

Abstract
Purpose
Glycogen Storage Disease (GSD) type III, glycogen debranching enzyme deficiency, causes accumulation of glycogen in liver, skeletal, and cardiac muscle. Some patients develop increased left ventricular (LV) thickness by echocardiography, but the rate of increase and its significance remain unclear.
Methods
We evaluated 33 patients with GSD type III, 23 with IIIa and 10 with IIIb, ages 1 month – 55.5 yrs, by echocardiography for wall thickness, LV mass, shortening and ejection fractions, at 1 time point (n = 33) and at 2 time points in patients with more than 1 echocardiogram (13 of the 33).
Results
Of 23 cross-sectional patients with type IIIa, 12 had elevated LV mass, 11 had elevated wall thickness. One type IIIb patient had elevated LV mass but 4 had elevated wall thickness. For those with multiple observations, 9 of 10 with type IIIa developed increased LV mass over time, with 3 already increased at first measurement. Shortening and ejection fractions were generally normal.
Conclusion
Elevated LV mass and wall thickness is more common in patients with type IIIa but develops rarely in type IIIb, though ventricular systolic function is preserved. This suggests serial echocardiograms with attention to LV thickness and mass are important for care of these patients.
Keywords: Glycogen Storage Disease Type III, left ventricular hypertrophy, ventricular function, left ventricular mass
INTRODUCTION
Glycogen Storage Diseases are disorders of metabolism caused by enzyme defects that affect glycogen synthesis or degradation within muscles, liver, heart and other cell types. Glycogen Storage Disease type III (GSD III, also called Cori-Forbes Disease) is caused by a deficiency in glycogen debranching enzyme (GDE).1 GDE is a bi-functional enzyme with both glucosidase activity and transferase activity encoded by the AGL gene located on chromosome 1p21.2.2 Deficiency of GDE results in accumulation of glycogen in the liver, skeletal, and cardiac muscle. Clinical features, particularly in young patients, include hypoglycemia and short stature. There is much variability in the clinical symptoms in these patients. GSD III is further subdivided as type IIIa with deficiency of the enzyme in both the liver and the muscle, type IIIb with deficiency of the enzyme in the liver only, or in rarer cases, selective loss of only one of the two GDE activities resulting in type IIIc or type IIId.3 Differences in tissue specific expression of GDE can explain the existence of various subtypes of GSD III.4 The extent of organ involvement is variable even within the subtypes.
Cardiac involvement with glycogen deposition in the myocardium has been identified in patients with GSD III, particularly patients with GSD IIIa since early descriptions of the disease.5,6 A subset of patients with GSD III develop left ventricular (LV) hypertrophy similar in appearance to hypertrophic cardiomyopathy when analyzed by echocardiography (Figure 1), although on a histological basis there is no myocyte disarray as seen in hypertrophic cardiomyopathy.7,8
Figure 1. Left Ventricular Hypertrophy on Echocardiography.
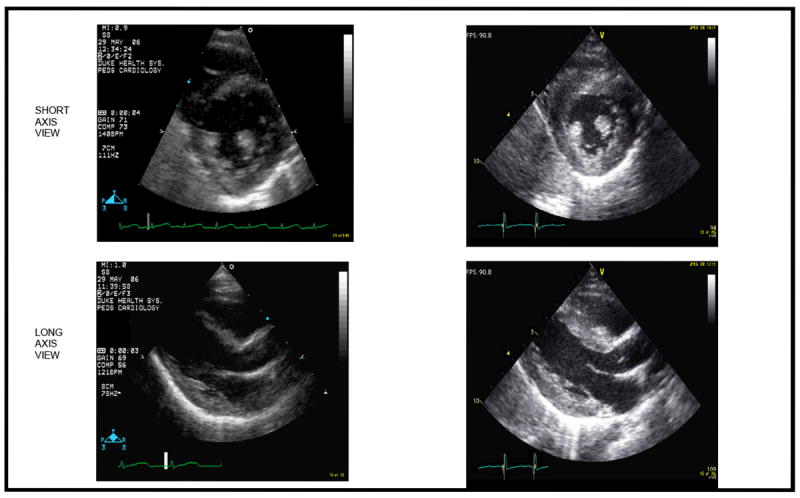
Two dimensional echocardiogram images from one patient with GSD IIIa who experienced marked increase in measures of wall thickness as well as increased LV mass. The left upper panel is the short axis view of the left ventricle at baseline; the left lower panel is the long axis view of the left ventricle at the same time. On the right, in the upper panel is the short axis view of the left ventricle one year later; the left lower panel is the long axis view of the left ventricle one year later.
Previous literature regarding cardiac involvement with GSD III has been limited to case reports and cross sectional studies precluding descriptions of cardiac involvement over time in individual patients. There is the suggestion that there is more hypertrophy in older patients, although there is no confirmation due to a lack of longitudinal data.9 Furthermore, many early studies base the diagnosis of GSD on liver enzyme levels, and therefore do not distinguish between IIIa and IIIb. Most authors note that there are no clinical cardiac symptoms that exist even in the presence of cardiac hypertrophy by echocardiogram, yet there are a few reports of sudden cardiac deaths and one reported case of heart, liver, and kidney transplant in a GSD III patient.6,10 Patients with GSD III develop LV hypertrophy by echocardiography, but the prevalence and clinical significance remain unknown.
Given the lack of longitudinal data in GSD III patients, subdivided by type, especially with regard to the extent and rate of progression of LV hypertrophy over time, we examined a cohort of GSD III patients to describe their natural history. We implemented a cross – sectional approach for all patients with at least one echocardiogram, and a longitudinal approach in patients with more than one echocardiogram to review changes that occur over time in this population. Since there have been no previous reports that used LV mass as an index of LV hypertrophy, this parameter was measured on all echocardiograms. LV mass measurements are based on information from two dimensional imaging taking into account myocardial density.11 LV mass has advantages over measurements of LV thickness in determining the presence of LV hypertrophy since it uses two dimensional imaging, it does not assume that the ventricle is spherical and it does not assume that the entire LV has the same thickness. In addition, there are normative data for LV mass in adults as well as children and the value is indexed to body surface area.11,12 Indices of wall thickness such as Interventricular Septal Dimension (IVSD), as well as Left Ventricular Posterior Wall Dimension (LVPWD), measured at the end of diastole, have been measured previously in studies of GSD III patients, but these measurements are most accurate in normally shaped ventricles, which may not be the case if asymmetric hypertrophy is present as can sometimes be seen in these patients.13,29 We performed wall thickness measurements as well as LV mass in all patients to determine if LV mass might be more useful in following disease progression over time in these patients. In addition, we compared our study results to prior reports in the literature.
MATERIALS AND METHODS
Patients with GSD III who attend the Metabolic Diseases Clinic at Duke University Medical Center consented for participation in a GSD III natural history study which was approved by the Duke Institutional Review Board. Our study population was a cohort of biochemically and genetically well characterized GSD III patients followed at Duke University Medical Center Metabolic Disease Clinic over a mean time period of 4.8 yrs (1.1 – 9.1 yrs). The diagnosis of GSD III was made based on clinical symptoms as well as biochemical measurement of elevated glycogen content, an altered ratio of Glucose-1-phosphate/glucose and evidence of GDE deficiency in biopsies of liver (n=16) or muscle (n=5) or both tissues (n=5). GSD III subtype was determined based on presence or absence of GDE activity in muscle tissue sample and/or the presence of one of the two GSD IIIb specific pathogenic mutations [(c16C>T(pGln6X) and c17_18delAG(pGln6HisfsX25)] in a patient (n=8), as has been described before.3, 4, 14 Genomic mutation analysis was done on DNA isolated from whole blood using Pure Gene DNA isolation kit (Gentra systems) and mutations were studied in detail using the ABI 3130xl Genetic Analyzer. Sequences were compared to the reference DNA sequence (GenBank Accession: NM_000028.2).
Of the 42 GSD III patients who consented for the study, 33 subjects had echocardiograms available and of adequate quality for review. No patients were excluded due to other causes of left ventricular hypertrophy such as hypertension. Each echocardiogram was reviewed in its entirety by three of the authors (KF, SV, SBW). Most echocardiograms were performed at Duke University Medical Center and a few at outside institutions. Several specific features were measured including: IVSD in diastole, LVPWD in diastole, LV mass, shortening fraction (SF), and ejection fraction (EF). The most recent echocardiogram was analyzed for the cross-sectional analysis. The first and last echocardiograms were utilized for the longitudinal data analysis. For measurements of ventricular thickness, M – mode was preferentially selected if available and of good quality. Parasternal views were used for ventricular thickness if M – Mode measurements were not available or were not of good quality. Two-dimensional views were obtained and the area length method was used for the LV mass measurements.11 Two-dimensional parasternal short axis views were selected for shortening fraction. Descriptive plots of each parameter indexed to BSA were analyzed versus Z score standards for these measurements in the general population (SAS v 9.0 - SAS Institute Inc, Cary, NC).11,15
RESULTS
Patient demographics are summarized in Table 1. Of the patients with echocardiograms available for review, 23 patients were classified as type IIIa, and 10 were type IIIb. The mean age of patients was 16.5 years (range1.0 through 55.5 years). Of the 33 subjects with echocardiograms, 19 were females and 14 were males. Eighteen were children under 18 years of age, and 15 were adults (>18 years of age). Twenty patients had a single echocardiogram for review, 13 with type IIIa and 7 with type IIIb. Thirteen patients had multiple echocardiograms for review, 10 with type IIIa and 3 with type IIIb. The average time between echocardiograms was 4.3 years (minimum of 0.09 years to a maximum of 11.6 years). Patients were followed over a mean time period of 4.8 yrs (1.1 – 9.1 yrs). No alternate cause of increased LV mass or wall thickness such as hypertension was identified in any patient.
Table 1.
Patient Demographics
| Type IIIa | Type IIIb | ||
|---|---|---|---|
| Children (<18 yrs) | Adults (>18 yrs) | Children (<18 yrs) | Adults (>18 yrs) |
| 12 | 11 | 6 | 4 |
| (4 males, 7 females) | (3 males, 1 female) | ||
| Echos available for review | Echos available for review | ||
| Single echo | 13 | Single echo | 7 |
| >1 echo | 10 | >1 echo | 3 |
Mutation analysis
Genomic sequence analysis of the AGL gene carried out in our patient cohort showed a wide heterogeneity of pathogenic mutations. In all, 23 different disease causing mutations were identified in our group of 33 patients, in various combinations of compound heterozygotes for 2 different pathogenic mutations (n=20) or homozygous mutations (n=10). The majority of the mutations included nonsense/frameshift nonsense mutations (n=14) with few missense (n=2), donor/acceptor splice site (n=3) and intronic mutations (n=1). A novel exon 3 deletion mutation was identified in a homozygous state in a patient with severe disease phenotype. Gene mutation analysis could not be performed on few patients due to unavailability of DNA sample.
Two previously reported exon 3 mutations associated with GSD IIIb phenotype (p.Gln6stop and p.Gln6HisfsX25) were observed in all of our GSD IIIb patients (n=10), mostly in combination with other pathogenic mutations located in the AGL gene. Additionally, muscle biopsy testing on 3 of these patients showed normal GDE activity; thus, proving them to have a type IIIb phenotype. The p.Gln6stop was seen in 4 patients and a common frameshift mutation, p.Gln6HisfsX25 was seen in 6 GSD IIIb patients showing a clear genotype-phenotype correlation with these 2 mutations, as has been reported before.3, 4, 14
Cross Sectional Analysis of Ventricular Thickness in Children
For LVDPW in children, the cross sectional analysis demonstrated that 6 out of 12 children (50%) with GSD type IIIa were abnormal with a Z score of ≥2 at their last observation point (Figure 2a). Of the 6 pediatric patients with GSD IIIb, 1 patient had a Z score of 2; the remaining patients had normal measurements. For the cross sectional analysis of IVSD in children, 5 out of 12 patients with GSD IIIa were abnormal with a Z score of ≥2 at most recent observation (Figure 2b). GSD IIIa patients with Z scores of ≥2 for LVDPW were usually also the patients with elevated IVSD. Of the GSD IIIb patients with IVSD measurements, all were normal. LV mass was utilized as an additional method of assessing LV hypertrophy. Seven children with GSD IIIa had LV mass that was above the upper limit of normal for age, in some cases markedly above normal (Figure 3a). Each GSD IIIa patient with increased LV mass also had LVDPW and/or IVSD ≥2. In the GSD IIIa patients with increased LV mass, both measurements of LV thickness were abnormal, but not in all; thus, there was not complete correlation of wall thickness measurements and LV mass. There was 1 patient with GSD IIIb with an LV mass measurement above the upper limit of normal for age (Figure 3a), though this patient’s LVDPW and IVSD were normal. For children with GSD IIIa with LV mass available (n = 11), the median observed LV mass was 82 g/m2, (range 44 to110 g/m2), exceeding the upper limit of normal of 70 g/m2. The GSD IIIa patients with elevated LV mass were those who also had increased wall thickness indices. In the case of the 5 children with GSD IIIb for whom LV mass was available, the median LV mass was 56 g/m2 (range 55 to 73 g/m2).
Figure 2. Wall thickness parameters in children with GSD III.
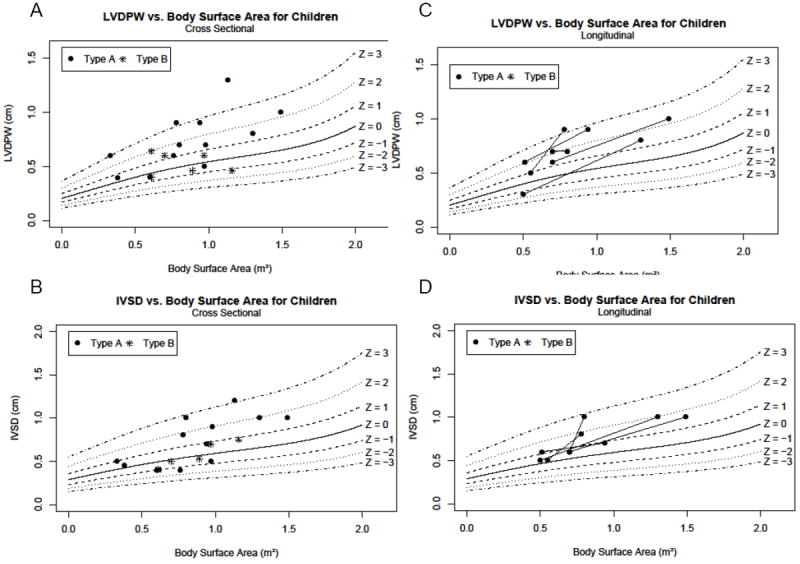
Measurements are indexed for body surface area and shown with reference lines for Z-scores for these parameters in children.15 In all panels, GSD IIIa patients are noted with closed circles. GSD IIIb patients are noted with stars. A) LVDPW indexed for body surface area. B) IVSD indexed for body surface area. C) Longitudinal assessment of LVDPW in children over time indexed for body surface area. D) Longitudinal assessment of IVSD in children over time indexed for body surface area.
Figure 3. LV mass for age in children and adults.
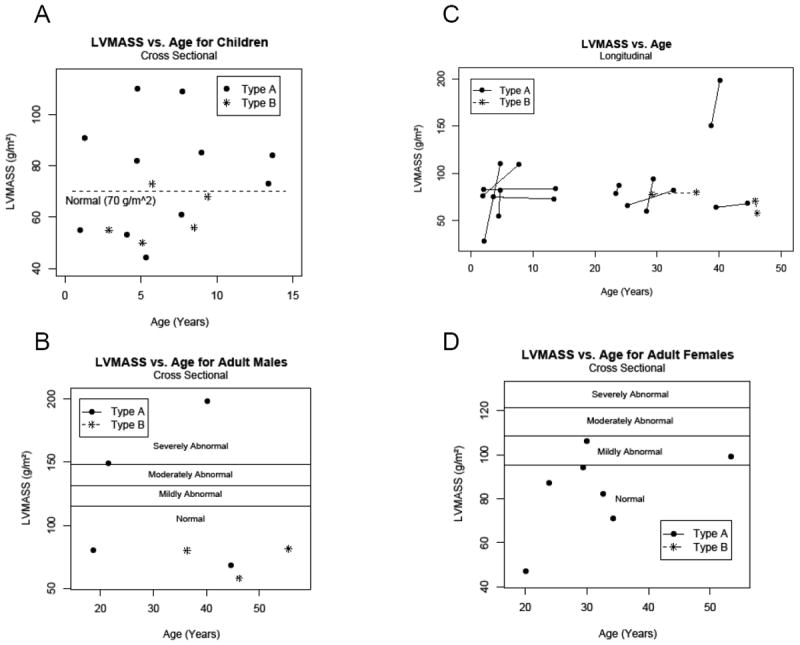
In all panels, GSD IIIa patients are noted with closed circles; GSD IIIb patients are noted with stars. A) Cross-sectional LV mass values for children. The upper limit of normal for LV mass for children (70 g/m2) is shown as a dotted line for reference.12 B) LV mass for adult male patients; reference lines for severity of abnormal value are noted.11 C) Longitudinal LV mass measurements for patients over time and shown by age. D) LV mass for adult female patients with reference lines for severity of abnormal value noted.11
Longitudinal Analysis of Ventricular Thickness in Children
For the children with multiple LVDPW assessments normalized for body surface area, we found that 2 out of the 5 patients with GSD IIIa had initial posterior wall measurements that were abnormal at a Z score of ≥2 at the time of their first echocardiogram, and 4 of the 5 patients developed an increase in Z scores from the first to the last measurement (Figure 2c). Three of the 5 patients had measurements with a Z score of ≥2 by their last echocardiogram (Figure 2c). In 1 patient, this increase took place in as short a period as 2 years and 6 months, while in another the increase was over 9 years and 7 months. For the longitudinal analysis of IVSD, all 5 children with GSD IIIa demonstrated an increased Z score from the first to the last observation (Figure 2d), and 2 of 5 patients had Z score ≥2 at their last evaluation. For the 5 children with GSD IIIa with multiple observations, the mean LV mass at baseline was 63.4 g/m2 (range 28 to 83 g/m2) with the mean final observed LV mass was 91.6 g/m2 (range 73 to 110 g/m2). Four children with GSD IIIa showed an increase in LV mass between measurements, 3 with a marked increase over time (Figure 3c). For example, 1 patient more than doubled her LV mass over time (Figure 1, 3c).
Cross Sectional Analysis of Ventricular Thickness in Adults
When performing the cross sectional analysis in adults we stratified by gender as well as age since the normal values are different both by age as well as gender in adults. We found that 3 out of 4 adult male patients with GSD IIIa had an LVDPW that fell in the abnormal range with 2 mildly abnormal and 1 severely abnormal (Figure 4a). Interestingly, of the 3 adult male patients with GSD IIIb, 2 had mildly abnormal values for LVDPW (Figure 4a). For the cross sectional view of LVDPW in adult women with GSD IIIa, we found that 2 out of 7 were in the abnormal range with both mildly abnormal (Figure 5a). There was only 1 adult female patient with GSD IIIb; her LVDPW was in the mildly abnormal range (Figure 5a). In the cross sectional look at IVSD in adult males with GSD IIIa, there were 3 of 4 in the abnormal range, 2 who were severely abnormal (Figure 4b). For IVSD in adult females, 2 of 7 GSD IIIa female patients fell in the abnormal range, with both only mildly abnormal (Figure 5b). For LV mass measurements in adult male patients with GSD IIIa, 2 of 4 had severely abnormal LV mass measurements (Figure 3b). Both patients with abnormal LV mass also had severely abnormal IVSD and one also had severely abnormal LVDPW. LV mass measurement in adult females showed that 3 of 7 GSD IIIa women had LV mass that was abnormal, 2 in the mildly abnormal range and 1 in the moderately abnormal range (Figure 3d). There was only 1 adult female GSD IIIb patient and an LV mass was not available for her. The 2 GSD IIIa women with the highest LV mass measurements actually had normal or only mildly abnormal wall thickness measurements (Figure 5a, b).
Figure 4. Wall thickness parameters for adult males with GSD III.
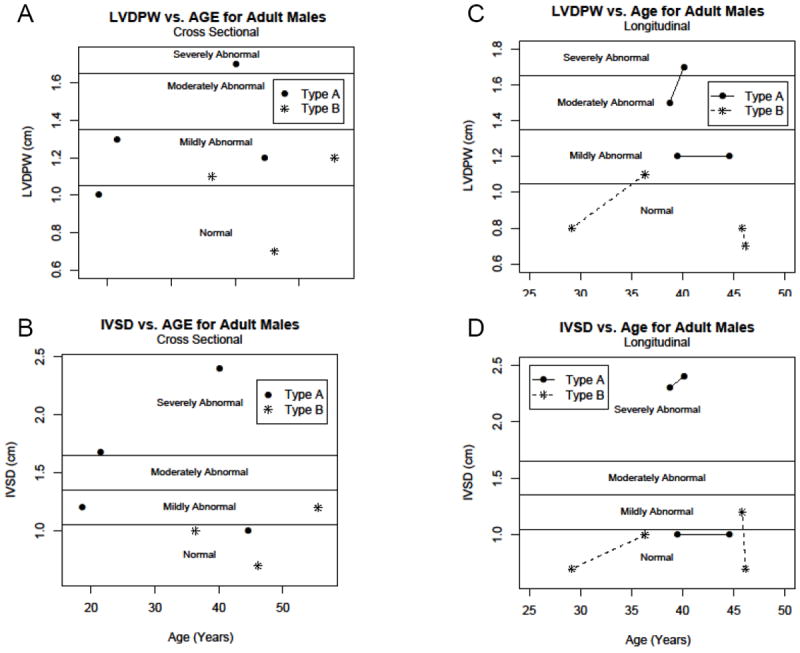
In all panels, GSD IIIa patients are noted with closed circles. GSD IIIb patients are noted with stars. A) LVDPW for adult males shown by age with reference lines denoting mild, moderate and severely abnormal values.11 B) IVSD for adult males shown by age with reference lines denoting mild, moderate and severely abnormal values.11 C) LVDPW for adult male patients with more than one echocardiogram. D) IVSD for adult male patients with more than one echocardiogram.
Figure 5. Wall thickness parameters for adult females with GSD III.
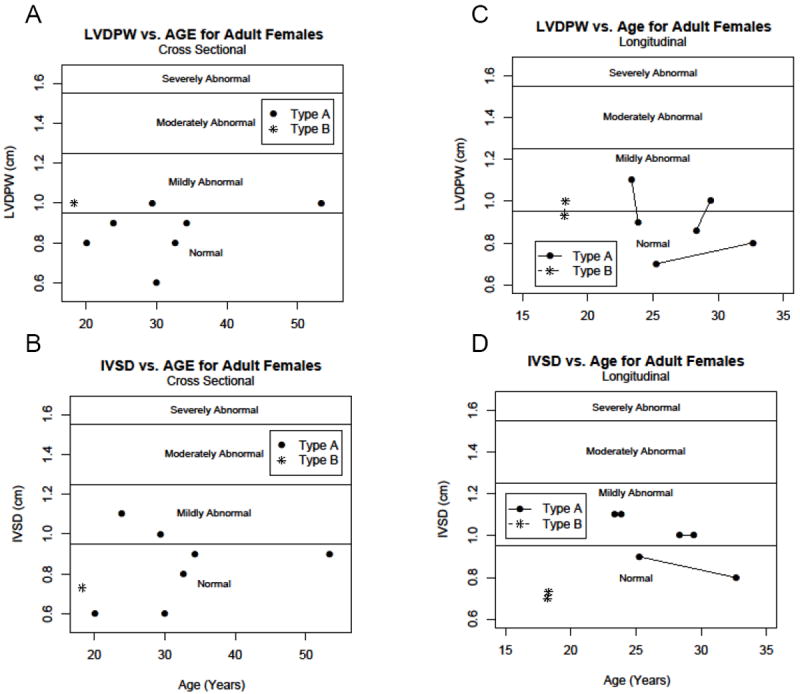
In all panels, GSD IIIa patients are noted with closed circles. GSD IIIb patients are noted with stars. A) LVDPW for adult females shown by age with reference lines denoting mild, moderate and severely abnormal values.11 B) IVSD for adult females shown by age with reference lines denoting mild, moderate and severely abnormal values.11 C) LVDPW for adult female patients with more than one echocardiogram. D) IVSD for adult female patients with more than one echocardiogram.
Longitudinal Analysis of Ventricular Thickness in Adults
There were only 2 male adult patients with GSD IIIa with more than one echocardiogram available for review (Figure 4c). One of these men was mildly abnormal for LVDPW at the time of the first echo and remained mildly abnormal at the time of the second echocardiogram. The other patient was severely abnormal and remained severely abnormal (Figure 4c). Two male patients with GSD IIIb had more than one echocardiogram. At each time point, the LVDPW was normal for age and gender. There were 3 female patients with GSD IIIa with more than 1 echocardiogram (Figure 5c). In 1 female GSD IIIa patient, the LVDPW was normal at both measurements; 1 patient was mildly abnormal on the first echocardiogram but returned to the normal range for the last echocardiogram. The final patient was normal initially but became mildly abnormal on the final measurement (Figure 5c). The only GSD IIIb female patient with >1 echocardiogram had a mildly abnormal LVDPW at both time points (Figure 5c). For IVSD in the 2 adult male patients with GSD IIIa, 1 patient was normal at the first echocardiogram and remained normal. The other patient was severely abnormal and remained so (Figure 4d). For IVSD in female adults with GSD IIIa, the IVSD was mildly abnormal and remained so at both time points for 2 of 3 patients. The other patient had a normal IVSD that remained normal (Figure 5d). There was only 1 female patient with GSD IIIb whose IVSD was normal at the first measurement and remained normal (Figure 5d). For LV mass measurements in adults over time, the LV mass increased in both male GSD IIIa patients as well as in all 3 female GSD IIIa patients (Figure 3c). LV mass was normal for both male GSD IIIb patients and actually decreased over time in 1 case (Figure 3c).
Longitudinal Analysis of Function in Adults and Children
Shortening fraction (SF) and ejection fraction (EF) are measures of LV performance that assess systolic function. SF and EF remained normal for almost all adult and children at all points examined over the entire period of follow up (Data not shown). The only exceptions were 1 adult male patient with GSD type IIIa with severely decreased SF and EF who also had marked LV hypertrophy by all parameters and 1 adult female patient with GSD type IIIb with one slightly low measurement of ejection fraction that normalized by the time of her second echocardiogram.
Cardiac Symptoms and Outcomes
Most patients remain asymptomatic from a cardiovascular standpoint; however there were a few patients with reported cardiovascular symptoms (Table 2). Notably, 6 patients in this cohort of 42 patients died. Two patients who did not have echocardiograms died of hepatocellular carcinoma, a recently reported long term risk in this patient group.16 There was 1 death in the patients with GDS IIIb; this patient died at age 18 years due to pulmonary hypertension (confirmed by cardiac catheterization) after a 2 year history of progressive dyspnea on exertion as well as occasional cyanosis. Her echocardiogram showed mildly abnormal LVDPW but normal IVSD; no LV mass was available for this patient. She had one measurement of EF that was mildly diminished but then her EF was normal on a later echocardiogram. Of the patients with GSD IIIa who had echocardiograms for review, there were 3 patients who died. In each case, these patients experienced sudden death at young ages (15 years, 25 years and 36 years). The patient who died at age 15 years of age had only 1 echocardiogram for review which showed significantly elevated LVDPW and IVSD with a Z score > 3 for both parameters; his LV mass was elevated as well. The 25 year old patient who died was mildly abnormal for LVDPW but severely abnormal for IVSD with severely increased LV mass. The 36 year old patient who died had normal LVDPW, IVSD and LV mass, though over time her LVDPW and her LV mass did increase. By autopsy, she had extensive glycogen deposition in the cells of the cardiac conduction system. This patient had a poorly controlled seizure disorder which could have contributed to her death; however, she did have a history of chest pain with exertion. In addition, there was 1 patient who required heart, liver, and kidney transplant who presumably would have died without this intervention. This patient was a 39 year old man with GSD IIIa and demonstrated clinical symptoms of heart failure and diminished LV systolic function as evidenced by an EF of 20%, moderate mitral regurgitation and pulmonary hypertension.10 His LVDPW, IVSD and LV mass were all severely abnormal and increased over time.
Table 2.
Clinical Characteristics
| Patient | IIIa vs IIIb | Major Cardiac Event | Cardiac Symptoms at Last Clinic Visit |
|---|---|---|---|
| A-1 | A | None | None |
| A-2 | A | None | Tired after activity |
| A-3 | A | Sudden death @ age 36 yrs | Chest pain w/exercise; referred for cardiology evaluation |
| A-4 | A | None | None |
| A-5 | A | None | None |
| A-6 | A | Liver, Kidney, Heart Transplant @ age 40 yrs | Congestive heart failure pretransplant |
| A-7 | A | None | None |
| A-8 | A | None | None |
| A-9 | A | None | None |
| A-10 | A | Sudden death @ age 15 yrs | Intermittent chest pain sometimes after exercise |
| A-11 | A | None | None |
| A-12 | A | None | Pleuritic right sided chest pain |
| A-13 | A | None | None |
| A-14 | A | None | Occasional orthostatic dizziness |
| A-15 | A | None | None |
| A-16 | A | None | None |
| A-17 | A | None | None |
| A-18 | A | None | Squeezing chest pain sometimes during strenuous exertion |
| A-19 | A | None | None |
| A-20 | A | None | Dyspnea on exertion; atypical chest pain; peripheral edema |
| A-21 | A | None | Chest pain on one occasion (associated w/diabetic ketoacidosis) |
| A-22 | A | Sudden death @ age 25 yrs | None |
| A-23 | A | None | None |
| B-1 | B | None | None |
| B-2 | B | None | None |
| B-3 | B | None | None |
| B-4 | B | None | None |
| B-5 | B | None | None |
| B-6 | B | None | None |
| B-7 | B | None | Fatigue; Dyspnea w/ exertion; Chest pain w/ and w/o exercise |
| B-8 | B | None | None |
| B-9 | B | Death @ age18 yrs due to pulmonary hypertension | Not available |
| B-10 | B | None | None |
DISCUSSION
Glycogen Storage Disease III is a rare but potentially serious disease that has variable cardiac involvement. In this study we examined a cohort of patients on both a cross – sectional and longitudinal basis for measurements of LV wall thickness and mass in order to assess the natural history of LV hypertrophy in this patient group. One limitation of previous research on these subjects is that it does not distinguish between the subtypes of GSD III. Patients with GSD IIIb are thought to have only liver involvement; thus, inclusion of these patients could cause an underestimation of the number of type IIIa patients who develop cardiac changes over time. With the availability of both biochemical analysis and detailed gene mutations in our patient cohort, we are able to describe the differences in trends of ventricular hypertrophy noted between type IIIa and type IIIb subtypes. Previous reports noted measurements of ventricular wall thickness as the main assessment of ventricular hypertrophy.17,18,9, 19 We analyzed ventricular wall thickness as well as LV mass, which has not previously been reported in GSD III patients, to determine if LV mass measurement provided a superior method for measuring LV hypertrophy. Monitoring of LV mass has been very useful in monitoring patients with Pompe Disease (GSD II.)12 Our overall goal was to determine a best-practice method for following patients with GSD III to determine who is at greater risk for developing cardiovascular complications of their disease. Our study demonstrates that both groups can develop ventricular hypertrophy, but it is much more common and more severe in type IIIa than type IIIb.
Initial reports of GSD III noted some cardiac involvement in GSD III patients (Table 3). Several case series between 1989 and 1997 suggested that ventricular hypertrophy is a relatively common finding in GSD III patients, though the exact incidence was quite variable, between 30 and 80% depending in the study. 17,20,18,19,9,21 Moses’ case series reported in 1989 that 13 of 16 patients (81.25%) had evidence of concentric LV hypertrophy by echocardiography, but only 1 patient in their series had any clinical symptoms.21 Labrune, et al reported 5 of 16 (31.25%) young patients (8 months to 18 years) with LV hypertrophy by echocardiography, but no clinical symptoms.18 The most comprehensive study of cardiac involvement in GSD III patients was by Lee, et al.19 In this study, 32 patients, including children and adults, were systematically evaluated by echocardiography. Indexing LV wall thickness to body surface area, 37.5% of their patient population (33% children and 50% of adults) had evidence of LV hypertrophy.9 Other cardiac manifestations in GSD III patients included myocardial fibrosis and dysrhythmias that have been reported in isolated cases.22,8 There have also been isolated cases of sudden death where gross cardiac involvement has been noted on autopsy.6 There has been no obvious relationship between degree of skeletal myopathy and cardiomyopathy. Therefore, the amount of peripheral muscle involvement does not dictate cardiac involvement or vice versa.18
Table 3.
Literature Review
| Date | First Author | # pts | Ages (yrs) | GSD Type | Type report | Cardiac Clinical Sx | Echocardiogram Findings |
|---|---|---|---|---|---|---|---|
|
| |||||||
| 2007 | Cochrane10 | 1 | 39 | IIIa | Case report | Yes (CHF; transplant) | LV ejection fraction 20% |
| Pulm HTN | |||||||
|
| |||||||
| 2007 | Ogimoto25 | 1 (mentions 6 others) | 42 | IIIa | Case report | Yes (exertional chest pain; dyspnea) | “Marked hypertrophy” |
| Abnormal diastolic function | |||||||
|
| |||||||
| 1997 | Akazawa26 | 1 | 38 | IIIa | Case report | Yes (dyspnea/fatigue w/ exertion; sudden death) | Akinetic & thin portions of LV wall |
| Abnormal systolic function (SF24%) | |||||||
| LVH present (LVmass 225 gm/m2 and septum 18 mm) | |||||||
|
| |||||||
| 1997 | Cuspidi13 | 1 | 30 | IIIa | Case report | Yes (tachypnea, dyspnea) | Severe asymmetric septal hypertrophy, systolic anterior motion of the mitral valve, LV outflow tract obstruction |
|
| |||||||
| 1997 | Shen27 | 1 | 4 | III | Case report | Yes (sudden death) | LVH present |
|
| |||||||
| 1997 | Lee9 | 32 | 0.6 to 31.6 (12 >18 yr) | III | Cross sectional | No symptoms in 5 patients | 12/32 w/ LVH (37.5%) (by LV posterior free wall thickness) |
|
| |||||||
| 1995 | Lee28 | 32 | 0.6 to 31.6 (12>18 yr) | III | Cross sectional | LV posterior free wall thickness | |
| SF | |||||||
| 6/18 children w/ LVH (33%) | |||||||
| 6/12 post pubertal (50%) | |||||||
| 12/32 overall w/ LVH (37.5%) | |||||||
|
| |||||||
| 1995 | Tada8 | 1 | 23 | III | Case report | Palpitations | LV dilation and LV concentric hypertrophy; LV wall thinning |
| Ventricular tachycardia | Abnormal systolic function (SF9%) | ||||||
|
| |||||||
| 1995 | Kobayashi29 | 1 | 23 | III | Case report | “Marked thickening of interventricular septum” | |
|
| |||||||
| 1994 | Talente30 | 9 | 18-57 | III | Case series | 4/6 abnormal echocardiograms (67%) | |
| LVH in 3, BVH in 1 | |||||||
|
| |||||||
| 1993 | Carvalho17 | 23 | 9 mos to 28 | III | Case series | Symptoms uncommon | 11/23 (~50%) w/ increased |
| 2 w/ dyspnea w/ exertion; 3 w/ fatigue; 1 w/ chest pain | LV septal and posterior wall thickness; LV dimensions | ||||||
| SF all normal | |||||||
|
| |||||||
| 1992 | Coleman20 | 13 | 4 to 57 | IIIa, b, d | Case series | None mentioned | IIIa: 3/7 abnormal echocardiograms (43%) including LVH in one |
|
| |||||||
| 1991 | Labrune18 | 18 | 8 mos to 18 | III | Case series | None symptomatic | 5/16 (30%) w/ increased LV septal and posterior wall thickness |
|
| |||||||
| 1990 | Smit31 | 50 | >10 | III | Cross sectional | Not reported | “Cardiomyopathy” in 22/44; no echo details reported |
|
| |||||||
| 1989 | Moses21 | 20 | 3-30 | III | Cross sectional | 19/20 asymptomatic | LVH 13/16 |
| 1/20: Dyspnea, weakness | Mild LV dilation 2/20 | ||||||
| LA dilation 7/20 | |||||||
|
| |||||||
| 1984 | Olson7 | 1 | 25 | III | Case report | Yes; orthopnea due to pulmonary edema during pregnancy | “Marked thickening of ventricular myocardium” |
| Normal systolic function | |||||||
|
| |||||||
| 1972 | Miller6 | 1 | 3 mos | III | Case report | Yes (sudden death at 3 mos) Marked LVH at autopsy | No echocardiogram |
| “Gross myocardial thickening” | |||||||
CHF – Congestive Heart Failure; LV – Left ventricle; HTN – Hypertension: LVH – Left Ventricular Hypertrophy; LA – Left atrium
Similar to other reports, we also find that LV thickness increases in many of the patients with GSD III. Specifically, in children with GSD IIIa, approximately half had increase in wall thickness in the cross sectional analysis of LVDPW and IVSD. Similarly, the LV mass was increased in 7 of 11 young GSD IIIa patients. In the longitudinal analysis of LV mass in children, LV mass increases over time, even when indexed for body surface area. In our longitudinal follow up, 1 patient had doubling of LV mass over time and 2 patients had rapid increases in LV mass over a short period of time. For adult patients with GSD IIIa, increased wall thickness measurements were more common in men than in women. LV mass increased over the time in men and women though our numbers for study were small. We observed that the same patients who exhibited pronounced increase in posterior and interventricular wall thickness, demonstrated similarly pronounced changes in their LV mass over time. Though this significant increase in LV mass was seen, measured aspects of systolic function including SF and EF remained normal over time in the large majority of cases. In adults with GSD IIIa, some patients do have increased LV thickness and mass; however, with our small sample size, we did not identify a pattern in progression of ventricular hypertrophy over time. Overall, except for the known association of cardiac involvement in GSD IIIa patients and generally little cardiac involvement in GSD IIIb patients, our findings showed no correlation between the type of mutation and LV wall thickness or LV mass, though our sample size is limited. Interestingly, we did find that a few GSD IIIb patients also had measurements of LV hypertrophy, both wall thickness and LV mass, that were at the upper limit of normal or even slightly greater than normal, though the GSD IIIb patients had much milder echocardiographic findings than the GSD IIIa patients.
There are several limitations to our study. First, as a retrospective review, this analysis is dependent on the accuracy of the analysis of previously collected images. We performed our own measurements of LV parameters, but we were limited to the echocardiography images that were available. Nine of the patients who consented for study either did not have echocardiograms available for review or else had poor quality echocardiograms which we could not accurately assess. Our longitudinal assessment was limited because several patients only had 1 echocardiogram available for analysis. Our study is limited in its examination of ventricular function. There are two components of ventricular function, systolic and diastolic. In some forms of cardiomyopathy, including hypertrophic cardiomyopathy, diastolic dysfunction precedes systolic dysfunction. Echocardiographic parameters of diastolic function have been less commonly measured and followed over time, though such parameters increasingly are part of complete echocardiography. Since diastolic parameters were not obtained in most of the echocardiograms we had available for assessment, we cannot describe the diastolic function in this cohort. Finally, our clinical symptom data are also dependent on that recorded at the time of clinic visits and were not prospectively obtained. Despite these limitations we do report a patient group as large as the largest cross- sectional cohort previously reported and our patient group is accurately stratified into type IIIa and type IIIb by biochemical and/or genetic analysis. We are able to add natural history information on a group of 13 patients with echocardiograms over time.
We found that patients who demonstrated increases in LV mass also had increases in LV wall thickness indicating that LV mass did not identify LV hypertrophy in patients that were not already identified by other means in this cohort. It is possible that in a prospective fashion, LV mass will be a more sensitive measure of ventricular hypertrophy. Certainly LV mass can be another useful way of following ventricular hypertrophy and it is more quantitative as a measurement method than wall thickness alone. The SF and EF measurements of systolic function were almost all normal in our patient cohort even though some of these patients had significant cardiac events. Since diastolic dysfunction may precede systolic dysfunction in some forms of cardiomyopathy, it is possible that despite preservation of systolic function there was concomitant gradual diastolic dysfunction, which was not detected. Prospective evaluation of both diastolic and systolic measurements of ventricular function may be quite helpful in determining if there are more subtle changes in function that are clinically important for patients with GSD III. In our study, we have evaluated only echocardiograms, but in some instances, the echocardiographic images were quite poor and precluded accurate assessment of ventricular thickness and mass. If inadequate echocardiographic images are noted in GSD III patients, consideration for using an alternative imaging modality such as cardiac MRI would seem indicated. There is one brief report in the literature noting myocardial fibrosis in a GSD IIIa patient by cardiac MRI.22
Despite the initially reassuring nature of the echocardiographic data regarding the preservation of systolic function and the lack of severely progressive hypertrophy in most patients as they aged, our analysis showed that there were several patients who died in our study group. Strikingly, there were several sudden deaths in relatively young patients, mostly in GSD IIIa patients. This occurrence of sudden death raises the possibility of arrhythmia as the cause of death. The possibility of arrhythmia is also suggested by the observation at autopsy of glycogen accumulation in the conduction system in one GSD IIIa patient in our series who died. In other forms of Glycogen Storage Disease such as Pompe Disease (GSD II) and PRKAG2-associated hypertrophic cardiomyopathy, abnormalities on electrocardiograms and clinical arrhythmias are well described and are thought to be due to glycogen accumulation involving the conduction system of the heart. 23, 24
Clinical cardiac symptoms are not commonly reported in our patient group as noted in Table 2. Of the three GSD IIIa patients who died, two had reported chest pain, though there were reports of chest pain in other patients who are alive and well. Complaints of palpitations were not noted; however, it is possible that patients do not spontaneously mention other cardiac symptoms unless specifically asked. Given the musculoskeletal features of GSD III, it may be difficult clinically to differentiate the etiology of some symptoms such as fatigue and exercise intolerance, since these symptoms could be due to muscle fatigue as well as poor cardiac function. Thus, it may be a clinical challenge to determine if there are specific cardiovascular symptoms in this patient group.
Based on our findings, we recommend regularly screening patients with GSD III with echocardiograms, including measurements of ventricular thickness as well as LV mass and both systolic and diastolic function. We suggest annual echocardiograms since the increase in LV thickness and mass was quite rapid in some of our GSD IIIa patients; however, this time interval may be extended for individual patients if there are several normal echocardiograms over a few years. Patients with GSD IIIb may also be screened by echocardiography less frequently, but given that a few do develop LV hypertrophy, serial echocardiograms every few years seem advisable. If poor echocardiographic windows make the acquisition of this information difficult, consideration for cardiac MRI is warranted. Evaluation of heart rhythm by electrocardiogram seems a reasonable screening test as well, given the patients with sudden death in our series. In patients who are potentially symptomatic and/or may have severe hypertrophy by imaging, consideration for more detailed evaluation of heart rhythm and conduction should be considered. The variability of clinical outcomes, some of them serious, in GSD III patients underscores the importance of echocardiographic screening in this patient population. Partnering with a cardiologist who is familiar with patients with GSD III and other Glycogen Storage Diseases may be the best way to provide optimal care for these patients. By performing good cardiac evaluations of these patients, including detailed serial echocardiograms, we will be better able to identify patients with ventricular hypertrophy who seem most at risk for developing poor outcomes.
Acknowledgments
This publication was made possible in part by Grant Number 5UL1RR024128-03 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH, and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NCRR or NIH. Information on NCRR is available at http://www.ncrr.nih.gov/. Information of Re-engineering the Clinical research Enterprise can be obtained from http://nihroadmap.nih.gov/clinicalresearch/overview-translational.asp/. We wish to thank Wendy Chung, MD, PhD for her help in including clinical information about one of the patients with GSD IIIb.
Funding for this project was provided in part by The Association of Glycogen Storage Disease, USA as well as the National Institutes of Health through the Duke Clinical Research Center.
Footnotes
Conflicts of Interests
The authors of this paper have no conflicts of interest to disclose.
References
- 1.Kishnani PS, Koeberl D. Chen YT Chapter 71: Glycogen Storage Diseases. In: Valle DBA, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, editors. The Online Metabolic and Molecular Bases of Inherited Diseases. New York: McGraw Hill Professional; 2009. [Google Scholar]
- 2.Yang-Feng TL, Zheng K, Yu J, Yang BZ, Chen TY, Kao FT. Assignment of the human glycogen debrancher gene to chromosome 1p21. Genomics. 1992 Aug;13(4):931–934. doi: 10.1016/0888-7543(92)90003-b. [DOI] [PubMed] [Google Scholar]
- 3.Bao Y, Dawson TL, Jr, Chen YT. Human glycogen debranching enzyme gene (AGL): Complete structural organization and characterization of the 5’ flanking region. Genomics. 1996 Dec;38(2):155–165. doi: 10.1006/geno.1996.0611. [DOI] [PubMed] [Google Scholar]
- 4.Bao Y, Yang B-Z, Dawson TL, Jr, Chen YT. Isolation and nucleotide sequence of human liver glycogen debranching enzyme mRNA: Identification of multiple tissue-specific isoforms. Gene. 1997 Sep 15;197(1-2):389–398. doi: 10.1016/s0378-1119(97)00291-6. [DOI] [PubMed] [Google Scholar]
- 5.Pearson C. Glycogen metabolism and storage diseases of types III, IV and V. American Journal of Clinical Pathology. 1968 Jul;50(1):29–43. doi: 10.1093/ajcp/50.1.29. [DOI] [PubMed] [Google Scholar]
- 6.Miller CG, Alleyne GA, Brooks SEH. Gross cardiac involvement in glycogen storage disease type III. British Heart Journal. 1972;34(8):862–864. doi: 10.1136/hrt.34.8.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson LJ, Reeder GS, Noller KL, Edwards WD, Howell RR, Michels VV. Cardiac Involvement in Glycogen Storage Disease III: Morphologic and Biochemical Characterization with Endomyocardial Biopsy. The American Journal of Cardiology. 1984 Mar 15;53:980–981. doi: 10.1016/0002-9149(84)90551-4. [DOI] [PubMed] [Google Scholar]
- 8.Tada H, Kurita T, Ohe T, Shimomura K, Ishihara T, Yamad Y, Osawa N. Glycogen storage disease type III associated with ventricular tachycardia. American Heart Journal. 1995 Oct;130(4):911–912. doi: 10.1016/0002-8703(95)90097-7. [DOI] [PubMed] [Google Scholar]
- 9.Lee PJ, Deanfield JE, Burch M, Baig K, McKenna WJ, Leonard JV. Comparison of the Functional Significance of Left Ventricular Hypertrophy in Hypertrophic Cardiomyopathy and Glycogenosis Type III. The American Journal of Cardiology. 1997 Mar 15;79:834–838. doi: 10.1016/s0002-9149(96)00885-5. [DOI] [PubMed] [Google Scholar]
- 10.Cochrane AB, Fedson SE, Cronin DC., II Nesiritide as Bridge to Multi-Organ Transplantation: A Case Report. Transplantation Proceedings. 2007;39:308–310. doi: 10.1016/j.transproceed.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, St John Sutton M, Stewart WJ. Recommendations for Chamber Quantification: A Report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, Developed in Conjunction with the European Association of Echocardiography, a Branch of the European Society of Cardiology. Journal of the American Society of Echocardiography. 2005 Dec;18(12):1440–1463. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 12.Levine JC, Kishnani PS, Chen YT, Herlong JR, Li JS. Cardiac Remodeling After Enzyme Replacement Therapy with Acid a-Glucosidase for Infants wtih Pompe Disease. Pediatric Cardiology. 2007;29(6):1033–1042. doi: 10.1007/s00246-008-9267-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuspidi C, Sampieri L, Pelizzoli S, Pontiggia G, Zanchetti A, Nappo A, Caputo V, Matturri L. Obstructive Hypertrophic Cardiomyopathy in Type III Glycogen-Storage Disease. Acta Cardiologica. 1997;52(2):117–123. [PubMed] [Google Scholar]
- 14.Shen J, Bao Y, Liu HM, Lee P, Leonard JV, Chen YT. Mutations in Exon 3 of the Glycogen Debranching Enzyme Gene Are Associated with Glycogen Storage Disease Type III That Is Differentially Expressed in Liver and Muscle. Journal of Clinical Investigation. 1996;98:352–357. doi: 10.1172/JCI118799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pettersen MD, Du W, Skeens ME, Humes RA. Regression Equations for Calculation of Z Scores of Cardiac Structures in a Large Cohort of Healthy Infants, Children, and Adolescents: an Echocardiographic Study. Journal of the American Society of Echocardiography. 2008 Aug;21(8):922–934. doi: 10.1016/j.echo.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Demo E, Frush D, Gottfried M, Koepke J, Boney A, Bali D, Chen YT, Kishnani PS. Glycogen storage disease type III-hepatocellular carcinoma a long-term complication? Journal of Hepatology. 2007 Mar;46(3):492–498. doi: 10.1016/j.jhep.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carvalho JS, Matthew EE, Leonard JV, Deanfield J. Cardiomyopathy of glycogen storage disease type III. Heart and Vessels. 1993;8:155–159. doi: 10.1007/BF01744800. [DOI] [PubMed] [Google Scholar]
- 18.Labrune P, Huguet P, Odievre M. Cardiomyopathy in Glycogen-Storage Disease Type III: Clinical and Echographic Study of 18 Patients. Pediatric Cardiology. 1991;12(3):161–163. doi: 10.1007/BF02238523. [DOI] [PubMed] [Google Scholar]
- 19.Lee P, Burch M, Leonard JV. Plasma creatine kinase and cardiomyopathy in glycogen storage disease type III. Journal of Inherited Metabolic Diseases. 1995;18:751–752. doi: 10.1007/BF02436768. [DOI] [PubMed] [Google Scholar]
- 20.Coleman RA, Winter HS, Wolf B, Gilchrist JM, Chen YT. Glycogen Storage Disease Type III (Glycogen Debranching Enzyme Deficiency): Correlation of Biochemical Defects with Myopathy and Cardiomyopathy. Annals of Internal Medicine. 1992;116:896–900. doi: 10.7326/0003-4819-116-11-896. [DOI] [PubMed] [Google Scholar]
- 21.Moses SW, Wanderman KL, Myroz A, Frydman M. Cardiac involvement in glycogen storage disease type III. European Journal of Pediatrics. 1989;148:764–766. doi: 10.1007/BF00443106. [DOI] [PubMed] [Google Scholar]
- 22.Moon JCC, Mundy HR, Lee PJ, Mohiaddin RH, Pennell DJ. Myocardial Fibrosis in Glycogen Storage Disease Type III. Circulation. 2003;107:e47. doi: 10.1161/01.cir.0000050691.73932.cb. [DOI] [PubMed] [Google Scholar]
- 23.Arad M, Maron BJ, Gorham JM, Johnson WH, Jr, Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, Seidman JG. Glycogen Storage Diseases Presenting as Hypertrophic Cardiomyopathy. New England Journal of Medicine. 2005 Jan;352(4):362–372. doi: 10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 24.Kishnani PS, Wechsler SB, Li JS. Enzyme-deficiency metabolic cardiomyopathies and the role of enzyme replacement therapy. Progress in Pediatric Cardiology. 2007;23:39–48. [Google Scholar]
- 25.Ogimoto A, Okubo M, Okayama H, Shin YS, Endo Y, Ebara T, Inoue K, Ohtsuka T, Tahara H, Murase T, Higaki J. A Japanese Patient With Cardiomyopathy Caused by a Novel Mutation R285X in the AGL Gene. Circulation Journal. 2007 Oct;71:1653–1656. doi: 10.1253/circj.71.1653. [DOI] [PubMed] [Google Scholar]
- 26.Akazawa H, Kuroda T, Kim S, Mito H, Kojo T, Shimada K. Specific heart muscle disease associated wtih glycogen storage disease type III: clinical similarity to the dilated phase of hypertrophic cardiomyopahty. European Heart Journal. 1997 Mar;18(18):532–533. doi: 10.1093/oxfordjournals.eurheartj.a015283. [DOI] [PubMed] [Google Scholar]
- 27.Shen J, Bao Y, Chen YT. A Nonsense Mutation Due to a Single Base Insertion in the 3’-Coding Region of the Glycogen Debranching Enzyme Gene Associated With a Severe Phenotype in a Patient With Glycogen Storage Disease Type IIIa. Human Mutation. 1997;9:37–40. doi: 10.1002/(SICI)1098-1004(1997)9:1<37::AID-HUMU6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 28.Lee PJ, Leonard JV. The hepatic glycogen storage diseases-problems beyond childhood. Journal of Inherited Metabolic Diseases. 1995;18:462–472. doi: 10.1007/BF00710057. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi A, Nishinomiya F, Fukamachi Y, Ohtaka M, Yamamoto J, Takagi K, Tanaka S, Takizawa, Imadachi H, Fukase M, Shimizu Y, Hayasaka K. A Case of Glycogen Storage Disease Type III (Glycogen Debranching Enzyme Deficiency) with Liver Cirrhosis and Hypertrophic Cardiomyopathy. Tohoku Journal of Experimental Medicine. 1995;176:181–185. doi: 10.1620/tjem.176.181. [DOI] [PubMed] [Google Scholar]
- 30.Talente GM, Coleman RA, Alter C, Baker L, Brown BI, Cannon RA, Chen YT, Crigler JF, Ferreira P, Haworth JC, Herman GE, Issenman RM, Keating JP, Linde R, Roe TF, Senior B, Wolfsdorf JI. Glycogen Storage Disease in Adults. Annals of Internal Medicine. 1994 Feb;120(3):218–226. doi: 10.7326/0003-4819-120-3-199402010-00008. [DOI] [PubMed] [Google Scholar]
- 31.Smit GP, Fernandes J, Leonard JV, Matthews EE, Moses SW, Odievre M, Ullrich K. The long-term outcome of patients with glycogen storage diseases. The Journal of Inherited Metabolic Diseases. 1990;13(4):411–418. doi: 10.1007/BF01799498. [DOI] [PubMed] [Google Scholar]