

Abstract
The first protein kinase structure, solved in 1991, revealed the fold that is shared by all members of the eukaryotic protein kinase superfamily and showed how the conserved sequence motifs cluster mostly around the active site. This structure of the PKA catalytic (C) subunit showed also how a single phosphate integrated the entire molecule. Since then the EPKs have become a major drug target, second only to the G-protein coupled receptors. Although PKA provided a mechanistic understanding of catalysis that continues to serve as a prototype for the family, by comparing many active and inactive kinases we subsequently discovered a hydrophobic spine architecture that is a characteristic feature of all active kinases. The ways in which the regulatory spine is dynamically assembled is the defining feature of each protein kinase. Protein kinases have thus evolved to be molecular switches, like the G-proteins, and unlike metabolic enzymes which have evolved to be efficient catalysis. PKA also shows how the dynamic tails surround the core and serve as essential regulatory elements. The phosphorylation sites in PKA, introduced both co- and post-translationally, are very stable. The resulting C-subunit is then package as an inhibited holoenzyme with cAMP-binding regulatory (R) subunits so that PKA activity is regulated exclusively by cAMP, not by the dynamic turnover of an activation loop phosphate. We could not understand activation and inhibition without seeing structures of R:C complexes; however, to appreciate the structural uniqueness of each R2:C2 holoenzyme required solving structures of tetrameric holoenzymes. It is these tetrameric holoenzymes that are localized to discrete sites in the cell, typically by A Kinase Anchoring Proteins where they create discrete foci for PKA signaling. Understanding these dynamic macromolecular complexes is the challenge that we now face.
1. Introduction
As we look back over the past two decades since the first protein kinase structure was solved, we find that we are entering a new era in our understanding not only of cAMP-dependent Protein Kinase (PKA) but also of all protein kinases. Although our most comprehensive understanding of the protein kinases at the molecular level has come from crystal structures, we are now coming to appreciate that these are surprisingly dynamic molecules that have specifically evolved to be highly regulated molecular switches and not efficient catalysts[1]. Thus our conventional ways of looking at enzymes may have to be revisited when we think about these regulated switches that work on proteins, not peptides, and are part of dynamic and typically oscillating macromolecular complexes. Furthermore, although the eukaryotic protein kinases (EPKs) all share a conserved kinase core, they are regulated in highly dynamic ways by the tails and linker segments that flank that kinase core. These dynamic flanking regions are frequently an integral part of the active kinase; however, unlike the conserved core, they differ in each kinase. Almost every protein kinase is also regulated in some way by phosphorylation whether it is mediated by auto-phosphorylation or by a heterologous kinase. Some of these phosphorylation sites lie in the core, specifically in the activation loop, while other sites are in the tails and linkers. These phosphates can play very different roles.
So at this point in time, just over twenty years since the first protein kinase structure was solved, we find ourselves with an enormous amount of information about this enzyme family, which has also become a major drug target for the pharmaceutical and biotechnology industries. We are also now entering a new era of computation where we can carry out simulations of proteins that reach μsec to msec time scales, which lie in the range where most catalytic and regulatory functions occur. This allows us for the first time to actually experimentally validate the simulations. Advances in NMR spectroscopy where we can explore the residue-specific dynamics of a protein in solution are also revealing new insights about the dynamic properties of this enzyme family [2–4]. Instead of being limited to the static structures that are defined in a crystal lattice, we can now begin to explore in a far more comprehensive way how these proteins behave in solution. Most importantly we are coming to appreciate that these molecules have evolved to do something different from metabolic enzymes that we have studied so intensely in the past. Whereas the metabolic enzymes have evolved to be efficient catalysts that turn over large amounts of small molecule substrates, the protein kinases, like the GTPases, have evolved to be molecular switches that initiate a cascade of downstream signaling events. Often they function as part of a macromolecular complex under single turnover conditions where the substrate and kinase are in a 1:1 complex. While efficient catalysis is important for a metabolic enzyme, dynamic and precise regulation is essential for a switch. Oscillating between off and on states is essential for almost every protein kinase.
2. Evolution of the eukaryotic protein kinases as dynamic and regulated molecular switches
It is remarkable how our definition of the protein kinase family has remained so stable over the past 20 years. Although the potential for the covalent addition of a phosphate to regulate the function of a protein and, in particular, to mediate the equilibrium between active and inactive conformations, was first recognized by the pioneering work of Krebs and Fischer with glycogen phosphorylase[5], we did not initially appreciate the vastness of this enzyme family nor did we appreciate the full complexity of the biological networks and cascades that are regulated by the simple addition of a phosphate. Nor did we appreciate the complex ways that phosphorylation can regulate the kinases themselves. In 1988 Hanks and Hunter carried out a sequence analysis based on only a handful of protein kinase sequences and mapped a set of 12 conserved motifs that were scattered throughout what they defined to be the “kinase core” [6]. Those motifs came from different parts of the protein, similar to the catalytic triads that characterize many proteases. However, these widely dispersed sequence motifs revealed a complexity that was far greater than the catalytic triad of the proteases, and the fact that there were phosphorylation sites scattered throughout the core perhaps gave us our first clues that these proteins were also highly regulated. Unlike the proteases where there are many examples of convergent evolution where the position of the triad is conserved but not the fold of the protein [7], the protein kinases have conserved the fold as a highly dynamic and regulated scaffold for a switch. In this regard they also differ from the protein phosphatases, which come in several different flavors that each display unique folds and active sites [8].
PKA, the first protein kinase structure to be solved [9], revealed the conserved protein kinase fold and led to the immediate prediction that this fold, consisting of an N-terminal lobe (N-Lobe) dominated by a β sheet and a C-terminal mostly helical lobe (C-Lobe), was conserved in every protein kinase. The function of many of the conserved sequence motifs, which are scattered throughout both lobes, was also revealed by the three dimensional template of the PKA catalytic subunit. Most of the conserved residues clustered around the active site, in particular, around the site of phosphoryl transfer. It should be noted that although an essential feature of the ATP binding site is a highly dynamic glycine rich loop, this loop is quite distinct from the Walker motif or P-Loop that is found in most metabolic enzymes and ATPases [10]. Instead of positioning the γ-phosphate at the base of a catalytic cleft where closing of the cleft brings together the two substrates and poises them for transfer, as in the case of hexokinase[11], the protein kinases position the adenine ring at the base of the cleft in a deep hydrophobic pocket between the two lobes. The γ-phosphate is then positioned at the edge of the active site cleft between two critical elements - the glycine rich loop and the catalytic loop while the Activation Loop positions the C Helix and the substrate docking surfaces and helps to control opening and closing of the active site cleft.
3. Molecular features that distinguish the EPKs from the ELKs
The eukaryotic protein kinases (EPKs) evolved from the eukaryotic like kinases (ELKs) and are distinguished from the ELKs by two structural elements that contribute to their function and regulation [1, 12]. The EPKs have a large and highly dynamic activation segment that is inserted between β strand 9 and the αF Helix. In most kinases this loop exists in an inactive conformation or sometimes a disordered state when the kinase is not active. Activation is typically achieved by phosphorylation of this activation loop. In addition the EPKs have a helical subdomain that consists of the αG, αH, and αI helices. This helical domain is involved mostly in docking of substrates. These two structural elements that distinguish the EPKs from the ELKs (Figure 1) are linked by an electrostatic interaction between an acidic residue in the APE motif at the end of the Activation Segment and an arginine that lies between the αH and αI helices (Glu208 and Arg280 in PKA). The overall conformation of the activation segment when the kinase is in an active conformation is conserved as shown in Figure 1 where many EPK activation loops are compared. The position of the activation segment differs significantly when the kinase is in an inactive conformation. Several examples are shown in Figure 1. In most cases a significant portion of the segment is simply disordered whereas in other cases such as Rsk and Msk, the segment is ordered very differently. Since most protein kinase structures represent only the kinase core, we most likely do not yet appreciate the extent to which the activation segment can be re-ordered in the inactive kinase and to what extent it may actually contribute to interactions with flanking inhibitory domains which are associated with so many kinases. This will require having structures of full-length protein kinases.
Fig 1. Structural features that distinguish the Eukaryotic Protein Kinases (EPKs) from the Eukaryote-Like Protein Kinases (ELKs).
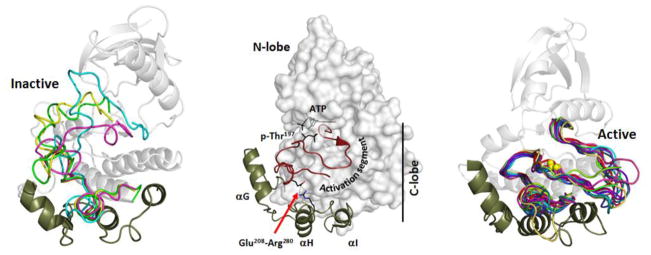
The N and C-lobe architecture is maintained in both the ELKs and the EPKs; however, there are two features that are unique to the EPKs. There is a large and highly regulated activation segment (shown in red) that is inserted between β strand 9 and the α F-Helix in the C-Lobe. The conformation of this segment is typically regulated by phosphorylation. The activation loop phosphorylation site for PKA, Thr197, is indicated. The second structural feature that is unique to the EPKs is a helical bundle that includes the αG, αH and αI Helices. This serves most often as a docking site for protein substrates and is thought to be coupled allosterically to the active site [43]. The helix bundle is directly linked to the activation segment by a conserved ion pair between Arg280 between the αH and αI helices and Glu208 in the activation segment.
4. Internal architecture of the EPKs is described by dynamically assembled hydrophobic spines
Because the protein kinases are so biologically important and because protein kinase mutations are associated with so many diseases, they have become major targets for drug discovery. We thus have many protein kinase structures that serve as templates for drug discovery. Having a structural kinome as well as a sequence-based kinome allows us to delve more deeply into the structural features that define this enzyme family. By comparing many protein kinase structures and asking what was different in active vs. inactive kinases, we discovered a hydrophobic spine architecture that defines the internal architecture of every protein kinase [13]. As seen in Figure 2, there are two “spines” that consist of a stack of nonlinear hydrophobic residues that span both lobes and allow the N- and C-lobes to move in a fluid way as the cleft opens and closes [14, 15]. The regulatory spine (R-spine) is assembled in a dynamic way, often as a consequence of phosphorylation of the activation loop as described below, and this correctly aligned R-spine is essential for every active kinase. These R-spine residues, two from the N-Lobe and two from the C-Lobe, are non linear and would never be apparent based on sequence comparisons alone. The catalytic spine (C-spine) is assembled by the binding of ATP, which then commits the kinase to catalysis [3]. Both spines are anchored to the hydrophobic αF-Helix, which penetrates through the middle of the C-Lobe. The R-spine is typically broken in inactive kinases, and it can be broken in a variety of ways. The “DFG-out” conformation, for example, represents a broken R-spine; however, “DFG in” conformations can also be inactive if another spine residue is incorrectly aligned (Figure 3). Four residues comprise the R-spine. At the base of the catalytic loop is the HRD motif. The first residue is either a His or a Tyr in every protein kinase (Tyr164 in PKA), and its backbone is positioned by a conserved Asp at the beginning of the αF-Helix. Stacked against the His/Tyr is the DFG phenylalanine (Phe185 in PKA). In addition to these two C-lobe residues there are two hydrophobic residues from the N-Lobe – one at the end of the C-Helix (Leu95 in PKA) and the other at the beginning of β strand 4 (Leu106 in PKA). There are many ways that the spine can be broken, but these four residues are aligned in a perpendicular manner in every active kinase. As seen in Figure 2, assembly of the R-spine brings together most of the essential motifs of the kinase core and leaves the enzyme poised for catalysis.
Fig. 2. Architecture of the kinase core is defined by two hydrophobic spines.
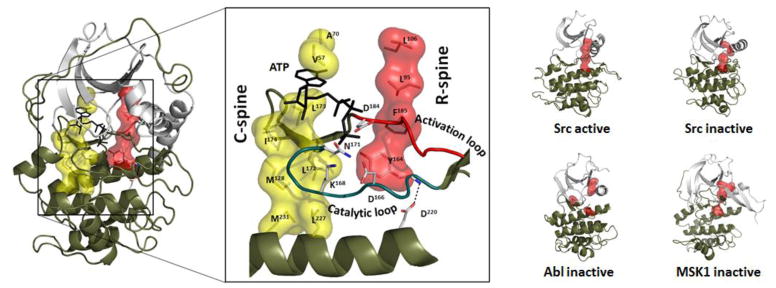
The two lobes of the protein kinase core are linked by two hydrophobic spines, a catalytic spine (C-spine; yellow) there the spine is completed by the adenine ring of ATP and a regulatory spine (R-spine; red() that is assembled in a dynamic manner, typically by phosphorylation of the activation loop. The two spines are intact in every active kinase whereas in inactive kinases the R-spine is broken. Examples showing how the R-spine is assembled in an active kinase (Src:pdb1y57) and broken in an inactive kinase are shown on the right for Src (PDB2src), Abl (PDB1Opj) and Msk (PDB1vzO).
Fig. 3. N-terminal and C-terminal tails flank the kinase core of PKA.
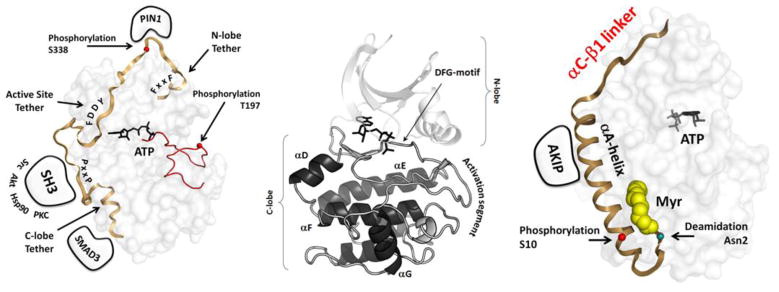
The conserved N and C-lobes of the PKA kinase core are shown in the center. This core is flanked by dynamic tails that serve as both cis and trans regulatory elements. On the right is the N-terminal tail which is myristylated at the N-terminal Glycine. On the left is the C-tail which is a conserved feature of all AGC kinases.
4. Linkers, tails and inserts
While the kinase core is conserved in every protein kinase, it is important to recognize that the core alone is not sufficient to define an active kinase. The tails and linkers that flank the core, although often dynamic in nature, are also often an essential part of an active kinase, as is the activation segment which is inserted between β strand 8 and the αF-Helix. In many cases there are also inhibitor domains or motifs that are embedded within the same gene as the kinase core as in the case of Src with its SH2 and SH3 domains. Release of inhibition, such as the dephosphorylation of the C-tail in Src or the binding of Ras in the case of BRaf and CRaf, is a fundamental mechanism for activation of most kinases. The activation loop is also a defining feature that distinguishes the EPKs from the ELKs. This loop tends to be very short in the ELKs and is not regulated whereas in the EPKs the activation loop is large and frequently regulated by phosphorylation. Like the linkers, it is often classified as an “” (IDS) although it seems more appropriate to define such segments as “dynamically ordered” regions. In crystal structures the activation segment is often disordered in the absence of phosphorylation; however, it can also be ordered in a different conformation in the inactive kinase. When the kinase is in an active conformation, the arginine in the HRD motif is anchored to the activation loop phosphate. Assembly of the activation loop thus represents an order/disorder transition and is a key feature that defines most kinases as a regulated switch.
In addition to the activation segment, the PKA catalytic subunit is flanked by two dynamic tails (Figure 4). The N-terminal glycine is myristylated and recent crystal structures and NMR studies are beginning to define the dynamic properties of the myristylation motif and the following amphipathic A-helix [16]. The C-terminal tail wraps around both the C-lobe and the N-lobe and is a conserved feature of all protein kinases that belong to the AGC sub-family. The first segment, defined as the C-lobe Tether (CLT), is stably anchored to the C-lobe, while the N-terminal segment, referred to as the N-lobe Tether (NLT), is anchored by a conserved Hydrophobic (HF) motif to the C-helix in the N-lobe. The intervening segment (residues 323-336 in PKA) is disordered in the absence of nucleotide but anchored through another conserved motif (FDDY) to the nucleotide in the active site cleft. A critical phosphorylation site, referred as the Turn Motif, is located in the C-tail (Ser338 in PKA). Most other AGC kinases have a third phosphorylation site that directly follows the HF motif. The C-tail functions as a cis-regulatory element and is an essential part of the active kinase [17, 18]. Replacement of the Phe 327, for example, creates an inactive enzyme [19]. The phosphorylation of these sites C-Tail sites, as well as the Activation Loop, is highly regulated in unique ways in every AGC kinase and frequently requires multiple heterologous kinases [20, 21].
Fig. 4. Assembly of an active PKA catalytic subunit.
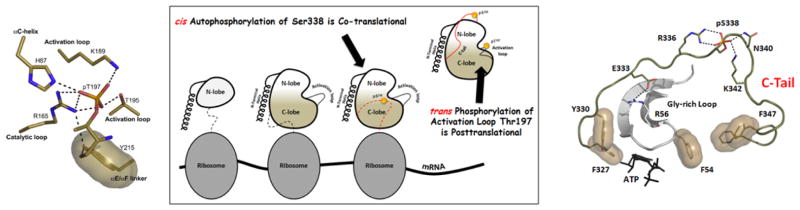
The PKA catalytic subunit is assembled as an active kinase by the phosphorylation of two residues, Thr197 in the activation loop and Ser338 in the C-terminal tail. The two phosphates serve essential but different roles in the mature enzyme. The activation loop phosphate (left) interacts with many residues and is essential for the fully active enzyme and for stability. Ser338 is part of the loop that wraps around the N-lobe and controls the C-Helix. The C-tail is anchored to the N-Lobe by a hydrophobic motif at the C-terminus (Phe-X-X-Phe) that binds to the C-Helix in the N-lobe while another C-Tail motif (FDDY) is anchored to the ATP. Both motifs as well as a phosphorylation site between the two sites is a conserved feature of all AGC kinases. Ser338 is phosphorylated co-translationally while the protein is still on the ribosome while the phosphate is added to Thr 197 by a trans-phosphorylation event after the protein leaves the ribosome.
5. Regulation by phosphorylation
Phosphates play an exceptional role in biology. The importance of phosphates for energy (ATP) and for information transfer (DNA and RNA) was reviewed comprehensively by Westheimer in 1987 [22], but the major role that phosphates play in regulation was completely ignored. The unique chemical features of a phosphate that allow it to play such an important role in signal transduction are captured in the recent review by Hunter [23]. A phosphate can play many different roles. In some cases it can serve as a simple reversible electrostatic switch whereas in other cases it can serve as a docking site and mediate localization and/or the assembly of a large macromolecular signaling complex. Many signaling proteins are modular and phosphorylation sites can modulate domain interactions. A single phosphate can also play an integral role in regulating the entire structure and function of a protein. Phosphorylation of the activation loop is just such an example.
The PKA catalytic subunit has two phosphorylation sites, one near the C-terminus (Ser338) and one in the Activation Loop (Thr197), and these two phosphates play very different roles. Since the C-subunit is auto-phosphorylated in E. coli, that first structure in 1991 [9] also revealed an active and fully phosphorylated kinase. We now recognize that each phosphate is essential, but only recently have we come to appreciate how different these phosphates are for regulation and function. The importance of the activation loop phosphate was revealed when we were able to comprehensively characterize the dephosphorylated protein. The C-tail phosphorylation site, however, has a different but equally important role. As seen in Figure 5, the first phosphate to be added is the C-Tail site at Ser338. This phosphorylation actually takes place while the protein is still on the ribosome. It occurs by a cis-auto-phosphorylation mechanism and is essential for the maturation of the kinase. When this step is blocked, no active kinase is synthesized. The subsequent phosphorylation of the activation loop Thr197 occurs by a trans- phosphorylation mechanism. In E. coli this is achieved by trans-auto-phosphorylation whereas in cells it is likely carried out by a heterologous protein kinase such as PDK1 or another activating kinase [24, 25]. What are the structural consequences of these two phosphates?
Fig. 5. Effects of activation loop phosphorylation on the activity and structure of the PKA catalytic subunit.
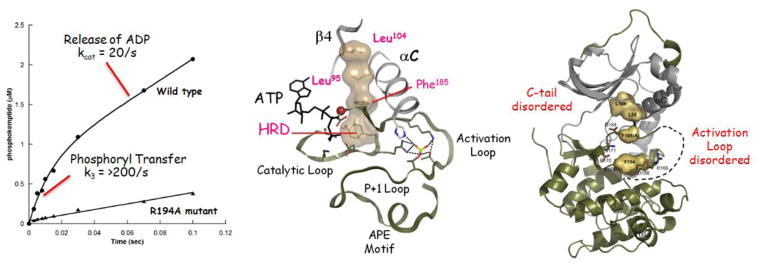
Phosphorylation of the activation loop assembles the R-spine (center) while removal of the activation loop phosphate eliminates the pre-steady state burst o catalytic activity (left) and leads to major disorder in the structure. Specifically the activation loop becomes disordered and the portion of the C-Tail that binds to ATP and is referred to as the active site tether is also disordered.
To characterize the dephosphorylated protein we first mutated the Arg that precedes Thr197. Without an Arg at the P-3 position the C-subunit cannot be auto-phosphorylated. Initial characterization of this dephosphorylated C-subunit showed that it was defective in its kinetic properties and also was much less stable than the fully phosphorylated protein. When the activation loop was phosphorylated by PDK1, a heterologous activating kinase, the properties of the mutant resembled those of the wild type protein [26]. Hydrogen/deuterium exchange coupled with mass spectrometry showed that the entire molecule, when it lacked the phosphate on the activation loop, was much more accessible to deuterium exchange, especially the N-Lobe. The specific structural consequences were revealed when the crystal structure was solved. As seen in Figure 5, not only was the R-spine broken, but also the entire activation loop was disordered. In addition the C-Tail was only partially ordered, and the entire N-Lobe seemed to be quite dynamic based on the high temperature factors. This is a dramatic example of the difference that a single phosphate can make.
To understand more fully the kinetic consequences of removing this phosphate we carried out a quenched flow analysis. In addition to its steady state catalytic rate, PKA has a much faster pre-steady state rate that reflects the actual rate of phosphoryl transfer. The steady state rate, on the other hand, is limited by the release of the nucleotide and does not represent the chemical transfer step. If we think about how protein kinases function in the cell, it is important to realize that they do not function typically under conditions that we routinely use for our assays where the enzyme is at low levels compared to the peptide substrates. Protein kinases, however, do not work on peptides; they work on proteins. Often they are working as part of a 1:1 complex with a protein substrate that is tethered in close proximity through some distal site. What limits the phosphorylation process in vivo? In many cases it could be the off-rate of the phosphorylated protein as well as the off-rate of the ADP. Kinases, in general, are also very poor enzymes, and this also highlights that their substrates are likely tethered close by and not limited by diffusion as is the case for metabolic enzymes that work on small molecules. Kinases are not, in general, efficient catalysts. As seen in Figure 5 de-phosphorylation of the activation segment abolishes the rapid pre-steady state burst.
The C-terminal phosphorylation site is not essential for activity but instead is essential for synthesis of the active enzyme in mammalian cells. This highlights again the importance of regulation in mammalian cells. It is also important to realize that Ser338 appears to be phosphorylated before the fully active enzyme is assembled. If we assume that the protein folds up into the N- and C-lobes as the protein comes off the ribosome, the last segment containing Ser338 would be positioned near the active site cleft. How it docks into the active site cleft where we predict that it will undergo cis-auto-phosphorylation is not known; however, once it is phosphorylated, this segment will be ejected from the active site cleft. The isolated C-tail is not a substrate for PKA due presumably to the string of acidic residues that precede the PKA recognition motif (DDYEEEE;residues 328-334).
6. Assembly of a tetrameric holoenzyme
While we have learned much about PKA and about the protein kinase family in general from the analysis of the isolated C-subunit, in cells PKA exists primarily as an inactive tetrameric holoenzyme. The regulatory subunits are constitutive dimers due to the N-terminal four-helix bundle that is referred to as the dimerization/docking domain. The name reflects its bi-functional properties where, in addition to dimerization, it serves as a docking site for A Kinase Anchoring Proteins (AKAPs)[27]. The AKAPs are scaffold proteins that target PKA in close proximity to dedicated substrates such as the tails of many receptors and ion channels. Each AKAP contains an amphipathic helix that binds with high affinity to the hydrophobic groove that is created by the D/D domain at the N-terminus of the R-subunit[28, 29]. Most AKAPs are specific for RII subunits whereas some are dual specific [30] and a few are specific for RI subunits [31, 32]. The D/D domain is joined by a flexible linker to two tandem cyclic nucleotide binding (CNB) domains that lie at the C-terminus of each R-subunit. Embedded within the linker is an inhibitor sequence that resembles a peptide substrate. In the absence of cAMP the inhibitor site docks to the active site cleft of the C-subunit and keeps the enzyme in an inactive state. The IS in the RI subunits is a pseudo-substrate where the P-site is either an Ala or a Gly. In the RII subunits the P-site is a Ser so that the RII subunits are both substrates and inhibitors.
By solving structures of tetrameric holoenzymes we have come to appreciate the unique features of each PKA isoform and also to better understand why each or the four isoforms (RIα, RIβ, RIIα, and RIIβ) are functionally non-redundant. The R:C hetero-dimers where one C-subunit is bound to a monomeric form of the R-subunit all appear to be very similar. However, when the two heterodimers, linked to the D/D domains by the flexible linkers are assembled into tetrameric holoenzymes each has a unique quaternary structure that is largely dictated by the linker sequences that lie N-terminal to the IS (Figure 7). These tetrameric holoenzymes are allowing us for the first time to appreciate the complexity of the allosteric networks and the uniqueness of each isoform (RIα, RIβ, AND RIIβ). The RIIβ holoenzyme demonstrates some of the unique features of the full-length holoenzyme where the IS can be phosphorylated or not.
Figure 7. Structure of the RIIb holoenzyme traps both products in the crystal lattice.

The tetrameric RIIb holoenzyme is assembled as two R:C heterodimers . the dimer interface in the holoenzyme is created by the b4-b5 loop in the CNB-A domain of the R-subunit and the FDDY motif in the C-terminal tail of the C-subunit. The C-Tail is in a fully closed conformation in the tetramer even though no nucleotide is present. When MgATP was added to the crystals, the phosphate was transferred and both products, ADP and the phosphorylated RIIb subunit including two Mg2+ ions were trapped in the crystal lattice.
The two phosphates in each PKA C-subunit subunit have another unusual feature. Once added, they are extremely resistant to phosphatases [25]. This is in contrast to most other protein kinases where the phosphates turnover rapidly. In the case of PKA, the C-subunits are rapidly assembled into an inactive tetrameric holoenzyme where their activity is now regulated exclusively by cAMP, not by the dynamic turnover of the activation loop phosphate. In contrast to the tetrameric RIIβ holoenzyme, which is allosterically activated by cAMP with a Hill Coefficient of 1.7, the RIIβ heterodimer shows little cooperativity. The Hill Coefficient for activation by cAMP is 0.82 in contrast to 1.6 for the holoenzyme [33]. In addition, the Ka(cAMP) for activation is 10-fold greater for the tetramer vs. the heterodimer (584 nM vs. 65 nM). The tetramer reveals a two-fold symmetry that is one could not see in the heterodimer, and symmetry is a requirement for allostery [34]. The RIIβ tetramer shows another unusual feature. We find that the C-subunits in the tetramer are in a closed conformation even though there is no bound ATP. In all of our previous structures MgATP was required to lock the enzyme into a closed conformation, but in the RIIβ holoenzyme the β4-β5 loop from the opposite tetramer pushes on the FDDY motif in the opposite C-subunit and forces it into a closed conformation (Figure 8).
7. Reaction products are trapped in the RIIβ holoenzyme
Given that we have a perfectly formed cavity for ATP to bind, we decided to add MgATP to the crystals hoping to trap a transition state intermediate. Surprisingly, we found that ADP and the phosphorylated RIIβ subunit, along with two Mg2+ ions, were trapped in the crystal lattice. Typically the phosphorylated peptide is released rapidly leaving release of the bound ADPMg2 as the slow and rate-limiting step of catalysis. In the case of the RIIβ holoenzyme, the off-rate of the substrate is controlled by cAMP. Phosphorylation of the IS reduces the on-rate for re-association of the RII subunit with the C-subunit but it does not appear to significantly affect the dissociation rate. Thus the Kd is still approximately 1 nM. The temperature factors of the backbone residues around the phosphorylated P site also remain very low. This is the first example where the two reaction products are trapped in the active site cleft, and this provides us with another important step in the catalytic cycle.
If we compare structure of the RIIα heterodimer with the RIIβ tetramer, we can see striking differences in the C-subunit simply by comparing the temperature factors. Unlike the RI subunits, the RII subunits do not require MgATP to form a high affinity holoenzyme complex. They have the same high affinity (Kd = 0.1 nM) as the RIα subunits even in the absence of Mg2ATP. In contrast the RIα subunits have a Kd of 300nM vs. 0.1 nM when the nucleotide is missing [35, 36]. Even the removal of one Mg ion is sufficient to reduce the affinity to 300 nM. The temperature factors for the RIIα:C heterodimer indicate that the N-Lobe of the C-subunit is highly disordered, and the AST segment of the C-Tail cannot be traced. In contrast, the temperature factors for the N-Lobe and the C-Tail are very low for the RIIβ, tetrameric holoenzyme and confirm the stability of the ATP binding pocket. The re-association of the RII subunit and the C-subunit is 5-fold lower when the RII subunit is phosphorylated and it is very unlikely that one could form the product complex by re-associating the phosphorylated RIIβ, subunit and ADP. This suggests that the phosphorylation of the RII subunit occurs subsequent to the formation of the holoenzyme, which often takes place in the presence of calcium and the calcium-activated phosphatase, calcineurin. However, once the reaction chamber has been formed and ATP can bind easily and the energy barrier for transferring the phosphate is very low. All of the critical residues are perfectly positioned. Once transfer has occurred the phosphorylated holoenzyme is poised to be activated by cAMP, which rapidly promotes dissociation of the phosphorylated RIIβ-subunit. Although this mechanism needs to be rigorously confirmed, it is consistent with the binding by Bond that the RII subunits in cells are phosphorylated in the resting state [37].
8. Targeted Holoenzymes
The structures of the tetrameric PKA holoenzymes are causing us to reconsider our ideas about PKA signaling in cells where localization of the holoenzyme is so important for achieving specificity. Furthermore, we cannot think of the kinase alone without considering that scaffold proteins such as AKAPs that bring together a community of signaling proteins that are in close proximity to a dedicated substrate such as the tail of a voltage gated ion channel. Calcineurin, a calcium activated phosphatase, is often nearby in the complex so that there is the opportunity for oscillating signaling between calcium and cAMP that control the addition and release of the phosphate. Under these conditions there is not time for full dissociation of the C-subunit and several lines of evidence including SAXS and fluorescence anisotropy suggest that the C-subunit does not fully dissociate from the R subunit in solution in the presence of cAMP[38, 39]. In some cases, a phosphodiesterase is also localized in the community [40] or even interacting directly with the R-subunit[41] so that there is a very precise mechanism for terminating the signal. In the cell, we therefore need to think of localized foci of PKA signaling, where the generation and termination of the signal is regulated in customized ways at each site. In this context, PKA signaling at the mitochondria might be quite segregated and timed differently from PKA signaling at an ion channel that is at the plasma membrane. The recognition of a soluble cyclase in the matrix of the mitochondria and in nuclei makes compartmentalized PKA signaling even more diverse [42]. We typically study PKA signaling in cells by adding forskolin, which will activate most cyclases, or isoproterenol which will activate the β-adrenergic receptors or by adding a cell permeant analog of cAMP. In all cases a phosphodiesterase inhibitor is also required. Under these conditions a sustained wave of cAMP is generated that can radiate throughout the cell. Under physiological conditions, however, PKA signaling is much more localized. Our new appreciation of the novel structural features of each PKA holoenzyme sheds additional light on the ways that selectivity and specificity in PKA signaling can be achieved.
9. Conclusions
In this review we highlight the features that define the protein kinase superfamily as being unique and distinct from metabolic enzymes. We review first how the eukaryotic protein kinases (EPKs) have evolved from the eukaryotic-like kinases (ELKs) to be regulated and highly dynamic molecular switches that phosphorylate proteins as opposed to small peptides. In addition to the conserved kinase core, the EPKs are regulated by flanking linkers, and, like the activation loop that is inserted into the core, these regions are highly dynamic and exist in ordered/disordered states. To show how phosphates control the protein kinase structure and function in different ways, we use protein kinase A as the prototypical kinase. We show finally how full length complexes are essential if one is to fully understand and appreciate how kinases are regulated and allosterically activated. Recent structures of tetrameric PKA holoenzymes have shown us how isoform specificity is achieved at the level of the quaternary structure. In the case of the RII holoenzymes, where the RII subunit is both a substrate and an inhibitor, we show how a stable catalytic chamber can be created in a manner that is strictly dependent on the tetrameric holoenzyme. Kinases such as PKA typically exist as part of highly dynamic molecular complexes that are localized to specific sites in close proximity to dedicated substrates. This localization is essential for PKA signaling in cells and is forcing us to think in new ways about catalysis and localized protein substrates in contrast to our classical ways of thinking about Michaelis-Menton catalysis.
Fig. 6. Tetrameric holoenzyme structures of RIa, RIb and RIIb.
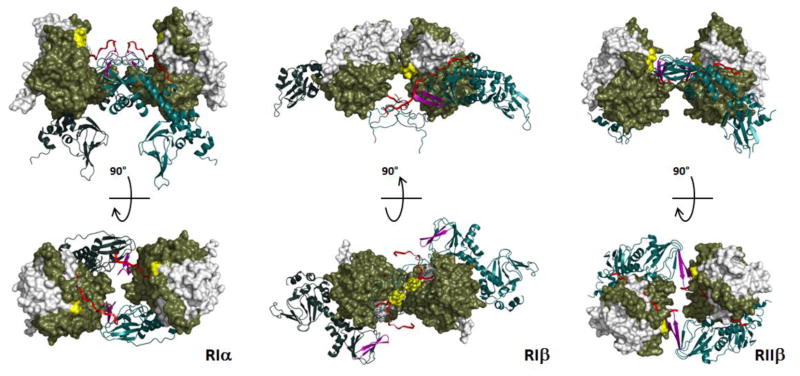
The structures all reveal a twofold symmetry that was not seen in earlier structures of R:C heterodimers; however, the symmetry is different for each holoenzyme. Although the domain organization of each R-subunit is conserved, the quaternary structures are quite different.
We discovered a hydrophobic spine architecture in protein kinases
This architecture define highly dynamic nature of protein kinases
We report diverse quaternary structures for different isoforms of protein kinase A
Acknowledgments
Support for this research was funded by grants from the National Institutes of Health (GM19301, GM34921 and DK54441) and by University of California San Diego Graduate Training Program in Cellular and Molecular Pharmacology through National Institutes of Health Institutional Training Grant T32 GM007752 from the NIGMS and by Ruth L. Kirschstein National Research Service Award NIH/NCI T32 CA009523 (to J. M. S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Taylor SS, Keshwani MM, Steichen JM, Kornev AP. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos T R Soc B. 2012;367:2517–2528. doi: 10.1098/rstb.2012.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masterson LR, Cembran A, Shi L, Veglia G. Allostery and binding cooperativity of the catalytic subunit of protein kinase A by NMR spectroscopy and molecular dynamics simulations. Adv Protein Chem Struct Biol. 2012;87:363–389. doi: 10.1016/B978-0-12-398312-1.00012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masterson LR, Cheng C, Yu T, Tonelli M, Kornev A, Taylor SS, Veglia G. Dynamics connect substrate recognition to catalysis in protein kinase A. Nat Chem Biol. 2010;6:821–828. doi: 10.1038/nchembio.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Masterson LR, Shi L, Metcalfe E, Gao J, Taylor SS, Veglia G. Dynamically committed, uncommitted, and quenched states encoded in protein kinase A revealed by NMR spectroscopy. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1102701108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krebs EG, Graves DJ, Fischer EH. Factors affecting the activity of muscle phosphorylase B kinase. Journal of Biological Chemistry. 1959;234:2867–2873. [PubMed] [Google Scholar]
- 6.Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 7.Dodson G, Wlodawer A. Catalytic triads and their relatives. Trends Biochem Sci. 1998;23:347–352. doi: 10.1016/s0968-0004(98)01254-7. [DOI] [PubMed] [Google Scholar]
- 8.Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 10.Ramakrishnan C, Dani VS, Ramasarma T. A conformational analysis of Walker motif A [GXXXXGKT (S)] in nucleotide-binding and other proteins. Protein Eng. 2002;15:783–798. doi: 10.1093/protein/15.10.783. [DOI] [PubMed] [Google Scholar]
- 11.Steitz TA, Shoham M, Bennett WS., Jr Structural dynamics of yeast hexokinase during catalysis. Philos Trans R Soc Lond B Biol Sci. 1981;293:43–52. doi: 10.1098/rstb.1981.0058. [DOI] [PubMed] [Google Scholar]
- 12.Kannan N, Neuwald AF. Did protein kinase regulatory mechanisms evolve through elaboration of a simple structural component? J Mol Biol. 2005;351:956–972. doi: 10.1016/j.jmb.2005.06.057. [DOI] [PubMed] [Google Scholar]
- 13.Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kornev AP, Haste NM, Taylor SS, Ten Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. P Natl Acad Sci USA. 2006;103:17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proc Natl Acad Sci U S A. 2008;105:14377–14382. doi: 10.1073/pnas.0807988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bastidas AC, Deal MS, Steichen JM, Keshwani MM, Guo Y, Taylor SS. Role of N-Terminal Myristylation in the Structure and Regulation of cAMP-Dependent Protein Kinase. J Mol Biol. 2012;422:215–229. doi: 10.1016/j.jmb.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kannan N, Haste N, Taylor SS, Neuwald AF. The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proc Natl Acad Sci U S A. 2007;104:1272–1277. doi: 10.1073/pnas.0610251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romano RA, Kannan N, Kornev AP, Allison CJ, Taylor SS. A chimeric mechanism for polyvalent trans-phosphorylation of PKA by PDK1. Protein Sci. 2009;18:1486–1497. doi: 10.1002/pro.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J, Kennedy EJ, Wu J, Deal MS, Pennypacker J, Ghosh G, Taylor SS. Contribution of non-catalytic core residues to activity and regulation in protein kinase A. J Biol Chem. 2009;284:6241–6248. doi: 10.1074/jbc.M805862200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keshwani MM, von Daake S, Newton AC, Harris TK, Taylor SS. Hydrophobic Motif Phosphorylation Is Not Required for Activation Loop Phosphorylation of p70 Ribosomal Protein S6 Kinase 1 (S6K1) J Biol Chem. 2011;286:23552–23558. doi: 10.1074/jbc.M111.258004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dettori R, Sonzogni S, Meyer L, Lopez-Garcia LA, Morrice NA, Zeuzem S, Engel M, Piiper A, Neimanis S, Frodin M, Biondi RM. Regulation of the interaction between protein kinase C-related protein kinase 2 (PRK2) and its upstream kinase, 3-phosphoinositide-dependent protein kinase 1 (PDK1) J Biol Chem. 2009;284:30318–30327. doi: 10.1074/jbc.M109.051151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westheimer FH. Why nature chose phosphates. Science. 1987;235:1173–1178. doi: 10.1126/science.2434996. [DOI] [PubMed] [Google Scholar]
- 23.Hunter T. Why nature chose phosphate to modify proteins. Philos Trans R Soc Lond B Biol Sci. 2012;367:2513–2516. doi: 10.1098/rstb.2012.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steichen JM, Kuchinskas M, Keshwani MM, Yang J, Adams JA, Taylor SS. Structural basis for the regulation of protein kinase A by activation loop phosphorylation. J Biol Chem. 2012;287:14672–14680. doi: 10.1074/jbc.M111.335091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keshwani MM, Klammt C, von Daake S, Ma Y, Kornev AP, Choe S, Insel PA, Taylor SS. Cotranslational cis-phosphorylation of the COOH-terminal tail is a key priming step in the maturation of cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 2012;109:E1221–1229. doi: 10.1073/pnas.1202741109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steichen JM, Iyer GH, Li S, Saldanha SA, Deal MS, Woods VL, Jr, Taylor SS. Global consequences of activation loop phosphorylation on protein kinase A. J Biol Chem. 2010;285:3825–3832. doi: 10.1074/jbc.M109.061820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 28.Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, Xuong NH, Jennings PA, Taylor SS. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol Cell. 2006;24:397–408. doi: 10.1016/j.molcel.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarma GN, Kinderman FS, Kim C, von Daake S, Chen L, Wang BC, Taylor SS. Structure of D-AKAP2:PKA RI complex: insights into AKAP specificity and selectivity. Structure. 2010;18:155–166. doi: 10.1016/j.str.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J Biol Chem. 1997;272:8057–8064. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- 31.Means CK, Lygren B, Langeberg LK, Jain A, Dixon RE, Vega AL, Gold MG, Petrosyan S, Taylor SS, Murphy AN, Ha T, Santana LF, Tasken K, Scott JD. An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc Natl Acad Sci U S A. 2011;108:E1227–1235. doi: 10.1073/pnas.1107182108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burgers PP, Ma Y, Margarucci L, Mackey M, van der Heyden MA, Ellisman M, Scholten A, Taylor SS, Heck AJ. A small novel A-kinase anchoring protein (AKAP) that localizes specifically protein kinase A-regulatory subunit I (PKA-RI) to the plasma membrane. J Biol Chem. 2012;287:43789–43797. doi: 10.1074/jbc.M112.395970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang P, Smith-Nguyen EV, Keshwani MM, Deal MS, Kornev AP, Taylor SS. Structure and Allostery of the PKA RIIbeta Tetrameric Holoenzyme. Science. 2012;335:712–716. doi: 10.1126/science.1213979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 35.Herberg FW, Taylor SS. Physiological inhibitors of the catalytic subunit of cAMP-dependent protein kinase: effect of MgATP on protein-protein interactions. Biochemistry. 1993;32:14015–14022. doi: 10.1021/bi00213a035. [DOI] [PubMed] [Google Scholar]
- 36.Whitehouse S, Walsh DA. Mg X ATP2-dependent interaction of the inhibitor protein of the cAMP-dependent protein kinase with the catalytic subunit. J Biol Chem. 1983;258:3682–3692. [PubMed] [Google Scholar]
- 37.Manni S, Mauban JH, Ward CW, Bond M. Phosphorylation of the cAMP-dependent protein kinase (PKA) regulatory subunit modulates PKA-AKAP interaction, substrate phosphorylation, and calcium signaling in cardiac cells. J Biol Chem. 2008;283:24145–24154. doi: 10.1074/jbc.M802278200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin BR, Deerinck TJ, Ellisman MH, Taylor SS, Tsien RY. Isoform-specific PKA dynamics revealed by dye-triggered aggregation and DAKAP1alpha-mediated localization in living cells. Chem Biol. 2007;14:1031–1042. doi: 10.1016/j.chembiol.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 39.Anand G, Taylor SS, Johnson DA. Cyclic-AMP and pseudosubstrate effects on type-I A-kinase regulatory and catalytic subunit binding kinetics. Biochemistry. 2007;46:9283–9291. doi: 10.1021/bi700421h. [DOI] [PubMed] [Google Scholar]
- 40.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- 41.Moorthy BS, Gao Y, Anand GS. Phosphodiesterases Catalyze Hydrolysis of cAMP-bound to Regulatory Subunit of Protein Kinase A and Mediate Signal Termination. Mol Cell Proteomics. 2011;10:M110 002295. doi: 10.1074/mcp.M110.002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tresguerres M, Levin LR, Buck J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int. 2011;79:1277–1288. doi: 10.1038/ki.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deminoff SJ, Ramachandran V, Herman PK. Distal recognition sites in substrates are required for efficient phosphorylation by the cAMP-dependent protein kinase. Genetics. 2009;182:529–539. doi: 10.1534/genetics.109.102178. [DOI] [PMC free article] [PubMed] [Google Scholar]