

Abstract
BACKGROUND
We have recently demonstrated that injured patients in hemorrhagic shock shed syndecan-1 and that the early use of fresh frozen plasma (FFP) in these patients is correlated with improved clinical outcomes. As the lungs are frequently injured after trauma, we hypothesized that hemorrhagic shock-induced shedding of syndecan-1 exposes the underlying pulmonary vascular endothelium to injury resulting in inflammation and hyperpermeability, and that these effects would be mitigated by FFP.
METHODS
In vitro, pulmonary endothelial permeability, endothelial monolayer flux, transendothelial electrical resistance (TER), and leukocyte-endothelial binding were measured in pulmonary endothelial cells after incubation with equal volumes of FFP or lactated Ringers (LR). In vivo, using a coagulopathic mouse model of trauma and hemorrhagic shock, pulmonary hyperpermeability, neutrophil infiltration, and syndecan-1 expression and systemic shedding were assessed after three hours of resuscitation with either 1XFFP or 3XLR and compared to shock alone and shams.
RESULTS
In vitro, endothelial permeability and flux were decreased, TER was increased, and leukocyte-endothelial binding was inhibited by FFP compared to LR treated endothelial cells. In vivo, hemorrhagic shock was associated with systemic shedding of syndecan-1 which correlated with decreased pulmonary sydnecan-1 and increased pulmonary vascular hyperpermeability and inflammation. FFP resuscitation, compared to LR resuscitation, abrogated these injurious effects.
CONCLUSIONS
After hemorrhagic shock, FFP resuscitation inhibits endothelial cell hyperpermeability and inflammation and restores pulmonary syndecan-1 expression. Modulation of pulmonary syndecan-1 expression may mechanistically contribute to the beneficial effects FFP.
Keywords: glycocalyx, endothelial injury, transendothelial resistance, leukocyte binding, mouse model of hemorrhagic shock and trauma
Introduction
A dysfunctional endothelium is increasingly recognized as a key component of a wide variety of pathologies, including hemorrhagic shock. The endothelium is the platform on which adhesion of leukocytes and the activation of coagulation occur after shock. Additionally, it is the surface to which membrane-bound syndecan-1 is attached. Syndecan-1 is a cell surface heparan sulfate proteoglycan that forms the structural backbone of a protective network of plasma components referred to as the glycocalyx. The glycocalyx is thought to consist primarily of membrane-bound proteoglycans with glycosaminoglycan side-chains, membrane-bound glypicans, and adsorbed plasma proteins. Syndecan-1 is one of the proteoglycans found on the luminal surface of endothelial cells.1
Syndecan-1 plays an important role in inflammation.2 In noninfectious states of inflammation, it facilitates resolution of inflammation by inhibiting leukocyte adhesion onto the endothelium, mitigating the expression and activity of pro-inflammatory mediators, and confining leukocytes to the site of injury.3 On the other hand, it serves primarily a pathologic role in infectious conditions by promoting attachment of bacteria and inhibiting host defense systems.4 Hemorrhagic shock is a pro-inflammatory state, but the function of syndecan-1 in the resolution of inflammation after hemorrhagic shock is unknown.
Vascular hyperpermeability is being recognized as an increasingly important component of hemorrhagic shock, though the “endotheliopathy” of hemorrhagic shock is poorly characterized and not well understood.5–6 Perturbations of the glycocalyx with shedding of syndecan-1 is postulated to alter endothelial integrity and lead to enhanced permeability and subsequent edema, though this is primarily based on clinical associations rather than mechanistic studies. We hypothesize that hemorrhagic shock-induced shedding of the syndecan-1 ectodomain is injurious, resulting in the exposure of the injured endothelium to pro-inflammatory leukocytes and in alterations to the structural integrity of the endothelium with resultant hyperpermeability.
We have been interested in fresh frozen plasma (FFP) as a therapeutic modality to hasten repair of the injured endothelium after hemorrhagic shock. Recent retrospective and prospective data suggest that the early and empiric use of FFP may decrease mortality in hemorrhagic shock patients, though the mechanism of this protection is unknown.7–8 Based on our prior finding that FFP aided in restoration of the glycocalyx after hemorrhagic shock9, we further hypothesized that FFP would decrease shedding of the syndecan-1 thereby protecting the endothelium from inflammatory insults and vascular hyperpermeability. We tested this hypothesis both in vitro using pulmonary endothelial cells and in vivo using a clinically relevant coagulopathic mouse model of hemorrhagic shock and trauma.
MATERIALS AND METHODS
In vitro studies
Human pulmonary endothelial cells (PECs) were purchased from Lonza (Walkersville, MD). Cells were maintained in EGM-2MV media on 96-well plates at 37°C and 5% CO2 in a humidified incubator. For experiments, cells were treated with either 10% – 30% concentrations of lactated Ringers (LR) or fresh frozen plasma (FFP). Donor units of FFP used in both in vitro and in vivo studies were obtained from Gulf Coast Regional Blood Center Houston, Texas and as per standard procedure, were frozen within eight hours of being drawn. It was kept frozen until the day of the experiment and was used within one to two hours of thaw.
Endothelial Cell Permeability
Two different parameters of permeability were assessed, flux and transendothelial electrical resistance (TER). Collagen-coated 0.4μm pore size inserts were seeded with PECs at 104 cells/insert. When confluency was reached, monolayers were pre-treated for one hour with either 10% FFP or lactated Ringers and compared to media alone.10 Permeability was then induced with VEGF-A165 (50ng/mL; R&D, Minneapolis, MN), administered simultaneously with the addition of FITC-Dextran- 70 kDa. Permeability was then assessed by adding 50μl of 1mg/mL 70 kDa FITC-Dextran to the upper chamber of each well (final concentration).
To calculate the baseline flux of FITC-Dx 70, we used Fick’s First Law of Diffusion [P= J/AΔC], where P is the permeability coefficient expressed as cm/min, J is the solute flux (or movement of FITC-Dx 70 per minute), and A is the area of the transwell at 0.3 cm2. ΔC the difference in concentration of FITC-Dx 70 between the transwell and the bottom well after FITC is added to the transwell. ΔC was assigned a value of 1 mg/mL, assuming that the concentration of FITC-Dx 70 in the bottom well is zero upon addition of FITC. Accumulated FITC-Dx 70 was regressed against time, using the quadratic regression method. Flux was calculated as the average slope from 5 min to 120 min for each regression model.11
In a separate set of experiments, transendothelial electrical resistance (TEER) was measured using an electric cell-substrate impedance sensing (ECIS) (Ztheta, Applied Biophysics, Troy NY) system. The ECIS system provides real-time quantitative measurements of vascular integrity using a change in impendance of the cell monolayer.12 TEER is a measurement of the resistance of the endothelial monolayer to the flow of charged ions through it. The higher the resistance the more energy it takes to propel an ion across it. While the permeability to small ions is not the same as the movement of proteins, experimental data shows that the inverse of TEER at 4000 Hertz is roughly equivalent to the permeability of an endothelial monolayer.13 Briefly, 8W10E+ plates (Applied Biophysics, Troy NY) were pretreated with cysteine and then media per the manufacturer’s instructions. PECs were seeded at a density of 80,000 cells per well. Resistance and impedance of the cell monolayer was measured continuously from the time of seeding. After 24 hours, half of media in each well was replaced with fresh media then one hour later half was replaced with fresh media containing 10U/ml heparin and either 10% FFP or LR and compared to media alone (controls). This treatment is considered time zero. Changes in TEER at 4000 Hertz were monitored as a measure permeability as demonstrated by Tiruppathi et al.13 Resistance traces are normalized to a point −0.5 hours before treatment, the top of the peak produced by the addition of fresh media.
Leukocyte binding
Pulmonary endothelial cells (104 cells/well) were seeded and incubated at 37°C until confluent then treated for one hour with 5, 10 or 30% FFP or LR and compared to controls. Adhesion molecule expression was then stimulated by the addition of TNFα (50ng/ml) for 4 hours, which increases binding of U937, a monocytoid cell line that we have previously used to study leukocyte-endothelial interactions.14 U937 cells were fluorescently labeled with Calcein-AM (Invitrogen Carlsbad, CA) (1μg/ml) then 104 cells were added to wells and allowed to adhere for one hour. Non-adherent cells were washed in PBS and labeled cells that remained bound to the endothelial cells were quantified by fluorescent readings on the Biotek Analyzer (Biotek, Winooski, VT) at 490 nm wavelength excitation and 520 nm emission.
In vivo studies
Coagulopathic mouse model of hemorrhagic shock and trauma
All procedures performed were protocols approved by the University of Texas Houston Medical School Animal Welfare Committee. The experiments were conducted in compliance with the National Institutes of Health guidelines on the use of laboratory animals. All animals were housed at constant room temperature with a 12:12-h light-dark cycle with access to food and water ad libitum. Male C57BL/6J mice, 8–10 weeks of age were used for all experiments. To mimic the clinical scenario of trauma-induced coagulopathy in patients in shock, the coagulopathic mouse model of trauma-hemorrhagic shock described by Cohen et al was employed.15 In brief, under isoflurane anesthesia, a 2cm midline laparotomy incision was made, organs inspected, and the incision closed. The bilateral femoral arteries were cannulated for continuous hemodynamic monitoring and blood withdrawal or resuscitation, respectively. After 10-minute period of equilibration, mice were bled to a mean arterial pressure (MAP) of 35±5 mmHg and maintained for 90 minutes. Shams underwent anesthesia and placement of catheters but were not subjected to hemorrhagic shock. Similar to Cohen et al, mouse were coagulopathic with a PT 12.1± 0.6 after hemorrhagic shock vs. 7.5 ± 0.2 sham, p=0.02. Mice were resuscitated over the next 15 minutes with either lactated Ringer’s at 3X shed blood volume16 or fresh frozen plasma at 1X shed blood volume and compared to animals that underwent shock alone. At the conclusion of resuscitation, vascular catheters were removed, incisions closed, and the animals were awoken from anesthesia. After three hours, animals were sacrificed by exsanguination under isoflurane anesthesia. Blood was obtained at the time of sacrifice and lungs harvested for further analysis. The three hour time point was chosen based on our previous investigation showing that the endothelial glycocalyx was being restored by three hours of resuscitation.9
Lung permeability
Vascular permeability was assessed in intact organs by measuring intravenous dye extravasation into the lung using a Caliper Lumina XR imaging system (IVIS).17 Animals received an intravenous bolus of the infra-red sensitive dye Alexafluor 680 (0.2 ml of 10 mg/ml, MW 10,000, Invitrogen, D34689) prior to the onset of shock.18 Upon completion of the three hour shock resuscitation period, animals were perfused via right ventricle with 4ºC PBS for 10 minutes to remove intravascular dye followed by 4% paraformaldehyde at 4ºC. Then lungs were excised and placed in the Lumina XR, from which the images were taken and quantified. Dye extravasation was measured as a function of signal intensity in response to applied fluorescent light at 675 nm. Permeability was quantified by measuring overall fluorescence intensity via Caliper software.19
In a separate set of animals, Evans blue dye extravasation was measured to confirm lung permeability. Animals received an intravenous injection of 3% Evans blue (4 ml/kg) one hour prior to sacrifice then were perfused via right ventricle with 4ºC PBS for 10 minutes to remove intravascular dye followed by 4% paraformadehyde at 4ºC. Lungs were incubated in N-methyl-formamide for 24 hour at 55°C to allow for dye extraction. After centrifugation, absorbance was measured in the supernatant at 620 nm using the VersaMax plate reader. (Molecular Devices Inc., Sunnyvale, CA). Tissues were then dried at 50°C for 72 hour to obtain dry weight to normalize the absorbance.
Myeloperoxidase (MPO)
Infiltration of neutrophils into the lung tissue was assessed by MPO immunofluorescence staining and MPO activity. For immunofluorescence, paraffin embedded tissue was cut into 5 μm-thick sections then incubated with MPO primary antibody (1:100 MPO mouse monoclonal antibody, Abcam, Cambridge, MA ) followed by incubation with secondary antibody (goat anti-mouse, Alexa Fluor 568, Invitrogen, Carlsbad, CA). Two random images were taken from each lung section with a fluorescent microscope (Nikon Eclipse Ti, Melville, NY) at 200X and quantified using ImageJ software (NIH). MPO activity was measured using a commercially available kit per manufacturer’s instructions (Enzo Life Sciences, ADI-907-029).
Syndecan-1
Plasma was stored at −80°C until use for measurement of syndecan-1 shedding by ELISA (ELISA Kit, UCL-E1966m, Hölzel Diagnostika). Rat anti-mouse syndecan-1 primary antibody was used with rabbit anti-rat HRP conjugated secondary antibody. Presence of secondary antibody was assessed with Magellen software to measure absorbance at 570nm. Absorbance was corrected by comparison to wells without added plasma.
To detect syndecan-1, lungs were sectioned and stained with rat anti-mouse syndecan-1 antibody (BD Biosciences, San Diego, CA) and green fluorescent goat anti-rat secondary antibody (Life Technologies, Grand Island, NY) following deparaffinization and antigen retrieval with citrate buffer. Two random images were taken from each lung section with a fluorescent microscope at 200X and quantified using ImageJ software (NIH).9
Statistical Analysis
Data was analyzed by one way analysis of variance and individual group means compared using Tukey’s post hoc test or repeated-measure ANOVA, followed by Tukey’s post-hoc tests and Bonferroni correction for multiple comparisons; p < 0.05 was considered significant. Data are expressed as mean ± SEM, n=8 per group for animal experiments.
RESULTS
In vitro
FFP compared to lactated Ringers reduces pulmonary endothelial hyperpermeability
Endothelial cell hyperpermeability is a hallmark characteristic of the injured vasculature. This was studied in an in vitro model using VEGF-A (165aa), a well-known inducer of hyperpermeability.20 In Fig. 1A, FFP significantly decreased endothelial cell paracellular permeability at all timepoints measured compared to LR and control. Calculation of the flux and permeability coefficients indicates that overall FFP is superior to LR and control in inhibiting EC permeability (Fig 1B). In a separate set of experiments, real-time quantitative measurements of vascular integrity were obtained over 24 hours using an electric cell-substrate impedance sensing system (Figure 1C). FFP treatment resulted in a significant increase in endothelial resistance (TEER) which translates to decreased endothelial cell paracellular permeability when compared to LR and control treated wells
Figure 1. FFP decreases pulmonary endothelial cell permeability in vitro.

A. Fluorescence intensity over time in a transwell endothelial cell permeability assay for cells treated with 10% LR or FFP diluted in basal medium. At all time points, fluorescence in FFP treated cells was significantly lower than in LR and control treated cells (*=p<0.05), while there was no significant difference at any time point between LR and controls. B. Permeability coefficients for each group at 10% concentration. All permeability coefficients are expressed as cm/min. Error bars represent the standard error of the mean. C. TEER measured at 4000 hertz in cells treated with 10% FFP, 10% LR or control media. FFP significantly increased transendothelial resistance compared to controls and LR (p < 0.05 by ANOVA with Tukey post hoc tests) indicating decreased endothelial permeability. Each trace represents the mean trace from three replicates. Dots appear with error bars indicating standard error of the mean at 1 hour intervals.
Leukocyte binding is reduced by FFP but not lactated Ringers in pulmonary endothelial cells
The purpose of these next studies was to determine and compare in vitro the effects of FFP to LR on inflammatory leukocyte-pulmonary endothelial cell adhesion. To stimulate inflammatory cell (U937) binding, endothelial cells were treated with TNF-α and the relative binding of calcein-labeled cells to treated endothelial cells was quantified by fluorimetry. Leukocyte-endothelial cell binding studies revealed that treatment of pulmonary endothelial cells with 5–30% concentrations of FFP inhibited U937 binding to the endothelial cells compared to LR and controls (Figure 2). Control and LR treated cells did not affect leukocyte binding at any concentration tested while there was a significant decrease in leukocyte binding in FFP treated cells at all concentrations.
Figure 2. FFP decreases leukocyte adhesion in vitro.
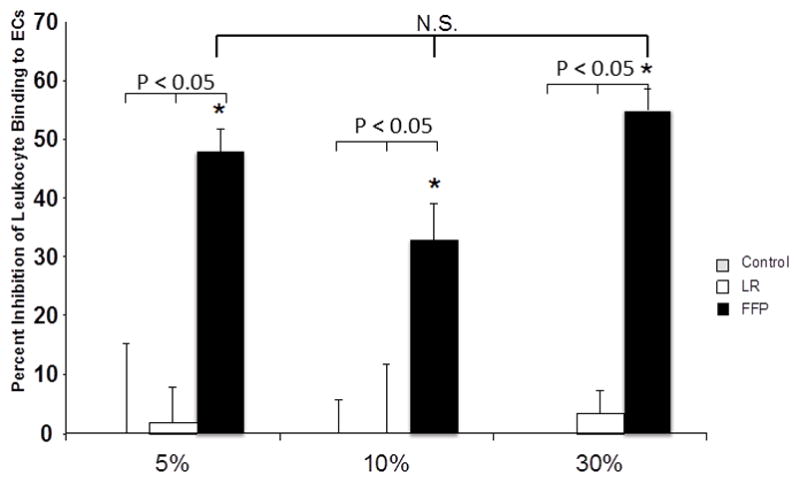
Percent decrease in the number of leukocytes bound to endothelial cells incubated with 10% LR or FFP diluted in basal medium. * indicated significance from both control and LR groups (p<0.05), NS= not significant.
In vivo
FFP and lactated Ringers similarly restore mean arterial pressure
To determine if our findings in vitro would translate to similar effects in vivo, we sought to determine if FFP would alter pulmonary inflammation and endothelial dysfunction in an established mouse model of HS and trauma. All mouse groups had a similar baseline weight and mean arterial blood pressure (MAP) as well as similar hemorrhage volume during the shock period (Table 1). By study design, the lactated Ringers group received a larger volume of resuscitation while the sham and shock alone groups received no resuscitation. During the early resuscitation period, there was no significant difference in MAP between the LR and FFP groups, while the HS group had a significantly lower MAP and the sham group a significantly higher MAP than the two resuscitation groups (Figure 3).
Table 1.
Characteristics of animals undergoing hemorrhagic shock
| Parameter | Shams | HS alone | HS + LR | HS + FFP | p value |
|---|---|---|---|---|---|
| Weight (g) | 21.0 ± 0.4 | 20.8 ± 0.4 | 21.9 ± 0.4 | 20.4 ± 0.4 | >0.05 |
| Baseline MAP | 77.0 ± 2.1 | 76.3 ± 2.4 | 77.4 ± 2.0 | 80.7 ± 2.3 | >0.05 |
| Hemorrhage% | 0 ± 0 | 37.2 ± 2.2 | 41.1 ± 0.5 | 39.8 ± 0.9 | >0.05* |
g=grams; MAP=mean arterial pressure; ETBV=; HS=hemorrhagic shock + trauma; LR= lactated Ringers; FFP= fresh frozen plasma. Hemorrhage is reported as % of estimated total blood volume.
p>0.05 for the three HS groups
Figure 3. Mean arterial pressure during early resuscitation.
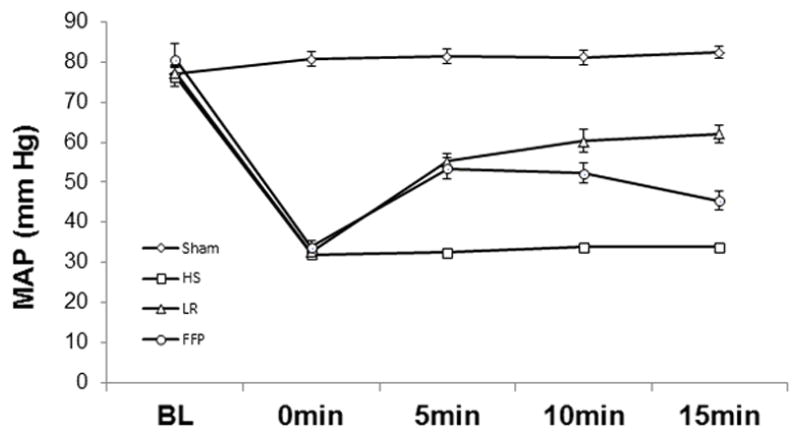
Mean arterial pressure (MAP) for shams was significantly higher compared to all other groups (p<0.01). There was no significant difference between 1X FFP and 3XLR while hemorrhagic shock (HS) was significantly lower than both FFP and LR (p<0.01). Data was analyzed using repeated measure ANOVA, followed by Tukey’s post-hoc tests and Bonerroni correction, n=8/group.
FFP mitigates lung hyperpermeability compared to lactated Ringers resuscitation
Using a novel technique, hemorrhagic shock-induced hyperpermeability was quantified in intact organs by measuring extravasation of an intravenously delivered fluorescently conjugated dextran into the lung using an In Vivo Imaging System (IVIS) Lumina XR. Lung permeability was significantly increased after hemorrhagic shock (9.5 × 1010 ± 1.9 × 1010 ) compared to shams (2.4 × 1010 ± 2.7 × 109). Permeability was reduced by 3× lactated Ringers (6.7 × 1010 ± 5.0 × 109) compared to hemorrhagic shock but further reduced by 1xFFP (4.1 × 1010 ± 3.2 × 109) (Figure 4). To validate our IVIS imaging and quantification, permeability was also assessed by Evan’s blue extravasation and findings were consistent. Evan’s blue extravasation is an indicator of lung permeability to circulating albumin (~65 KDa) was minimal in shams (4.1 ± 0.4) but was significantly increased after hemorrhagic shock (15.1 ± 1.9), lessened by 3xLR (9.7 ± 1.1) but further significantly decreased by 1xFFP (5.7 ± 0.5).
Figure 4. FFP mitigates pulmonary hyperpermeability in vivo.
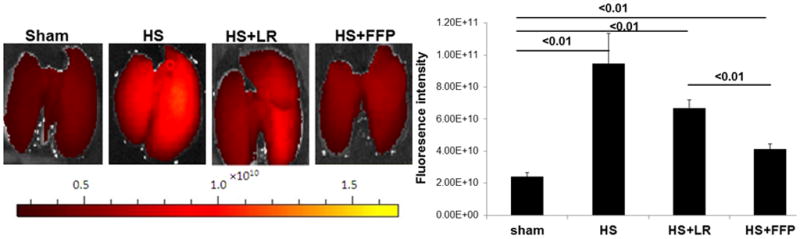
Vascular permeability was assessed in intact organs by measuring dye extravasation the infrared sensitive dye Alexafluor 680 into the lung using a Caliper Lumina XR imaging system (IVIS). Lung permeability was significantly increased after trauma and hemorrhagic shock and 3X lactated Ringers but was reduced by 1X FFP. Data are expressed as mean ± SEM, n=8 per group, p<0.05 considered statistically significant. HS= trauma and hemorrhagic shock; FFP= fresh frozen plasma; and LR= lactated Ringers.
Lung inflammation is reduced by FFP compared to lactated Ringers resuscitation
Myeloperoxidase (MPO) is a measure of neutrophil infiltration. Lung MPO immunofluorescence demonstrated a significant increase in MPO staining in lungs after hemorrhagic shock (1172±207 RFUs) compared to shams (287±32 RFUs) and 3xlactated Ringers resuscitation (815±126 RFUs) that was lessened by 1xplasma resuscitation (485±65 RFUs) (Figure 5A and B). MPO activity was consistent with changes in MPO immunofluorescence (Figure 5C).
Figure 5. FFP lessens neutrophil infiltration.
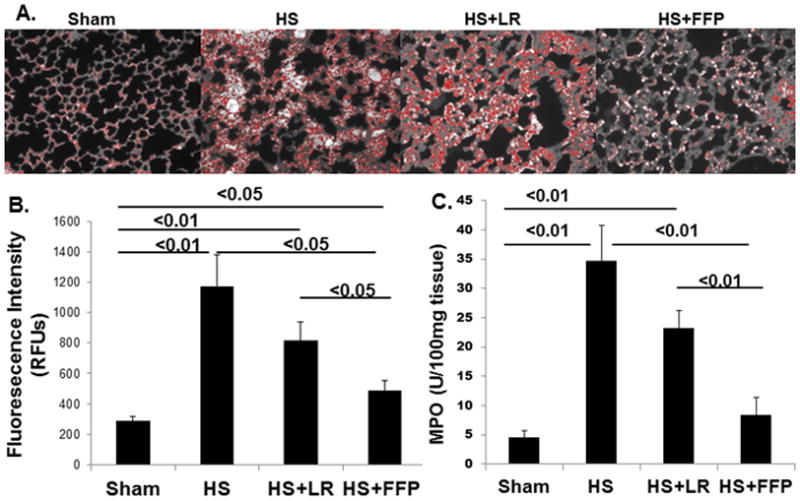
Infiltration of neutrophils into the lung tissue was assessed by myeloperoxidase (MPO) immunofluorescence staining (A and B) and MPO activity (C). Both MPO immunofluorescence and activity demonstrated HS significantly increased MPO compared to shams and 3XLR but was further significantly decreased by 1XFFP. Data are expressed as mean ± SEM, n=8 per group; p<0.05 considered statistically significant. HS= trauma and hemorrhagic shock; FFP= fresh frozen plasma; and LR= lactated Ringers
Pulmonary syndecan-1 is increased and shed syndecan-1 reduced by FFP compared to lactated Ringers resuscitation
After hemorrhagic shock alone (329 ± 48 RFUs) or 3xlactated Ringers resuscitation (402 ± 44 RFUs), pulmonary syndecan-1 immunostaining was significantly decreased compared to 1xplasma resuscitation (688 ± 69 RFUs) and shams (724 ± 76 RFUs) (Figure 6A and B). We have previously demonstrated similar results in pulmonary syndecan-1 biology using a rat model of pressure controlled hemorrhagic shock, thus highlighting the generalizability of this these findings.9
Figure 6. Pulmonary syndecan-1 is increased by FFP compared to lactated Ringers resuscitation.
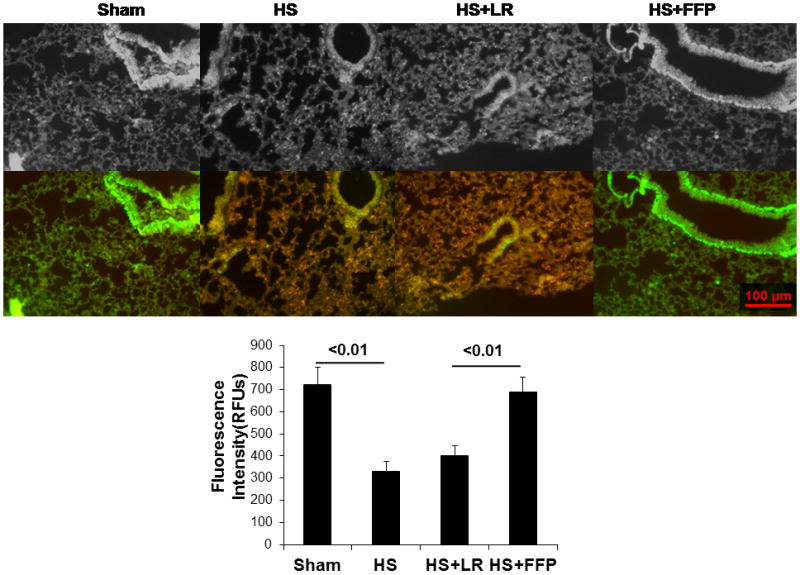
After trauma and HS alone or HS with 3XLR resuscitation, pulmonary syndecan-1 immunostaining was significantly decreased compared to 1XFFP and shams. Data are expressed as mean ± SEM, n=8 per group, p<0.05 considered statistically significant. HS= trauma and hemorrhagic shock; FFP= fresh frozen plasma; and LR= lactated Ringers
Pulmonary syndecan-1 inversely correlated with an increase in systemic syndecan-1 shedding after hemorrhagic shock. Shams had low levels of detectable shed syndecan-1 (14 ± 4 ng/ml) that was significantly increased after hemorrhagic shock (150 ± 23 ng/ml), reduced by 3xLR (103 ± 17 ng/ml) but further lessened by 1xplasma (55 ± 8 ng/ml) (Figure 7).
Figure 7. Shedding of the syndecan-1 ectodomain is lessened by FFP.
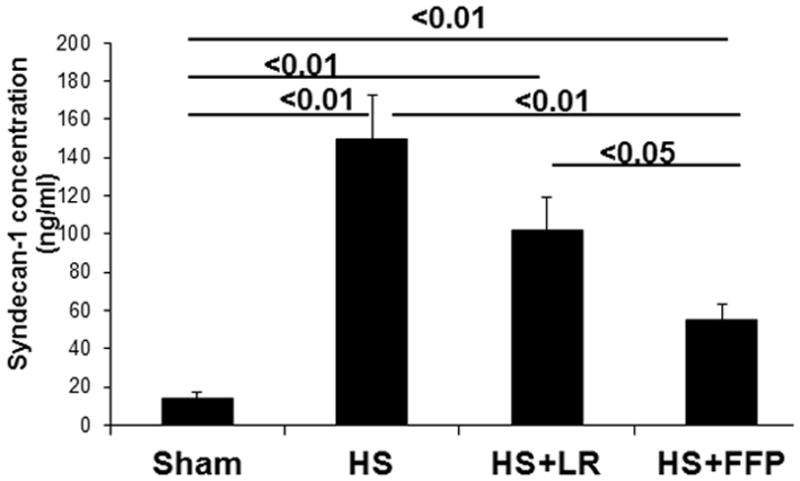
Systemic levels of syndecan-1 ectodomain were quantified by ELISA. Shedding was increased by trauma and hemorrhagic shock (HS), lessened by 3X lactated Ringers (LR) resuscitation, but further decreased by 1X fresh frozen plasma (FFP). Data are expressed as mean ± SEM, n=8 per group, p<0.05 considered statistically significant.
DISCUSSION
In vitro and in vivo we demonstrated that FFP-based resuscitation mitigated lung hyperpermeability and reduced lung inflammation compared to lactated Ringers resuscitation. These findings were not due to differences in mean arterial pressure in the early post resuscitation period but were associated with enhanced pulmonary syndecan-1 immunostaining and reduced systemic syndecan-1 ectodomain shedding.
We expanded on our previous in vitro findings of shock-induced hyperpermeability10 by now assessing flux, and extended permeability studies to examine changes over time with the use of ECIS. ECIS can assess barrier function over time with measurements being performed directly in the cell culture medium.12 FFP lessened flux and maintained reduced permeability compared to LR for the 24 hour study period. Additionally, FFP reduced leukocyte binding compared to LR based resuscitation. This is important as FFP has been associated with transfusion-related acute lung injury21 and suggests that when evaluating leukocyte mediated transfusion-related complications that all components, including crystalloid, be considered. As Middleburg et al have shown, there are additional factors that are not well understood.22 Ex vivo we employed an innovative technique to image and quantify hyperpermeability in intact organs by measuring extravasation of a fluorescent intravenous dye into the lung using IVIS. This technique will permit whole animal imaging in vivo with serial visual and quantitative analysis of hyperpermeability.17 As a first step, we imaged intact lungs ex vivo and compared our results to the well-established technique of Evan’s blue extravasation. Results were consistent between assays.
We chose three hours after the hemorrhagic shock as our experimental endpoint. This time point was based on a prior study where we demonstrated partial restoration of the endothelial glycocalyx at three hours.9 Importantly, at three hours we are not studying acute lung injury or adult respiratory distress syndrome as part of multiple organ dysfunction syndrome. Rather, we are looking at changes in pulmonary vascular hyperpermeability and inflammation as a result of hemorrhagic shock, at a clinically relevant time.8
Vascular hyperpermeability is being recognized as an increasingly important component of hemorrhagic shock. Alterations of the adherens junction complex (via VE-cadherin) with the underlying cytoskeleton (through the catenins) have been implicated as an etiology of hyperpermeability after hemorrhagic shock.23,24 We have shown that VE-cadherin is disrupted in an in vitro model of hemorrhagic shock and is associated with hyperpermeability.25 Though the clinical sequela of organ injury from hyperpermeability is well-appreciated, the contribution of hyperpermeability to death after hemorrhagic shock is not known. Interestingly, there is rare disorder termed systemic capillary leak syndrome that may provide some important clues.26 This disorder is characterized by transient episodes of hypotensive shock and death which may be linked to alterations in VE-cadherin.27 This at least suggests that hyperpermeability alone may be contributing to mortality by reducing intravascular volume. If reductions in hyperpermeability by FFP demonstrated in the current study translate into the clinical arena, this may partially explain the decrease in mortality in hemorrhagic patients receiving early empiric FFP.7–8,28
We have shown in a small pilot study that syndecan-1 is shed at the time of injury in severely injured patients in hemorrhagic shock.25 Our data also suggested that patients with higher post resuscitation sdc-1 levels had a higher mortality. More recently, Johansson et al demonstrated increased mortality in patients with high admission syndecan-1 shedding and that shedding correlated with inflammation and coagulopathy.29 The etiology of this association, however, is unclear. It is also uncertain as to the biologic role, if any, of circulating syndecan. Shedding is regulated by multiple intracellular signaling pathways converging on a diverse group of activated matrix metalloproteinases, known as sheddases.30 While shedding is known to be activated by pathologic stimuli, little is known about how ectodomain release from the cell surface is regulated, and there is no information on regulation after hemorrhagic shock. The shed ectodomains have been shown to facilitate resolution of inflammation by removing chemokines from the lung in the mouse model of endotoxemia.3 We hypothesize the hemorrhagic shock-induced shedding results in exposure of the injured endothelium to pro-inflammatory mediators and in alterations to the structural integrity of the endothelium with resultant hyperpermeability. We demonstrated this in the lung, showing that loss of syndecan-1 was associated with pulmonary permeability and inflammation. How we resuscitate the injured endothelium can either propagate injury or hasten repair. Our results suggest that FFP based resuscitation can hasten repair by restoring syndecan-1 on the cell surface and reducing syndecan-1 shedding. Syndecan-1 provides the structural backbone for the the endothelial glycocalyx and the glycocalyx has been implicated as a key modulator of permeability and inflammation in a wide variety of models and clinical diseases. In models of cardiac ischemia, shedding of the glycocalyx was associated with vascular hyperpermeability. This effect was mitigated by pre-treatment with antithrombin, which can lower susceptibility to enzymatic degradation, or hydrocortisone, which can inhibit mast cell degranulation and thus inflammation.31,32 Grundman et al have shown that alterations in the endothelial glycocalyx may be responsible for vascular leakage and leukocyte adhesion seen after cardiac arrest33 and Rehm et al have demonstrated shedding in patients undergoing abdominal aortic aneurysm repair.34 Chappell et al have highlighted the importance of the microcirculation in patients with systemic inflammatory response syndrome and sepsis and suggested targeting the glycocalyx as a therapy to improve oxygen distribution in these patients.35 A dysfunctional glycocalyx has also been implicated in tissue edema seen with diabetes and is a key initiator of atherothrombosis.36
Our data at least suggests that targeting the endothelial glycocalyx may be beneficial in severely injured patients in shock. Regulation of syndecan-1 post-injury could potentially serve as a viable therapeutic target for novel drug discovery. As FFP is composed of thousands of circulating proteins, investigation of the specific component(s) of FFP responsible for its protective effects may warrant further investigation. It would also be interesting to investigate the effects of FFP in other models of vascular instability such as pulmonary hypertension, venous insufficiency, and acute renal failure or in models of organ edema such as burns or pancreatitis.
There are several limitations to this study. One difficulty in evaluating systemic syndecan-1 levels is that ectodomain shedding may occur from multiple sites, making it possible that its loss from the lungs may not directly correlate with the levels detected in the plasma. However, our results suggest that there is an association between loss of syndecan-1 in the lungs and the corresponding increase in systemic levels. Additionally, syndecan-1 is expressed on both epithelial and endothelial cells, thus we cannot differentiate the cell type of shed ectodomain. This is a flaw of most published reports of systemic syndecan shedding. Shed syndecan is frequently attributed to the endothelial glycocalyx, but the contribution of epithelial syndecan is not discussed. To address this problem, we are planning on generating an endothelial overexpressing syndecan-1 null mouse. Although we assume that endothelial syndecan-1 is shed, we do know that loss of endothelial syndecan-1 and the glycocalyx is associated with vascular hyperpermeability.25 Lastly, we cannot rule out the possibility that enhanced perfusion by FFP may be an explanation for its beneficial effects. Our results in the early resuscitation period do not support this, as there was no difference in MAP between FFP and LR. However, as animals were awoken as soon as volume resuscitation was complete, we only have early hemodynamic data. A recent study by Jin et al in a large animal model of hemorrhagic shock and traumatic brain injury reported MAP for six hours post shock.37 They also found that FFP did not augment blood pressure or heart rate compared to crystalloid resuscitation. Interestingly, they also demonstrated decreased brain injury size and brain edema with FFP. A related limitation is that different volumes of resuscitation were used between experimental groups in vivo. Resuscitation with crystalloids at 3× shed blood volume or even 4× shed blood is common in models of hemorrhagic shock. This practice is based on the laboratory findings that demonstrated that extracellular fluid was redistributed during shock into both the intravascular and intracellular spaces. To correct this extracellular fluid deficit, infusion of isotonic crystalloid fluid in a ratio of three volumes of crystalloid to one of blood was required.38 However, using crystalloid in excess of FFP may have affected our results, particularly permeability. Our in vitro experiments utilized equal volumes between groups and results were consistent.
In conclusion, in vitro endothelial cell permeability and flux were decreased, TER was increased, and leukocyte-endothelial binding was inhibited by FFP compared to LR treated endothelial cells. In a coagulopathic mouse model of hemorrhagic shock and trauma, FFP resuscitation inhibited endothelial cell hyperpermeability and inflammation and restored pulmonary syndecan-1 expression. Modulation of pulmonary syndecan-1 expression may mechanistically contribute to the beneficial effects FFP.
Acknowledgments
This grant was funded in part by NIH P50 GM038529 and Department of Defense W81XWH-11-2-006
Footnotes
Conflict of Interest: The authors declare no competing conflicts of interest.
The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or reflecting the views of the US Department of Defense or the US Government.
References
- 1.Savery MD, Jiang JX, Park PW, Damiano ER. The endothelial glycocalyx in syndecan-1 deficient mice. Microvascular Research. 2013;87:83–91. doi: 10.1016/j.mvr.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teng YH, Aquino RS, Park PW. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012;31(1):3–16. doi: 10.1016/j.matbio.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayashida K, Parks WC, Park PW. Syndecan-1 shedding facilitates the resolution of neutrophilic inflammation by removing sequestered CXC chemokines. Blood. 2009;114:3033–3043. doi: 10.1182/blood-2009-02-204966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayashida A, Bartlett AH, Foster TJ, Park PW. Staphylococcus aureus beta-toxin induces lung injury through syndecan-1. Am J Pathol. 2009;174:509–518. doi: 10.2353/ajpath.2009.080394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Childs EW, Tharakan B, Byrge N, Tinsley JH, Hunter FA, Smythe WR. Angiopoietin-1 inhibits intrinsic apoptotic signaling and vascular hyperpermeability following hemorrhagic shock. Am J Physiol Heart Circ Physiol. 2008;294:2285–2295. doi: 10.1152/ajpheart.01361.2007. [DOI] [PubMed] [Google Scholar]
- 6.Childs EW, Tharakan B, Hunter FA, Tinsley JH, Cao X. Apoptotic signaling induces hyperpermeability following hemorrhagic shock. Am J Physiol Heart Circ Physiol. 2007;292:3179–3189. doi: 10.1152/ajpheart.01337.2006. [DOI] [PubMed] [Google Scholar]
- 7.Holcomb JB, Wade CE, Michalek JE, Chisholm GB, Zarzabal L, Schreiber MA, Gonzalez EA, Pomper GJ, Perkins JG, Spinella PC, Williams KL, Park MS. Increased plasma and platelet to red blood cell ratios improves outcome in 466 massively transfused civilian trauma patients. Ann Surg. 2008;248:447–458. doi: 10.1097/SLA.0b013e318185a9ad. [DOI] [PubMed] [Google Scholar]
- 8.Holcomb JB, del Junco DJ, Fox EE, et al. for the PROMMTT Study Group. The Prospective, Observational, Multicenter, Massive Transfusion Study, PROMMTT: Comparative Effectiveness of a Time-varying Treatment and Competing Risks. Arch Surg. 2012;15:1–10. doi: 10.1001/2013.jamasurg.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kozar RA, Peng Z, Zhang R, Holcomb JB, Pati S, Park P, Ko TC, Paredes A. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth Analg. 2011;112:1289–1295. doi: 10.1213/ANE.0b013e318210385c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pati S, Matijevic N, Doursout MF, Ko T, Cao Y, Deng X, Kozar RA, Hartwell E, Conyers J, Holcomb JB. Protective effects of fresh frozen plasma on vascular endothelial permeability, coagulation, and resuscitation after hemorrhagic shock are time dependent and diminish between days 0 and 5 after thaw. J Trauma. 2010;69 (Suppl 1):S55–63. doi: 10.1097/TA.0b013e3181e453d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pati S, Wataha K, Menge T, Deng X, Bode A, Holcomb JB, Potter D, Kozar R, Spinella RC, Pati S. Sprayed dried plasma and fresh frozen plasma modulate permeability and inflammation in vitro in vascular endothelial cells. Transfusion. 2013;53 (Suppl 1):80S–90S. doi: 10.1111/trf.12040. [DOI] [PubMed] [Google Scholar]
- 12.Bagnaninchi PO, Drummond N. Real-time label-free monitoring of adipose-derived stem cell differentiation with electric cell-substrate impedance sensing. PNAS. 2011;108(16):6462–67. doi: 10.1073/pnas.1018260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. PNAS. 1992;89:7919–23. doi: 10.1073/pnas.89.17.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Letourneau PA, Menge TD, Wataha KA, Wade CE, Cox CS, Holcomb JB, Pati S. Human Bone Marrow Derived Mesenchymal Stem Cells Regulate Leukocyte-Endothelial Interactions and Activation of Transcription Factor NF-Kappa B. Journal of Tissue Science & Engineering. 2011;0:S3–001. doi: 10.4172/2157-7552.S3-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chesebro BB, Rahn P, Carles M, Esmon CT, Xu J, Brohi K, Frith D, Pittet JF, Cohen MJ. Increase in activated Protein C mediates acute traumatic coagulopathy in mice. Shock. 2009;32(6):659–665. doi: 10.1097/SHK.0b013e3181a5a632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiang M, Yuan Y, Fan L, Li Y, Li A, Yin L, Scott MJ, Xiao G, Billiar TR, Wilson MA, Fan J. Role of macrophages in mobilization of hematopoietic progenitor cells from bone marrow after hemorrhagic shock. Shock. 2012;37(5):518–23. doi: 10.1097/SHK.0b013e318249b81d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costantini TW, Eliceiri BP, Peterson CY, Loomis WH, Putnam JG, Baird A, Wolf P, Bansal V, Coimbra R. Quantitative assessment of intestinal injury using a novel in vivo, near-infrared imaging technique. Mol Imaging. 2010;9(1):30–39. [PMC free article] [PubMed] [Google Scholar]
- 18.Childs EW, Tharakan B, Hunter FA, Tinsley JH, Cao X. Apoptotic signaling induces hyperpermeability following hemorrhagic shock. Am J Physiol Heart Circ Physiol. 2007;292:3179–3189. doi: 10.1152/ajpheart.01337.2006. [DOI] [PubMed] [Google Scholar]
- 19.Peng Z, Ban K, Sen A, Grill R, Park PY, Costantini TW, Lin W, Kozar RA. Syndecan-1 plays a novel role in enteral glutamine’s gut protective effects of the post ischemic gut. Shock. 2012;38(1):57–62. doi: 10.1097/SHK.0b013e31825a188a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menge T, Zhao Y, Zhao J, Wataha K, Geber M, Zhang J, Letourneau P, Redell J, Shen K, Wang J, Peng Z, Xue H, Kozar R, Cox C, Khakoo AY, Holcomb H, Dash P, Pati S. Mesenchymal stem cells regulate blood brain barrier integrity in traumatic brain injury through production of the soluble factor TIMP3. Sci Transl Med. 2012;21;4(161):161ra150. doi: 10.1126/scitranslmed.3004660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vamvakas EC, Blajchman MA. Blood still kills: six strategies to further reduce allogeneic blood transfusion-related mortality. Transfus Med Rev. 2010;24(2):77–124. doi: 10.1016/j.tmrv.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Middelburg RA, van Stein D, Atsma F, Wiersum-Osselton JC, Porcelijn L, Beckers EA, Briët E, van der Bom JG. Alloexposed blood donors and transfusion-related acute lung injury: a case-referent study. Transfusion. 2011;51(10):2111–7. doi: 10.1111/j.1537-2995.2011.03118.x. [DOI] [PubMed] [Google Scholar]
- 23.Tharakan B, Hellman J, Sawant DA, Tinsley JH, Hunter FA, Smythe WR, Childs EW. β-catenin dynamics in the regulatin of microvascular endothelial cell hyperpermeability. Shock. 2012;37(3):306–311. doi: 10.1097/SHK.0b013e318240b564. [DOI] [PubMed] [Google Scholar]
- 24.Sawant DA, Tharakan B, Hunter FA, Smythe WR, Childs EW. Role of β-catenin in regulating microvascular endothelial cell hyperpermeability. J Trauma. 2011;70(2):487–488. doi: 10.1097/TA.0b013e31820b3ed7. [DOI] [PubMed] [Google Scholar]
- 25.Haywood-Watson RJ, Holcomb JB, Gonzalez EA, Peng Z, Pati S, Park PW, Wang WW, Zaske AM, Menge T, Kozar RA. Modulation of syndecan-1 shedding after hemorrhagic shock and resuscitation. PLoS ONE. 2011;6(8):e23530. doi: 10.1371/journal.pone.0023530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Druey KM, Greipp PR. Narrative review: the systemic capillary leak syndrome. Ann Intern Med. 2010;153(2):90–98. doi: 10.1059/0003-4819-153-2-201007200-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiw Z, Ghosh CC, Patel R, Iwaki S, Gaskins D, Nelson C, Jones N, Greipp PR, Druey KM. Vascular endothelial hyperpermeability induces the clinical symptoms of Clarkson disease (the systemic capillary leak syndrome) Blood. 2012;119(18):4321–4332. doi: 10.1182/blood-2011-08-375816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spinella PC, Perkins JG, Grathwohl KW, Beekley AC, Niles SE, McLaughlin DF, Wade CE, Holcomb JB. Effect of plasma and red blood cell transfusions on survival in patients with combat related traumatic injuries. J Trauma. 2008;64(2):S69–S78. doi: 10.1097/TA.0b013e318160ba2f. [DOI] [PubMed] [Google Scholar]
- 29.Johansson PI, Stensballe J, Rasmussen LS, Ostrowski SR. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, Protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann Surg. 2011;254(2):194–200. doi: 10.1097/SLA.0b013e318226113d. [DOI] [PubMed] [Google Scholar]
- 30.Fitzgerald ML, Wang Z, Park PW, Murphy G, Bernfield M. Shedding of syndecan-1 and −4 ectodomain is regulated by multiple signaling pathways and mediated by a TIMP-3 sensitive metalloproteinase. J Cell Biol. 2000;148:811–824. doi: 10.1083/jcb.148.4.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chappell D, Jacob M, Hofmann-Kiefer K, Rehm M, Welsch U, Conzen P, Becker BF. Antithrombin reduces shedding of the endothelial glycocalyx following ischaemia/reperfusion. Cardiovasc Res. 2009;83(2):388–96. doi: 10.1093/cvr/cvp097. [DOI] [PubMed] [Google Scholar]
- 32.Chappell D, Jacob M, Hofmann-Kiefer K, Bruegger D, Rehm M, Conzen P, Welsch U, Becker BF. Hydrocortisone preserves the vascular barrier by protecting the endothelial glycocalyx. Anesthesiology. 2007;107(5):776–84. doi: 10.1097/01.anes.0000286984.39328.96. [DOI] [PubMed] [Google Scholar]
- 33.Grundmann S, Fink K, Rabadzhieva L, Bourgeois N, Schwab T, Moser M, Bode C, Busch HJ. Perturbation of the endothelial glycocalyx in post cardiac arrest syndrome. Resuscitation. 2012;83(6):715–20. doi: 10.1016/j.resuscitation.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 34.Rehm M, Bruegger D, Christ F, Conzen P, Thiel M, Jacob M, Chappell D, Stoeckelhuber M, Welsch U, Bruno R, Peter K, Becker BF. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation. 2007;116:1896–1906. doi: 10.1161/CIRCULATIONAHA.106.684852. [DOI] [PubMed] [Google Scholar]
- 35.Chappell D, Westphal M, Jacob M. The impact of the glycocalyx on microcirculatory oxygen distribution in critical illness. Curr Opin Anaesthesiol. 2009;22:155–62. doi: 10.1097/ACO.0b013e328328d1b6. [DOI] [PubMed] [Google Scholar]
- 36.Drake-Holland AJ, Noble MI. Update on the important new drug target in cardiovascular medicine - the vascular glycocalyx. Cardiovasc Hematol Disord Drug Targets. 2012;12(1):76–81. doi: 10.2174/187152912801823183. [DOI] [PubMed] [Google Scholar]
- 37.Jin G, deMoya MA, Duggan M, Knightly T, Mejaddam AY, Hwabejire J, Lu J, Smith WM, Kasotakis G, Velmahos GC, Socrate S, Alam HB. Traumatic brain injury and hemorrhagic shock evaluation of different resuscitation strategies in a large animal model of combined insults. Shock. 2012;38(1):49–56. doi: 10.1097/SHK.0b013e3182574778. [DOI] [PubMed] [Google Scholar]
- 38.Moore FA, McKinley BA, Moore EE. The next generation of shock resuscitation. Lancet. 2004;363(9425):1988–96. doi: 10.1016/S0140-6736(04)16415-5. [DOI] [PubMed] [Google Scholar]