

Abstract
Context:
Activation of the unfolded protein response (UPR) is emerging as an important molecular signature of cardiometabolic diseases associated with obesity. However, despite the well-established role of the vascular endothelium in obesity-related cardiometabolic dysfunction, it is unclear whether the UPR is activated in endothelial cells of obese adults.
Objective:
The objective of the study was to determine whether markers of UPR activation are increased in endothelial cells (ECs) of nondiabetic obese adults with impaired endothelial function.
Design, Setting, and Participants:
Endothelial cells were obtained from antecubital veins of the nondiabetic obese adults [body mass index (BMI) ≥ 30 kg/m2, n = 12] with impaired endothelial function and from their nonobese peers (BMI < 30 kg/m2, n = 14).
Main Outcome Variables:
UPR activation via expression (quantitative immunofluorescence) of the proximal UPR sensors, inositol-requiring endoplasmic reticulum (ER)-to-nucleus signaling protein 1 (IRE1), RNA-dependent protein kinase-like ER eukaryotic initiation factor-2α kinase (PERK), and activating transcription factor 6 (ATF6), were the main outcome variables.
Results:
IRE1 expression was greater in obese vs nonobese individuals (0.84 ± 0.09 vs 0.47 ± 0.02 IRE1 intensity/human umbilical vein EC (HUVEC) intensity (n = 10/8, P < .01). Obese individuals also had greater EC activation of UPR stress sensors PERK and ATF6, indicated by increased expression of phosphorylated PERK [p-PERK; 0.49 ± 0.05 vs 0.36 ± 0.03, p-PERK (threonine 981) intensity/HUVEC intensity, n = 10 men, 13 women, P < .05] and nuclear localization of ATF6 (0.38 ± 0.05 vs 0.23 ± 0.02, nuclear ATF6 intensity/HUVEC intensity, n = 5 men, 9 women, P < .01), respectively. Stepwise linear regression analysis revealed that indices of body fat (BMI and waist circumference) were the strongest independent predictors of all 3 UPR mediators, explaining between 18% and 59% of the variance in endothelial cell expression of IRE1, p-PERK, and nuclear ATF6 localization.
Conclusion:
These results provide novel evidence for UPR activation in the endothelial cells of nondiabetic obese adults with vascular endothelial dysfunction.
The endoplasmic reticulum (ER) plays an integral role in various cellular processes, including the folding of membrane and secretory proteins (1). Various cellular insults can disrupt ER homeostasis and lead to the accumulation of unfolded proteins within the ER lumen. This homeostatic dysregulation, termed ER stress, evokes the unfolded protein response (UPR), an adaptive pathway that aims to restore equilibrium within the ER (2). Upon ER stress, the UPR is initiated by activation of 3 ER transmembrane proteins: RNA-dependent protein kinase-like ER eukaryotic initiation factor-2α kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring ER-to-nucleus signaling protein 1 (IRE1). These proteins elicit 3 distinct signaling cascades that cooperatively act to reduce protein translation and increase the folding capacity (3).
Although the UPR is the first line of defense against ER stress, chronic activation of the UPR has been implicated in the pathophysiology of cardiometabolic disorders associated with obesity (4, 5). The ability of ER stress to facilitate cardiovascular disease processes likely occurs via its activation in various tissues, including the vascular endothelium (16). Indeed, ER stress-inducing agents cause endothelial cell dysfunction and death, and activation of the UPR was demonstrated in atherosusceptible regions of the vascular endothelium of swine (6, 7). More recent preclinical studies suggest that ER stress impairs endothelium-dependent dilation, an independent predictor of future cardiovascular events in humans (8, 9).
Despite substantial in vitro and preclinical evidence implicating chronic UPR activation in the pathogenesis of obesity-related cardiometabolic diseases, there remains a paucity of supporting data in humans, and to our knowledge, no studies have examined UPR activation in vascular endothelial cells biopsied from humans. This lack of data is due largely to the limited access to human endothelial cells. Using a novel translational research technique, we sought to establish whether endothelial UPR activation occurs in the setting of human obesity. To do so, we assessed IRE1 expression and PERK and ATF6 activation in endothelial cells obtained from nondiabetic obese and nonobese adults.
Materials and Methods
Subjects
A total of 26 adults (22 men and 4 women) were studied. Subjects were free of clinical diseases as assessed by medical history, physical examination, and blood chemistries. Subjects were nonsmokers between the ages of 31–74 years who were not taking medications and who refrained, for 2 weeks prior to participation, from all dietary supplements that could influence study results. Subjects were characterized as obese [body mass index (BMI) ≥ 30 kg/m2, n = 12] or nonobese (BMI < 30 kg/m2, n = 14). All procedures were approved by the University of Colorado Institutional Review Board. The nature, benefits, and risks were explained to the volunteers, and their written informed consent was obtained prior to participation.
Study procedures and blood analyses
All measurements were performed at the University of Colorado at Boulder Clinical Translational Research Center after an overnight fast and a 24-hour abstention from alcohol and exercise. Fasting plasma metabolic factors were determined by the Clinical Translational Research Center core laboratory using standard assays (10). White blood cell count was measured by the Coulter counter technique (Ac·T 5diff CP; Beckman Coulter). An ELISA was used to measure the serum concentrations of the oxidized low-density lipoprotein (ALPCO), TNF-α, and IL-6 (R&D Systems). C-reactive protein (CRP) was measured using a high-sensitivity chemistry immunoanalyzer (AU400e; Olympus).
Endothelium-dependent dilation
Brachial artery flow-mediated dilation (FMD) was determined using duplex ultrasonography (Power Vision 6000; Toshiba) with a linear array transducer as previously described (10). Responses were expressed as millimeters and percentage change from the baseline diameter (11). Measurements were performed within 2 weeks of establishing baseline subject characteristics.
Endothelial cell protein expression
Endothelial cell collection and protein expression measurements were performed as previously described (12, 13). Briefly, J-wires were advanced into an antecubital vein and cells were recovered by washing and centrifugation. Cells were then rehydrated and incubated with primary antibodies against IRE1 (1:100; Cell Signaling), phosphorylated PERK (p-PERK; threonine 981, 1:50; Santa Cruz Biotechnology), or ATF6 (1:100; Abcam) and with Alexaflour 555 fluorescent secondary antibody (1:300; Invitrogen Corp).
Slides were viewed using a fluorescence microscope (Eclipse Ni-U; Nikon) and whole-cell fluorescence was assessed using Metamorph Software (Universal Imaging). Nuclear ATF6 expression was estimated by quantifying Alexaflour 555 intensity within the 4′,6′-diamino-2-phenylindole region of the cell. The technician was blinded to the identity of the subject during the staining and analysis procedures.
Statistics
Statistical analyses were performed with SPSS, and all data are presented as mean ± SE. Group differences were determined by t tests for independent sample comparisons. Bivariate Pearson correlation analyses were used to assess relations of interest and stepwise linear regression analyses were performed to identify independent predictors of endothelial cell expression of UPR mediators. Statistical significance for all analyses was set at P < .05.
Results
Subject characteristics
Subject characteristics are shown in Table 1. The obese subjects demonstrated endothelial dysfunction as indicated by lower brachial artery FMD compared with normal-weight subjects (P < .05). Obese individuals also had higher fasting plasma triglyceride and lower plasma high-density lipoprotein (HDL) cholesterol levels (both P < .05). Consistent with chronic, low-grade inflammation, obese subjects demonstrated higher white blood cell count, TNF-α, and log-normalized CRP concentrations (all P < .05).
Table 1.
Subject Characteristics
| Characteristic | Nonobese | Obese |
|---|---|---|
| Age, y | 59 ± 2 | 52 ± 3 |
| Gender, men/women | 11/3 | 11/1 |
| BMI, kg/m2 | 24 ± 1 | 33 ± 1a |
| Waist circumference, cm | 84 ± 3 | 109 ± 2a |
| Fasting glucose, mg/dL | 89 ± 2 | 96 ± 3 |
| Triglycerides, mg/dL | 131 ± 23 | 258 ± 41b |
| Total cholesterol, mg/dL | 203 ± 10 | 207 ± 8 |
| LDL, mg/dL | 122 ± 10 | 117 ± 10 |
| HDL, mg/dL | 55 ± 4 | 38 ± 2a |
| White blood cell count, 109 cells/L | 5.1 ± 0.3 | 6.3 ± 0.3a |
| Log CRP, mg/L | −0.14 ± 0.10 | 0.21 ± 0.11b |
| TNF-α, pg/mL | 1.1 ± 0.13 | 1.6 ± 0.10a |
| FMD, % | 6.16 ± 0.68 | 3.86 ± 0.45b |
| FMD, mm | 0.23 ± 0.02 | 0.15 ± 0.02a |
Abbreviation: LDL, low-density lipoprotein. Data are mean ± SE.
P < .01 vs nonobese.
P < .05 vs nonobese.
Evidence of UPR activation in endothelial cells of obese individuals
Expression of the proximal UPR stress sensors are shown in Figure 1. Expression of IRE1 was greater in obese vs nonobese subjects (0.84 ± 0.09 vs 0.47 ± 0.02, IRE1 intensity/human umbilical vein endothelial cell (HUVEC) intensity, P < .01). Obese subjects also had greater endothelial cell activation of PERK, as indicated by the increased expression of the phosphorylated protein (0.49 ± 0.05 vs 0.36 ± 0.03, p-PERK intensity/HUVEC intensity, P < .05). ATF6 activation, as indicated by its nuclear localization, was also increased in obese individuals compared with lean controls (0.38 ± 0.05 vs 0.23 ± 0.02, nuclear ATF6 intensity/HUVEC intensity, P < .01).
Figure 1.
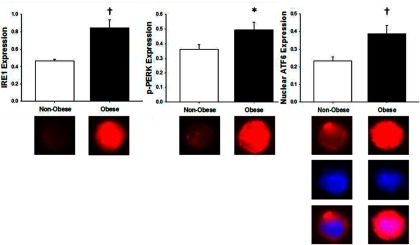
Endothelial cell IRE1 (left panel), p-PERK (center panel), and nuclear ATF6 (right panel) protein expression in nonobese and obese subjects. For all proteins, values are endothelial cell protein expression relative to human umbilical vein endothelial cell control. Below, Representative images of IRE1 (left panel), p-PERK (center panel), and nuclear ATF6 (right) panel expression (red); nuclear (4′,6′-diamino-2-phenylindole) fluorescence (blue) and merged images in nonobese and obese subjects. Data are mean ± SE. *, P < .05 vs nonobese; †, P < .01 vs nonobese.
Relation between UPR activation and variables of interest
Endothelial expression of the UPR stress sensors were positively related to measures of body fat and markers of inflammation. Specifically, nuclear ATF6 expression was related to BMI (r = 0.76), waist circumference (r = 0.66), age (r = −0.66), and circulating TNF-α (r = 0.63; all P < .05). BMI (r = 0.54, P < .01) and waist circumference (r = 0.48, P < .05) remained independent predictors of nuclear ATF6 expression when controlling for age (multiple regression). IRE1 expression was positively related to waist circumference (r = 0.69), log-corrected CRP (r = 0.50), and HDL (r = −0.54, all P < .05), whereas p-PERK was related to BMI (r = 0.42, P < .05). There was a positive relation between endothelial cell expression of p-PERK and nuclear ATF6 (r = 0.76, P < .01); IRE1 was not related to ATF6 or p-PERK expression.
Stepwise linear regression was performed to determine independent relations between subject characteristics and endothelial cell expression of IRE1, p-PERK, and nuclear ATF6. Fasting glucose and log CRP were excluded from all models due to multicollinearity with other subject characteristics. Criteria for entry into and removal from the model was set at P < .05 and P > .1, respectively. Because no subject characteristics met these criteria for the p-PERK model, inclusion and exclusion criteria were adjusted to P < .1 and P > .2, respectively. Stepwise linear regression analysis revealed that indices of body fat (BMI and waist circumference) were the strongest and most consistent independent predictors of UPR activation, explaining between 18% and 59% of the variance in endothelial expression of IRE1, p-PERK, and nuclear ATF6 localization.
Discussion
We used a novel translational approach to determine whether endothelial cells are a site of UPR activation in the obese state. The results provide the first direct evidence that, relative to their nonobese peers, nondiabetic obese individuals exhibit increased endothelial activation of the UPR. These results are consistent with, and extend to the vascular endothelium, previously published data indicating obesity-associated UPR activation in tissues involved in the regulation of metabolism (14, 15).
In the present study, we found evidence for the up-regulation of all 3 UPR-initiating pathways in the vascular endothelial cells of obese adults. This is in contrast to previous studies reporting less uniform activation of the 3 UPR branches in metabolically active tissues (14, 15). For example, Puri et al (15) demonstrated an up-regulation of the PERK pathway (ie, phosphorylated eukaryotic initiation factor-2α) in the liver of patients with nonalcoholic fatty liver disease, whereas the IRE1 branch (ie, spliced X-box binding protein-1 mRNA) was not altered. Thus, the present study extends these findings by demonstrating increased protein expression or activation of all 3 proximal sensors of the UPR within vascular endothelial cells.
Kassan et al (9) recently demonstrated that endothelium-dependent dilation was impaired by a chemical inducer of ER stress in experimental animals, suggesting that ER stress directly participates in endothelial damage and increased cardiovascular risk. Although we cannot confirm such a direct relation in the present study, UPR activation in the obese subjects was accompanied by endothelial dysfunction (reduced FMD).
As is commonly observed, obese individuals in the current study demonstrated several metabolic abnormalities (eg, reduced HDL-cholesterol and elevated triglycerides, CRP, and TNF-α), and these metabolic characteristics were selectively related, albeit inconsistently, to the expression of UPR sensors. Although the current study was not designed to definitively determine whether the increase in endothelial UPR activation is driven by increased adiposity per se or to these metabolic abnormalities associated with obesity, we used stepwise linear regression analyses to gain some insight into this matter. We found that indices of body fat were the strongest and most consistent independent predictors of UPR activation, highlighting the importance of excess adiposity in UPR activation.
Age was negatively associated with nuclear ATF6 expression. These data are consistent with some (17, 18), but not all, data demonstrating that UPR activation is impaired with aging (19). Although this relation represents a potential confound, regression analyses suggest that indices of body fat are stronger independent predictors of UPR activation. Still, the inverse relation between age and markers of UPR activation is an interesting finding that warrants further investigation.
It is important to note that we measured only the proximal UPR sensors and thus must be cautious in extrapolating the data to downstream UPR effectors. The method by which endothelial cells were extracted yields a small number of cells, and thus, we are limited on the number of proteins that can be assessed. The 3 proximal UPR sensors were selected because they most broadly reflect each arm of UPR activation.
Finally, protein expression measurements were performed on cells obtained from veins as opposed to arteries due to the invasiveness of arterial cell sampling. Although the latter would be optimal, we (20) and others (12) have shown that cardiovascular disease-related differences in arterial endothelial cell protein expression are consistently reflected in endothelial cells obtained from veins.
In summary, the results of the present study provide the first evidence that endothelial cells are a site of UPR activation in individuals at elevated risk for cardiovascular disease. These novel translational data lend clinical relevance to the large body of literature in cell culture models and experimental animals implicating ER stress in the pathology of obesity-related metabolic dysfunction.
Acknowledgments
This work was supported by National Institutes of Health Grants TR000154 (to D.R.S.), AG000279 (to D.R.S.), AG13038 (to D.R.S.), DK087777 (to C.G.), and P30DK048520 (to C.G.; principal investigator: J.O. Hill).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ATF6
- activating transcription factor 6
- BMI
- body mass index
- CRP
- C-reactive protein
- ER
- endoplasmic reticulum
- FMD
- flow-mediated dilation
- HDL
- high-density lipoprotein
- HUVEC
- human umbilical vein endothelial cell
- IRE1
- inositol-requiring ER-to-nucleus signaling protein 1
- PERK
- RNA-dependent protein kinase-like ER eukaryotic initiation factor-2α kinase
- p-PERK
- phosphorylated PERK
- UPR
- unfolded protein response.
References
- 1. Pagliassotti MJ. Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annu Rev Nutr. 2012;32:17–33 [DOI] [PubMed] [Google Scholar]
- 2. Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–S109 [DOI] [PubMed] [Google Scholar]
- 4. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Civelek M, Manduchi E, Riley RJ, Stoeckert CJ, Jr, Davies PF. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ Res. 2009;105:453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sanson M, Auge N, Vindis C, et al. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 expression. Circ Res. 2009;104:328–336 [DOI] [PubMed] [Google Scholar]
- 8. Galan M, Kassan M, Choi SK, et al. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. 2012;60:71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kassan M, Galan M, Partyka M, et al. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol. 2012;32:1652–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pierce GL, Lesniewski LA, Lawson BR, Beske SD, Seals DR. Nuclear factor-κB activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation. 2009;119:1284–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donald AE, Halcox JP, Charakida M, et al. Methodological approaches to optimize reproducibility and power in clinical studies of flow-mediated dilation. J Am Coll Cardiol. 2008;51:1959–1964 [DOI] [PubMed] [Google Scholar]
- 12. Colombo PC, Ashton AW, Celaj S, et al. Biopsy coupled to quantitative immunofluorescence: a new method to study the human vascular endothelium. J Appl Physiol. 2002;92:1331–1338 [DOI] [PubMed] [Google Scholar]
- 13. Eskurza I, Kahn ZD, Seals DR. Xanthine oxidase does not contribute to impaired peripheral conduit artery endothelium-dependent dilatation with ageing. J Physiol. 2006;571:661–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharma NK, Das SK, Mondal AK, et al. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J Clin Endocrinol Metab. 2008;93:4532–4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Puri P, Mirshahi F, Cheung O, Natarajan R, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–576 [DOI] [PubMed] [Google Scholar]
- 16. Zhang C, Cai Y, Adachi MT, et al. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J Biol Chem. 2001;276:35867–35874 [DOI] [PubMed] [Google Scholar]
- 17. Erickson RR, Dunning LM, Holtzman JL. The effect of aging on the chaperone concentrations in the hepatic, endoplasmic reticulum of male rats: the possible role of protein misfolding due to the loss of chaperones in the decline in physiological function seen with age. J Gerontol A Biol Sci Med Sci. 2006;61:435–443 [DOI] [PubMed] [Google Scholar]
- 18. Naidoo N, Ferber M, Master M, Zhu Y, Pack AI. Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J Neurosci. 2008;28:6539–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Das SK, Chu WS, Mondal AK, et al. Effect of pioglitazone treatment on endoplasmic reticulum stress response in human adipose and in palmitate-induced stress in human liver and adipose cell lines. Am J Physiol Endocrinol Metab. 2008;295:E393–E400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Donato AJ, Eskurza I, Silver AE, et al. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-κB. Circ Res. 2007;100:1659–1666 [DOI] [PubMed] [Google Scholar]