. [DOI] [PubMed] [Google Scholar]
- 80.Csoma Z, Huszar E, Vizi E, et al. Adenosine level in exhaled breath increases during exercise-induced bronchoconstriction. Eur Respir J. 2005;25:873–8. doi: 10.1183/09031936.05.00110204. [DOI] [PubMed] [Google Scholar]
- 81.Gulliksson M, Palmberg L, Nilsson G, et al. Release of prostaglandin D2 and leukotriene C4 in response to hyperosmolar stimulation of mast cells. Allergy. 2006;61:1473–9. doi: 10.1111/j.1398-9995.2006.01213.x. [DOI] [PubMed] [Google Scholar]
- 82.Uozumi N, Kume K, Nagase T, et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390:618–22. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 83.Bowton DL, Seeds MC, Fasano MB, et al. Phospholipase A2 and arachidonate increase in bronchoalveolar lavage fluid after inhaled antigen challenge in asthmatics. Am J Respir Crit Care Med. 1997;155:421–5. doi: 10.1164/ajrccm.155.2.9032172. [DOI] [PubMed] [Google Scholar]
- 84.Chilton FH, Averill FJ, Hubbard WC, et al. Antigen-induced generation of lysophospholipids in human airways. J Exp Med. 1996;183:2235–45. doi: 10.1084/jem.183.5.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stadel JM, Hoyle K, Naclerio RM, et al. Characterization of phospholipase A2 from human nasal lavage. Am J Respir Cell Mol Biol. 1994;11:108–13. doi: 10.1165/ajrcmb.11.1.8018333. [DOI] [PubMed] [Google Scholar]
- 86.Singer AG, Ghomashchi F, Le Calvez C, et al. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases a2. J Biol Chem. 2002;277:48535–49. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- 87.Henderson WR, Jr, Chi EY, Bollinger JG, et al. Importance of group X-secreted phospholipase A2 in allergen-induced airway inflammation and remodeling in a mouse asthma model. J Exp Med. 2007;204:865–77. doi: 10.1084/jem.20070029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Munoz NM, Meliton AY, Arm JP, et al. Deletion of secretory group V phospholipase A attenuates cell migration and airway hyperresponsiveness in immunosensitized mice. J Immunol. 2007;179:4800–7. doi: 10.4049/jimmunol.179.7.4800. [DOI] [PubMed] [Google Scholar]
- 89.Hendersen WR, Jr, Oslund RC, Bollinger JG, et al. Blockade of human group X secreted phospholipase A-induced airway inflammation and hyperresponsiveness in a mouse asthma model by a selective group X secreted phospholipase A inhibitor. J Biol Chem. 2011;286:28049–55. doi: 10.1074/jbc.M111.235812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hallstrand TS, Lai Y, Ni Z, et al. Relationship between levels of secreted phospholipase A groups IIA and X in the airways and asthma severity. Clin Exp Allergy. 2011;41:801–10. doi: 10.1111/j.1365-2222.2010.03676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lai Y, Oslund RC, Bollinger JG, et al. Eosinophil cysteinyl leukotriene synthesis mediated by exogenous secreted phospholipase A group X. J Biol Chem. 2010;285:41491–500. doi: 10.1074/jbc.M110.153338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cookson W. The immunogenetics of asthma and eczema: a new focus on the epithelium. Nat Rev Immunol. 2004;4:978–88. doi: 10.1038/nri1500. [DOI] [PubMed] [Google Scholar]
- 93.Kim DY, Park BS, Hong GU, et al. Anti-inflammatory effects of the R2 peptide, an inhibitor of transglutaminase 2, in a mouse model of allergic asthma, induced by ovalbumin. Br J Pharmacol. 2011;162:210–25. doi: 10.1111/j.1476-5381.2010.01033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim Y, Eom S, Kim K, et al. Transglutaminase II interacts with RAC1, regulates production of reactive oxygen species, expression of snail, secretion of Th2 cytokines and mediates in vitro and in vivo allergic inflammation. Mol Immunol. 2010;47:1010–22. doi: 10.1016/j.molimm.2009.11.017. [DOI] [PubMed] [Google Scholar]