

Abstract

On the basis of synergism observed between a selective c-Src kinase inhibitor with an HDAC inhibitor, the development of the first chimeric c-Src kinase and HDAC inhibitor is described. The optimized chimeric inhibitor is shown to be a potent c-Src and HDAC inhibitor. Chimeric inhibitor 4 is further shown to be highly efficacious in cancer cell lines and significantly more efficacious than a dual-targeting strategy using discrete c-Src and HDAC inhibitors.
Keywords: c-Src, kinase, chimera, HDAC, combination therapy
The nonreceptor tyrosine kinase c-Src plays an important role in many aspects of cell physiology, regulating diverse cellular processes including division, motility, adhesion, angiogenesis, and survival.1,2 c-Src was the first proto-oncogene identified and is frequently overexpressed in cancer, and the extent of overexpression of c-Src correlates with malignant potential.1,2 Furthermore, c-Src expression levels inversely correlate with patient survival.1,2 Recently, c-Src activity was shown to be a main mode of resistance to Herceptin, a first line therapy for Her2+ breast cancer.3 Therefore, c-Src kinase is an attractive therapeutic target in cancer.
We recently reported the first highly selective inhibitor of c-Src (Figure 1).4 Despite potent biochemical activity against c-Src, our selective c-Src inhibitor (1) is only modestly potent in cellular proliferation assays using breast cancer cell lines.4 Following the success of combinatorial drug therapies in the treatment of HIV,5 tuberculosis,6 and other microbial infections,7 the use of multiple targeted drugs for cancer chemotherapy is increasingly being pursued.8 We reasoned that multitarget inhibition using our selective c-Src inhibitor might lead to improved cellular efficacy.
Figure 1.

Structures of highly selective c-Src inhibitor 1, vorinostat, and panobinostat.
To identify drug combinations that would be synergistic with c-Src inhibition, we examined a small library of targeted inhibitors in combination with our selective c-Src inhibitor 1. These studies were performed in SK-BR-3 cells, a Her2+ breast cancer cell line previously shown to be growth dependent upon c-Src kinase activity.4,9 From these experiments, we identified that panobinostat, a histone deacetylase (HDAC) inhibitor in clinical trials,10 was highly synergistic with c-Src inhibitor 1 (Figure 2). HDAC inhibitors have been shown to promote the growth arrest and apoptosis of cancer cells with minimal toxicity.11 We believe that the observed synergy is due to previously reported mechanisms whereby HDAC inhibitors can down-regulate c-Src levels through repression of SRC transcription.12
Figure 2.
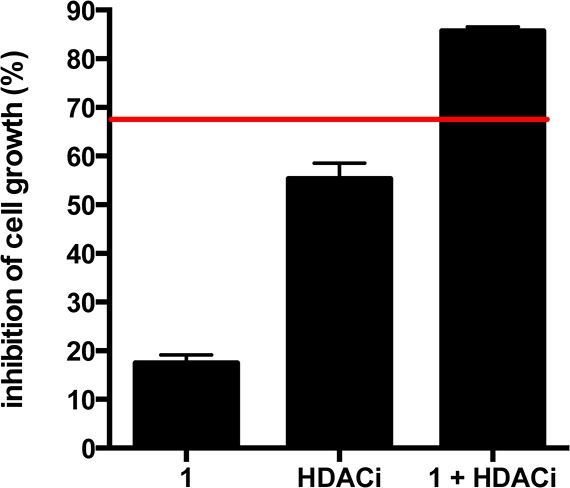
Synergy studies of selective c-Src inhibitor 1 (2 μM), panobinostat (HDACi, 10 nM), and combination (1 + HDACi, 2 μM 1, 10 nM panobinostat) in SK-BR-3 cell line. Red line denotes predicted additivity [(eA+eB)-(eA*eB)] of 1 + panobinostat. The higher level of inhibition than the predicted additivity indicated synergism between 1 and panobinostat.
To determine whether the synergy observed with c-Src inhibition and panobinostat was general for any HDAC inhibitor, we performed combination experiments with vorinostat,13 an FDA approved HDAC inhibitor, and c-Src inhibitor 1 (Table 1). c-Src inhibitor 1 and vorinostat have a GI50 of 4.8 and 1.2 μM, respectively, for SK-BR-3 proliferation. In combination, c-Src inhibitor 1 + vorinostat (1:1) has a GI50 for SK-BR-3 proliferation of 0.8 μM, which is an improvement over either inhibitor dosed alone.14 Next, as a measure of cellular toxicity, we examined each compound’s ability to inhibit proliferation of primary human mammary epithelial cells (HMEC). c-Src inhibitor 1 and vorinostat have a GI50 of 4.3 and 5.8 μM, respectively, for HMEC proliferation. The combination of 1 + vorinostat (1:1) has a GI50 of 5.4 μM against primary mammary epithelial cells.
Table 1. Cellular Efficacy of Selective c-Src Inhibitor 1, Vorinostat, 1/Vorinostat (1:1), and Chimera 4.
| GI50 (μM), SK-BR-3 | GI50 (μM), HMEC | therapeutic index | |
|---|---|---|---|
| compound 1 | 4.8 ± 0.2 | 4.3 ± 0.4 | 0.9 |
| vorinostat | 1.2 ± 0.1 | 5.8 ± 0.2 | 4.8 |
| 1 + vorinostat | 0.80 ± 0.03 | 5.4 ± 0.2 | 6.8 |
| chimera 4 | 0.20 ± 0.03 | 4.7 ± 0.3 | 23.5 |
Using the SK-BR-3 and HMEC data, we calculated a therapeutic index (GI50 HMEC/GI50 SK-BR-3) for c-Src inhibitor 1, vorinostat, and the combination of 1 + vorinostat (Table 1).15 c-Src inhibitor 1 has a poor therapeutic index of 0.9, while vorinostat’s therapeutic index is 4.8. Disappointingly, the combination of 1 + vorinostat has an insignificant improvement in therapeutic index (6.8) relative to vorinostat alone (4.8).
We wondered whether there would be any advantage for a chimeric inhibitor, where a single molecule could serve as both a c-Src kinase and HDAC inhibitor, rather than using two separate agents in combination. For example, we thought that we might obtain improved cellular efficacy. In addition, using a single agent to inhibit both c-Src and HDAC does not lead to the additive toxicity that is often observed with combination therapy.14 Chimeric kinase-HDAC inhibitors have previously been developed; however, no Src-HDAC chimeric compounds have been reported.16−18 In addition, previously reported studies of kinase-HDAC chimeras lack a comparison of therapeutic indices between combination therapy and chimeric inhibition.16−18
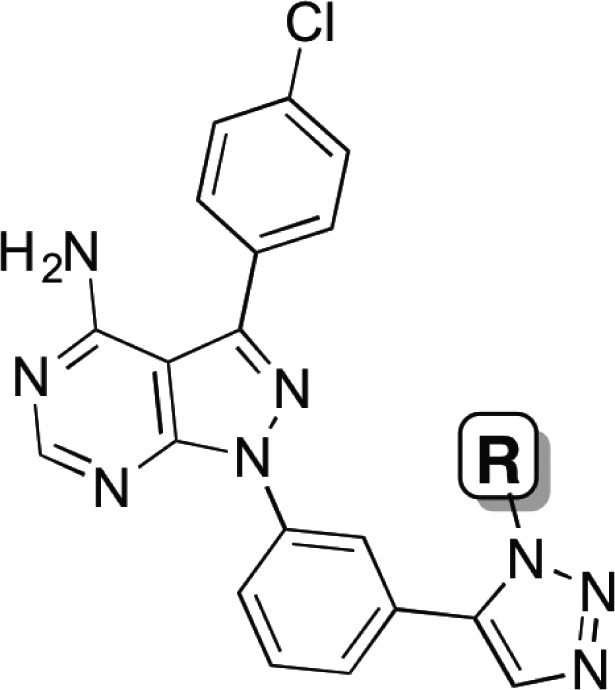
We previously reported PP2∼alkyne (2), a modular and selective c-Src inhibitor scaffold.4 We envisioned using this kinase inhibitor scaffold to append HDAC pharmacophores. The classic pharmacophore for HDAC inhibitors consists of a zinc-binding motif, a hydrophobic linker, and a recognition cap.19 Using PP2∼alkyne, HDAC elements can readily be appended using “click” chemistry.20 Importantly, the use of a triazole ring as the recognition cap in HDAC inhibitors has previously been reported and shown to be highly efficacious both in vitro and in cellulo.21 Previous reports with triazole-based HDAC inhibitors have demonstrated that a 6-carbon hydrophobic linker will provide potent HDAC inhibition.21 While only 1,4-[1,2,3]-triazoles have been reported as HDAC inhibitors,21 we reasoned that because our selective c-Src inhibitor 1 contains a 1,5-[1,2,3]-triazole,4 we would synthesize and evaluate both regioisomers.
We synthesized compounds 3 and 4 as chimeric Src/HDAC inhibitors. Compound 3 has a 1,4-triazole and was synthesized using a copper-mediated cycloaddition reaction,20 while compound 4 has a 1,5-triazole synthesized using a ruthenium-mediated cycloaddition reaction (Chart 1, see Supporting Information for synthetic details).22 Using a previously reported fluorescence assay for c-Src kinase activity,23 we found that 3 and 4 were competent c-Src kinase inhibitors (Ki = 371 and 138 nM, respectively). We next examined the ability of 3 and 4 to inhibit HDAC1 using a Fluor de Lys based-assay24 and found both compounds were potent inhibitors of HDAC1 (Ki = 0.62 and 0.26 nM, respectively). In our assays, compound 4 was a better inhibitor of both c-Src and HDAC1. Thus, the 1,5-triazole regiochemistry was used exclusively for subsequent linker optimization.
Chart 1. Structure of PP2∼Alkyne (2) and Chimeric Inhibitors 3 and 4.

In an effort to optimize potency for both c-Src and HDAC1, we synthesized a series of chimeric HDAC-Src inhibitors containing varied hydrophobic linkers (Table 2). This series included alkyl linkers of varied length as well as styrene-containing linkers that are found in panobinostat.10 The six-carbon alkyl linker (compound 4) was found to be optimal for inhibition of both c-Src kinase and HDAC1. Of note, we found the styrene linkers (compounds 7 and 8) were ineffective as c-Src inhibitors and only modest inhibitors of HDAC1 compared to the n-alkyl linkers.
Table 2. SAR of Linker.
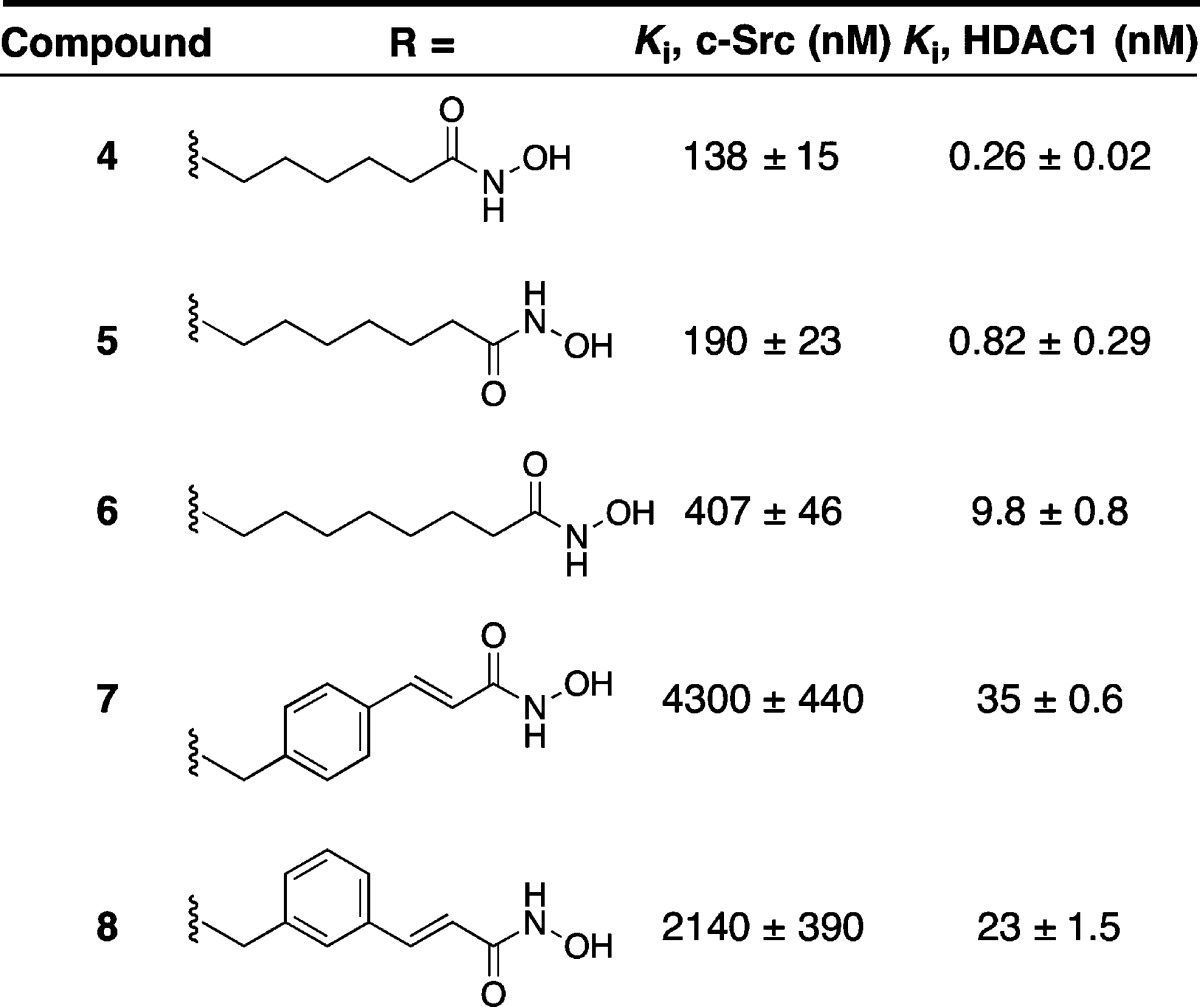
Chimeric inhibitor 4 is one of the most potent HDAC1 inhibitors reported to date (Ki = 260 pM) and is also a potent c-Src inhibitor (Ki = 138 nM). To decipher the binding contributions for each half of the chimera, two fragments of inhibitor 4 were synthesized (Chart 2). Compound 10 contains only the HDAC inhibitor pharmacophore, while compound 9 includes the c-Src kinase binding elements. Interestingly, we observe a marked decrease in affinity for both c-Src and HDAC1 when both elements are not present. Specifically, compound 10, which retains all of the HDAC inhibitor pharmacophore elements, has a Ki for HDAC1 that is >170-times higher than found with chimeric inhibitor 4. These data imply that the c-Src binding elements enhance HDAC1 inhibition observed with compound 4. Likewise, the c-Src inhibitor fragment 9 has nearly 10× less affinity for c-Src than chimera 4, suggesting that the addition of the HDAC fragment is important for c-Src inhibition. Together, these data demonstrate that chimera 4 is not simply two inhibitors linked together, but rather represents a merged inhibitor where both elements are required for affinity against each target.
Chart 2. c-Src Inhibitor 9 and HDAC Inhibitor 10.

Our chimeric inhibitor was initially optimized for HDAC inhibition using HDAC1; however, we assumed it could be a promiscuous inhibitor of HDACs. Profiling of compound 4 against a panel of 11 HDACs was performed by Reaction Biology (Malvern, PA). The HDAC profiling revealed that our chimera is a potent and nonselective inhibitor against class I, IIa, and IV HDACs (Table 3). Consistent with vorinostat’s selectivity, chimera 4 is not an effective inhibitor of class IIa HDACs (Table 3). Relative to vorinostat, chimera 4 has improved affinity to all HDACs except HDAC8 and HDAC11.
Table 3. HDAC Profiling of Chimera 4 and Vorinostat.
| HDAC class | IC50 (nM), Chimera 4 | IC50 (nM), vorinostata | |
|---|---|---|---|
| HDAC1 | I | 86 ± 11 | 306 |
| HDAC2 | I | 231 ± 39 | 232 |
| HDAC3 | I | 19 ± 1 | 132 |
| HDAC4 | IIa | 3982 ± 920 | 76000 |
| HDAC5 | IIa | 3891 ± 681 | 27200 |
| HDAC6 | IIb | 2.7 ± 0.5 | 20 |
| HDAC7 | IIa | 13220 ± 860 | 105000 |
| HDAC8 | I | 2311 ± 251 | 306 |
| HDAC9 | IIa | 28020 ± 1910 | 141000 |
| HDAC10 | IIb | 51 ± 3 | 432 |
| HDAC11 | IV | 224 ± 26 | 200 |
Data from Reaction Biology (Malvern, PA).
In previously published work, we found that c-Src inhibitiors that are selective for c-Src over c-Abl are more efficacious in cell culture with nonhematopoietic cancers.4 Thus, we wanted to determine whether chimera 4 has selectivity for c-Src over c-Abl. Gratifyingly, in our biochemical assay, chimera 4 was selective for c-Src over c-Abl (Ki for c-Src = 138 nM; Ki for c-Abl = 2350 nM). We next tested the ability of 4 to inhibit Hck, a SRC-family kinase with 85% similarity across the kinase domain to c-Src, and found it has a Ki = 504 nM. Together, these data suggest that chimera 4 is selective for c-Src over homologous kinases. Given that our compound shares many features with our highly selective c-Src inhibitor 1,4 it is likely that chimera 4 is also selective for c-Src.
In an effort to compare chimeric inhibition to dual-targeting c-Src and HDAC1, we examined the efficacy of chimera 4 in cellulo. Combination dosing of selective c-Src inhibitor 1 + vorinostat (1:1) was found to have a GI50 = 0.78 μM for SK-BR-3 cells and a GI50 = 5.4 μM for noncancer HME cells. This resulted in a therapeutic index of 6.8 (vide supra). In comparison, chimeric inhibitor 4 was more efficacious at inhibiting the growth of SK-BR-3 cells (GI50 = 0.2 μM) and has similar noncaner cellular toxicity (GI50 = 4.7 μM for HME cells), resulting in a cellular therapeutic index of 23.5 (Table 1). This corresponds to chimeric inhibitor 4 having an improvement in therapeutic index significantly higher than dual targeting c-Src and HDACs with two distinct compounds (23.5 versus 6.8, respectively). These results highlight an important advantage for chimeric inhibition over dual-agent targeting: we observe synergistic activity against cancer cells while not increasing the cellular toxicity relative to the single agent counterparts.
To better characterize the cellular efficacy of our chimeric c-Src/HDAC inhibitor, compound 4 was submitted to the National Cancer Institute for screening in the NCI-60 panel (see Supporting Information for full NCI-60 data).25 From this panel, chimera 4 has an average GI50 = 0.26 μM. Significantly, the efficacy of chimera 4 across the NCI-60 is better than vorinostat (NCI-60 average GI50 = 0.53 μM) and an FDA-approved c-Src inhibitor (dasatinib, NCI-60 average GI50 = 5.7 μM). In addition to the improved efficacy across the NCI-60 panel, chimera 4 does not have increased toxicity relative to primary human mammary cells (chimera 4, HMEC GI50 = 4.7 μM; vorinostat, HMEC GI50 = 5.8 μM; dasatinib, HMEC GI50 = 1.8 μM).
Analysis of the NCI-60 data demonstrates that chimera 4 is a highly efficacious agent in cell lines where vorinostat and dasatinib are ineffective alone (Table 4a). Furthermore, chimera 4 is more efficacious than vorinostat when c-Src inhibition is shown to be efficacious (Table 4b). For example, chimera 4 is an efficacious inhibitor of Hs 578T, a triple negative breast cancer cell line, cell growth (GI50 = 0.17 μM), while vorinostat is not (GI50 = 4.83 μM), due to c-Src inhibition having an important role in Hs 578T cell proliferation (dasatinib GI50 = 0.03 μM). This dramatic increase in efficacy demonstrates that chimera 4 is acting as more than a HDAC inhibitor alone in cellulo. Finally, chimera 4 is observed to be more effective than dasatinib in cell lines where cellular proliferation is dependent upon HDAC1 activity (Table 4c). Together, these data demonstrate that compound 4’s impressive cellular efficacy in the NCI-60 panel is inherent in its chimeric nature, and the ability to inhibit both c-Src kinase and HDAC1 is required for the cellular potency observed.
Table 4. NCI-60 Panel Data for Chimera 4, Vorinostat, and Dasatinib against Select Cell Lines.
| cell line | GI50 (μM), chimera 4 | GI50 (μM), vorinostat | GI50 (μM), dasatinib | |
|---|---|---|---|---|
| a | KM12 | 0.47 | 1.88 | 7.44 |
| MCF7 | 0.35 | 2.19 | 8.32 | |
| U251 | 0.28 | 1.53 | 2.81 | |
| b | DU-145 | 0.39 | 1.36 | 0.16 |
| HS 578T | 0.17 | 4.83 | 0.03 | |
| MDA-MB-231 | 0.39 | 2.32 | 0.02 | |
| c | HCT-116 | 0.22 | 0.37 | 3.70 |
| MALME-3M | 0.07 | 0.37 | 6.61 | |
| SW-620 | 0.25 | 0.54 | 8.43 |
In summary, we have reported the first chimeric c-Src kinase and HDAC inhibitor. Furthermore, we have performed detailed studies that demonstrate that chimera 4 is a potent and selective c-Src kinase inhibitor as well as a potent and nonselective HDAC inhibitor. We demonstrated that our chimeric inhibitor has improved efficacy in cellular experiments compared to dosing two individual inhibitors targeting c-Src and HDACs. Chimera 4 has significant efficacy in the NCI-60 panel, while not possessing significant toxicity to primary human cells, and represents a novel small molecule probe that can provide simultaneous inhibition of c-Src and HDACs. Our approach to constructing kinase-HDAC inhibitor hybrids using a triazole linkage should be general and readily adapted to any kinase and/or HDAC pair of interest.
Acknowledgments
We would like to thank Christel Fox for preparation of c-Src, c-Abl, and Hck kinase domain proteins.
Supporting Information Available
Synthetic procedures and characterization data, assay conditions, and cell data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors, and all authors have given approval to the final version of the manuscript.
Funding of this research was provided in part by NIH grant R01GM088546 to M.B.S. and the University of Michigan College of Pharmacy.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Thomas S. M.; Brugge J. S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [DOI] [PubMed] [Google Scholar]
- Martin G. S. The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2001, 2, 467–475. [DOI] [PubMed] [Google Scholar]
- Zhan S.; Huang W.-C.; Li P.; Guo H.; Poh S.-B.; Brady S. W.; Xiong Y.; Tseng L.-M.; Li S.-H.; Ding Z.; Sahin A. A.; Esteva F. J.; Hortobagyi G. N.; Yu D. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandvold K. R.; Steffey M. E.; Fox C. C.; Soellner M. B. Development of a highly selective c-Src kinase inhibitor. ACS Chem. Biol. 2012, 7, 1393–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrone V.; Thakkar N.; Jacobson J. M.; Wingdahl B.; Krebs F. C. Combinatorial approaches to the prevention and treatment of HIV-1 infection. Antimicrob. Agents Chemother. 2011, 55, 1831–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harries A. D.; Dye C. Tuberculosis. Ann Top. Med. Parasitol. 2006, 5–6, 415–431. [DOI] [PubMed] [Google Scholar]
- Bouza E.; Muñoz P. Monotherapy versus combination therapy for bacterial infections. Med. Clin. North Am. 2000, 84, 1357–1389. [DOI] [PubMed] [Google Scholar]
- Dancey J. E.; Chen H. X. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat. Rev. Drug Discovery 2006, 5, 649–659. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Resnick R. J.; Shalloway D. Apoptosis of estrogen-receptor negative breast cancer and colon cancer cell lines by PTPα and Src RNAi. Int. J. Cancer 2008, 122, 1999–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atadja P. Development of the pan-HDAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [DOI] [PubMed] [Google Scholar]
- Bolden J. E.; Peart M. J.; Johnstone R. W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discovery 2006, 5, 769–784. [DOI] [PubMed] [Google Scholar]
- Kostyniuk C.; Dehm S.; Batten D.; Bonham K. The ubiquitous and tissue specific promoters of the human SRC gene are repressed by inhibitors of tissue deacetylases. Oncogene 2002, 21, 6340–6347. [DOI] [PubMed] [Google Scholar]
- Marks P. A.; Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [DOI] [PubMed] [Google Scholar]
- Zimmermann G. R.; Lehár J.; Keith C. T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discovery Today 2007, 12, 34–42. [DOI] [PubMed] [Google Scholar]
- Muller Y.; Milton M. The determination and interpretation of the therapeutic index in drug development. Nat. Rev. Drug Discovery 2012, 11, 751–761. [DOI] [PubMed] [Google Scholar]
- Cai X.; Zhai H.-X.; Wang J.; Forrester J.; Qu H.; Yin L.; Lai C.-J.; Bao R.; Qian C. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 2000–2009. [DOI] [PubMed] [Google Scholar]
- Mahboobi S.; Dove S.; Sellmer A.; Winkler M.; Eichhorn E.; Pongratz H.; Ciossek T.; Baer T.; Maier T.; Beckers T. Design of chimeric histone deacetylase- and tyrosine kinase-inhibitors: A series of imatinib hybrides as potent inhibitors of wild-type and mutant BCR-ABL, PDGFRβ, and histone deacetylases. J. Med. Chem. 2009, 52, 2265–2344. [DOI] [PubMed] [Google Scholar]
- Mahboobi S.; Dove S.; Sellmer A.; Winkler M.; Eichhorn E.; Pongratz H.; Ciossek T.; Baer T.; Maier T.; Beckers T. Novel chimeric histone deacetylase inhibitors: a series of lapatinib hybrides as potent inhibitors of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and histone deacetylase activity. J. Med. Chem. 2010, 53, 8546–8601. [DOI] [PubMed] [Google Scholar]
- Paris M.; Porcelloni M.; Binaschi M.; Fattori D. Histone deacetylase inhibitors: From bench to clinic. J. Med. Chem. 2008, 51, 1505–1534. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- Chen P.; Patil V.; Guerrant W.; Green P.; Oyelere A. Synthesis and structure–activity relationship of histone deacetylase (HDAC) inhibitors with triazole-linked cap group. Bioorg. Med. Chem. 2008, 16, 4839–4892. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Chen X. G.; Xue P.; Sun H. Y. Y.; Williams I. D.; Sharpless K. B.; Fokin V. V.; Jia G. C. Ruthenium-catalyzed cycloaddition of alkynes and organic azides. J. Am. Chem. Soc. 2005, 127, 15998–15999. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Cahill S. M.; Blumenstein M.; Lawrence D. S. Self-reporting fluorescent substrates of protein tyrosine kinases. J. Am. Chem. Soc. 2006, 128, 1808–1809. [DOI] [PubMed] [Google Scholar]
- Shultz B. E.; Misialek S.; Wu J.; Tang J.; Conn M. T.; Tahilramani R.; Wong L. Kinetics and comparative reactivity of human class I and IIb histone deacetylases. Biochemistry 2004, 43, 11083–11091. [DOI] [PubMed] [Google Scholar]
- Shoemaker R. H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.