

Abstract
Vascular pathology is a major feature of Alzheimer's disease (AD) and other dementias. We recently showed that chronic administration of the target-of-rapamycin (TOR) inhibitor rapamycin, which extends lifespan and delays aging, halts the progression of AD-like disease in transgenic human (h)APP mice modeling AD when administered before disease onset. Here we demonstrate that chronic reduction of TOR activity by rapamycin treatment started after disease onset restored cerebral blood flow (CBF) and brain vascular density, reduced cerebral amyloid angiopathy and microhemorrhages, decreased amyloid burden, and improved cognitive function in symptomatic hAPP (AD) mice. Like acetylcholine (ACh), a potent vasodilator, acute rapamycin treatment induced the phosphorylation of endothelial nitric oxide (NO) synthase (eNOS) and NO release in brain endothelium. Administration of the NOS inhibitor L-NG-Nitroarginine methyl ester reversed vasodilation as well as the protective effects of rapamycin on CBF and vasculature integrity, indicating that rapamycin preserves vascular density and CBF in AD mouse brains through NOS activation. Taken together, our data suggest that chronic reduction of TOR activity by rapamycin blocked the progression of AD-like cognitive and histopathological deficits by preserving brain vascular integrity and function. Drugs that inhibit the TOR pathway may have promise as a therapy for AD and possibly for vascular dementias.
Keywords: age-associated, Alzheimer's disease, cerebral amyloid angiopathy, nitric oxide synthase, rapamycin, target-of-rapamycin (TOR), vascular dysfunction
Introduction
Vascular pathology causes or contributes to dementia in a substantial portion of patients.1 Cerebral microbleeds are present in patients with vascular dementia and with Alzheimer's disease (AD).2 Cerebral microbleeds and macroscopic hemorrhage are frequently consequences of cerebral amyloid angiopathy (CAA),3 which arises from the deposition of amyloid-β peptide (Aβ) in blood vessels. The vast majority of AD patients show CAA.3 Cerebral amyloid angiopathy is also associated with Parkinson's disease and with dementia with Lewy bodies, and is strongly linked to cognitive decline in these disorders. The relevance of microbleeds in dementias became evident with the availability of improved functional imaging technologies.4, 5, 6 Although CAA is recapitulated in various mouse models of AD,3, 7, 8, 9 there is a paucity of functional imaging studies in AD models.10
Brain endothelial cells (EC) signal to vascular smooth muscle cells to regulate cerebral blood flow (CBF). Available evidence indicates that failure of nitric oxide (NO)-induced, endothelium-dependent vasodilation is causal to vascular aging,11 which has been linked to age-associated dysfunction in various organs and tissues including brain.12, 13 Endothelial cells and vascular smooth muscle cells dysfunction are prominent in AD14, 15, 16 and have a negative impact on CBF. The target-of-rapamycin (TOR) is a major signaling hub that integrates nutrient and growth factor availability with cell metabolism.17 It was recently demonstrated18, 19, 20 that chronic reduction of mammalian target-of-rapamycin (mTOR) signaling by rapamycin extends lifespan in mice by delaying aging.19 Consistent with this observations, inhibition of mTOR delays EC senescence in vivo21 and in vitro,22 improves vascular outcomes of stroke,23, 24 and has been shown to have EC-dependent vasodilatory effects.25, 26 Although distal EC dysfunction has been observed with rapamycin-eluting stents implanted in coronary heart disease patients,27, 28 recent studies have shown that this effect may be due to delayed or absent re-endothelization of the stent,29 or to effects of the stent itself.30
We and others have shown that, consistent with its lifespan-extending effects, reduction of mTOR activity by chronic rapamycin treatment halts the progression of AD-like memory deficits and reduces Aβ accumulation in mouse models of the disease31, 32 when administered before the onset of robust cognitive deficits. Moreover, these studies showed that a mechanism of action of chronic TOR inhibition involves the enhancement of proteostasis though the activation of autophagy,31, 32 and we demonstrated the upregulation of heat shock proteins33 in rapamycin-treated AD mouse brain parenchyma. Chronic rapamycin treatment, however, did not ameliorate AD-like deficits when administered in the late stages of AD-like disease in the 3xTg-AD mouse model.34 Whether rapamycin would improve cognitive outcomes and ameliorate AD-like histopathological deficits in hAPP(J20) mice modeling AD when administered after establishment of robust AD-like memory impairments (equivalent to the time at diagnosis of possible AD), however, was not known. To answer this question, in the present study we used behavioral and biochemical tools and in vivo optical brain imaging in conjunction, for the first time, with multimodal magnetic resonance imaging (MRI) techniques in vivo to study the effects of rapamycin treatment on functional outcomes in a mouse model of AD.35, 36
Materials and methods
Mice
The derivation and characterization of hAPP(J20) mice (AD mice) has been described elsewhere.35, 36, 37 Human Aβ produced from the hAPP transgene is cleared across the blood–brain barrier (BBB) into the circulation (Supplementary Figure 2). hAPP(J20) mice were maintained by heterozygous crosses with C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME, USA). The hAPP transgene carrying the Swedish and Indiana familial AD mutations (hAPPSw,Ind) is driven by a neuron-specific promoter that is activated at embryonic day 14. Heterozygous crosses were set up such that the transgenic animal in was the dam or the sire in approximately 50% of the breeding pairs to minimize confounds related to potential effects of transgene expression during gametogenesis, or imprinting effects. hAPP(J20) mice were heterozygous with respect to the transgene. Non-transgenic littermates were used as controls. Experimental groups were: control-fed non-Tg, n=17; rapamycin-fed non-Tg, n=18; control-fed Tg, n=10; rapamycin-fed Tg, n=10, all animals were males and 11-month-old at the time of testing. Tg hAPP(J20) mice are referred to as ‘AD mice' and non-Tg littermates are called ‘WT' throughout the text. Rapamycin was administered for 16 weeks starting at 7 months of age. All animal experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at University of Texas Health Science Center at San Antonio (Animal Welfare Assurance Number: A3345-01).
Rapamycin and L-NAME Treatment
Mice were fed chow containing either microencapsulated rapamycin at 2.24 mg/kg or a control diet as described by Harrison et al.18 Rapamycin was used at 14 mg per kg food (verified by HPLC). On the assumption that the average mouse weighs 30 gm and consumes 5 gm of food/day, this dose supplied 2.24 mg rapamycin per kg body weight/day.18 All mice were given ad libitum access to rapamycin or control food and water for the duration of the experiment. Body weights and food intake were measured weekly. Food consumption remained constant and was comparable for control- and rapamycin-fed groups. Littermates were housed together, thus we could not distinguish effects of genotype on food consumption. Even though there were no differences in food consumption, body weights of rapamycin-fed WT, but not AD, females increased moderately during treatment, (6.8% increase for rapamycin-fed vs control-fed WT females). The higher increase in body weight for WT animals is not unexpected, since non-transgenic WT littermate animals of both genders tend to be slightly (1 to 3 g) heavier than AD transgenic mice. L-NAME was injected intraperitoneally every other day at 30 mg/kg.
Measurement of Rapamycin Using HPLC-tandem MS
Rapamycin and ascomycin were obtained from LC Laboratories (Woburn, MA, USA). HPLC grade methanol and acetonitrile were purchased from Fisher (Fair Lawn, NJ, USA). All other reagents were purchased from Sigma Chemical Company (St Louis, MO, USA). Milli-Q water was used for preparation of all solutions. Rapamycin and ascomycin super stock solutions were prepared in methanol at a concentration of 1 mg/mL and stored in aliquots at −80oC. A working stock solution prepared each day from the super stock solutions at a concentration of 10 μg/mL was used to spike the calibrators. The HPLC system consisted of a Shimadzu (Kyoto, Japan) SCL-10 A Controller, LC-10AD pump with a FCV-10AL mixing chamber, SIL-10AD autosampler, and an AB Sciex (Framingham, MA, USA) API 3200 tandem mass spectrometer with turbo ion spray. The analytical column was a Grace Alltima C18 (4.6 × 150 mm, 5 μ) purchased from Alltech (Deerfield, IL, USA) and was maintained at 60oC during the chromatographic runs using a Shimadzu CTO-10A column oven. Mobile phase A contained 10 mM ammonium formate and 0.1% formic acid dissolved in HPLC grade methanol. Mobile phase B contained 10 mM ammonium formate and 0.1% formic acid dissolved in 90% HPLC grade methanol. The flow rate of the mobile phase was 0.5 mL/minute. Rapamycin was eluted with a step gradient. The column was equilibrated with 100% mobile phase B. At 6.10 minutes after injection, the system was switched to 100% mobile phase A. Finally, at 15.1 minutes, the system was switched back to 100% mobile phase B in preparation for the next injection. The rapamycin transition was detected at 931.6 Da (precursor ion) and the daughter ion was detected at 864.5 Da. Ascomycin was detected at 809.574 Da and the daughter ion was 756.34 Da. Rapamycin was quantified in perfused mouse brain according to the following protocol. Briefly, 100 mg of calibrator and unknown brain samples were mixed by sonication (three 5 second bursts) with 10 μL of 0.5 μg/mL ascomycin (internal standard) and 300 μL of a solution containing 0.1% formic acid and 10 mM ammonium formate dissolved in 95% HPLC grade methanol. After sonication, the samples were vortexed vigorously for 2 minutes, and then centrifuged at 15,000 g for 5 minutes at 23oC (subsequent centrifugations were performed under the same conditions). Supernatants were transferred to 1.5 mL microfilterfuge tubes and spun at 15,000 g for 1 minute and then 40 μL of the final extracts were injected into the LC/MS/MS. The ratio of the peak area of rapamycin to that of the internal standard ascomycin (response ratio) for each unknown sample was compared against a linear regression of calibrator response ratios at 0, 1.78, 3.13, 6.25, 12.5, 50, and 100 pg/mg to quantify rapamycin. The concentration of rapamycin was expressed as pg/mg of brain tissue.
Animal Preparation for Functional Neuroimaging
Mice were anesthetized with 4.0% isoflurane for induction, and then maintained in a 1.2% isoflurane and air mixture using a face mask. Heart rate (90 to 130 b.p.m.), respiration rate, and rectal temperature (37±0.5°C) were continuously monitored. Heart rate and blood oxygen saturation level were recorded using a MouseOx system (STARR Life Science, Oakmont, PA, USA) and maintained within normal physiological ranges.
Cerebral Metabolic Rate of Glucose Measurements
Cerebral metabolic rate of glucose was measured using 18FDG PET methods (Focus 220 MicroPET, Siemens, Nashville, TN, USA). A quantity of 0.5 mCi of 18FDG dissolved in 1 mL of physiologic saline solution was injected through the tail vein. Forty minutes were allowed for 18FDG uptake before scanning. Animals were then moved to the scanner bed and placed in the prone position. Emission data were acquired for 20 minutes in a three-dimensional (3D) list mode with intrinsic resolution of 1.5 mm. For image reconstruction, three-dimensional positron emission tomography data was rebinned into multiple frames of 1-second duration using a Fourier algorithm. After rebinning the data, a three-dimensional image was reconstructed for each frame using a 2D filtered back projection algorithm. Decay and dead time corrections were applied to the reconstruction process. Cerebral metabolic rate of glucose was determined using the mean standardized uptake value equation: Standardized uptake value=(AxW)/Ainj, where A is the activity of the region of interest ( i.e., brain region in the study), W is the body weight of the mice, and Ainj is the injection dose of the 18FDG.38
Cerebral Blood Flow Measurements
Quantitative CBF (with units of mL/g per minute) was measured using MRI-based continuous arterial spin labeling techniques39, 40 on a horizontal 7T/30 cm magnet and a 40 G/cm BGA12S gradient insert (Bruker, Billerica, MA, USA). A small circular surface coil (ID=1.1 cm) was placed on top of the head and a circular labeling coil (ID=0.8 cm), built into the cradle, was placed at the heart position for continuous arterial spin labeling. The two coils were positioned parallel to each other, separated by 2 cm from center to center, and were actively decoupled. Paired images were acquired in an interleaved fashion with field of view=12.8 × 12.8 mm2, matrix=128 × 128, slice thickness=1 mm, 9 slices, labeling duration=2100 milliseconds, TR=3,000 milliseconds, and TE=20 milliseconds. Continuous arterial spin labeling image analysis employed codes written in Matlab (Natick, MA, USA) 39, 40 and STIMULATE software (University of Minnesota, Minneapolis, MN, USA) to obtain CBF.
Vascular Density Measurement
Magnetic resonance angiography was acquired using three-dimensional gradient-echo MR imaging with flow compensation with following parameters: repetition time/echo time=26/4.3 milliseconds; field of view =12.8 × 12.8 mm2; acquisition matrix=128 × 128 × 192; repetition n=4. The magnetic resonance angiography was taken before and after monocrystalline iron oxide nanoparticle (MION) injection. Monocrystalline iron oxide nanoparticle was injected through the tail vein at 30 mg/kg. Image analysis was performed using custom-written programs (Matlab; MathWorks, Natick, MA, USA). Blood vessel intensity was calculated as the change in transverse relaxivity between images obtained before and after monocrystalline iron oxide nanoparticle administration.
Apparent Diffusion Coefficient Measurements
The average MRI-based apparent diffusion coefficient was obtained by averaging three apparent diffusion coefficient maps acquired separately with diffusion-sensitive gradients applied along the x, y, or z direction. Data were acquired using four-segment, spin-echo, echo-planar images with field of view =12.8 × 12.8 mm2, 64 × 64 matrix; other parameters were spectral width=200 kHz, TR=2 second (90° flip angle), b=10 and 1269 seconds/mm2, TE=37.5 milliseconds, Δ=17.53 milliseconds, δ=5.6 milliseconds, diffusion gradient amplitude (G)=10 and 190 mT/m, and 16 averages.
In Vivo Optical Imaging Experiments
Details of experimental procedures were identical to our previously published protocols.41 Briefly, mice were anesthetized with volatile isofluorane through a nose cone (3% induction, 1.5% maintenance). The depth of anesthesia was monitored by regular checking of whisker movement and the pinch withdrawal reflex of the hind limb and tail. Also, during surgery and imaging, three main vital signs including heart rate, respiratory rate, and oxygen saturation were periodically assessed by use of the MouseOx system (STARR Life Sciences, Oakmont, PA, USA). Body temperature was maintained at 37oC by use of a feedback-controlled heating pad (Gaymar T/Pump, Stryker, Kalamazoo, MI, USA). Initially, the scalp was shaved, incised along the midline, and retracted to expose the dorsal skull. Then removal of periosteum by forceps and cleaning of skull by a sterile cotton swab were performed. A stainless steel head plate was glued (VetBond, 3M, St Paul, MN, USA) to the dorsal skull and screwed to a custom-made stereotaxic frame. To create a thin-skull cranial window over the somotosensory cortex, the skull was initially thinned with a high-speed electric drill (Fine Science Tools, Foster City, CA, USA) and subsequently thinned to approximate 50 μm by using a surgical blade under a dissecting microscope (Nikon SMZ800, Melville, NY, USA). The optimal thinness was indicated by the high transparency and flexibility of skull. Artificial cerebrospinal fluid was used to wash the thinned area and enable pial vasculature to be clearly visible through the window. In vivo imaging of cortical vasculature was performed by using an Olympus FV1000 MPE with a × 40 0.8 NA water-immersion objective (Nikon). To illuminate vasculature, FITC-dextran or Rhodamine-dextran dissolved in sterilized phosphate-buffered saline (300 μL, 10 mg/mL) was injected through the tail vein at the beginning of the experiments. To observe NO derived from blood vessels, the NO indicator dye DAF-FM (Molecular Probes, Invitrogen, Carlsbad, CA, USA) was dissolved in dimethyl sulfoxide, diluted in Rhodamine-dextran solution (250 μM), and introduced into blood vessels through tail vein injection. High-resolution z-stacks of cortical vasculature were sequentially acquired at different times. The NIH ImageJ plugins stackreg and turboreg were used to align the z-stacks or maximal intensity z-projections of z-stacks to facilitate identification and comparison of the same blood vessels. The diameter of blood vessels was analyzed with the Image J plugin vessel diameter. Rapamycin (250 μL, 10 mg/kg solution in phosphate-buffered saline) or the NO synthase inhibitor L-NAME (250 μL, 30 mg/kg solution in phosphate-buffered saline) were injected intraperitoneally. Acetylcholine (Ach) (300 μL, 7.5 μg/mL solution in phosphate-buffered saline), as a positive control for vasodilation, together with Rhodamine-dextran and DAF-FM was injected intravenously via tail vein.
Behavioral Testing
The Morris water maze42 was used to test spatial memory. All animals showed no deficiencies in swimming abilities, directional swimming, or climbing onto a cued platform during pretraining and had no sensorimotor deficits as determined with a battery of neurobehavioral tasks performed before testing. The procedure described by Morris42 was followed as described.32, 33, 35, 43 Experimenters were blind with respect to genotype and treatment. Briefly, mice were given a series of three trials in a light-colored tank filled with opaque water whitened by the addition of non-toxic paint at a temperature of 24.0±1.0°C. In the cued part of the protocol, mice were trained to find a 12 × 12 cm submerged platform (1 cm below water surface) marked with a colored pole that served as a landmark placed in different quadrants of the pool. Animals were released at different locations in each 60′ trial. If mice did not find the platform in 60 seconds, they were gently guided to it. After remaining on the platform for 15 to 20 seconds, the animals were removed and placed in a dry cage under a warm heating lamp. Five to 8 minutes later, each animal was given a second trial using a different release position. In the non-cued part of the protocol, the water tank was surrounded by opaque dark panels with geometric designs at approximately 30 cm from the edge of the pool, to serve as distal cues. The animals were trained to find the unmarked platform with three swims/day for 5 days after the same procedure described above but maintaining the position of the platform constant. At the end of training, a 45-second probe trial was administered in which the platform was removed from the pool. The number of times that each animal crossed the previous platform location was determined as a measure of platform location retention. During the course of testing, animals were monitored daily, and their weights were recorded weekly. Performance in all tasks was recorded by a computer-based video tracking system (Water2020, HVS Image, Buckingham, UK). Animals that spent more than 70% of trial time in thigmotactic swim were removed from the study. Data were analyzed offline by using HVS Image and processed with Microsoft Excel before statistical analyses.
Isolation of Brain Microvasculature
The brains from adult mice were dissected and rinsed with MCDB131 media with 2% fetal bovine serum (FBS) and 100 U/mL penicillin (P) and 100 μg/mL streptomycin (S; 1% P/S). The brain was transferred to a clean Petri dish and minced into small pieces with the same media. In a loose-fitting 7-mL dounce tissue grinder, the brain was homogenized with three strokes. An equal volume of MCDB131 media with 2% FBS and 1% P/S with 30% dextran (average molecular weight 68,800) was added to the homogenate for a final concentration of 15% dextran. The homogenate was centrifuged at 10,000 x g for 15 minutes using a swing bucket rotor. The pellet was collected and resuspended in 1x cell lysis buffer (Cell Signaling, Danvers, MA, USA). The lysate was sonicated briefly and centrifuged at 12,000 r.p.m. for 15 minutes. The supernatant was collected for western blot analysis.
Western Blotting and Aβ Determinations
Mice were killed by isoflurane overdose followed by cervical dislocation. Hemibrains were flash frozen. One hemibrain was homogenized in liquid N2 while the other was used in immunohistochemical determinations (5 to 7 per group). For Western blot analyses, proteins from soluble fractions of brain LN2 homogenates were resolved by SDS–PAGE (Invitrogen, Temecula, CA, USA) under reducing conditions and transferred to a polyvinylidene difluoride membrane, which was incubated in a 5% solution of non-fat milk or in 5% bovine serum albumin for 1 hour at 20°C. After overnight incubation at 4°C with anti-APP (6E10), anti-Ser1177 eNOS, and anti-eNOS (BD Transduction Laboratories, Franklin Lakes, NJ, USA, Cat #612392 and Cat#610296, 1:500 and 1:1,000, respectively) the blots were washed in TBS-Tween 20 (TBS-T) (0.02% Tween 20, 100 mmol/L Tris pH 7.5; 150 nmol/L NaCl) for 20 minutes and incubated at room temperature with appropiate secondary antibodies. The blots were then washed 3 times for 20 minutes each in TBS-T and then incubated for 5 minutes with Super Signal (Pierce, Rockford, IL, USA), washed again, and exposed to film or imaged with a Typhoon 9,200 variable mode imager (GE Healthcare, Waukesha, WI, USA).
Immunohistochemistry and Confocal Imaging of Fixed Tissues
Ten-micrometer coronal cryosections from snap-frozen brains were post-fixed in 4% paraformaldehyde and stained with Aβ-specific antibodies (6E10, 10 μg/mL) followed by AlexaFluor594-conjugated donkey anti-rabbit IgG (1:500, Molecular Probes, Invitrogen), and with biotinylated Lycopersicon esculentum (tomato) lectin (1:4,000, Vector Laboratories, Burlingame, CA) followed by strepdavidin-AlexaFluor488, conjugate (1:500, Molecular Probes, Invitrogen) and imaged with a laser scanning confocal microscope (Nikon Eclipse TE2000-U) using a 488 Argon laser and a 515/30 nm filter for the AlexaFluor488, fluorophore, and a 543.5 Helium-neon laser and a 590/50 nm filter for the AlexaFluor594 fluorophore. Stacks of confocal images for each channel were obtained separately at z=0.15 μm using a × 60 objective. Z-stacks of confocal images were processed using Volocity software (Perkin Elmer, MA, USA). Images were collected in the hilus of the dentate gyrus (or the stratum radiatum of the hippocampus immediately beneath the CA1 layer) at Bregma ∼−2.18. The MBL Mouse Brain Atlas was used for reference.
Microhemorrhages
Ten micrometer coronal cryosections from snap frozen brains postfixed in 4% paraformaldehyde were washed × 3 in Tris-buffered saline (TBS; Fisher BioReagents). Sections were then washed 3 times in distilled water and immersed in 2% potassium hexacyanoferrate(III) trihydrate (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and 2% hydrochloric acid (Sigma Life Sciences). After three washes in TBS, sections were coverslipped with ProLong Gold antifade reagent with 4',6-diamidino-2-phenylindole (Life Technologies, Carlsbad, CA, USA). The number of microhemorrhages per section was counted at Bregma approximately −2.18 using a × 40 objective on a Zeiss Axiovert 200M microscope (Carl Zeiss AG, Oberkochen, Germany) using four sections per animal, and numbers of microhemorrhages were averaged for each animal.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism (GraphPad, San Diego, CA, USA) and StatView. Individual data points from different immunoblot experiments were scaled by subtracting the experiment-wide mean and dividing by the experiment-wide standard error. In two-variable experiments, two-way analysis of variance followed by Bonferroni's post hoc tests were used to evaluate the significance of differences between group means. When analyzing one-variable experiments with more than 2 groups, significance of differences among means was evaluated using one-way analysis of variance followed by Tukey's post hoc test. Evaluation of differences between two groups was done using Student's t-test. Values of P<0.05 were considered significant.
Ethics and Guidelines Statement
All studies reported were in compliance with all rules and regulations for the humane use of animals in research and were approved by the University of Texas Health Science Center Institutional Animal Care and Use Committee. To minimize pain or discomfort, killing was by isoflurane overdose, which is significantly less stressful than other methods such as asfixiation by CO2 overdose. The Animal Research Reporting In Vivo Experiments (ARRIVE) guidelines as described by Kilkenny et al44 have been followed in the preparation of this manuscript.
Results
Improved Memory in Rapamycin-treated Alzheimer's Disease Mice
Alzheimer's disease mice and unaffected littermates were treated with rapamycin after the onset of robust AD-like impairments at 7 months of age32, 35, 36 for a total of 16 weeks. Average rapamycin levels in different brain regions of mice chronically fed with rapamycin ranged from 0.98 to 2.40 pg/mg tissue weight (Supplementary Figure 1), resulting in concentrations of rapamycin in brain tissues ranging from ∼1 to 3 pmol/L. To determine whether chronically treating AD mice with rapamycin after the onset of AD-like deficits would affect spatial learning and memory, we trained and tested control- and rapamycin-fed AD and WT littermate mice in the Morris water maze.35, 42, 45 As described,35, 45, 46 control-fed symptomatic AD mice showed significant deficits during spatial training (Figure 1A). Learning deficits of AD mice, however, were partially abrogated by rapamycin treatment (Figure 1A). Prominently, acquisition rates for rapamycin-treated AD mice were restored to levels indistinguishable from those of WT littermates' with increasingly improved performance as training progressed (Figure 1A). In contrast, control-fed AD mice showed worsening performance as training progressed, a behavioral pattern associated with increased anxiety levels in animals that do not learn well.45, 47 Moreover, while memory was significantly impaired in control-fed AD mice (Figure 1B), as previously described,35, 45, 46 memory of the trained location for the escape platform in rapamycin-treated mice was indistinguishable from that of WT littermates' and was significantly improved compared with control-fed AD mice (Figure 1B). Thus, chronic rapamycin treatment started after the onset of AD-like cognitive deficits ameliorated spatial learning and restored spatial memory in symptomatic AD mice.
Figure 1.

Improved memory and restored cerebral blood flow (CBF) in AD mice treated with rapamycin after the onset of disease. (A) Spatial learning. While learning in AD mice was impaired35, 45, 46 (*P<0.001 and P<0.01, Bonferroni's post hoc test applied to a significant effect of genotype and treatment, F(3,188)=6.04, P=0.0014, repeated measures (RM) two-way ANOVA), performance of rapamycin-fed AD mice was indistinguishable from WT littermates. No significant interaction was observed between day number and genotype (P=0.96), thus genotype and treatment had the same effect at all times during training. Overall, learning was effective in all groups (F(4,188)=3.36, P=0.01, RM two-way ANOVA). (B) Spatial memory is restored by rapamycin treatment. While memory in control-fed AD mice was impaired35, 45, 46 (P values as indicated, Tukey's test applied to a significant effect of genotype and treatment (P<0.0001), one-way ANOVA), memory in rapamycin-fed AD mice was indistinguishable from WT littermate groups and was significantly improved compared with control-fed AD mice (P=0.03). (C–G) Rapamycin restores CBF in AD mice. (C) CBF maps and regional CBF maps (E) of representative control- and rapamycin-treated WT and AD mice obtained by MRI. (D) Decreases in CBF in AD mice are abrogated by rapamycin treatment (P as indicated, Bonferroni's test on a significant effect of genotype and treatment on CBF, F(1,16)=14.54, P=0.0015, two-way ANOVA). (F and G) Decreased hippocampal (F) but not the thalamic (G) CBF in AD mice is restored by rapamycin treatment (P as indicated, Bonferroni's test on a significant effect of treatment on CBF, F(1,16)=13.62, P=0.0020, two-way ANOVA). Data are means±s.e.m. Panels (A–B), n=10–17 per group. Panels (C–G), n=6 per group.
Restored Cerebral Blood Flow in Rapamycin-treated Alzheimer's Disease Mice
Because cerebral hypoperfusion is an early event in the pathogenesis of AD,48, 49 we examined the effects of chronic rapamycin treatment on hemodynamic function in brains of AD mice using high-field magnetic resonance imaging (MRI). As shown in Figures 1C and D, control-fed AD animals had significantly lower global CBF compared with WT littermates. In contrast, global CBF in rapamycin-treated AD mice was indistinguishable from that of WT littermate groups (Figures 1C and D). At its early and moderate stages, AD is associated with synaptic dysfunction in the entorhinal cortex and hippocampus while other brain regions such as the thalamus are largely spared. In agreement with these observations, hippocampal, but not thalamic CBF was reduced in control-treated AD mice (Figures 1E–G). Chronic rapamycin treatment, however, restored hippocampal CBF to levels indistinguishable from those of WT littermates (Figures 1E–G).
Increased Vascular Density without Changes in Glucose Metabolism in Rapamycin-treated Alzheimer's Disease Mice
Because CBF can change as a consequence of changes in tissue metabolism, we next determined cerebral glucose uptake in control- and rapamycin-fed AD mice using positron emission tomography. In spite of the observed differences in CBF, cerebral metabolic rate of glucose (CMRGlc) was not significantly different between control- and rapamycin-treated groups (Figures 2A and B) as had been shown previously for APP23 or APP(Tg2576) mice with high Aβ loads.50, 51 Thus, differences in CBF among experimental groups could not be explained by changes in brain metabolism. Changes in CBF, however, can also be brought about by changes in cerebral vascularization. Thus, we next used high-resolution magnetic resonance angiography to measure vascular density in control- and rapamycin-fed AD mouse brains. Magnetic resonance angiography studies of control-treated AD mice showed a pronounced reduction in cerebral vessel density with respect to WT littermates. This reduction in brain vascularity, however, was completely abrogated by rapamycin treatment (Figures 2C and D). Thus, decreases in CBF in AD mice likely arise from cerebrovascular damage, and restored CBF reflects the preservation of vascular density as a result of rapamycin treatment.
Figure 2.

Increased vascular density without changes in glucose metabolism in rapamycin-treated brains of AD mice. (A) Cerebral metabolic rate of glucose (CMRGlc) maps of representative control- and rapamycin-treated WT and AD mice obtained by positron emission tomography. (B) Cerebral metabolic rate of glucose as standardized uptake values (SUV) for the region of interest were not different among experimental groups (F(1,20)=0.77, P=0.39 for the effect of genotype and F(1,20)=3.63, P=0.071 for the effect of treatment, two-way analysis of variance). (C) Magnetic resonance angiography images of brains of rapamycin-treated WT and AD mice. Representative regions showing loss of vasculature in control-treated AD mice and its restoration in rapamycin-treated animals are denoted by arrows. (D) Decreased cerebral vessel density in control-treated AD mice is abrogated by rapamycin treatment (P as indicated, Bonferroni's post hoc test applied to a significant effect of treatment on vascular density, F(1,16)=24.47, P=0.0001, two-way ANOVA). Data are means±s.e.m. n=6 per group.
Reduced Cerebrovascular Amyloid Angiopathy and Aß Plaques in Rapamycin-Treated Alzheimer's Disease Mice
Impaired clearance of Aß leads to its accumulation on blood vessels, ultimately resulting in CAA and plaque deposition.15 Thus, we next determined whether rapamycin affected Aβ plaques. As shown in Figures 3A–F, Aβ deposits present in control-fed symptomatic AD mice were significantly decreased in animals fed with a rapamycin-supplemented chow. Consistent with this observation, numbers of fibrillar, Thioflavin S-positive amyloid deposits were decreased in rapamycin-fed AD mouse brains (Figure 3G). In agreement with their high-plaque burden, control-fed AD mice showed decreased tissue integrity as evidenced by higher apparent water diffusion coefficients (Figures 3H and I). In contrast, rapamycin-treated AD mice showed apparent water diffusion coefficient values that were not significantly different from those of WT littermates. These results indicate that integrity of brain tissue was preserved (Figures 3H and I), likely as a result of decreased amyloid load, in rapamycin-treated AD mice.
Figure 3.

Reduced cerebral amyloid angiopathy (CAA) and Aß plaques in rapamycin-treated AD mice. (A–F). Reduced Aß plaques in rapamycin-treated AD mice. (A and B) Representative images of hippocampi of control- (A) and rapamycin-treated (B) AD mice incubated with an Aß-specific antibody. (C and D) secondary antibodies only. (D) 4',6-diamidino-2-phenylindole fluorescence of the field in C. (E and F) Quantitative analyses of Aß immunoreactivity (P as indicated). (G) Decreased numbers of Thioflavin S-positive amyloid deposits in rapamycin-treated AD mice. (H–I) Improved brain tissue integrity in rapamycin-treated AD mice. (H) Representative images of heat maps displaying apparent diffusion coefficients for water in brains of control- and rapamycin-treated mouse AD or WT brains. (I) Quantitative analyses of whole-brain average diffusivity values. (J–L) Reduced CAA in rapamycin-treated AD mouse brains. Representative maximum intensity projections of stacks of confocal images of control- (J) and rapamycin- (K) treated AD mouse brain sections reacted with Aß-specific antibodies and with tomato lectin to illuminate brain vasculature. (L) Quantitative analyses of colocalization of Aß immunoreactivity and tomato lectin labeling of brain vasculature indicate reduced Aß deposition on vessels in rapamycin-treated AD mice (P as indicated). (M and N) Reduced microhemorrhages in rapamycin-treated AD mouse brains. (M) Representative hemosiderin deposit. (N) Quantitative analyses of numbers of hemosiderin deposits (P as indicated). Significance of differences between group means was determined using two-tailed unpaired Student's t-test, or one-way ANOVA followed by Tukey's multiple comparisons test or two-way ANOVA followed by Bonferroni's test. Data are means±s.e.m. n=6 to 8 per experimental group.
The observed decrease in Aβ deposition and improved tissue integrity in rapamycin-treated AD mouse brains may reflect increased clearance of Aβ through preserved vasculature. To test this hypothesis, we quantified Aβ associated with brain blood vessels (CAA) in control- and rapamycin-treated brains. Cerebral amyloid angiopathy was pronouncedly reduced in rapamycin-treated AD mice (Figures 3J–L). Because CAA is accompanied by microhemorrhage,3, 52 we looked for hemosiderin deposits, indicative of previous microhemorrhage, in brains of control- and rapamycin-fed animals. Hemosiderin deposits (Figure 3M) were significantly increased in AD mouse brains (Figure 3N) as previously shown.3 In contrast, numbers of hemosiderin deposits in rapamycin-treated AD mice were not significantly different from those observed in WT littermates (Figure 3N), suggesting that decreased CAA prevented microvessel disruption in rapamycin-treated AD mouse brains.
Rapamycin-Induced Nitric Oxide Synthase Activity is Required for Restoration of Cerebral Blood Flow in Alzheimer's Disease Mouse Brains
It has been hypothesized that CBF reduction in AD results from loss of cholinergic innervation of cerebral blood vessels, resulting in hypercontractility, hypoperfusion, and CAA in AD brains.48 To examine the effects of rapamycin on cerebral vasomotion, we used in vivo 2-photon microscopy on cortical vessels of control- or rapamycin-treated mice. Rapamycin treatment induced a 23% to 35% increase in diameter of small and medium-sized cortical vessels (Figures 4A and B). This response was roughly equivalent to one-third of the response observed after treatment with ACh Figure 4C), a powerful vasodilator, and was comparable to that observed for other known vasodilators such as substance P.53 Endothelium-derived NO is the dominant effector by which EC signal to vascular smooth muscle cells to regulate blood flow. To determine whether rapamycin-induced dilation of cortical vessels was associated with NO release, we used an NO-sensitive fluorescent probe, DAF-FM, to monitor NO production in cortical vessels of control- and rapamycin-treated mice. Treatment with rapamycin resulted in local increases in NO production that reached a maximum at 5 to 7 minutes after treatment (Figure 4D) and were sustained for 18 minutes before returning to baseline (data not shown). Vessel segments that showed increases in NO release subsequently increased in diameter (Figure 4D). Treatment with ACh, however, resulted in an uniform increase in NO production along cortical vessels that resulted in subsequent uniform increases in vessel diameter (Figure 4F).Consistent with the observed local increases in NO production in brain vasculature after rapamycin or ACh treatment, both drugs induced phosphorylation of endothelial NO synthase (eNOS) at Ser1176, which is critical for its activation.54 Ser1176 phosphorylation of eNOS after rapamycin and after ACh treatment proceeded with a comparable time course and showed maximal Ser1176-eNOS phosphorylation 1 minute after administration (Figures 4G and H). To determine whether rapamycin-induced NO release and vasodilation were dependent on NO synthase (NOS) activity, we pretreated experimental animals with a NOS inhibitor (L-NAME, which becomes a functional inhibitor, L-NAA, after hydrolysis by esterases) immediately before the administration of rapamycin or ACh. Pretreatment with L-NAME abrogated both NO release at brain endothelium and vasodilation induced by rapamycin administration (Figure 4E) as well as by ACh (not shown) as previously demonstrated.54 The observed localization and time course of NO release (Figure 4D), which rule out an involvement of the inducible isoform of NOS (iNOS), and the phosphorylation of eNOS at Ser1176 (Figures 4G and H) suggest that NO release and vasodilation by acute rapamycin treatment may depend on eNOS activation.
Figure 4.

Rapamycin-induced nitric oxide (NO)-dependent vasodilation in brain. (A) Rapamycin-induced cortical vasodilation. In vivo imaging of cortical vasculature illuminated by FITC-Dextran (green). Arrows indicate areas of maximal vasodilatory effect 10 minutes after rapamycin administration (tabbed white lines). (B) Quantitative analyses of changes in diameter for cortical vessels of different sizes (P as indicated, Bonferroni's test applied to a significant effect of treatment, F(1,20)=154.12, P<0.0001, two-way ANOVA). (C) Quantitative analyses of changes in diameter for cortical vessels of different sizes 10 minutes after treatment with acetylcholine (ACh, P as indicated, Bonferroni's test applied to a significant effect of treatment, F(1,15)=2900.20, P<0.0001, two-way ANOVA). (D) Rapamycin-induced vasodilation is preceded by NO release. Arrowheads indicate regions of local NO release by DAF-FM fluorescence (green) followed by dilation of rhodamine-dextran labeled vasculature (red) in vivo. (E) Rapamycin-induced vasodilation requires NO synthase activity. L-NG-Nitroarginine methyl ester (L-NAME) administration abolished rapamycin-induced NO release (DAF-FM fluorescence) and dilation of cortical vasculature. (F) Acetylcholine-induced vasodilation is preceded by NO release. Uniform NO release (DAF-FM fluorescence, green) preceded vasodilation induced by ACh. (G and H) Ser1176 phosphorylation of endothelial nitric oxide synthase (eNOS) in brain vasculature. (G) eNOS and phospho-eNOS (p-eNOS, Ser1176) immunoreactivity in lysates of vasculature purified from brains at the indicated times after injection of rapamycin or ACh. (H) The relative ratio of p-eNOS(Ser1176) to total eNOS was quantified (P as indicated, Tukey's test applied to a significant effect of treatment, P<0.0052, one-way ANOVA). n=3 to 4 per group. (I) NO synthase activity is required for rapamycin-induced preservation of cerebral blood flow (CBF). Four weeks of intermittent L-NAME administration (once every other day) abolished rapamycin-mediated preservation of CBF in AD mice (P as indicated, Tukey's test applied to a significant effect of treatment, P<0.0001, one-way ANOVA). n=6 per group. (J and K) Chronic rapamycin treatment does not affect eNOS levels in AD and WT mouse brains. (J) Endothelial nitric oxide synthase and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) immunoreactivity in lysates of brains from control- and rapamycin-treated mouse brains. (K) Quantitative analyses of eNOS immunoreactivity normalized to GAPDH levels. n=4 per group. Data are means±s.e.m.
If the restoration of CBF (Figures 1C–G and Figures 2C and D) in AD mice chronically treated with rapamycin was dependent on rapamycin-induced NOS activation, then inhibition of NOS activity should abolish the restoration of brain perfusion in brains of rapamycin-treated AD mice. To test this hypothesis, we treated AD mice fed with rapamycin for 16 weeks with vehicle or with L-NAME for 4 additional weeks and measured CBF in both groups. In contrast to rapamycin-fed AD animals that were treated with vehicle (Figure 4I), rapamycin-fed mice that were treated with L-NAME showed CBF deficits comparable to control-fed AD mice, indicating that inhibition of NOS activity abolished the protective effects of chronic rapamycin treatment. Because L-NAME can inhibit eNOS as well neuronal NOS (nNOS) and iNOS, our data indicate that NO synthase activity is required for rapamycin-dependent preservation of CBF in AD mice, but cannot unequivocally identify the NOS isoform(s) involved. A significant role of nNOS in the restoration of CBF by chronic rapamycin may be unlikely, however, because the BBB permeability of L-NAME is a matter of debate.55 Endothelial nitric oxide synthase abundance was unchanged in rapamycin-treated AD mouse brains (Figures 4J and K), ruling out an effect of chronic rapamycin treatment on levels of eNOS expression in brain vasculature.
Discussion
Vascular dysfunction has a critical role in the pathogenesis of AD.2, 3 In addition to its well-defined deleterious effects on neuronal function and network regulation, abnormally high levels of Aβ damage the cerebral vasculature through its deposition and aggregation on brain vessels, leading to microhemorrhage and vascular disintegration in AD patients. Vascular damage diminishes CBF, and consequently the supply of oxygen and nutrients to the brain, causing further neuronal dysfunction and degeneration. Moreover, because low Aβ levels in normal brain are maintained by the continuous removal of the peptide through the vasculature, vascular damage drives further increases of Aβ in AD brain. It has been proposed that vascular dysfunction may be the first ‘hit' in a ‘two-hit' process that leads to AD,12 in which the second hit is the abnormal accumulation of Aβ.
Because immunotherapy approaches to AD have shown adverse effects consistent with breakdown of BBB (such as a redistribution of amyloid from parenchyma to vasculature and increased microhemorrhage in mouse models,56, 57, 58 as well as inflammation, vasogenic edema, and microhemorrhage in some AD patients,59, 60) it has been proposed61 that Aβ may have a protective role and may normally act to stabilize the BBB. In our studies, however, improved cerebral perfusion and vascular density were associated with decreased plaque deposition as well as with decreases in CAA and reduced microhemorrhage, which are not consistent with expected outcomes of chronic BBB destabilization.61
Our data indicate that chronically treating transgenic hAPP(J20) mice (AD mice) with rapamycin after the onset of moderate AD-like cognitive deficits is sufficient to improve memory and to ameliorate AD-like vascular damage. Consistent with these observations, restoration of CBF and vascular density in AD mice led to decreased Aβ deposition in brain vessels, lower Aβ plaque load, and reduced incidence of microbleeds, suggesting that decreasing Aβ deposition in vasculature preserves its functionality and integrity, enabling the continuing clearance of Aβ from brain, thus resulting in decreased plaque load (Figure 5). Because memory deficits were ameliorated in rapamycin-treated AD mice, our data suggest that preservation of CBF and vascular density may be sufficient to improve cognitive outcomes in AD mice. We and others had previously demonstrated, however, that chronic inhibition of TOR by rapamycin enhances autophagy,31, 32 and we have shown that it upregulates the chaperone response33 in AD mouse brains. Thus, we cannot rule out a contribution of rapamycin-induced enhanced proteostasis in the observed improvement in cognitive outcomes in rapamycin-treated symptomatic AD mice.
Figure 5.
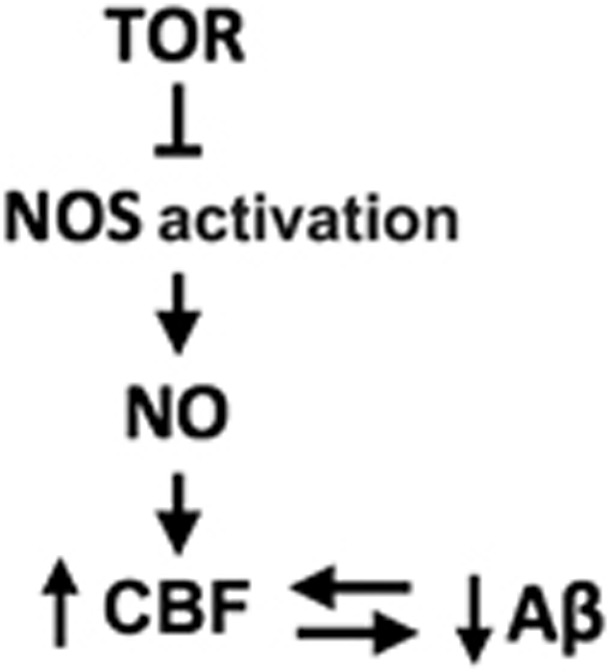
Proposed pathway for target-of-rapamycin (TOR) inhibition of nitric oxide synthase (NOS)-dependent regulation of cerebral blood flow (CBF) in AD mouse brains.
As had been shown previously for different mouse models of AD,50, 51 we did not observe significant changes in brain metabolism in AD mice. Other studies, however, have shown increased cerebral glucose uptake in mice modeling AD62 that was explained by increased glucose uptake at amyloid plaques, rather than in adjacent amyloid-free cerebral parenchyma. Because we did not observe increased cerebral metabolic rate of glucose in control-treated AD mice as compared with WT littermates, our results suggest that glucose metabolism is not overtly affected by the presence of plaques in brain tissues of hAPP(J20) mice.
It has been suggested that failure of endothelium-dependent vasodilation is a ‘hotspot' for aging.11 Acute rapamycin treatment induced relaxation of cortical vessels that was preceded by the phosphorylation of eNOS at Ser1176 and subsequent NO release, and rapamycin-induced vasodilation required NO synthase activity in a manner comparable to ACh. Thus, our results indicate that rapamycin acts as a vasodilator, in agreement with prior reports.63, 64 A link between the inhibition of mTOR and the activation of eNOS had been suggested by studies showing that Akt, which phosphorylates eNOS and increases NO production,65 can be activated by rapamycin treatment,66, 67 and conversely, that activation of mTOR results in Akt inhibition.66, 68 Restoration of vascular density in AD mice required NO synthase activity, suggesting that mTOR has a critical role in the inhibition of NO release in brain vascular endothelium during the progression of AD-like disease in mice (Figure 5). Of note, rapamycin did not affect vascular density or CBF in WT mice, suggesting that restoration of vascular integrity by chronic mTOR inhibition involves a pathway activated by chronic injury54 such as that associated with the progression of neurodegeneration in AD mice. Because mTOR controls key metabolic functions in most cell types, and pharmacologically inhibiting mTOR extends lifespan and healthspan,18, 19, 20 retarding multiple, but not all, aspects of aging in mice, it is possible that mTOR may be involved in several different, specific processes of complex disease mechanisms that enable neurodegeneration. The present studies suggest that one of these processes is endothelial cell dysfunction causally linked to vascular aging. mTOR-dependent vascular deterioration may thus be a critical feature of brain aging that enables AD.
In summary, our data indicate that chronic inhibition of mTOR by rapamycin, an intervention that extends lifespan in mice, negates vascular breakdown through the activation of NOS, likely eNOS, in brain vascular endothelium and improves cognitive function when administered after onset of AD-like deficits in transgenic mice modeling the disease. Although rapamycin or rapamycin analogs have side effects that are undesirable in the elderly, it is possible that therapies in which the drug is used in ‘on-off' schedules may be devised for the treatment of early or moderate AD. Also, studies using drugs different from rapamycin that inhibit the TOR pathway and are devoid of its side effects may be warranted.
Footnotes
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
This work was supported by an Ellison Medical Foundation New Scholar Award in Aging, a William & Ella Owens Medical Research Foundation Grant and a UTHSCSA University Research Council Award to VG, an UTHSCSA Institute for Integration of Medicine and Science Award to ALL, the NIA Interventions Testing Center (U01AG022307) to RS, RC2AG036613 NIH Recovery Act Grand Opportunities “GO” grant to AR, and by 5T32AG021890 NIA Biology of Aging Training Grant Fellowship to SH. Images were generated in the Core Optical Imaging Facility, which is supported by UTHSCSA and NIH-NCI P30 CA54174 (CTRC at UTHSCSA). We recognize the support of the San Antonio Nathan Shock Aging Center (P30AG-13319, AR) and the VA Neurodegeneration Research Center (REAP) from the Research and Development Service of the Department of Veterans Affairs to AR and RS.
Supplementary Material
References
- Breteler MM. Vascular involvement in cognitive decline and dementia. Epidemiologic evidence from the Rotterdam Study and the Rotterdam Scan Study. Ann N Y Acad Sci. 2000;903:457–465. doi: 10.1111/j.1749-6632.2000.tb06399.x. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Taylor JW, DeMattos RB, Bales KR, Paul SM, Parsadanian M, et al. Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci. 2003;23:7889–7896. doi: 10.1523/JNEUROSCI.23-21-07889.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Boyle PA, Arvanitakis Z, Bienias JL, Bennett DA. Subcortical infarcts, Alzheimer's disease pathology, and memory function in older persons. Ann Neurol. 2007;62:59–66. doi: 10.1002/ana.21142. [DOI] [PubMed] [Google Scholar]
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–817. [PubMed] [Google Scholar]
- Park JH, Seo SW, Kim C, Kim GH, Noh HJ, Kim ST, et al. Pathogenesis of cerebral microbleeds: in vivo imaging of amyloid and subcortical ischemic small vessel disease in 226 individuals with cognitive impairment Ann Neuroladvance online publication, 29 January 2013; doi: 10.1002/ana.23845e-pub ahead of print); PMID: 23495089. [DOI] [PubMed]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, et al. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, et al. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer's disease model. Ann Neurol. 2000;47:739–747. [PubMed] [Google Scholar]
- Tong XK, Nicolakakis N, Kocharyan A, Hamel E. Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer's disease. J Neurosci. 2005;25:11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Larionov S, Staufenbiel M, Zurbruegg S, Beckmann N. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol Aging. 2009;30:1936–1948. doi: 10.1016/j.neurobiolaging.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Marin J, Rodriguez-Martinez MA. Age-related changes in vascular responses. Exp Gerontol. 1999;34:503–512. doi: 10.1016/s0531-5565(99)00029-7. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prisby RD, Dominguez JM, 2nd, Muller-Delp J, Allen MR, Delp MD. Aging and estrogen status: a possible endothelium-dependent vascular coupling mechanism in bone remodeling. PLoS One. 2012;7:e48564. doi: 10.1371/journal.pone.0048564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin S, Villien M, Moreaud O, Tropres I, Keignart S, Chipon E, et al. Impaired cerebral vasoreactivity to CO2 in Alzheimer's disease using BOLD fMRI. Neuroimage. 2011;58:579–587. doi: 10.1016/j.neuroimage.2011.06.070. [DOI] [PubMed] [Google Scholar]
- Bell RD, Deane R, Chow N, Long X, Sagare A, Singh I, et al. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Guo H, Chow N, Sallstrom J, Bell RD, Deane R, et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat Med. 2005;11:959–965. doi: 10.1038/nm1287. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, et al. Rapamycin slows aging in mice. Aging Cell. 2012;11:675–682. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66:191–201. doi: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Kim HH, Hiroi Y, Sawada N, Salomone S, Benjamin LE, et al. Obesity increases vascular senescence and susceptibility to ischemic injury through chronic activation of Akt and mTOR. Sci Signal. 2009;2:ra11. doi: 10.1126/scisignal.2000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepuri G, Velagapudi S, Xiong Y, Rajapakse AG, Montani JP, Ming XF, et al. Positive crosstalk between arginase-II and S6K1 in vascular endothelial inflammation and aging. Aging Cell. 2012;11:1005–1016. doi: 10.1111/acel.12001. [DOI] [PubMed] [Google Scholar]
- Chauhan A, Sharma U, Jagannathan NR, Reeta KH, Gupta YK. Rapamycin protects against middle cerebral artery occlusion induced focal cerebral ischemia in rats. Behav Brain Res. 2011;225:603–609. doi: 10.1016/j.bbr.2011.08.035. [DOI] [PubMed] [Google Scholar]
- Zhang W, Khatibi NH, Yamaguchi-Okada M, Yan J, Chen C, Hu Q, et al. Mammalian target of rapamycin (mTOR) inhibition reduces cerebral vasospasm following a subarachnoid hemorrhage injury in canines. Exp Neurol. 2012;233:799–806. doi: 10.1016/j.expneurol.2011.11.046. [DOI] [PubMed] [Google Scholar]
- Parlar A, Can C, Erol A, Ulker S. Posttransplantation therapeutic rapamycin concentration protects nitric oxide-related vascular endothelial function: comparative effects in rat thoracic aorta and coronary endothelial cell culture. Transplant Proc. 2010;42:1923–1930. doi: 10.1016/j.transproceed.2010.03.134. [DOI] [PubMed] [Google Scholar]
- Corbin F, Blaise GA, Parent M, Chen H, Daloze PM. Effect of rapamycin on rat aortic ring vasomotion. J Cardiovascul Pharmacol. 1994;24:813–817. doi: 10.1097/00005344-199424050-00017. [DOI] [PubMed] [Google Scholar]
- Horigome M, Kumazaki S, Hattori N, Kasai H, Horigome M, Aizawa K, et al. Noninvasive evaluation of coronary endothelial function following sirolimus-eluting stent implantation by using positron emission tomography. Cardiology. 2009;114:157–163. doi: 10.1159/000226093. [DOI] [PubMed] [Google Scholar]
- Togni M, Windecker S, Cocchia R, Wenaweser P, Cook S, Billinger M, et al. Sirolimus-eluting stents associated with paradoxic coronary vasoconstriction. J Am Coll Cardiol. 2005;46:231–236. doi: 10.1016/j.jacc.2005.01.062. [DOI] [PubMed] [Google Scholar]
- Kotani J, Awata M, Nanto S, Uematsu M, Oshima F, Minamiguchi H, et al. Incomplete neointimal coverage of sirolimus-eluting stents: angioscopic findings. J Am Coll Cardiol. 2006;47:2108–2111. doi: 10.1016/j.jacc.2005.11.092. [DOI] [PubMed] [Google Scholar]
- Brugaletta S, Heo JH, Garcia-Garcia HM, Farooq V, van Geuns RJ, de Bruyne B, et al. Endothelial-dependent vasomotion in a coronary segment treated by ABSORB everolimus-eluting bioresorbable vascular scaffold system is related to plaque composition at the time of bioresorption of the polymer: indirect finding of vascular reparative therapy. Eur Heart J. 2012;33:1325–1333. doi: 10.1093/eurheartj/ehr466. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce A, Podlutskaya N, Halloran JJ, Hussong SA, Lin PY, Burbank R, et al. Overexpression of heat shock factor 1 phenocopies the effect of chronic inhibition of TOR by rapamycin and is sufficient to ameliorate Alzheimer's-like deficits in mice modeling the disease. J Neurochem. 2012;124:880–893. doi: 10.1111/jnc.12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S, Caccamo A, Medina DX, Benavides AD, Javors MA, Kraig E, et al. Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1beta and enhancing NMDA signaling. Aging Cell. 2012;11:326–335. doi: 10.1111/j.1474-9726.2011.00791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, et al. Reversal of Alzheimer's-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci USA. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, et al. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Chang CH, Ho CL, Chen LC, Wu YH, Chen JT, et al. Early detection of tumor response by FLT/microPET Imaging in a C26 murine colon carcinoma solid tumor animal model. J Biomed Biotechnol. 2011;2011:535902. doi: 10.1155/2011/535902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong TQ, Silva AC, Lee SP, Kim SG. Functional MRI of calcium-dependent synaptic activity: cross correlation with CBF and BOLD measurements. Magn Reson Med. 2000;43:383–392. doi: 10.1002/(sici)1522-2594(200003)43:3<383::aid-mrm10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Muir ER, Shen Q, Duong TQ. Cerebral blood flow MRI in mice using the cardiac-spin-labeling technique. Magn Reson Med. 2008;60:744–748. doi: 10.1002/mrm.21721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Watts LT, Holstein DM, Prajapati SI, Keller C, Grass EH, et al. Purinergic receptor stimulation reduces cytotoxic edema and brain infarcts in mouse induced by photothrombosis by energizing glial mitochondria. PLoS One. 2010;5:e14401. doi: 10.1371/journal.pone.0014401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Zhang J, Gorostiza OF, Tang H, Bredesen DE, Galvan V. Reversal of learning deficits in hAPP transgenic mice carrying a mutation at Asp664: a role for early experience. Behav Brain Res. 2010;206:202–207. doi: 10.1016/j.bbr.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS biology. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan V, Zhang J, Gorostiza OF, Banwait S, Huang W, Ataie M, et al. Long-term prevention of Alzheimer's disease-like behavioral deficits in PDAPP mice carrying a mutation in Asp664. Behav Brain Res. 2008;191:246–255. doi: 10.1016/j.bbr.2008.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Venero C, Tilling T, Hermans-Borgmeyer I, Herrero AI, Schachner M, Sandi C. Water maze learning and forebrain mRNA expression of the neural cell adhesion molecule L1. J Neurosci Res. 2004;75:172–181. doi: 10.1002/jnr.10857. [DOI] [PubMed] [Google Scholar]
- Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 2009;118:103–113. doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre JC. Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 2004;3:184–190. doi: 10.1016/S1474-4422(04)00683-0. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, et al. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci. 2006;26:1343–1354. doi: 10.1523/JNEUROSCI.4236-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntner C, Kesner AL, Bauer M, Kremslehner R, Wanek T, Mandler M, et al. Limitations of small animal PET imaging with [18F]FDDNP and FDG for quantitative studies in a transgenic mouse model of Alzheimer's disease. Mol Imag Biol. 2009;11:236–240. doi: 10.1007/s11307-009-0198-z. [DOI] [PubMed] [Google Scholar]
- Greenberg SM. Cerebral amyloid angiopathy: prospects for clinical diagnosis and treatment. Neurology. 1998;51:690–694. doi: 10.1212/wnl.51.3.690. [DOI] [PubMed] [Google Scholar]
- Champion HC, Kadowitz PJ. Vasodilator responses to acetylcholine, bradykinin, and substance P are mediated by a TEA-sensitive mechanism. Am J Physiol. 1997;273 (1 Pt 2:R414–R422. doi: 10.1152/ajpregu.1997.273.1.R414. [DOI] [PubMed] [Google Scholar]
- Schleicher M, Yu J, Murata T, Derakhshan B, Atochin D, Qian L, et al. The Akt1-eNOS axis illustrates the specificity of kinase-substrate relationships in vivo. Sci Signal. 2009;2:ra41. doi: 10.1126/scisignal.2000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirtz H.Letter regarding article by Kaufmann et al. ‘systemic inhibition of nitric oxide synthase unmasks neural constraint of maximal myocardial blood flow in humans' Circulation 2005111e278–e279.author reply e278-9. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, et al. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, et al. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RS, Sperling RA, Safirstein B, Motter RN, Pallay A, Nichols A, et al. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 2010;24:198–203. doi: 10.1097/WAD.0b013e3181c53b00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Jack CR, Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimer's Dement. 2011;7:367–385. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood CS, Bishop GM, Perry G, Smith MA. Amyloid-beta: a vascular sealant that protects against hemorrhage. J Neurosci Res. 2002;70:356. doi: 10.1002/jnr.10388. [DOI] [PubMed] [Google Scholar]
- Poisnel G, Herard AS, El Tannir El Tayara N, Bourrin E, Volk A, Kober F, et al. Increased regional cerebral glucose uptake in an APP/PS1 model of Alzheimer's disease. Neurobiol Aging. 2012;33:1995–2005. doi: 10.1016/j.neurobiolaging.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Tempel D, Oostlander A, Helderman F, Gijsen F, Wentzel J, et al. Rapamycin modulates the eNOS vs shear stress relationship. Cardiovasc Res. 2008;78:123–129. doi: 10.1093/cvr/cvm103. [DOI] [PubMed] [Google Scholar]
- Naoum JJ, Zhang S, Woodside KJ, Song W, Guo Q, Belalcazar LM, et al. Aortic eNOS expression and phosphorylation in Apo-E knockout mice: differing effects of rapamycin and simvastatin. Surgery. 2004;136:323–328. doi: 10.1016/j.surg.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Atochin DN, Wang A, Liu VW, Critchlow JD, Dantas AP, Looft-Wilson R, et al. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. J Clin Invest. 2007;117:1961–1967. doi: 10.1172/JCI29877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Gagnon A, Veilleux A, Sorisky A, Marette A. Activation of the mammalian target of rapamycin pathway acutely inhibits insulin signaling to Akt and glucose transport in 3T3-L1 and human adipocytes. Endocrinology. 2005;146:1328–1337. doi: 10.1210/en.2004-0777. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Ras CantleyLC. PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.