

Abstract
DNA methylation is an epigenetic modification involved in regulatory processes such as cell differentiation during development, X-chromosome inactivation, genomic imprinting and susceptibility to complex disease. However, the dynamics of DNA methylation changes between humans and their closest relatives are still poorly understood. We performed a comparative analysis of CpG methylation patterns between 9 humans and 23 primate samples including all species of great apes (chimpanzee, bonobo, gorilla and orangutan) using Illumina Methylation450 bead arrays. Our analysis identified ∼800 genes with significantly altered methylation patterns among the great apes, including ∼170 genes with a methylation pattern unique to human. Some of these are known to be involved in developmental and neurological features, suggesting that epigenetic changes have been frequent during recent human and primate evolution. We identified a significant positive relationship between the rate of coding variation and alterations of methylation at the promoter level, indicative of co-occurrence between evolution of protein sequence and gene regulation. In contrast, and supporting the idea that many phenotypic differences between humans and great apes are not due to amino acid differences, our analysis also identified 184 genes that are perfectly conserved at protein level between human and chimpanzee, yet show significant epigenetic differences between these two species. We conclude that epigenetic alterations are an important force during primate evolution and have been under-explored in evolutionary comparative genomics.
Author Summary
Differences in protein coding sequences between humans and their closest relatives are too small to account for their phenotypic differences. It has been hypothesized that these differences may be explained by alterations of gene regulation rather than primary genome sequence. DNA methylation is an important epigenetic modification that is involved in many biological processes, but from an evolutionary point of view this modification is still poorly understood. To this end, we performed a comparative analysis of CpG methylation patterns between humans and great apes. Using this approach, we were able to study the dynamics of DNA methylation in recent primate evolution and to identify regions showing species-specific methylation pattern among humans and great apes. We find that genes with alterations of promoter methylation tend to show increased rates of divergence in their protein sequence, and in contrast we also identify many genes with regulatory changes between human and chimpanzee that have perfectly conserved protein sequence. Our study provides the first global view of evolutionary epigenetic changes that have occurred in the genomes of all species of great apes.
Introduction
The genomic era is characterized by different comparative approaches to understand the effect of genomic changes upon phenotypes. In the context of human evolution, the genomes of all species of great apes have now been sequenced [1]–[4] allowing nucleotide resolution comparisons to understand the evolution of our genome. However, in contrast to these advances in comparative genomic analyses, there has been relatively little progress in the understanding of the evolution of genome regulation [5]–[9].
DNA methylation is an important epigenetic modification found in many taxa. In mammals, it is involved in numerous biological processes such as cell differentiation, X-chromosome inactivation, genomic imprinting and susceptibility to complex diseases [10]–[13]. Promoter hypermethylation is generally thought to act as a durable silencing mechanism [14]. However, the exact relationship between DNA methylation and gene expression is not clear since recent studies have also linked gene body methylation with transcriptional activity and alternative splicing [15]–[17]. At some loci DNA methylation patterns are influenced by the underlying genotype [18]–[20]. However, due to the fact that patterns of DNA methylation can change during development [16], [21], [22] or as a result of environmental factors [23], [24], the exact mechanisms governing DNA methylation states remain unclear.
Most efforts to understand DNA methylation changes in primates have focused on the comparison of human with chimpanzee or macaque [6], [7], [9], [25]. This is largely attributable to the difficulty of obtaining samples from endangered species and the lack of genome sequence for the great apes. The publication last year of draft sequences of the gorilla [2] and bonobo [3] genomes facilitates a more accurate characterization of the species-specific events in all the great ape phylogeny, and interrogation of this epigenetic modification from an evolutionary point of view. Studies to date have found that DNA methylation profiles are, in general, more similar between homologous tissues than between different tissues of the same species [9]. However, differentially expressed genes between human and chimpanzee are often associated with promoter methylation differences, regardless of tissue type, establishing that some differences in the expression rates of genes between the species are associated with differences in DNA methylation. It is estimated that around 12–18% (depending on the tissue) of interspecies differences in gene expression levels could be explained by changes in promoter methylation [9].
Here we present the first comparative analysis of DNA methylation patterns between humans and all great ape species, allowing us to recapitulate the evolution of CpG methylation over the last 15 million years in these species. We used Illumina Methylation450 BeadChips to profile DNA methylation genome-wide in blood-derived DNA from a total of 9 humans and 23 wild-born individuals of different species and sub-species of chimpanzee, bonobo, gorilla and orangutan. We observed that the methylation values recapitulate the known phylogenetic relationships of the species, and we were able to characterize methylation differences that have occurred exclusively in the human lineage and among different great apes species. We also identified a significant positive relationship between the rate of coding variation and alterations of methylation at the promoter level, indicative of co-occurrence between evolution of protein sequence and gene regulation
Results
Data filtering
We obtained cytosine methylation profiles of peripheral blood DNA isolated from a set of males and females of nine humans, five chimpanzees, six bonobos, six gorillas and six orangutans (Table S1) using the Illumina HumanMethylation450 DNA Analysis BeadChip assay. Because the probes on the array are designed using the human reference genome, we performed a set of strict filters to remove divergent probes that could bias our methylation measurements. The filtering was based on the number and location of mismatches with their target site in each species genome assembly tested [1]–[4] (Figure S1 and Figure S2, see Methods). This resulted in the retention of 326,535 probes (72%) in chimpanzee, 328,501 probes (73%) in bonobo, 274,084 probes (61%) in gorilla and 197,489 probes (44%) in orangutan, consistent with their evolutionary distance to human. We also applied a second filtering step to remove probes that overlapped with intra-species common variation (see Methods) [26].
Cell heterogeneity may also act as a confounder when measuring DNA methylation, particularly from whole blood [27]. Due to the difficulty of obtaining fresh blood samples from wild-born great apes, we were unable to either isolate a specific blood cell type or measure the cellular composition of the blood samples from which our DNA was extracted. To minimize false positives resulting from different cellular compositions or other confounders, we performed two filtering steps. First, we removed CpG sites that showed differential methylation in human between whole blood and each of the two most abundant subtypes of blood cell (CD4+ T-cells and CD16+ neutrophils, see Methods). Second, we required a minimum threshold of at least 10% change in mean methylation (mean β-value difference ≥0.1) at each CpG in order to define differential methylation between species. As a result of this threshold, differences in other cell types that account for <10% of the cellular composition of blood, are unlikely to affect our results (see Methods).
In this work we used two different datasets: i) we confined our analysis to 114,739 autosomal probes and 3,680 probes on the X chromosome that were directly comparable across all the species to facilitate an unbiased comparison of human and all great apes (32 individuals), and ii) we used 291,553 shared autosomal probes between humans and chimpanzees to compare these two species. We performed separate analyses of autosomal and sex-linked probes to prevent confounding effects of X chromosome inactivation on DNA methylation between males and females [13]. Unless specifically mentioned, all results presented below refer to analysis of autosomal probes only.
Phylogenetic relationships
To investigate the global correspondence of DNA sequence differences between species and the degree of methylation changes, we examined the Enredo-Pecan-Orthus (EPO) whole-genome multiple alignments of human, chimpanzee, gorilla, and orangutan [Ensemble Compara.6_primates_EPO] [28], [29] and we calculated pairwise distances between these four species. Upon comparison of these sequence distances and methylation data (see Methods), we observed a high global correlation between sequence substitution and methylation divergence (R2 = 0.98, p = 0.0003) (Figure 1A). We then constructed a neighbor-joining phylogenetic tree based on the methylation levels of the 114,739 autosomal CpGs measured in all individuals and species (Figure S3). This tree accurately recapitulates the known evolutionary relationships of great apes, including the separation at sub-species level of the Pan, Gorilla and Pongo genera. These results are also maintained when using only the subset of probes that have a perfect match (n = 31,853) to each of the primate reference genome and contain no common polymorphisms suggesting that that methylation levels are associated with the evolutionary history of these species (Figure 1B).
Figure 1. Methylation patterns mimic sequence based phylogenetic relationships.

(A) Methylation changes correlate with DNA sequence changes. x-axis shows the number of nucleotide substitutions between two species per kb, y-axis shows the changes in methylation based on β-values. (B) Neighbor-joining tree based on methylation data from probes with a perfect match in all reference genomes (31,853 autosomal CpGs). Bootstrap values (1,000 permutations) are shown for each node.
Lineage-specific methylation changes
Due to the relatively recent origin of all partitions within genera of great apes [2]–[4] and our sample size, we focused our analysis on changes at the genus taxonomic level. To identify only those methylation differences that represent fixed changes between these groups and to avoid possible artifacts due to intraspecific polymorphism, we retained only those CpGs with low methylation variance within each genus (intragenus standard deviation <0.1). This filtering step resulted in the removal of 1,377 CpGs in human, 5,224 in the Pan sp., 5,289 in Gorilla sp. and 5,740 in Pongo sp., with the resulting final set being 99,919 CpGs shared across all five species, covering 12,593 genes (≥2 probes within a 1 kb interval and overlapped with RefSeq genes, −1500 bp transcription start site (TSS) to 3′UTR). The proportion of sites removed in this step are consistent with the relative population diversity within each of these species [2]–[4], [30].
Approximately 22% of the sites tested (n = 21,884 CpGs) showed no significant changes among any of the species (conserved sites: Wilcoxon rank-sum test, FDR-adjusted p>0.05 and mean β-value difference all cases <0.1). Comparison of genes linked with these sites showed an enrichment of Gene Ontology (GO) categories for fundamental cellular processes. In contrast, we identified 2,284 human-specific (2.3%) differentially methylated CpGs, 1,245 specific to Pan species (1.2%), 1,374 specific to Gorilla species (1.4%). and 5,501 changes specific to Pongo species (5.5%) (Wilcoxon rank-sum test, FDR-adjusted p<0.05 and mean β-value difference ≥0.1, see Methods) (Figure 2 and Table S2). We clustered these sites into regions with at least two nearby differentially methylated CpGs (<1 kb interval) and overlapped with RefSeq genes (−1500 bp from TSS to 3′UTR). Doing this, we identified 171 genes that show human specific methylation patterns, 101 genes in Pan species, 101 genes in Gorilla species and 445 genes in Pongo species (Table S3). We observed that this spatial aggregation of differentially methylated sites is significantly non-random (random permutation compared to all 99,919 CpGs used in our analysis, p<0.0001, see Methods) and a simple Likelihood Ratio Test also suggested a non-homogenous rate of methylation changes in the human and great ape evolution (LRT, p<10−5, see Text S1).
Figure 2. Differentially methylated CpG sites in each great ape genus.
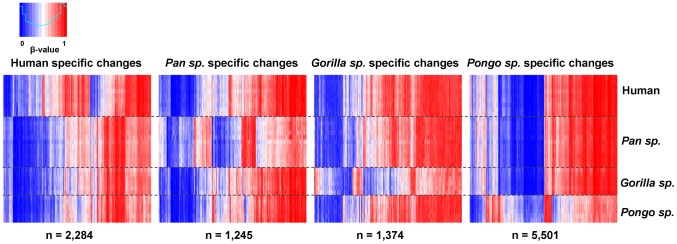
Heat maps showing genus specific differentially methylated CpG sites. We found 2,284 human-specific differentially methylated CpGs, 1,245 specific to Pan species, 1,374 specific to Gorilla species and 5,501 changes specific to Pongo species. Each vertical line represents a single CpG, with each row showing the β-value obtained in each individual tested.
Using the Genomic Regions Enrichment of Annotation Tool (GREAT) [31] (see Methods) we identified significant enrichments (FDR-corrected p<0.05) for several biological processes associated with lineage-specific differentially methylated genes. Within the human-specific differentially methylated regions most of the categories found were related with the circulatory system, as expected from testing blood-derived DNA. However, we also found enrichment for terms related to development and neurological functions, including semicircular canal formation and facial nucleus development (Table S4). The use of disease ontology terms showed that mutations in several of these genes are known to be associated with diseases including Möbius syndrome, Asperger's syndrome and malignant hyperthermia. In the Pan genus (chimpanzee and bonobo) we observed significant enrichments among genes involved in epithelial development and the respiratory system, while in Pongo species (orangutan) enriched categories included a variety of basic metabolic and reproductive processes (Table S4).
We found a particular set of genes with methylation changes specifically in the human lineage including examples such as ARTN, COL2A1 and PGAM2 (Figure 3). ARTN is a neurotrophic factor which supports the survival of sympathetic peripheral neurons and dopaminergic neurons. COL2A1 encodes the alpha-1 chain of type II collagen, which is found primarily in the cartilage, the inner ear and the vitreous humor of the eye. Mutations in this gene are associated with several developmental syndromes [32]. PGAM2 is an enzyme involved in the glycolytic pathway, mutations in which are associated with glycogen storage disease [MIM: 261670], a defect that causes muscle cramping, myoglobinuria and intolerance for strenuous exercise. In addition to the identification of regions showing changes in a single species, we also detected loci with more complex changes in methylation profiles among great apes. One example is the promoter region associated with different isoforms of the GABBR1 gene (Figure 3D). This gene encodes the GABAB receptor 1, a G protein-coupled receptor involved in synaptic inhibition, hippocampal long-term potentiation, slow wave sleep, muscle relaxation and sensitivity to pain. While human and gorilla have GABBR1 promoter methylation patterns that are broadly similar to each other, orangutan shows relative hypomethylation across this region. In contrast chimpanzee and bonobo show increased methylation specifically at the TSS of long GABBR1 isoforms, and intermediate methylation levels associated with the short isoform. These data suggest some epigenetic differences among primates are associated with isoform regulation.
Figure 3. Examples of differentially methylated genes.

Each data point represents the mean β-value of the group and whiskers show 2 standard deviations above and below the mean. (A) ARTN is a neurotrophic factor and it shows hypermethylation of 3 CpG sites associated with the long isoform specifically in human. (B) COL2A1 shows hypermethylation of 4 CpG sites at the promoter specifically in human. This gene encodes the alpha-1 chain of type II collagen, which is found primarily in cartilage, the inner ear and the vitreous humor of the eye. (C) PGAM2 shows hypomethylation of CpG sites at the promoter specifically in human. PGAM2 is an enzyme involved in the glycolitic pathway, mutations in which are associated with muscle cramping and intolerance for strenuous exercise. (D) GABBR1 shows a complex methylation pattern in which human and gorilla shows similar pattern of methylation, orangutan shows relative hypomethylation, while chimpanzees and bonobos show increased methylation levels at TSS and intermediate levels associated with the short isoform of this gene. GABBR1 is a neuronal receptor involved in synaptic inhibition, slow wave sleep, muscle relaxation and sensitivity to pain.
Functional context of differentially methylated sites in the genome
We observed a highly non-random distribution of the differentially methylated CpGs (Figure 4A and 4B) in relationship to gene annotations and CpG density. From the functional distribution standpoint, there was a significant excess of changes (p<0.0001, permutation test, see Methods) for sites located within 1,500 bp upstream of gene TSSs, gene bodies and intergenic regions, and from the CpG content standpoint, differential methylation occurred preferentially in CpG shores (±2 kb CpG island) and non-CpG island regions. These results highlight CpG shores as epigenetically variable regions, as it has been observed in human development and disease [12], [33]. In contrast, the regions immediately surrounding gene TSSs (−200 bp of the TSS and 1st exon) and CpG islands showed relative conservation of methylation.
Figure 4. Location of differential methylation in primate genomes.

(A) Distribution of 99,191 CpG sites interrogated in all great ape species. Left: Gene-centric functional distribution of methylation changes : 1,500 bp upstream of gene TSSs, 200 bp upstream of TSSs, 5′UTR, 1st exon, gene body, 3′ UTR and intergenic. Right: CpG-island centric distribution: CpG island, shore (±2 kb flanking CpG islands), shelf (2–4 kb from CpG islands). (B) A non-random distribution of methylation changes in recent primate evolution. We observe an excess of differential CpG methylation within the first 1,500 bp upstream of gene TSSs, gene bodies, intergenic regions, shore regions flanking CpG islands and non-CpG island regions. In contrast DNA methylation tends to be relatively conserved close to gene transcription start sites (−200 bp of TSS to 1st exon), and in the body of CpG islands. Each bar shows the relative enrichment for differential methylation versus that expected under a null distribution. * denotes p<0.0001 (permutation test). (C) Density plot showing the distribution of methylation levels of differentially methylated sites compared to that in the rest of the genome. Sites of evolutionary change among great apes have a significantly different distribution (p = 2.2×10−16 Kolmogorov-Smirnov test).
We also observed a significant difference in the distribution of methylation levels at differentially methylated sites compared to the rest of the genome (Figure 4C). While the overall genome-wide pattern of methylation levels shows a strongly bi-modal distribution, with most sites having either very high or very low methylation levels, sites of evolutionary change have a significantly different distribution to genome wide distribution (p = 2.2×10−16 Kolmogorov-Smirnov test), showing intermediate methylation levels, which has been shown to be a hallmark of distal regulatory elements. [34].
X-chromosome inactivation
In female mammals, X chromosome inactivation (XCI) is maintained via a number of epigenetic marks, including altered DNA methylation [35], [36]. Consistent with a role in XCI, the majority of sites we identified on the X chromosome in great apes showed relatively higher methylation levels in females versus males due to the contribution from the inactive X chromosome (63%, p = 0.005, Figure S4A). We searched for CpG sites on the X chromosome presenting no significant changes between males and females in a specific lineage (mean β-value difference <0.1) but showing significant gender differences in all the other species (see Methods). This analysis identified 22 CpGs in human, 59 in chimpanzee and bonobo, 72 in gorilla and 41 in orangutan (Table S5). Some regions are particularly interesting such as the MID1 gene which has been previously reported as a gene subject to X-inactivation in humans but not in mouse [37]. Our results suggest that this gene may escape XCI in the Pan lineage, but not in all other great apes. Similarly the HTR2C gene shows multiple probes upstream of the TSS with similar patterns of methylation in both male and female humans, potentially suggesting that this gene escapes XCI in the human lineage. In contrast, the same sites show significantly higher methylation levels in females compared to males in all other primate species, suggesting that in these species HTR2C may be subject to XCI (Figure S4B). Using published RNAseq data [38], we did not observe a female-specific increase in HTR2C gene expression for in humans, although we note that many genes escaping XCI show no clear sex differences in expression levels [39].
Pairwise comparison of human and chimpanzee
To maximize the identification of altered methylation patterns between human and our closest living relative, the chimpanzee, we performed a pairwise comparison of these two species using a larger dataset of 289,007 filtered probes common to human and chimpanzee. We used the chimpanzee species and not the whole genera to make use of the better annotation in the genome reference assembly for this species compared to the rest of non-human primate genome reference assemblies [1]. We identified 16,365 sites that showed significant hypermethylation in human, and 9,693 sites showing significant hypomethylation (FDR-adjusted p<0.05, β-value difference ≥0.1). This represents ∼9% of the total number of sites tested, and includes ∼2,500 genes (≥2 differentially methylated CpGs within a 1 kb interval and overlapped with RefSeq genes, −1500 bp TSS to 3′UTR).
Using this larger dataset, we then investigated the relationship between the evolution of protein coding sequences and epigenetic change at promoter level. Using a curated set of 7,252 human∶chimpanzee 1∶1 orthologs [1] we identified 745 genes (∼10% of those tested) that showed at least two differentially methylated sites at the promoter (−1500 bp from the TSS to 1st exon, see Methods). We then compared both the number of amino acid changes and the KA/KI ratio (the number of coding base substitutions that result in amino acid changes as a fraction of the local intergenic/intronic substitution rate) of these differentially methylated genes against the remainder [1] (Figure 5). We observed a significant difference in both the number and rate of non-synonymous amino acid changes between genes with altered promoter methylation compared to those without significant methylation differences (p<0.0001, permutation test) suggesting that rapid evolution at the protein coding level is frequently coupled with epigenetic changes in the promoter. We also observed similar results when using only those probes with a perfect match to the chimpanzee reference genome (Figure S5). An interesting example is the BRCA1 gene, which contains 32 amino acid changes between human and chimpanzee and has a KA/KI ratio of 0.69 (three times the average of all orthologous genes). This gene shows large differences in methylation ∼1–1.5 kb upstream of the TSS (Figure 6). Previous studies have shown that methylation changes of this same region are associated with altered BRCA1 expression [40].
Figure 5. A significant relationship between changes in promoter methylation and protein evolution between human and chimpanzee.

We performed a comparison of alterations in promoter methylation with (A) the frequency of amino-acid alterations and (B) the relative rate of coding to non-coding variation with genes (KA/KI) between human and chimpanzee. Using both metrics we observed a significant association between the rate of protein evolution and epigenetic regulatory changes. P-values are based on 1,000 permutations (Differentially methylated genes, n = 745; genes without significant changes in methylation, n = 6,507).
Figure 6. Example gene showing methylation differences between human and chimpanzee at promoter level.

BRCA1 provides an example of co-occurrence between protein sequence evolution and gene regulation. The BRCA1 gene shows changes in DNA methylation in a regulatory region upstream of the TSS [40] and a KA/KI ratio of 0.69 between human and chimpanzee.
In contrast, we also observed 184 genes that show perfect human:chimpanzee conservation at the amino acid level, yet they show significant epigenetic differences at their promoter (Table S6). Within this set of genes, we observed significant enrichments for categories related with gene expression (table S7) [41], [42].
As our survey of evolutionary changes in primate DNA methylation patterns utilized DNA derived from whole blood, we tested whether these changes are also present in other somatic tissues by comparing against an independent dataset. A previous study [9] utilized a similar array platform, although with a much reduced probe density, to compare DNA methylation levels in humans and chimpanzees using DNA isolated from heart, liver and kidney. Comparing the 457 sites common to both datasets that we defined as differentially methylated in blood samples versus these three other tissues, we observed a highly significant trend for methylation differences identified between human and chimpanzee to be conserved across all four tissue types (Figure 7).
Figure 7. Conservation of human-chimpanzee differentially methylated sites among multiple somatic tissues.

Differentially methylated sites we identified using whole blood of human and chimpanzee were compared to a previous study that used the Illumina HumanMethylation27 DNA Analysis BeadChip to study liver, kidney and heart tissue in an independent population of humans and chimpanzees [19]. We found a significant trend for sites that are differentially methylated in blood to also show higher human-chimpanzee divergence in these other tissues (yellow box plot, n = 457) suggesting a conservation across other somatic tissues compared to non-differentially methylated sites in blood (grey box plot, n = 7,942).
Discussion
The primary focus to date for understanding human evolution from a comparative genomic perspective has been the study of changes in DNA sequence and gene expression levels [43]–[45]. Our study of DNA methylation profiles among human and great apes adds to this wealth of information, reinforcing the view that epigenetic changes contribute significantly to species divergence, and therefore they should be considered in studies of human evolution.
In this study, one of the main challenges was the technical limitation stemming from the use of arrays designed against the human genome to profile methylation patterns in great ape species with divergent genomes. We utilized a set of filters to account for these differences, and were also able to replicate the results even after limiting our analysis to those probes with 100% identity in each of the non-human reference genome assemblies. Supporting a biological role for our findings, we observed that the clustering of differential methylation within each species was highly non-random, and showed significant enrichments within functional genomic elements.
From a biological perspective, it is conceivable that differences in the constitutive fractions of whole blood between species might introduce a bias due to the fact that different cell types possess distinct epigenomes [27]. This limitation is shared by nearly all comparative molecular studies of primary tissues from endangered species (i.e. great apes) due to the difficulty of obtaining relevant samples, especially in the case of wild-born individuals as the ones used in this study. However, in order to minimize this problem we removed all CpG sites that vary significantly between whole blood and the most abundant cell populations in blood. We further required a minimum threshold of 10% change in global methylation between sites in these species in order to identify differentially methylated sites, meaning that changes in the prevalence of minor cell fractions would not influence the results. Finally, while all samples were obtained from adult individuals, we could not match the ages perfectly among all samples, so in order to compensate for this effect, and to minimize the effects of intraspecific polymorphism, we focused our study on sites with low intragenus variance.
Our results show that ∼9% of the CpGs we assayed showed significant methylation differences between human and chimpanzee, including the promoter regions of 745 genes (10% of those tested). We estimate that over 2,500 genes present at least some methylation changes between human and chimpanzees (≥2 differentially methylated sites separated by ≤1 kb), a higher number than that known to be affected by copy number variation or under positive selection in the same species [46]–[49]. Although the arrays we used do not provide a complete and unbiased coverage of the genome, these data suggest that epigenetic changes have been frequent during recent primate evolution and represent an important substrate for adaptive modification of genome function. Underlining this idea, the changes we observed among primates are highly enriched for sites showing intermediate DNA methylation levels. Previous studies have shown that such methylation values are often a hallmark of distal regulatory elements [34], suggesting that many epigenetic changes occurring among human and great ape species impact transcriptional regulation. Consistent with these findings, we detected global enrichments for epigenetic change within known regulatory regions, including distal regions upstream of gene transcription start sites and regions flanking CpG islands (termed ‘CpG shores’).
We observed that the great ape phylogeny can be recapitulated from methylation data alone. Potential explanations for this are that methylation values could be driven by proximal DNA changes that were not controlled in this study, or that epigenetic changes have occurred independently of DNA sequence but are subject to similar rates of change either through selective pressures or neutral drift.
Interestingly, we also identified a significant positive relationship between the rate of coding variation within genes and alterations of promoter methylation, suggesting a co-occurrence between changes in protein sequence and gene regulation that may be related to expression changes in fast evolving genes [50]. In contrast, and consistent with previous analysis indicating the importance of regulatory changes in evolution [51], our study also identified scores of genes that are perfectly conserved at the amino acid level between human and chimpanzee, yet showing significant epigenetic change between these two species. Furthermore, gene ontology analysis of this set showed that they are significantly enriched for the functional category of gene expression. These observations highlight the evolutionary importance of epigenetic changes that affect gene regulation, and also demonstrate that sequence-based studies are insufficient to capture the full spectrum of evolutionary change.
Overall our analysis identified >800 genes with significantly altered methylation patterns specifically within each species of human and great apes, including 171 with a methylation pattern unique to humans. Analysis of these 171 genes identified interesting enrichments for a number of functional categories that could suggest a relationship to human-specific traits. For example, we observed that genes involved in the regulation of blood pressure and development of the semicircular canal of the inner ear among others, were all highly enriched for DNA methylation changes specifically in the human lineage. While major changes in circulatory physiology are required for upright locomotion, the inner ear provides sensory input crucial for maintaining balance. Furthermore, a previous study of primates and other mammals has shown that the size of the semicircular canals is correlated with locomotion and with relatively larger canals found in species that utilize fast or agile movement [52]. While these trends hint at the potential importance of epigenetic changes in the evolution of several human-specific features, we caution that at this stage they should be considered as preliminary, as our studies were performed using DNA derived from whole blood, and it is well known that epigenetic patterns often vary widely between different tissues of an organism [27]. Therefore further studies in physiologically relevant tissues will be required to confirm the significance of these findings. However, we note that comparison with previously published data [9] suggests that many of the changes in DNA methylation that we detected between blood of human and chimpanzee appear to be conserved across several other tissues, suggesting that inter-specific differences observed in blood can in some cases be informative for other tissues.
Although sequencing studies have undoubtedly provided major advances in our understanding of primate evolution, our analysis of primate epigenomes unveils many novel differences among the great apes that are not apparent from purely sequence-based approaches. Of particular note is the fact that we identify enrichments in multiple independent functional gene categories which suggests that regulatory changes may have played a key role in the acquisition of human-specific trait. Therefore, epigenetic alterations likely represent an important facet of evolutionary change in primate genomes. Future studies that integrate epigenetic data with recent detailed maps of functional elements, selective constraint and chromatin interactions in the human genome [53]–[55] will likely provide many novel insights into genomic and phenotypic evolution.
Methods
Ethics statement
The non-human research has been approved by the ethical committee of the European Research Union. No living animal has been used and DNA has been obtained during standard veterinary checks. Methylation profiling of human subjects was approved by the Institutional Review Board of the Icahn School of Medicine at Mount Sinai (HS#: 12-00567 HG).
Hybridization and normalization
We obtained methylation data from peripheral blood DNA extracted from 9 adult humans, 5 chimpanzees, 6 bonobos, 6 gorillas and 6 orangutans. All individuals were unrelated adults and the non-human primates were all wild born. DNA samples were bisulfite converted, whole-genome amplified, enzymatically fragmented, and hybridized to the Infinium HumanMethylation450 BeadChip which provides quantitative estimates of methylation levels at 482,421 CpG sites distributed genome-wide. The assay was performed according to the manufacturer's instructions. The BeadChip array data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE41782. Due to the low density of probes targeting non-CpG dinucleotides (<0.7%) on the array, we focused our study on CpG sites.
Since the 50 bp probes on the array were designed against the human reference genome but we performed hybridizations utilizing DNA from different great ape species, we first mapped the probe sequences to the chimpanzee (panTro3), bonobo (panPan1), gorilla (gorGor3) and orangutan (ponAbe2) reference genomes using BWA [56], allowing a maximum edit distance of 3. We then assessed probe performance as a function of the number and relative location of sequence differences at the probe binding site in each primate genome (Figure S1 and Figure S2). Based on this analysis, in each species we only retained those probes that had either a perfect match, or had 1 or 2 mismatches in the first 45 bp but no mismatches in the 3′ 5 bp closest to the CpG site being assayed. We also removed all probes that contained human SNPs with minor allele frequency ≥0.05 within the last 5 bp of their binding site closest to the CpG being assayed [57]. Using published SNP data [26] for each species we removed probes containing SNPs with minor allele frequency ≥0.15 within the last 5 bp of their binding site closest to the CpG being assayed. We also removed all probes that contained more than two SNPs with minor allele frequency ≥0.15 in the first 45 bp.
Methylation values for CpG sites in each sample were obtained as β-values, calculated as the ratio of the methylated signal intensity to the sum of both methylated and unmethylated signals after background subtraction (β-values range from 0 to 1, corresponding to completely unmethylated and fully methylated sites, respectively). Within each individual, probes with a detection p>0.01 were excluded. We performed a two color channel signal adjustment and quantile normalization on the pooled signals from both channels and recalculation of average β-values as implemented in “lumi” package of R [58]. The Illumina Infinium HumanMethylation450 BeadChip contains two assay types (Infinium type I and type II probes) which utilize different probe designs. As the data produced by these two assay types shows distinct profiles (Figure S6), to correct this problem we performed a BMIQ (beta mixture quantile method) [59] on the quantile normalized data sets.
Using a published human data set [27] we identified differentially methylated sites between whole blood and CD4+ T-cells, and between whole blood and CD16+ neutrophils, representing the two most abundant cell fractions of blood (comprising ∼13% and ∼65%, respectively) (Wilcoxon rank-sum test, FDR-adjusted p<0.05 and mean β-value difference in each case ≥0.1). These sites (n = 10,151) were removed to mitigate potential confounders due to differing proportions of blood cell types among primates, leaving for comparison only those sites that do not significantly vary among the most abundant cell types of blood. β-values can be interpreted as the percentage of methylation at a given site. A β-value of 0.1 indicates that there has been a change in methylation in 10% of the molecules tested. Because our analyses required a mean β-value difference >0.1 to achieve significance, this threshold means that changes in blood cell fractions representing <10% of whole blood will be unlikely to affect our results. The final dataset after all filtering steps comprised 114,739 probes shared across all great ape species, and 291,553 probes shared between human and chimpanzee.
Phylogenetic relationships
To investigate the global correspondence of DNA sequence differences between species and the degree of methylation changes, we examined the Enredo-Pecan-Orthus (EPO) whole-genome multiple alignments of human, chimpanzee, gorilla, and orangutan [Ensemble Compara.6_primates_EPO] [28], [29]. Considering only those blocks with alignments for all great apes, we first excluded regions containing gaps or indels and then calculated pairwise distances between these four species based on the frequency of single nucleotide substitutions. To calculate the global changes in methylation we used a distance matrix, we first averaged the β-values per probe within a species and then calculated the difference between two species using Euclidean distances.
We built phylogenetic trees based on the methylation states of 114,739 filtered probes (perfect match probes and probes containing 1 or 2 mismatches in the first 45 bp) (Figure S3). We used the “ape” R package to construct the phylogenetic tree using the Neighbor-Joining algorithm and 1,000 bootstraps of the resulting tree [60]. We repeated the analysis using only the subset of probes with a perfect match to each of the primate reference genome assemblies (n = 31,853) (Figure 1B).
Differentially methylated sites
To identify only those methylation differences that represent fixed changes between genera, we retained only those CpGs with low methylation variance within each genus (intragenus standard deviation <0.1). This filtering step resulted in the removal of 1,377 CpGs in human, 5,224 in the Pan genus, 5,289 in Gorilla and 5,740 in Pongo, with the resulting final set being 99,919 CpGs shared across all five species.
We performed six pairwise comparisons among groups (Human-Pan species/Human-Gorilla species/Human-Pongo species/Pan species-Gorilla species/Pan species-Pongo species/Gorilla species-Pongo species). We defined a site to be genus-specific differentially methylated if all three comparisons with other groups were significant (Wilcoxon rank-sum test, FDR-adjusted p<0.05) and mean β-value difference in each case ≥0.1. We also tried other statistical approaches (linear modeling, limma package, [59]) and obtained very similar results (concordance for 98% of the sites).
All coordinates quoted are based on hg19. We intersected human probe coordinates provided by Illumina with RefSeq genes, retaining CpG sites overlapping genes (−1500 bp from TSS to 3′UTR). We defined a gene to be differentially methylated if there were at least two differentially methylated CpG sites separated by ≤1 kb. To assess significance of these observations we performed a permutation test, as follows. Based on the number of differentially methylated sites detected in each species (Human = 2,284; Pan = 1,245; Gorilla = 1,374; Orangutan = 5,501) we randomly sampled from the 99,919 CpGs and then determined the number of clusters (at least two differentially methylated CpG sites separated by ≤1 kb), repeating this process 10,000 times to create the null distribution. The p-value corresponded to the number of times that differentially methylated clusters appeared within the null distribution divided by the number of permutations (n = 10,000).
The Genomic Regions Enrichment of Annotations Tool (GREAT version 2.0.1) [31] was utilized to identify significant enrichments (FDR-corrected p<0.05) for Gene Ontology biological processes. While tools for identifying enriched GO terms are usually based on genes, GREAT permits the assignment of biological function to non-coding genomic regions by analyzing the annotations of nearby genes. For this analysis regulatory regions were associated to the single nearest gene situated within 10 kb. The background data set was the 99,919 CpG sites interrogated in all great ape species. In order to evaluate the positional context of the differentially methylated sites, we compared the distribution of these 10,404 sites detected among the primate species with all 99,919 CpGs tested. Permutation p-values were calculated as described above using 10,000 iterations.
X-chromosome inactivation
We performed two color channel signal adjustment and quantile normalization on males and females separately. Due to the different methylation pattern in females no BMIQ normalization was done in this data set. For studies of DNA methylation on the X-chromosome that might be linked with XCI between species, we searched for CpG sites presenting no significant changes between males and females in a specific lineage (mean β-value difference <0.1) but showing significant changes in all the other species (mean β-value difference between sexes >0.1).
Human-chimpanzee analysis
The number of probes shared between human and chimpanzee after applying our mapping and SNP filters was 291,554. Based on this set of probes, we performed a separate two color channel signal adjustment and quantile normalization of the raw data using only human and chimpanzee samples. We performed a BMIQ normalization to correct the probe design bias. After excluding probes with a standard deviation within either species >0.1 we retained a total of 289,007 probes. Differentially methylated sites were those with p<0.05 (Wilcoxon rank-sum test, FDR-adjusted p<0.05) and a mean β-value difference ≥0.1.
From the total set of 13,454 human:chimpanzee orthologous genes [1], we removed genes with <150 or >1500 amino acids, and then compared the number of amino acid changes and the KA/KI ratio of genes with robust alterations of promoter methylation (mean β-value difference of top 2 probes within promoter ≥0.1, considering CpGs located ≤1,500 bp upstream of Refseq gene TSSs, in the 5′UTR or the 1st exon, n = 745) versus those without methylation changes (n = 6,507). The Gene Ontology enRIchment anaLysis and visuaLizAtion tool (GOrilla) [41], [42] was utilized to obtain the functional enrichments within the 184 genes conserved at amino acid level, yet having significant epigenetic differences at their promoter. The data set containing 7,252 human∶chimpanzee 1∶1 orthologs was used as a background.
Supporting Information
Effect of sequence mismatches on probe performance. Difference in mean β-values between human and chimpanzee (A), gorilla (B) and orangutan (C) for each category of probes. Probes with mismatches located in the last 5 bp at the 3′ end, C>T transitions of the CpG site being measured, or ≥3 bp of mismatch showed an excess of variation compared to probes with ≤2 mismatches, and were removed from further analysis.
(PDF)
Effect of sequence mismatches on probe performance. Distribution of differences in mean β-value between human and (A) chimpanzee, (B) gorilla and (C) orangutan. Dotted lines represent the probes used in this study (perfect match and 1–2 mismatches).
(PDF)
Neighbor-joining tree based on 114,739 autosomal CpGs measured in all individuals and species. Bootstrap values (1,000 permutations) are shown for each node.
(PDF)
(A) Density plot showing the distribution of methylation levels of CpG sites in males and females. (B) The HTR2C gene on the X chromosome shows a relative reduction in promoter methylation specifically in human females compared to other great ape species. Probes upstream of the TSS of HTR2C show similar patterns of methylation in both male and female humans. The same sites show significantly increased methylation in females versus males in all other primate species tested. These observations suggest evolutionary changes in the X chromosome inactivation status of HTR2C specifically in humans compared to other primates.
(PDF)
Comparison of alterations in promoter methylation with frequency of amino-acid changes and the relative rate of coding to non-coding variation within genes (KA/KI) between human and chimpanzee in two data sets. (A) Probes with perfect match to the chimpanzee genome. Differentially methylated genes: n = 334, genes without significant changes in methylation: n = 5,655. (B) Probes with 1 or 2 mismatches in the first 45 bp in the chimpanzee genome. Differentially methylated genes: n = 247, genes without significant changes in methylation: n = 4,840.
(PDF)
(A) β-value distribution of the 114,739 sites shared among the five species before and after BMIQ. (Infinium type I probe, n = 32,216. Infinium type II probes, n = 82,523). (B) β-value distribution of the 291,553 sites shared among human and chimpanzee before and after BMIQ, (Infinium type I probe, n = 83,528. Infinium type II probes, n = 208,025).
(PDF)
Sample information. NA: Information not available for these samples.
(XLS)
10,404 CpG sites showing species-specific differential methylation.
(XLS)
815 genes associated with species-specific methylation changes.
(XLS)
Results of GREAT analysis showing GO terms significantly associated with methylation changes in human, Pan species, and Pongo species. All GO terms shown have p<0.05 (5% FDR).
(XLS)
CpG sites on the X chromosome showing gender-specific methylation changes.
(XLS)
184 genes that show perfect conservation of amino acid sequence between human and chimpanzee, but which have significant epigenetic changes within their promoter regions.
(XLS)
184 genes that show perfect conservation of amino acid sequence between human and chimpanzee, but which have significant epigenetic changes within their promoter regions.
(XLSX)
Supplementary methods.
(DOCX)
Acknowledgments
We wish to thank G. Santpere for constructive comments and useful discussions.
Funding Statement
TMB is supported by the European Research Council (ERC Starting Grant, StG_20091118) and the Spanish Government (BFU2011-28549). AJS is supported by NIH grants 1R01DA033660, 1R01HG006696, and a grant from the Alzheimer's Association (2012ALZNIRG69983). IHH is supported by the European Social Fund, AGAUR (Generalitat de Catalunya, Spain) and the Spanish National Research Council (CSIC). We also thank the Spanish Government for the grant BFU2009-13409-C02-02 to AN and the Barcelona Zoo (Ajuntament de Barcelona) for an award to JPM. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. The Chimpanzee Sequencing AC (2005) Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437: 69–87 doi:10.1038/nature04072 [DOI] [PubMed] [Google Scholar]
- 2. Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, et al. (2012) Insights into hominid evolution from the gorilla genome sequence. Nature 483: 169–175 doi:10.1038/nature10842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Prüfer K, Munch K, Hellmann I, Akagi K, Miller JR, et al. (2012) The bonobo genome compared with the chimpanzee and human genomes. Nature 486: 527–31 doi:10.1038/nature11128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Locke DP, Hillier LW, Warren WC, Worley KC, Nazareth L V, et al. (2011) Comparative and demographic analysis of orang-utan genomes. Nature 469: 529–533 doi:10.1038/nature09687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cain CE, Blekhman R, Marioni JC, Gilad Y (2011) Gene expression differences among primates are associated with changes in a histone epigenetic modification. Genetics 187: 1225–1234 doi:10.1534/genetics.110.126177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martin D, Singer M, Dhahbi J, Mao G (2011) Phyloepigenomic comparison of great apes reveals a correlation between somatic and germline methylation states. Genome 2049–2057 doi:10.1101/gr.122721.111.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Molaro A, Hodges E, Fang F, Song Q, McCombie WR, et al. (2011) Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 146: 1029–1041 doi:10.1016/j.cell.2011.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, et al. (2012) DNA methylation signatures in development and aging of the human prefrontal cortex. American journal of human genetics 90: 260–272 doi:10.1016/j.ajhg.2011.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pai A a, Bell JT, Marioni JC, Pritchard JK, Gilad Y (2011) A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS genetics 7: e1001316 doi:10.1371/journal.pgen.1001316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sado T, Fenner MH, Tan SS, Tam P, Shioda T, et al. (2000) X inactivation in the mouse embryo deficient for Dnmt1: distinct effect of hypomethylation on imprinted and random X inactivation. Developmental biology 225: 294–303 doi:10.1006/dbio.2000.9823 [DOI] [PubMed] [Google Scholar]
- 11. Reik W (2007) Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447: 425–432 doi:10.1038/nature05918 [DOI] [PubMed] [Google Scholar]
- 12. Irizarry R a, Ladd-Acosta C, Wen B, Wu Z, Montano C, et al. (2009) The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature genetics 41: 178–186 doi:10.1038/ng.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharp AJ, Stathaki E, Migliavacca E, Brahmachary M, Montgomery SB, et al. (2011) DNA methylation profiles of human active and inactive X chromosomes. Genome research 21: 1592–1600 doi:10.1101/gr.112680.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jones PA, Takai D (2001) The role of DNA methylation in mammalian epigenetics. Science (New York, NY) 293: 1068–1070 doi:10.1126/science.1063852 [DOI] [PubMed] [Google Scholar]
- 15. Laurent L, Wong E, Li G, Huynh T, Tsirigos A, et al. (2010) Dynamic changes in the human methylome during differentiation. Genome research 20: 320–331 doi:10.1101/gr.101907.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, et al. (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322 doi:10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, et al. (2011) CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479: 74–79 doi:10.1038/nature10442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bell JT, Pai A a, Pickrell JK, Gaffney DJ, Pique-Regi R, et al. (2011) DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome biology 12: R10 doi:10.1186/gb-2011-12-1-r10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, et al. (2011) Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS genetics 7: e1002228 doi:10.1371/journal.pgen.1002228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gibbs JR, Van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, et al. (2010) Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS genetics 6: e1000952 doi:10.1371/journal.pgen.1000952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brunner AL, Johnson DS, Kim SW, Valouev A, Reddy TE, et al. (2009) Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome research 19: 1044–1056 doi:10.1101/gr.088773.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, et al. (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770 doi:10.1038/nature07107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H (2011) Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. American journal of human genetics 88: 450–457 doi:10.1016/j.ajhg.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gordon L, Joo JE, Powell JE, Ollikainen M, Novakovic B, et al. (2012) Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence. Genome Research 22: 1395–406 doi:10.1101/gr.136598.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zeng J, Konopka G, Hunt BG, Preuss TM, Geschwind D, et al. (2012) Divergent Whole-Genome Methylation Maps of Human and Chimpanzee Brains Reveal Epigenetic Basis of Human Regulatory Evolution. The American Journal of Human Genetics 91: 455–465 doi:10.1016/j.ajhg.2012.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, et al. (2013) Great ape genetic diversity and population history. Nature 499: 471–475 doi:10.1038/nature12228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén S-E, et al. (2012) Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PloS one 7: e41361 doi:10.1371/journal.pone.0041361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paten B, Herrero J, Beal K, Fitzgerald S, Birney E (2008) Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome research 18: 1814–1828 doi:10.1101/gr.076554.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paten B, Herrero J, Fitzgerald S, Beal K, Flicek P, et al. (2008) Genome-wide nucleotide-level mammalian ancestor reconstruction. Genome research 18: 1829–1843 doi:10.1101/gr.076521.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Becquet C, Patterson N, Stone AC, Przeworski M, Reich D (2007) Genetic structure of chimpanzee populations. PLoS genetics 3: e66 doi:10.1371/journal.pgen.0030066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, et al. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nature biotechnology 28: 495–501 doi:10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuivaniemi H, Tromp G, Prockop DJ (1997) Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Human mutation 9: 300–315 doi:;10.1002/(SICI)1098-1004(1997)9:4<300::AID-HUMU2>3.0.CO;2-9 [DOI] [PubMed] [Google Scholar]
- 33. Doi A, Park I-H, Wen B, Murakami P, Aryee MJ, et al. (2009) Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature genetics 41: 1350–1353 doi:10.1038/ng.471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, et al. (2011) DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480: 490–495 doi:10.1038/nature10716 [DOI] [PubMed] [Google Scholar]
- 35. Payer B, Lee JT (2008) X chromosome dosage compensation: how mammals keep the balance. Annual review of genetics 42: 733–772 doi:10.1146/annurev.genet.42.110807.091711 [DOI] [PubMed] [Google Scholar]
- 36. Lyon MF (1961) Gene Action in the X-chromosome of the Mouse (Mus musculus L.). Nature 190: 372–373 doi:10.1038/190372a0 [DOI] [PubMed] [Google Scholar]
- 37. Dal Zotto L, Quaderi Na, Elliott R, Lingerfelter Pa, Carrel L, et al. (1998) The mouse Mid1 gene: implications for the pathogenesis of Opitz syndrome and the evolution of the mammalian pseudoautosomal region. Human molecular genetics 7: 489–499 doi:10.1093/hmg/7.3.489 [DOI] [PubMed] [Google Scholar]
- 38. Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, et al. (2011) The evolution of gene expression levels in mammalian organs. Nature 478: 343–348 doi:10.1038/nature10532 [DOI] [PubMed] [Google Scholar]
- 39. Johnston CM, Lovell FL, Leongamornlert D a, Stranger BE, Dermitzakis ET, et al. (2008) Large-scale population study of human cell lines indicates that dosage compensation is virtually complete. PLoS genetics 4: e9 doi:10.1371/journal.pgen.0040009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rice JC, Futscher BW (2000) Transcriptional repression of BRCA1 by aberrant cytosine methylation, histone hypoacetylation and chromatin condensation of the BRCA1 promoter. Nucleic acids research 28: 3233–3239 doi:10.1093/nar/28.17.3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48 doi:10.1186/1471-2105-10-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eden E, Lipson D, Yogev S, Yakhini Z (2007) Discovering Motifs in Ranked Lists of DNA Sequences. PLoS Computational Biology 3: e39 doi:10.1371/journal.pcbi.0030039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O'Bleness M, Searles VB, Varki A, Gagneux P, Sikela JM (2012) Evolution of genetic and genomic features unique to the human lineage. Nature reviews Genetics 13: 853–866 doi:10.1038/nrg3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Whitehead A, Crawford DL (2006) Neutral and adaptive variation in gene expression. Proceedings of the National Academy of Sciences of the United States of America 103: 5425–5430 doi:10.1073/pnas.0507648103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gilad Y, Oshlack A, Rifkin S a (2006) Natural selection on gene expression. Trends in genetics: TIG 22: 456–461 doi:10.1016/j.tig.2006.06.002 [DOI] [PubMed] [Google Scholar]
- 46. Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, et al. (2005) A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS biology 3: e170 doi:10.1371/journal.pbio.0030170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, et al. (2010) Diversity of human copy number variation and multicopy genes. Science (New York, NY) 330: 641–646 doi:10.1126/science.1197005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marques-Bonet T, Kidd JM, Ventura M, Graves T a, Cheng Z, et al. (2009) A burst of segmental duplications in the genome of the African great ape ancestor. Nature 457: 877–881 doi:10.1038/nature07744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dumas L, Kim YH, Karimpour-Fard A, Cox M, Hopkins J, et al. (2007) Gene copy number variation spanning 60 million years of human and primate evolution. Genome research 17: 1266–1277 doi:10.1101/gr.6557307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kosiol C, Vinar T, Da Fonseca RR, Hubisz MJ, Bustamante CD, et al. (2008) Patterns of positive selection in six Mammalian genomes. PLoS genetics 4: e1000144 doi:10.1371/journal.pgen.1000144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. King MC, Wilson AC (1975) Evolution at two levels in humans and chimpanzees. Science 188: 107–116 doi:10.1126/science.1090005 [DOI] [PubMed] [Google Scholar]
- 52. Spoor F, Garland T, Krovitz G, Ryan TM, Silcox MT, et al. (2007) The primate semicircular canal system and locomotion. Proceedings of the National Academy of Sciences of the United States of America 104: 10808–10812 doi:10.1073/pnas.0704250104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Burrows AM (2008) The facial expression musculature in primates and its evolutionary significance. Bio Essays: news and reviews in molecular, cellular and developmental biology 30: 212–225 doi:10.1002/bies.20719 [DOI] [PubMed] [Google Scholar]
- 54. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, et al. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380 doi:10.1038/nature11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ward LD, Kellis M (2012) Evidence of Abundant Purifying Selection in Humans for Recently Acquired Regulatory Functions. Science 337: 1675–8 doi:10.1126/science.1225057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 25: 1754–1760 doi:10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sherry ST, Ward M, Sirotkin K (1999) dbSNP — Database for Single Nucleotide Polymorphisms and Other Classes of Minor Genetic Variation dbSNP — Database for Single Nucleotide Polymorphisms and Other Classes of Minor Genetic Variation. 677–679 doi:10.1101/gr.9.8.677 [PubMed] [Google Scholar]
- 58. Du P, Kibbe W a, Lin SM (2008) lumi: a pipeline for processing Illumina microarray. Bioinformatics (Oxford, England) 24: 1547–1548 doi:10.1093/bioinformatics/btn224 [DOI] [PubMed] [Google Scholar]
- 59. Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, et al. (2013) A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics (Oxford, England) 29: 189–196 doi:10.1093/bioinformatics/bts680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Paradis E, Claude J, Strimmer K (2004) APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20: 289–290 doi:10.1093/bioinformatics/btg412 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of sequence mismatches on probe performance. Difference in mean β-values between human and chimpanzee (A), gorilla (B) and orangutan (C) for each category of probes. Probes with mismatches located in the last 5 bp at the 3′ end, C>T transitions of the CpG site being measured, or ≥3 bp of mismatch showed an excess of variation compared to probes with ≤2 mismatches, and were removed from further analysis.
(PDF)
Effect of sequence mismatches on probe performance. Distribution of differences in mean β-value between human and (A) chimpanzee, (B) gorilla and (C) orangutan. Dotted lines represent the probes used in this study (perfect match and 1–2 mismatches).
(PDF)
Neighbor-joining tree based on 114,739 autosomal CpGs measured in all individuals and species. Bootstrap values (1,000 permutations) are shown for each node.
(PDF)
(A) Density plot showing the distribution of methylation levels of CpG sites in males and females. (B) The HTR2C gene on the X chromosome shows a relative reduction in promoter methylation specifically in human females compared to other great ape species. Probes upstream of the TSS of HTR2C show similar patterns of methylation in both male and female humans. The same sites show significantly increased methylation in females versus males in all other primate species tested. These observations suggest evolutionary changes in the X chromosome inactivation status of HTR2C specifically in humans compared to other primates.
(PDF)
Comparison of alterations in promoter methylation with frequency of amino-acid changes and the relative rate of coding to non-coding variation within genes (KA/KI) between human and chimpanzee in two data sets. (A) Probes with perfect match to the chimpanzee genome. Differentially methylated genes: n = 334, genes without significant changes in methylation: n = 5,655. (B) Probes with 1 or 2 mismatches in the first 45 bp in the chimpanzee genome. Differentially methylated genes: n = 247, genes without significant changes in methylation: n = 4,840.
(PDF)
(A) β-value distribution of the 114,739 sites shared among the five species before and after BMIQ. (Infinium type I probe, n = 32,216. Infinium type II probes, n = 82,523). (B) β-value distribution of the 291,553 sites shared among human and chimpanzee before and after BMIQ, (Infinium type I probe, n = 83,528. Infinium type II probes, n = 208,025).
(PDF)
Sample information. NA: Information not available for these samples.
(XLS)
10,404 CpG sites showing species-specific differential methylation.
(XLS)
815 genes associated with species-specific methylation changes.
(XLS)
Results of GREAT analysis showing GO terms significantly associated with methylation changes in human, Pan species, and Pongo species. All GO terms shown have p<0.05 (5% FDR).
(XLS)
CpG sites on the X chromosome showing gender-specific methylation changes.
(XLS)
184 genes that show perfect conservation of amino acid sequence between human and chimpanzee, but which have significant epigenetic changes within their promoter regions.
(XLS)
184 genes that show perfect conservation of amino acid sequence between human and chimpanzee, but which have significant epigenetic changes within their promoter regions.
(XLSX)
Supplementary methods.
(DOCX)