

Abstract
Osteoarthritis (OA) is a debilitating disease of the joints characterized by cartilage degradation but to date there is no available pharmacological treatment to inhibit disease progression neither is there any available biomarker to predict its development. In the present study, we examined the expression level and possible involvement of novel cell-ECM adhesion-related molecules such as Iintegrin Linked Kinase (ILK), PINCH, parvin, Mig-2 and Migfilin in OA pathogenesis using primary human articular chondrocytes from healthy individuals and OA patients. Our findings show that only ILK and Migfilin were upregulated in OA compared to the normal chondrocytes. Interestingly, Migfilin silencing in OA chondrocytes rather exacerbated than ameliorated the osteoarthritic phenotype, as it resulted in even higher levels of catabolic and hypertrophic markers while at the same time induced reduction in ECM molecules such as aggrecan. Furthermore, we also provide a link between Migfilin and β-catenin activation in OA chondrocytes, showing Migfilin to be inversely correlated with β-catenin. Thus, the present study emphasizes for the first time to our knowledge the role of Migfilin in OA and highlights the importance of cell-ECM adhesion proteins in OA pathogenesis.
Keywords: osteoarthritis, Migfilin, β-catenin, chondrocytes, extracellular matrix
INTRODUCTION
Osteoarthritis (OA), a chronic degenerative disease of the joints, is the most common form of arthritis in older adults and a major cause of morbidity, activity limitation, physical disability, pain and reduction in quality of life. OA etiology is practically unknown although it is believed to be multifactorial. Mechanical load, joint injuries, obesity[1], and genetic predisposition are among the most serious risk factors for disease development. It is estimated that annually 15 million people are diagnosed with OA in the European Union while in the United States estimates indicate a rise in OA prevalence to 27 million in 2008[1, 2]. Although analgesics are used, to date there is no available pharmacological treatment to inhibit or reverse disease progression and thus, most patients end up undergoing joint replacement surgery. Moreover, to date there is no available biomarker to predict OA development, leaving room for research in the field towards early diagnosis.
OA is generally characterized by progressive degeneration of the articular cartilage, which is accompanied by synovitis, formation of subchondral cysts, subchondral bone attrition, formation of osteophytes[3] and decrease in the content of extracellular matrix (ECM) proteoglycans leading to loss of cartilage elasticity and reduction of its ability to withstand mechanical load resulting in pain and difficulty during movement. Articular cartilage in healthy adults consists of chondrocytes with low metabolic activity[4] that do not undergo maturation, proliferation or apoptosis. In OA, however, the affected cartilage cannot maintain these characteristics and chondrocytes lose their differentiation, start expressing collagen 10A1 instead of collagen 2A1, enter the process of maturation, and acquire the ability to propagate[5] thus becoming hypertrophic cells[6]. Furthermore, to repair damage in the articular cartilage, osteoarthritic chondrocytes exhibit increased anabolic activity composing ECM components. However, this increased anabolic activity cannot compensate for the degradation taking place at the same time. Thus, osteoarthritic chondrocytes demonstrate at the same time dramatically elevated levels of catabolic enzymes such as matrix metalloproteases (MMP) and MMP-13 in particular[5], and significantly reduced levels of ECM proteins such as aggrecan.
Although OA is considered to be a disease that affects the entire joint, the articular cartilage[7] remains the best studied component of the joint. Therefore, the investigation of osteoarthritic chondrocytes’ characteristics is paramount. Considering the fact that chondrocytes are practically embedded in ECM, cell-ECM communication is of utmost significance for their homeostasis. This communication is achieved via a network of protein complexes with structural and functional role in transmitting signals[8], such as integrins and their binding partners. Thus, many cell-ECM adhesion proteins play important role in chondrocyte biology, as it has been shown that integrin alpha1 knockout mice leads to OA[9], and β1 integrin knock-out in mice results in decreased chondrocyte proliferation[10]. In addition, Integrin-Linked Kinase (ILK) conditional knock-out in chondrocytes also causes decreased chondrocyte proliferation, impaired adhesion to ECM and chondrodysplasia[11]. Thus, inactivation or knock-out of integrin-associated genes and the ECM, leads to reduced chondrocyte proliferation and associated disorders, such as OA. Further evidence showing the importance of ECM/integrin-associated proteins in chondrocytes is the fact that ILK and its binding partners play an important role in the development and growth of tumors of chondrogenic origin[12].
It has been shown that when activated[13], ILK regulates three major survival signaling pathways; protein kinase B/Akt[13, 14], Extracellular signal-Regulated Kinase (Erk)[13], and Glycogen Synthase Kinase 3- β (GSK3β) pathway[13, 15]. In fact, inactivation of GSK3β by ILK leads to induction of the ranscription complex β-catenin/Lymphoid Enhancer Factor-1 (LEF1)[16], which in turn leads to increased regulation of multiple pro-survival genes. It should be noted that ILK forms a stable ternary complex with two other cell-ECM adhesion proteins, namely PINCH and a-parvin[17–20], which has been shown to be involved in cell shape regulation, migration, proliferation, and ECM synthesis[21]. Interestingly, however, ILK has been also shown to interact with multiple other novel ECM-adhesion related proteins including Mitogen inducible gene-2 (Mig-2, which is also known as kindlin-2)[22], which in turn, binds to Migfilin (also known as FBLP-1, Filamin-binding LIM protein 1), a novel interactor of filamin[23] confirming the association between cell-ECM adhesion proteins and actin cytoskeleton.
Interestingly, similar type of proteins related to cellular organization, actin cytoskeleton and adhesion to the ECM have been identified by proteomic analysis to be expressed in chondrocytes[24, 25] with elevated expression levels in OA patients compared to controls[26–28]. Nevertheless, none of the above-described ILK-related proteins has been studied in chondrocytes and their role in OA is completely unknown.
In the present study, to add on the association between ECM/integrin-associated proteins, we investigated, for the first time to our knowledge, the expression levels and possible involvement of ILK, PINCH, parvin, Mig-2 and Migfilin in OA pathogenesis.
MATERIALS AND METHODS
Articular cartilage samples
OA articular cartilage samples were obtained from femoral heads, femoral condyles and tibial plateaus of patients with primary OA undergoing knee replacement surgery at the Orthopaedics Department of University Hospital of Larissa. A total of 18 patients were included in the study. Each sample was categorized according to its gross morphology, as either severely damaged (OA=max), which was taken from the main defective area of maximal load, or mildly affected (OA=min) taken from areas with no obvious surface defects. Macroscopic findings were validated by histological studies and graded using the Mankin score (Mankin score for mild OA: 1–4 and for severe OA: 10–14). Radiographs were obtained before surgery and graded using the Kellgrene-Lawrence system[29]. All patients had a Kellgren-Lawrence score >2. Patients with rheumatoid arthritis and other autoimmune diseases as well as chondrodysplasias, infection-induced OA and post-traumatic OA were excluded from the study.
Normal cartilage was obtained from 10 individuals with 0 Mankin score, undergoing fracture repair surgery, with no history of joint disease and no clinical manifestations of OA when specifically explored by radiographs.
Cartilage samples from both patients and healthy individuals were obtained following verbal informed consent. The method for obtaining verbal consent was approved by the Institutional Review Board of the University Hospital of Larissa. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the Local Ethical Committee of the University Hospital of Larissa.
Primary chondrocyte isolation from articular cartilage
Primary chondrocytes were isolated from articular cartilage as described previously[29]. In brief, articular cartilage was dissected and subjected to digestion with 1 mg/ml pronase followed by overnight digestion with 1 mg/ml collagenase P(Roche). Chondrocytes were counted and checked for viability using trypan blue staining. Chondrocytes were cultured in Dulbecco’s Modified Eagles Medium/Ham’s F-12 (DMEM/F-12) supplemented with 5% fetal bovine serum and 1% penicillin-streptomycin for only 2 passages, to avoid dedifferentiation events occurring over time in culture.
RNA isolation and Real Time PCR
Total cellular RNA was extracted from cultured chondrocytes using Trizol reagent (Invitrogen). Transcription of 1mg RNA to complementary DNA (cDNA) was performed using Superscript III (Invitrogen). Quantification of ILK, PINCH, α-parvin, mig-2, Migfilin, β-catenin, MMP-13, aggrecan, collagen 10A1 and collagen 2A1 expression was performed by real-time PCR (MiniOpticon, BioRad). Actin was used as housekeeping gene for α-parvin mRNA quantification while porphobilinogen deaminase (PBGD) was used as a housekeeping gene for all the other genes. Reactions were done in triplicate and at least 3 independent experiments were performed. All primers used are shown in Table 1. To quantify the relative expression of each gene, Ct values were normalized against the endogenous reference (ΔCt =Ct target − Ct PBGD) and were compared with a calibrator using the ΔΔCt method.
Table 1.
primer sequences for Real Time PCR analysis.
| Target gene | Sequence |
|---|---|
| ILK | Forward 5′-GAC ATG ACT GCC CGA ATT AG-3′ Reverse 5′-CTG AGC GTC TGT TTG TGT CT-3′ |
| PINCH | Forward 5′ CCG CTG AGA AGA TCG TGA AC 3′ Reverse 5′ GGG CAA AGA GCA TCT GAA AG 3′ |
| a-parvin | Forward 5′ CAATTCGACTCCCAGACCAT 3′ Reverse 5′TGGTCGAACAAGGTGTCAAA 3′ |
| Mig-2 | Forward 5′AGCTTTATGAGCAGGCCAAA 3′ Reverse 5′ GAAAGGGCAGCATCAACTTC 3′ |
| Migfilin | Forward 5′ CGAATGCATGGGAAGAAACT 3′ Reverse 5′ GCAGGTTAGGAAGGGAAACC 3′ |
| β-catenin | Forward 5′ TCATGCGTTCTCCTCAGATG 3′ Reverse 5′ AATCCACTGGTGAACCAAGC 3′ |
| MMP-13 | Forward 5′ TGGCATTGCTGACATCATGA 3′ Reverse 5′ GCCAGAGGGCCCATCAA 3′ |
| Collagen 10A1 | Forward 5′ CAGGCATAAAAGGCCCACTA 3′ Reverse 5′GTGGACCAGGAGTACCTTGC 3′ |
| Collagen 2A1 | Forward 5′ATGACAATCTGGCTCCCAACACTGC 3′ Reverse 5′ GACCGGCCCTATGTCCACACCGAAT 3′ |
| Aggrecan | Forward 5′TGAGGAGGGCTGGAACAAGTACC 3′ Reverse 5′ GGAGGTGGTAATTGCAGGGAACA 3′ |
Protein Extraction
Normal and osteoarthritic chondrocytes were lysed using RIPA lysis buffer containing 10mM Tris(pH 7.5), 150mM NaCl, 1% Triton X-100, 1% Sodium Deoxycholated, 0.1% SDS, 1mM EDTA, and a cocktail of protease and phosphatase inhibitors. Protein concentration was quantified using the BCA method (PIERCE).
Western Blot Analysis
Cell lysates from normal and OA chondrocytes were run in 10% acrylamide gels and transferred to PVDF membranes (Millipore). Signals were detected using suitable horseradish peroxidase-conjugated immunoglobulin. Anti-β-actin antibody (Sigma-Aldrich) was used as loading control. When necessary, western blot bands from several different blots were quantified using the NIH Scion Image Software according to the guidelines.
Antibodies
Antibodies against ILK[30], Mig-2[23], and Migfilin[23] were described previously. MMP-13 antibody was purchased from Abcam and β-actin was purchased from Sigma.
Transfection with siRNAs
Primary osteoarthritic chondrocytes were treated for 24h with 100 nM siRNA non-specific control or siRNA against Migfilin using the Lipofectamine 2000 transfection reagent (Invitrogen) according to the company’s guidelines. Migfilin siRNA sequence was 5′GAAGAGGGTGGCATCGTCTGTCTTT3′, while the sequence 5′AAACUCUAUCUGCACGCUGAC3′ was used as Non Specific Control (NSC).
Statistical analysis
A paired Student’s t-test was used for statistical analyses of the results. P values <0.05 were considered statistically significant. The Graphpad Prism software was used for the generation of all graphs.
RESULTS
MMP-13 protein and mRNA expression is elevated in OA chondrocyte samples
Primary normal and osteoarthritic chondrocytes isolated from normal and OA articular cartilage, respectively, were first tested for the expression level of MMP-13, a basic catabolic marker used to molecularly identify OA samples. As shown in Figure 1A and B, OA chondrocytes showed dramatically elevated MMP-13 mRNA (Figure 1A) and protein (Figure 1B) expression level compared to normal chondrocytes indicating that they were indeed taken from patients with advanced OA and increased catabolism of ECM. It should be noted however, that although all OA chondrocytes were isolated from patients with advanced OA undergoing joint replacement surgery, they do exhibit differences in MMP-13 protein and mRNA expression level, which is indicative of the variation observed in human samples due to other unidentified factors that may play a role such as genetic predisposition, physical activity, age, and obesity. Thus, for the present study we used chondrocytes which exhibited high mRNA and protein MMP-13 expression at a level that is proportional to one another.
Figure 1. MMP-13 is dramatically elevated in OA chondrocytes.

A) Indicative Real Time PCR result for MMP-13 mRNA expression using cDNA isolated from normal and OA chondrocytes. For normal chondrocytes n=7, and for OA chondrocytes n=18. Asterisk indicates a statistically significant difference (p <0.05). B) Indicative western blot showing MMP-13 protein expression in normal and OA primary chondrocytes. β-actin is used as loading control.
ILK expression level is elevated in OA chondrocytes but PINCH, a-parvin and Mig-2 are not affected
In order to test whether ILK and its associated proteins are involved in OA pathogenesis, we used normal and OA primary chondrocytes to examine the expression of ILK and its binding partners in the complex ILK-PINCH-parvin. As shown in Figure 2, ILK protein (Figure 2A and 2B) expression was significantly elevated in OA chondrocytes compared to their normal counterparts. In addition, the protein expression level of ILK was also tested in cells taken from a minimally (min) and a maximally (max) affected area of the cartilage tissue as described in the material and method’s section. Figure 2B shows a clear difference in the expression level between less affected and severely affected cells further suggesting that ILK is also associated with the stage of the disease. Interestingly, however, the mRNA expression of ILK (Figure 2C) and that of its binding partners PINCH-1 and α-parvin (Figure 2D), was found to be unaffected in OA, clearly indicating that ILK may exert its action through an alternative pathway in human chondrocytes.
Figure 2. Expression profile of ILK, PINCH-1, α-parvin and Mig-2 in human chondrocytes.

A) Indicative western blot showing ILK protein expression in normal and OA chondrocytes. B) Western blot showing ILK protein expression in OA chondrocytes isolated from minimally and maximally affected areas of the same cartilage tissue as described in the materials and methods section. C) Real Time PCR result for ILK. D) Real Time PCR result for PINCH-1 and α-parvin E) Indicative western blot showing Mig-2 protein expression in normal and OA chondrocytes. F) Real Time PCR result for Mig-2 mRNA expression in RNA isolated from normal and OA chondrocytes. Diagrams are the result of three independent experiments. For normal chondrocytes n=8, and for OA chondrocytes n=18. Asterisk indicates a statistically significant difference (p <0.05).
To test whether ILK acts through its interaction with Mig-2 which is a relatively novel ECM-adhesion protein also demonstrated to be associated with it[22], we performed expression analysis for Mig-2 in normal and OA primary human chondrocytes. As shown in Figure 2E and 2F, Mig-2 mRNA (Figure 2F) as well as protein (Figure 2E) expression level remained unaltered in normal and OA samples.
Migfilin is upregulated in OA chondrocytes
Although Mig-2 expression was not altered in OA, we moved on to test the expression level of Migfilin which is a Mig-2 and filamin-interacting protein[23]. Migfilin, was an especially promising candidate as recent findings indicated that Migfilin is essential for proper control of bone remodeling[31] while Migfilin knock-out mice exhibited a severe osteopenic phenotype[31]. As shown in Figure 3A Migfilin protein expression is indeed dramatically elevated in OA samples compared to normal and in fact this difference is also significant between minimally and maximally affected OA samples (Figure 3B and 3D), indicating a potentially important role of Migfilin in OA pathogenesis. In addition, Migfilin expression is also elevated at the mRNA level as well (Figure 3C).
Figure 3. Migfilin is upregulated in OA chondrocytes.

A) Indicative western blot showing Migfilin protein expression in normal and OA chondrocytes. B) Western blot showing Migfilin protein expression in OA chondrocytes isolated from minimally and maximally affected areas of the same cartilage tissue as described in the materials and methods section. C) Real Time PCR result for Migfilin mRNA expression in RNA isolated from normal and OA chondrocytes. Three independent experiments in duplicate were performed using n=8 normal chondrocytes, and n=18 OA chondrocytes (p=0.044). D) Diagram showing Migfilin protein expression normalized to β-actin using the NIH Scion Image Software. The result shown represents the mean from three different blots (p=0.048). Asterisk indicates a statistically significant difference (p <0.05).
Migfilin silencing in OA chondrocytes exacerbates osteoarthritic phenotype
Being intrigued by the finding that Migfilin is elevated in OA chondrocytes, we further investigated its role in human OA by gene silencing. More specifically, primary OA chondrocytes were subjected to siRNA trasfection using a non-specific control (NSC) siRNA or Migfilin siRNA. As shown in Figure 4, Migfilin silencing was successful both at the mRNA (Figure 4A) and at the protein level (Figure 4B, compare lanes 1 and 2). Interestingly, Migfilin silencing resulted in significant changes in all the markers normally used to define osteoarthritic state. Thus, Migfilin elimination from osteoarthritic chondrocytes resulted in increased expression of the catabolic marker MMP-13, increased expression of the hypertrophic marker collagen 10A1 and a simultaneous decrease in aggrecan which is abundant in normal chondrocytes. Collagen 2A1 characteristic of normal chondrocytes was also found significantly reduced in OA chondrocytes treated with Migfilin siRNA (Figure 4A).
Figure 4.
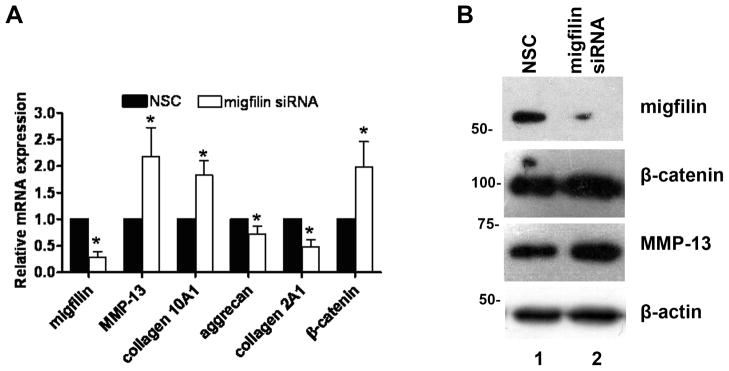
A) Real Time PCR results in OA chondrocytes 24h post-transfection with NSC or Migfilin-specific siRNA. The diagram shows results for the genes: Migfilin (p= 0.00018), MMP-13 (p=0.027), collagen 10A1 (p=0.001), aggrecan (p=0.03), collagen 2A1 (p=0.0014) and β-catenin (p=0.033). Diagrams are the result of 4 independent experiments performed in ducplicate from 5 different OA samples. Asterisk indicates a statistically significant difference (p <0.05). B) Indicative western blot showing Migfilin, β-catenin, and MMP-13 protein expression in OA chondrocytes following Migfilin depletion. β-actin is used as loading control.
Migfilin silencing leads to β-catenin upregulation
Taking into account recent work by He et al[32] which showed that Migfilin is inversely correlated with β-catenin, promoting its degradation and combining this piece of information with previous work from our group showing elevated β-catenin expression in OA[33, 34], we hypothesized that β-catenin may also be affected by Migfilin silencing. Indeed, as shown in Figure 4A and 4B Migfilin depletion led to upregulation of free β-catenin both at the mRNA (Figure 4A) and the protein (Figure 4B) level.
DISCUSSION
In the present study we evaluated the possible involvement of cell-ECM adhesion-related proteins, such as ILK, PINCH, parvin, Mig-2 and Migfilin in OA pathogenesis. Our findings show that from the proteins examined, only ILK and Migfilin’s expression levels were upregulated in OA samples. The observed upregulation could indicate a possible involvement of both proteins in OA pathogenesis.
Considering the fact that Migfilin knock-out animals exhibit osteopenia[31], and that severe OA is characterized by the formation of new bone in the form of osteophytes[3, 5], it makes perfect sense that Migfilin is upregulated in advanced OA. In fact, in the present study we have shown that there is differential protein expression of Migfilin in chondrocytes isolated from severely damaged (OA max) and mildly affected (OA min) areas of the cartilage. As severely damaged cartilage was taken from the main defective areas of maximal load it is considered to depict the most advanced form of the disease. The mildly affected cartilage (OA min) however, was taken from an adjacent macroscopically and histological intact area of the same joint that was clearly involved in the disease process, and thus cannot be completely comparable to normal cartilage nor to severely damaged one. This places the OA min cartilage and chondrocytes in an intermediate state and could be considered as an earlier stage of the disease. Thus, the findings of the present study indicate that Migfilin’s expression is correlated with the disease stage. This is in accordance with previous studies from our group[35], in which several proteins considered to be important for OA pathogenesis (such as leptin) are differentially expressed between maximally and minimally affected chondrocytes.
Since upregulation of Migfilin was more dramatic and consistent than that of ILK among samples tested, and since it occurred in both mRNA and protein level, we decided to further pursue it. Thus, we proceeded with Migfilin silencing in OA chondrocytes and we found that Migfilin elimination rather exacerbates than ameliorates osteoarthritic phenotype. Following Migfilin ablation, OA chondrocytes exhibited even higher expression levels of catabolic and hypertrophic markers (MMP-13, Collagen 10A1 respectively) while exhibiting reduction in ECM molecules (aggrecan and collagen 2A1). This could be explained by a negative feedback-loop mechanism activated by Migfilin in OA chondrocytes.
Furthermore, in the present study we showed that migfillin silencing leads to β-catenin upregulation. This finding is in accordance with reports in cancer cells showing Migfilin to be inversely correlated with β-catenin[31]. Moreover, β-catenin signaling pathway has been recently shown to be involved in OA, as conditional activation of β-catenin in mouse articular chondrocytes leads to premature chondrocytes differentiation and the development of an OA-like phenotype[36]. Recent work from our group demonstrated that β-catenin signaling pathway activated by BMP-2 enhances ECM degradation and promotes hypertrophy as evidenced by collagen 10 expression, suggesting a major role played by β-catenin in OA progreesion [34]. However, this is the first time that a connection between Migfilin and β-catenin activation is highlighted in OA chondrocytes, adding to the complexity of the underlining mechanisms.
This work highlights the importance of cell-ECM adhesion proteins in OA pathogenesis and provides a rough network of proteins involved. Furthermore, it emphasizes for the first time to our knowledge the role of Migfilin in OA, pointing towards a negative regulatory mechanism of action. However, there are still a few questions that need to be further investigated in relation to the exact molecular mechanism involved. Nevertheless, the present work paves the way for a more thorough investigation of the role of cell-ECM adhesion proteins in OA pathogenesis.
Highlights.
We studied expression of ILK, PINCH, parvin, Mig2 and Migfilin in osteoarthritis
We show that ILK and Migfilin were upregulated in osteoarthritic chondrocytes.
We show for the first time that migfilin silencing exacerbates osteoarthritis.
We provide a link between migfilin and β-catenin activation in OA chondrocytes.
In osteoarthritic chondrocytes migfilin is inversely correlated with β-catenin.
Acknowledgments
This study was supported by the Cooperation I action of the General Secretariat for Research and Technology (GSRT) of the Greek Ministry of Education, Religion and Lifelong Learning (to A. Tsezou) and by the NIH grant GM65188 (to C. Wu).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Vasiliki Gkretsi, Email: vasso.gkretsi@gmail.com.
Vassilis Papanikolaou, Email: papanikolaou.vassilis@gmail.com.
Stephanie Dubos, Email: dubos.stephanie@hotmail.com.
Ioanna Papathanasiou, Email: ioanna_papathanasiou@yahoo.gr.
Nikolina Giotopoulou, Email: linagiotopoulou@gmail.com.
Vaia Valiakou, Email: venymay@hotmail.com.
Chuanyue Wu, Email: carywu@pitt.edu.
Konstantinos N. Malizos, Email: kmalizos@otenet.gr.
Aspasia Tsezou, Email: atsezou@med.uth.gr.
References
- 1.Neogi T, Zhang Y. Osteoarthritis prevention. Curr Opin Rheumatol. 2011;23:185–191. doi: 10.1097/BOR.0b013e32834307eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawrence RC, Felson DT, Helmick CG, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58:26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Felson DT. Developments in the clinical understanding of osteoarthritis. Arthritis Res Ther. 2009;11:203. doi: 10.1186/ar2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mc Anulty R, Laurent GJ. In vivo measurement of collagen metabolism in cartilage and bone. Academic press Inc; 1990. [Google Scholar]
- 5.Sandell LJ, Aigner T. Articular cartilage and changes in arthritis. An introduction: cell biology of osteoarthritis. Arthritis Res. 2001;3:107–113. doi: 10.1186/ar148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aigner T, Reichenberger E, Bertling W, et al. Type X collagen expression in osteoarthritic and rheumatoid articular cartilage. Virchows Arch B Cell Pathol Incl Mol Pathol. 1993;63:205–211. doi: 10.1007/BF02899263. [DOI] [PubMed] [Google Scholar]
- 7.Abramson SB, Attur M. Developments in the scientific understanding of osteoarthritis. Arthritis Res Ther. 2009;11:227. doi: 10.1186/ar2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Chen K, Tu Y, et al. Distinct roles of two structurally closely related focal adhesion proteins, alpha-parvins and beta-parvins, in regulation of cell morphology and survival. J Biol Chem. 2004;279:41695–41705. doi: 10.1074/jbc.M401563200. [DOI] [PubMed] [Google Scholar]
- 9.Zemmyo M, Meharra EJ, Kuhn K, et al. Accelerated, aging-dependent development of osteoarthritis in alpha1 integrin-deficient mice. Arthritis Rheum. 2003;48:2873–2880. doi: 10.1002/art.11246. [DOI] [PubMed] [Google Scholar]
- 10.Aszodi A, Hunziker EB, Brakebusch C, et al. Beta1 integrins regulate chondrocyte rotation, G1 progression, and cytokinesis. Genes Dev. 2003;17:2465–2479. doi: 10.1101/gad.277003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terpstra L, Prud’homme J, Arabian A, et al. Reduced chondrocyte proliferation and chondrodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J Cell Biol. 2003;162:139–148. doi: 10.1083/jcb.200302066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papachristou DJ, Gkretsi V, Rao UN, et al. Expression of integrin-linked kinase and its binding partners in chondrosarcoma: association with prognostic significance. Eur J Cancer. 2008;44:2518–2525. doi: 10.1016/j.ejca.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu C, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol. 2001;155:505–510. doi: 10.1083/jcb.200108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Persad S, Attwell S, Gray V, et al. Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells. Proc Natl Acad Sci U S A. 2000;97:3207–3212. doi: 10.1073/pnas.060579697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan C, Costello P, Sanghera J, et al. Inhibition of integrin linked kinase (ILK) suppresses beta-catenin-Lef/Tcf-dependent transcription and expression of the E-cadherin repressor, snail, in APC−/− human colon carcinoma cells. Oncogene. 2001;20:133–140. doi: 10.1038/sj.onc.1204052. [DOI] [PubMed] [Google Scholar]
- 16.Novak A, Hsu SC, Leung-Hagesteijn C, et al. Cell adhesion and the integrin-linked kinase regulate the LEF-1 and beta-catenin signaling pathways. Proc Natl Acad Sci U S A. 1998;95:4374–4379. doi: 10.1073/pnas.95.8.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukuda T, Guo L, Shi X, et al. CH-ILKBP regulates cell survival by facilitating the membrane translocation of protein kinase B/Akt. J Cell Biol. 2003;160:1001–1008. doi: 10.1083/jcb.200212113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukuda T, Chen K, Shi X, et al. PINCH-1 Is an Obligate Partner of Integrin-linked Kinase (ILK) Functioning in Cell Shape Modulation, Motility, and Survival. J Biol Chem. 2003;278:51324–51333. doi: 10.1074/jbc.M309122200. [DOI] [PubMed] [Google Scholar]
- 19.Tu Y, Huang Y, Zhang Y, et al. A new focal adhesion protein that interacts with integrin-linked kinase and regulates cell adhesion and spreading. J Cell Biol. 2001;153:585–598. doi: 10.1083/jcb.153.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Chen K, Tu Y, et al. Assembly of the PINCH-ILK-CH-ILKBP complex precedes and is essential for localization of each component to cell-matrix adhesion sites. J Cell Sci. 2002;115:4777–4786. doi: 10.1242/jcs.00166. [DOI] [PubMed] [Google Scholar]
- 21.Guo D, Tan W, Wang F, et al. Proteomic analysis of human articular cartilage: identification of differentially expressed proteins in knee osteoarthritis. Joint Bone Spine. 2008;75:439–444. doi: 10.1016/j.jbspin.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Mackinnon AC, Qadota H, Norman KR, et al. C. elegans PAT-4/ILK functions as an adaptor protein within integrin adhesion complexes. Curr Biol. 2002;12:787–797. doi: 10.1016/s0960-9822(02)00810-2. [DOI] [PubMed] [Google Scholar]
- 23.Tu Y, Wu S, Shi X, et al. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell. 2003;113:37–47. doi: 10.1016/s0092-8674(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 24.Ruiz-Romero C, Lopez-Armada MJ, Blanco FJ. Proteomic characterization of human normal articular chondrocytes: a novel tool for the study of osteoarthritis and other rheumatic diseases. Proteomics. 2005;5:3048–3059. doi: 10.1002/pmic.200402106. [DOI] [PubMed] [Google Scholar]
- 25.Vincourt JB, Lionneton F, Kratassiouk G, et al. Establishment of a reliable method for direct proteome characterization of human articular cartilage. Mol Cell Proteomics. 2006;5:1984–1995. doi: 10.1074/mcp.T600007-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Ruiz-Romero C, Carreira V, Rego I, et al. Proteomic analysis of human osteoarthritic chondrocytes reveals protein changes in stress and glycolysis. Proteomics. 2008;8:495–507. doi: 10.1002/pmic.200700249. [DOI] [PubMed] [Google Scholar]
- 27.Wu J, Liu W, Bemis A, et al. Comparative proteomic characterization of articular cartilage tissue from normal donors and patients with osteoarthritis. Arthritis Rheum. 2007;56:3675–3684. doi: 10.1002/art.22876. [DOI] [PubMed] [Google Scholar]
- 28.Iliopoulos D, Malizos KN, Oikonomou P, et al. Integrative microRNA and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS One. 2008;3:e3740. doi: 10.1371/journal.pone.0003740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kostopoulou F, Gkretsi V, Malizos KN, et al. Central role of SREBP-2 in the pathogenesis of osteoarthritis. PLoS One. 2012;7:e35753. doi: 10.1371/journal.pone.0035753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li F, Zhang Y, Wu C. Integrin-linked kinase is localized to cell-matrix focal adhesions but not cell-cell adhesion sites and the focal adhesion localization of integrin-linked kinase is regulated by the PINCH-binding ANK repeats. J Cell Sci. 1999;112(Pt 24):4589–4599. doi: 10.1242/jcs.112.24.4589. [DOI] [PubMed] [Google Scholar]
- 31.Xiao G, Cheng H, Cao H, et al. Critical role of filamin-binding LIM protein 1 (FBLP-1)/migfilin in regulation of bone remodeling. J Biol Chem. 2012;287:21450–21460. doi: 10.1074/jbc.M111.331249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He H, Ding F, Li Y, et al. Migfilin regulates esophageal cancer cell motility through promoting GSK-3beta-mediated degradation of beta-catenin. Mol Cancer Res. 2012;10:273–281. doi: 10.1158/1541-7786.MCR-11-0419. [DOI] [PubMed] [Google Scholar]
- 33.Papathanasiou I, Malizos KN, Tsezou A. Low-density lipoprotein receptor-related protein 5 (LRP5) expression in human osteoarthritic chondrocytes. J Orthop Res. 2010;28:348–353. doi: 10.1002/jor.20993. [DOI] [PubMed] [Google Scholar]
- 34.Papathanasiou I, Malizos KN, Tsezou A. Bone morphogenetic protein-2-induced Wnt/beta-catenin signaling pathway activation through enhanced low-density-lipoprotein receptor-related protein 5 catabolic activity contributes to hypertrophy in osteoarthritic chondrocytes. Arthritis Res Ther. 2012;14:R82. doi: 10.1186/ar3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simopoulou T, Malizos KN, Iliopoulos D, et al. Differential expression of leptin and leptin’s receptor isoform (Ob-Rb) mRNA between advanced and minimally affected osteoarthritic cartilage; effect on cartilage metabolism. Osteoarthritis Cartilage. 2007;15:872–883. doi: 10.1016/j.joca.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 36.Zhu M, Tang D, Wu Q, et al. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J Bone Miner Res. 2009;24:12–21. doi: 10.1359/JBMR.080901. [DOI] [PMC free article] [PubMed] [Google Scholar]