

Summary
As a complementary approach to positional cloning, we used in vivo complementation with bacterial artificial chromosome (BAC) clones expressed in transgenic mice to identify the circadian Clock gene. A 140 kb BAC transgene completely rescued both the long period and the loss-of-rhythm phenotypes in Clock mutant mice. Analysis with overlapping BAC transgenes demonstrates that a large transcription unit spanning “100,000 base pairs is the Clock gene and encodes a novel basic–helix-loop-helix–PAS domain protein. Overexpression of the Clock transgene can shorten period length beyond the wild-type range, which provides additional evidence that Clock is an integral component of the circadian pacemaking system. Taken together, these results provide a proof of principle that “cloning by rescue” is an efficient and definitive method in mice.
Introduction
Circadian (~24 hour) rhythms regulate the function of living systems at virtually every level of organization from molecular to organismal (Pittendrigh, 1993; Turek, 1994; Takahashi, 1995). Significant progress has been made in two different arenas of the circadian field. At the physiological level, there is now a general understanding of the neural organization of circadian pacemaking systems in both vertebrate and invertebrate model systems (Meijer and Rietveld, 1989; Takahashi et al., 1989, 1993; Klein et al., 1991; Block et al., 1993). In mammals, circadian pacemakers have been localized in the hypothalamic suprachiasmatic nucleus (SCN) and the retina (Ralph et al., 1990; Moore, 1996; Tosini and Menaker, 1996). The SCN is both necessary and sufficient for the generation of circadian rhythms at the organismal level (Ralph et al., 1990; Klein et al., 1991; Moore, 1995). At the molecular level, genetic analysis of circadian rhythms has led to the identification and cloning of three genes (period and timeless in Drosophila and frequency in Neurospora) that are essential for the generation of circadian rhythms in these organisms (reviewed in Dunlap, 1996; Hall, 1995; Rosbashet al.,1996). These genes have circadian expression patterns that define molecular oscillations of transcription and translation forming autoregulatory feedback loops thought to constitute the core elements of the circadian clock mechanism (Hardin et al., 1990, 1992; Aronson et al., 1994; Zeng et al., 1994; Sehgal et al., 1995; Dunlap, 1996).
Despite mutual interest, the two approaches of physiology and genetics have, for the most part, remained on separate paths because the organisms in which physiological approaches have been most fruitful (e.g., rats, hamsters, chick, Xenopus, Aplysia, Bulla) have not been amenable to genetics; conversely, the genetically tractable organisms, Drosophila and Neurospora, have not been optimal for physiological studies (Dunlap, 1993, 1996; Takahashi, 1995). Moreover, physiological approaches have thus far failed to identify molecular components of the circadian clock, while, at the same time, genetic approaches have not led to the cloning of orthologs of the canonical clock genes (per, tim, frq) in vertebrates (Takahashi, 1995).
For these reasons, we have undertaken a systematic genetic approach to elucidate the molecular mechanisms of circadian rhythms in mammals using the mouse. Our strategy, outlined in Takahashi et al. (1994), is to use forward genetics (from phenotype to gene) as a tool for gene discovery in the mouse. First, phenotype-driven N-ethyl-N-nitrosourea (ENU) mutagenesis screens (Dove, 1987) are used to isolate circadian rhythm mutants in the mouse. Second, the genes defined by mutations are identified using candidate gene and positional cloning methods. Finally, the functions of the identified genes are studied and used to unravel the pathways in which these genes act. Ultimately, the essential set of genes underlying the circadian mechanism can be defined and analyzed to explain the circadian clock system at a mechanistic level.
In the initial phase of our mutagenesis program, we identified a novel mutation named Clock that defines a gene essential for normal circadian behavior (Vitaterna et al., 1994). In the work presented here and in the accompanying paper (King et al., 1997b, this issue of Cell), we report the completion of the second phase for the Clock mutation—the elucidation of its molecular identity. We have used two different but complementary approaches to find Clock: positional cloning and in vivo complementation via BAC transgenesis.
In a number of genetically tractable model organisms, such as Caenorhabditis and Arabidopsis, genes defined by mutation are routinely cloned by rescue of the mutant phenotype using in vivo complementation with genomic clones mapping to the non-recombinant interval (Gibson and Somerville, 1993; Mello and Fire, 1995). The development of transgenic mice expressing large insert YAC and P1 clones raised the possibility of using in vivo complementation to clone genes in the mouse as well (Schedl et al., 1993; Smith et al., 1995 Peterson et al., 1997). In this paper, we show that transgenic expression of BAC clones in mice can be used for in vivo complementation to identify a novel gene underlying the Clock mutation. These results demonstrate complete rescue of a complex behavior and identification of the gene underlying a mutation in mice. The method of cloning by rescue in mice is an important approach with wide application because it is both efficient and definitive.
Results
The Clock mutation maps to the midportion of chromosome 5, approximately 0.7 cM distal of the Kit locus (King et al., 1997a). High resolution genetic and physical mapping of the Clock mutation described in the accompanying paper (King et al., 1997b) defines a 0.2 cM non-recombinant interval that is flanked by the markers D5Mit307 and D5Nwu2 and that corresponds to a physical distance of approximately 200–250 kb (Figure 1). The closest distal (relative to the centromere) recombinant marker, D5Nwu2, is located on a 40 kb NotI fragment from BAC 54, while the non-recombinant marker D5Nwu1 is located on the 100 kb NotI fragment from BAC 54. This analysis showed that BAC 54 covers the largest physical interval within the distal portion of the critical region containing Clock. A second BAC clone (BAC 52) extends an additional ~60 kb proximal to BAC 54; these two BAC clones form a ~200 kb contig that covers most of the non-recombinant region of the Clock locus (Figure 1). Within this non-recombinant interval, there were no previously identified genes or expressed sequences. However, long-range restriction mapping of YAC clones and genomic sequencing of BAC clones covering this interval show there are two distinct CpG islands (Figure 1), which are characteristic features of the promoter regions of many constitutively expressed and tissue-specific genes (Bird, 1992).
Figure 1.
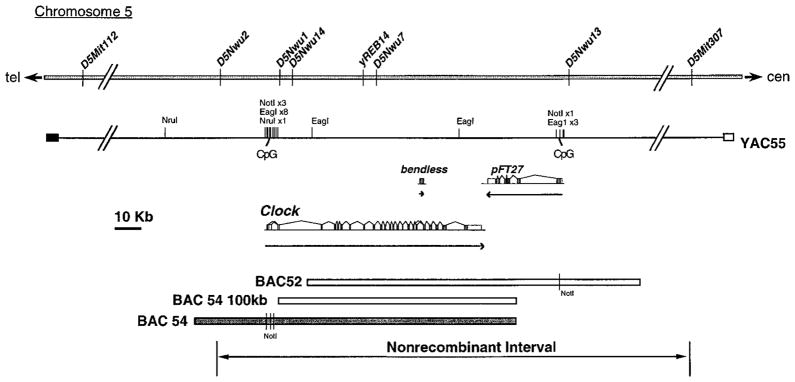
Physical Map of the Mouse Clock Locus Clock is localized to the mid-portion of mouse chromosome 5; SSLP markers D5Nwu2 and D5Mit307 define the nonrecombinant interval containing Clock. The relative positions of various sequence-tagged sites, SSLP markers, and restriction enzyme sites are indicated. D5Nwu2, D5Nwu1, D5Nwu14, D5Nwu7, and D5Nwu13 are SSLP markers described in King et al. (1997b). YAC55 appears to be a nonchimeric clone based on STS content mapping and long-range restriction mapping of overlapping YAC clones (data not shown). NotI, EagI, and NruI restriction profiles reveal two CpG islands. The three transcription units identified in this region, bendless, pFT27, and Clock, define the possible candidate genes; arrows indicate the direction of transcription and genomic extent, and introns and exons are indicated above the arrows. The BAC 54 rescuing clone is shaded. The two clones that fail to rescue, BAC 52 and the 100 kb NotI BAC 54 fragment, are shown as open bars. Telomeric (tel) and centromeric (cen) directions are indicated.
Generation and Analysis of BAC Transgenic Mouse Lines
As an alternative approach to the positional cloning methods described in the accompanying paper (King et al., 1997b), we used in vivo complementation with BAC clones expressed in transgenic mice as a functional assay for the Clock gene. Because previous work has shown that the mutant Clock allele is antimorphic (a “competitive” type of dominant-negative mutation) (King et al., 1997a), we reasoned that it should be possible to rescue the Clock mutant phenotype by overexpression of the wild-type gene.
We generated transgenic mice by pronuclear injection of BAC DNA using clones that mapped to the critical region containing Clock. Three sets of DNA preparations were used: (1) a 140 kb circular BAC 54 clone, (2) a 100 kb linear NotI fragment from BAC 54, and (3) a 160 kb circular BAC 52 clone (Figure 1). Fertilized oocytes were obtained from two different crosses. In one cross, wild-type CD1 females were mated with wild-type CD1 males to produce wild-type embryos for pronuclear injection. In the other cross, wild-type CD1 females were mated with (BALB/cJ x C57BL/6J)F2 Clock/Clock males to produce Clock/+ heterozygous embryos for injection. As a consequence, the transgenic founder animals were either wild-type or heterozygous at the Clock locus. Transgenic mice were identified by PCR of the BAC vector–insert junctions and by Southern blot analysis using probes specific for either the BAC vector or for genomic sequences within the clone. Of the mice born from the BAC 54–injected embryos, 5 of 64 were positive for the transgene by both methods, 3 of 54 were positive for the 100 kb linear fragment of BAC 54, and 2 of 12 were positive for BAC 52 DNA integration. A summary of the BAC transgenic lines and their characteristics is presented in Table 1.
Table 1.
Summary of BAC Transgenic Lines
| Transgenic Line | Founder Genotype | DNA Injected | Transgene Copy Number | Transmittance |
|---|---|---|---|---|
| TG14 | Clock/+ | BAC 54, 140 kb circular | 2–3 | 23/45 |
| TG36 | Clock/+ | BAC 54, 140 kb circular | 3–4 | 50/99 |
| TG55 | +/+ | BAC 54, 140 kb circular | 8–10 | 17/45 |
| TG60 | +/+ | BAC 54, 140 kb circular | 1–2 | 10/35 |
| TG48 | +/+ | BAC 54, 140 kb circular | ND | 6/46 |
| TG80 | +/+ | BAC 54, 100 kb linear | 2–3 | 27/57 |
| TG97 | +/+ | BAC 54, 100 kb linear | 10–12 | 15/26 |
| TG91 | Clock/+ | BAC 54, 100 kb linear | ND | 2/21 |
| TG121 | Clock/+ | BAC 52, 160 kb circular | 1 | 20/75 |
| TG126 | Clock/+ | BAC 52, 160 kb circular | 4–5 | 10/24 |
Each transgenic line was derived independently from one founder mouse. Founder animals were either wild-type (+/+) or heterozygous (Clock/+) at the Clock locus as determined by SSLP genotype. Three different DNA constructs were used to produce transgenic mice. Transgene copy numbers were estimated from densitometry analysis of Southern blots (Figure 2). ND, not determined. Transmittance indicates the ratio of the number of transgene positive progeny over the total number of progeny. TG48 and TG91 founder animals appeared mosaic and were not analyzed further.
To establish different transgenic lines, each founder was crossed either to Clock/+ females or to Clock/Clock males. We used Clock/+ rather than Clock/Clock females to produce F1 progeny because matings with homozygous mutant females are, in general, less successful than those with heterozygous females (M. H. V. et al., unpublished data). The F1 progeny were analyzed by PCR and Southern blotting for the presence of the integrated transgene and were genotyped for the Clock locus by flanking SSLP markers. In the majority of cases, founder animals transmitted the transgene to ~50% of their progeny, which is consistent with nonmosaic germ-line transmission. Two founder animals (TG48 and TG91) produced few transgenic progeny and appeared to be germline mosaics (Table 1). These lines were not analyzed further.
Transgene copy number was estimated by Southern blot analysis of genomic DNA from F1 transgenic animals and their non-transgenic littermates (Figure 2, Table 1). The range of transgene copy numbers varied from 1–2 copies/genome (line TG60) to 10–12 copies/genome (line TG97). Similar estimates were obtained from analysis of BamHI-digested genomic DNA hybridized with a 1.3 kb probe from the pBeloBAC11 vector (data not shown). Integrity of the incorporated transgene was assessed by Southern blot analysis using genomic DNA probes that map to different regions of BAC 52 BAC 54 (data not shown). The hybridization pattern for transgenic lines was consistent with that observed for both non-transgenic mouse genomic DNA and BAC DNA, with one exception: the probe 052A3AA01, which over laps yREB14 (Brunkow et al., 1995), identified a BamHI band (“2 kb) in line TG55 transgenic animals that was not present in non-transgenic animals. In addition, PCR analysis of BAC 54 transgenic lines showed the presence of both BAC vector–insert junctions in DNA of all positive animals. Taken together, these data suggest that the BAC transgenes, with the exception of line TG55 appear to be intact without obvious rearrangements.
Figure 2.
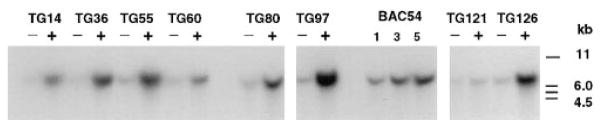
Transgene Copy Number Quantification
BgIII-digested genomic DNA from transgenic and non-transgenic F1 progeny from each founder was probed with cDNA clone L4, which originates from the 3′untranslated region of the Clock gene. A 7kb fragment, which represents the endogenous Clock gene sequence) 1 copy/genome), was detected in all samples and was of much higher intensity in transgene-positive animals. A standard amount of BglII-digested BAC 54 DNA equivalent to 1, 3, and 5 copies per genome was mixed with 5 μg of control mouse DNA and loaded on the same gel. Band intensity was quantitated by densitometry, and transgene copy number was estimated (Table 1). (−) and (+) indicate mice negative or positive for transgene integration, respectively. The blot was normalized for DNA loading by hybridization with a mouse rhodopsin cDNA probe (a single copy copy gene [Al-Ubaidi et al., 1990]) (data not shown).
BAC 54 Rescues the Clock Mutation
Transgenic expression of the 140 kb BAC 54 clone completely rescued the long period and loss-of-rhythm phenotypes in Clock mutant mice. Among the four BAC 54 transgenic lines included in this analysis, line TG36 is of particular interest, owing to the genetic background of the founder and the breeding protocol that was used to produce F1 offspring (Figure 3). The founder male, a Clock heterozygote, was mated with (BALB/cJ x C57BL/6J)F2 Clock heterozygous females, which allowed us to examine the effect of the BAC 54 transgene on circadian behavior in mice of all three Clock genotypes. Figure 4 shows representative activity records of wild type, Clock heterozygous and Clock homozygous mice in the presence and absence of the BAC 54 transgene. As shown in Figures 4B and 4C, non-transgenic progeny from this founder express all the phenotypic characteristics described for the Clock mutation. Clock/+ animals express longer circadian period (mean = 24.18 hours) as compared to their wild-type littermates (mean = 23.48 hours). After initial transfer to constant darkness, Clock/Clock mice express even longer circadian periods (mean = 27.36 hours). In the presence of the BAC 54 transgene, both Clock/+ and Clock/Clock mice expressed circadian periodicity comparable to wild-type mice (Figure 4D). Similar results were obtained for all four BAC 54 lines tested (Table 2). Interestingly, in the wild-type mice, the BAC 54 transgene shortened period beyond the wild-type range (Figure 4D).
Figure 3.
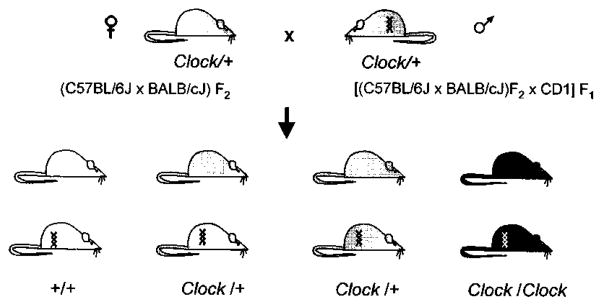
Genetic Cross Used to Produce Line TG36.
Clock genotypes are depicted by shading, with wild-type open, heterozygotes shaded, and homozygotes closed. The presence of the 140kb BAC54 transgene is indicated by a double helix symbol. A Clock/+ transgenic founder male was bred with Clock heterozygous females so that mice of six genotypic classes (the three Clock genotypes, both with and without the transgene) were represented in the progeny.
Figure 4.

The BAC 54 Transgene Rescues Mutant Circadian Phenotype and Affects Wild-Type Circadian Activity Wheel running activity records and period estimates are shown for F1 transgenic and control mice from transgenic line TG36, which carries the BAC 54 transgene. All animals were maintained on an LD 12:12 cycle (as indicated by the light/dark bar) for 5–7 days and transferred into constant darkness at a time indicated by the arrow.
(A) Activity records of the wild-type mice with (right) and without (left) the BAC 54 transgene (tg).
(B) Activity records of Clock/+ mice with (right) and without (left) the BAC 54 transgene.
(C) Activity records of Clock/Clock mice with (right) and without (left) the BAC 54 transgene.
(D) Histogram of period estimates for 83 F1 progeny from line TG36. Individual data points are indicated by open circles. Not all individual data points can be seen due to their overlap. Numbers on the bottom of the bars indicate the number of animals in each group that were wheel tested for their circadian behavior. Within this line, significant effects of Clock genotype (2 DF, F = 239, p < 0.00001), transgene presence (1 DF, F = 645, p < 0.00001), as well as genotype by transgene interactions (2 DF, F = 205, p < 0.00001) were detected (GLM ANOVA). Wild-type transgenic mice had significantly shorter periods than all non-transgenic groups, Clock/+ and Clock/Clock transgenic mice had significantly shorter periods than Clock/+ and Clock/Clock non-transgenic mice, and Clock/Clock non-transgenic mice had significantly longer periods than all other groups as determined by Tukey’s post hoc tests (p < 0.05).
Table 2.
Period Estimates for Different BAC Transgenic Lines
| Transgenic Line1 | +/+ | +/+ tg | Clock/+ | Clock/+ tg | Clock/Clock | Clock/Clock tg |
|---|---|---|---|---|---|---|
| BAC 54 (140 kb circular)2 | ||||||
| TG14 | N/A | N/A | 24.22 ± 0.183 (n = 5) | 23.08 ± 0.146 (n = 7) | 27.06 ± 0.314 (n = 7) | 23.27 ± 0.099 (n = 9) |
| TG36 | 23.48 ± 0.048 (n = 11) | 22.89 ± 0.051 (n = 10) | 24.18 ± 0.053 (n = 20) | 23.21 ± 0.047 (n = 20) | 27.36 ± 0.282 (n = 8) | 23.18 ± 0.082 (n = 14) |
| TG55 | 23.41 ± 0.091 (n = 10) | 22.92 ± 0.137 (n = 8) | 24.12 ± 0.206 (n = 8) | 22.77 ± 0.099 (n = 7) | N/A | N/A |
| TG60 | N/A | N/A | 23.91 ± 0.1 (n = 13) | 23.13 ± 0.122 (n = 6) | N/A | N/A |
| BAC 54 (100 kb linear)3 | ||||||
| TG80 | 23.44 ± 0.101 (n = 8) | 23.50 ± 0.07 (n = 5) | 23.92 ± 0.125 (n = 9) | 23.64 ± 0.083 (n = 4) | N/A | N/A |
| TG97 | N/A | N/A | 23.93 ± 0.04 (n = 4) | 23.67 ± 0.065 (n = 7) | N/A | N/A |
| BAC 52 (160 kb circular)4 | ||||||
| TG121 | 23.50 ± 0.142 (n = 4) | 23.66 ± 0.125 (n = 2) | 23.99 ± 0.11 (n = 13) | 23.96 ± 0.032 (n = 5) | 26.83 ± 0.40 (n = 2) | 26.87 ± 0.161 (n = 4) |
| TG126 | N/A | N/A | 23.45 ± 0.155 (n = 5) | 23.57 ± 0.125 (n = 4) | 26.87 ± 0.569 (n = 3) | 25.54 ± 0.103 (n = 3) |
Period estimates of BAC transgenic (tg) animals and their nontransgenic littermates. The rhythm of locomotor activity was recorded from a total of 266 F1 progeny from 8 transgenic lines. Free-running circadian periods were calculated for the 20-day interval during exposure to DD (days 1–20) by a χ2 periodogram (Sokolove and Bushel, 1978) (see text). 21 animals were excluded from analysis due to missing data or lack of a single dominant or significant circadian periodicity being detected. The Clock genotype was assigned in all but 5 cases, which were recombinant, by SSLP genotype (see text); 5 recombinant non-transgenic animals were included in the analysis on the basis of their behavioral phenotype. Values shown are the mean ± standard error, with the number of animals that were analyzed from each group indicated beneath. N/A, not applicable.
A Generalized Linear Models (GLM) Analysis of Variance (ANOVA) was performed on the data presented here (NCSS 6.0). Significant effects of the transgenic construct used (DF = 2; F = 34.5; p < 0.00001), the Clock genotype (DF = 2; F = 246; p < 0.00001), and the presence of the transgene (DF = 1; F = 97.7; p < 0.00001) were detected. In addition, significant construct by genotype (DF = 4; F = 12.8; p < 0.00001), construct by transgene presence (DF = 2; F = 58.7; p < 0.00001), genotype by transgene presence (DF = 2; F = 40.5; p < 0.00001) and three-way interactions (DF = 4; F = 17.2; p < 0.00001) were detected. This indicates that circadian period is affected by Clock genotype, by transgenic BAC DNA, and by the transgenic construct used. The effect of the transgene depends on the construct used (full-length BAC 54 had an effect; the others did not), and on the Clock genotype (i.e., greater period shortening by the transgene is seen in Clock mutants than in wild-type mice).
Among the lines involving this transgenic construct, a 3-way GLM ANOVA was performed. Significant effects of Clock genotype (DF = 2; F = 68.58; p < 0.0001), transgene presence (DF = 1; F = 149.67; p < 0.005), and genotype by transgene presence interaction (DF = 2; F = 60.34; p = 0.000106) were detected. This transgenic construct is effective in shortening period, and does so differentially with respect to the Clock genotype. No significant effect of transgenic line was detected (DF = 3; F = 1.8; p = 0.149). The effects of the BAC 54 transgene on circadian period thus apparently do not depend on the position of incorporation or level of transgene expression in this analysis.
Among the lines involving this transgenic construct, a 2-way GLM ANOVA was performed. Clock genotype (DF = 1; F = 13.1; p < 0.001) was significant, but no significant effect of the transgene presence (DF = 1; F = 1.41; p = 0.244) was detected. The 100 kb fragment of BAC 54 fails to shorten circadian period significantly as a transgene.
Among the lines involving this transgenic construct, a 2-way GLM ANOVA was performed. Clock genotype (DF = 1; F = 130; p < 0.00001) was significant, but no significant effect of the transgene presence (DF = 1; F = 0.61; p = 0.438) was detected. Transgenic BAC 52 fails to shorten circadian period significantly.
A second phenotypic effect of the Clock mutation is the loss of circadian rhythmicity with time in constant darkness in homozygotes (Vitaterna et al., 1994). To quantitate the relative amplitude and persistence of circadian rhythms, Fourier analyses were performed on the wheel-running activity data from the first and second rescue of the Clock mutation. Because the different BAC 10-day intervals in constant darkness (Table 3). As expected, in both transgenic and non-transgenic wild-type and Clock/+ mice, a clear peak within the circadian range was present with no change in the amplitude of the power spectral density (PSD) between the two time intervals. In Clock/Clock mice, a circadian peak with reduced amplitude was found in most animals during the first 10-day interval, while for the second 10-day interval it was significantly reduced (Table 3). The BAC 54 transgene completely rescued the loss-of-rhythm phenotype in Clock/Clock mice (lines TG14 and TG36, Table 3). Overall, mice from all of the BAC 54 transgenic lines showed complete rescue of the Clock mutant phenotype. These results demonstrate that the 140 kb genomic interval within BAC 54 is sufficient for complete rescue of the Clock mutation. Because the different BAC54 transgenes must be acting in trans (the Clock mutation and transgene segregate independently), these results strongly suggest that the gene and its requisite regulatory sequences are contained within BAC 54.
Table 3.
Amplitude of the Circadian Peak for Different BAC Transgenic Lines
| Transgenic Line1 | +/+ | +/+ tg | Clock/+ | Clock/+ tg | Clock/Clock2 | Clock/Clock tg |
|---|---|---|---|---|---|---|
| BAC 54 140 kb | ||||||
| TG14 | ||||||
| DD day 1–10 | N/A | N/A | 3.20 ± 0.103 | 3.28 ± 0.204 | 2.84 ± 0.136 | 3.38 ± 0.112 |
| DD day 11–20 | 3.21 ± 0.136 (n = 5) | 3.30 ± 0.080 (n = 6) | 2.31 ± 0.192 (n = 7) | 3.43 ± 0.063 (n = 9) | ||
| TG36 | ||||||
| DD day 1–10 | 3.28 ± 0.151 | 3.24 ± 0.113 | 2.94 ± 0.114 | 2.94 ± 0.128 | 2.79 ± 0.117 | 3.30 ± 0.064 |
| DD day 11–20 | 3.29 ± 0.150 (n = 11) | 3.37 ± 0.102 (n = 10) | 2.93 ± 0.146 (n = 21) | 3.10 ± 0.122 (n = 18) | 2.27 ± 0.217 (n = 14) | 3.24 ± 0.089 (n = 13) |
| TG55 | ||||||
| DD day 1–10 | 3.44 ± 0.083 | 3.31 ± 0.103 | 3.46 ± 0.094 | 3.53 ± 0.094 | N/A | N/A |
| DD day 11–20 | 3.49 ± 0.075 (n = 10) | 3.36 ± 0.088 (n = 8) | 3.53 ± 0.078 (n = 8) | 3.53 ± 0.057 (n = 7) | ||
| TG60 | ||||||
| DD day 1–10 | N/A | N/A | 3.02 ± 0.099 | 3.09 ± 0.120 | N/A | N/A |
| DD day 11–20 | 3.21 ± 0.078 (n = 13) | 3.31 ± 0.132 (n = 6) | ||||
| BAC 54 100 kb | ||||||
| TG80 | ||||||
| DD day 1–10 | 3.35 ± 0.108 | 3.31 ± 0.161 | 3.19 ± 0.122 | 3.47 ± 0.216 | N/A | N/A |
| DD day 11–20 | 3.59 ± 0.107 (n = 8) | 3.52 ± 0.050 (n = 4) | 3.24 ± 0.197 (n = 8) | 3.33 ± 0.280 (n = 4) | ||
| TG97 | ||||||
| DD day 1–10 | N/A | N/A | 2.98 ± 0.157 | 3.03 ± 0.124 | N/A | N/A |
| DD day 11–20 | 2.90 ± 0.106 (n = 4) | 3.02 ± 0.134 (n = 8) | ||||
| BAC 52 160 kb | ||||||
| TG121 | ||||||
| DD day 1–10 | 3.32 ± 0.086 | 3.36 | 3.08 ± 0.103 | 3.22 ± 0.085 | 2.50 ± 0.197 | 2.83 ± 0.099 |
| DD day 11–20 | 3.20 ± 0.134 (n = 4) | 3.56 (n = 1) | 3.06 ± 0.099 (n = 13) | 3.20 ± 0.117 (n = 5) | 2.15 ± 0.130 (n = 3) | 2.44 ± 0.302 (n = 4) |
| TG126 | ||||||
| DD day 1–10 | N/A | N/A | 3.42 ± 0.053 | 3.50 ± 0.071 | 2.46 ± 0.224 | 2.62 ± 0.188 |
| DD day 11–20 | 3.32 ± 0.212 (n = 5) | 3.55 ± 0.088 (n = 4) | 2.19 ± 0.219 (n = 5) | 2.12 ± 0.141 (n = 4) | ||
Circadian rhythm amplitude was estimated for BAC transgenic mice (tg) and their nontransgenic littermates from the rhythm of locomotor activity. Amplitude was measured as the logarithm of the power spectral density (log PSD) of the circadian (18 to 30 hrs) peak from Fourier analysis (Bracewell, 1986) of two 10-day intervals in DD (days 1–10 and 11–20) as described in the text. Values shown are the mean ± standard error. Of a total of 266 F1 progeny behaviorally tested, 16 were excluded from analysis due to low levels of activity or missing data. The number of animals included in the analysis for each group is indicated. In some cases, Clock homozygous mice did not exhibit significant circadian rhythms upon release in constant darkness on days 1–20. In these cases, it was not possible to estimate period for inclusion in Table 2, but Fourier analysis was valid so these values were included in this Table.
A Generalized Linear Models (GLM) Analysis of Variance (ANOVA) was performed on the data presented here (NCSS 6.0). Significant effects of the Clock genotype (DF = 2; F = 4.19, p < 0.05) and the presence of the transgene (DF = 1; F = 14.6; p < 0.0005) were detected. In addition, significant construct by genotype (DF = 4; F = 8.30; p < 0.000005), genotype by transgene presence (DF = 2; F = 5.29; p < 0.01) and 3-way interactions (DF = 4; F = 2.86; p < 0.05) were detected. No significant effects of the transgenic construct used (DF = 2; F = 2.87; p = 0.0578) or of the data interval analyzed (first versus second 10 days) (DF = 1; F = 2.11; p = 0.147) were detected. Because the Clock mutation affects this measure only in the homozygous condition (Vitaterna et al., 1994), transgenic lines lacking Clock homozygotes confounded the analysis. The effects of the Clock mutation could only be detected in those lines in which homozygotes were examined, hence the significant construct by genotype interaction. Further, the effects of the transgene could only be detected in homozygotes, hence the significant genotype by transgene presence interaction.
To examine the effects of the transgenic constructs on the loss of rhythmicity in Clock homozygotes, a separate 3-way GLM ANOVA was performed on the data of this Clock genotype. Significant effects of the construct (DF = 1; F = 6.94; p < 0.05), the presence of the transgene (DF = 1; F = 10.1; p < 0.005), and the interval (DF = 1; F = 31.5; p < 0.00001) were detected. Transgene presence by construct (DF = 1; F = 11.4; p < 0.005) and presence by construct by interval (DF = 1; F = 10.17; p < 0.005) interactions were significant. BAC 54 transgenes prevented the decline in circadian amplitude in Clock homozygotes with time in DD, while BAC 52 transgenes had no effect.
To reduce the rescuing interval further, we used the same functional assay to test two other DNA segments: a 100 kb linear NotI fragment of BAC 54 and the full-length circular 160 kb BAC 52 DNA that overlaps BAC 54 by ~90 kb (Figure 1). Analysis of circadian phenotype was performed for the animals from two BAC 54 100 kb lines (TG80 and TG97) and two BAC 52 lines (TG121 and TG126). Figure 5 shows examples of the activity records for Clock/+ animals from each set of DNA constructs used to generate transgenic mice: TG55 (BAC54), TG80 (100 kb fragment of BAC 54), and TG121 (BAC 52). In all cases, the 100 kb fragment from BAC 54 and BAC 52 failed to rescue both the long period and loss-of-rhythm phenotypes in transgenic Clock mutant mice (Tables 2 and 3). These results show that functional rescue of the Clock mutation is achieved only with the full-length BAC 54 transgene.
Figure 5.

The 100 kb BAC 54 and BAC 52 Transgenes Do Not Affect Circadian Phenotype Wheel running activity records and period estimates of Clock/+ F1 progeny from different BAC transgenic lines are shown. Both transgenic and control animals were behaviorally tested using the experimental protocol described in Figure 4 and the text.
(A) Activity records of line TG55 Clock/+ progeny, in which the full-length BAC 54 was the transgenic construct. (tg) indicates transgenic animals’ records.
(B) Activity records of line TG80 Clock/+ progeny, in which the 100 kb BAC 54 fragment was the transgenic construct.
(C) Activity records of line TG121 Clock/+ progeny, in which the full-length BAC 52 was the transgenic construct.
(D) Histogram of period estimates for 46 F1 animals from lines TG55, TG80, and TG121. Individual data points are indicated by open circles.
Not all individual points can be seen due to overlapping values. Number of animals from each line tested is indicated on the bottom of the bar. Among the Clock/+ mice from these lines, significant effects of transgenic construct (2 DF, F = 9.60, p < 0.0005), transgene presence (1DF, F = 16.55, p < 0.0005), as well as construct by transgene interactions (2 DF, F = 10.8, p < 0.0001) were detected (GLM ANOVA). TG55 transgenic progeny had significantly shorter periods than all other groups as determined by Tukey’s post hoc tests (p < 0.05). There were no significant differences between transgenic and non-transgenic Clock/+ progeny in lines TG80 and TG121.
Transcription unit analysis of the Clock region (King et al., 1997b) reveals that there are only three candidate genes that lie within or overlap BAC 54 (Figure 1). Of these three candidates, only one transcription unit that spans ~100,000 base pairs and contains 24 exons is consistent with the pattern of rescue in the different transgenic constructs (Figure 1). This entire transcription unit is contained within full-length BAC 54, and, interestingly, it begins near a CpG island. By this analysis, the 100 kb NotI fragment from BAC 54 is “promoterless”: it is missing all 5′ flanking regulatory sequences as well as exons 1a and 1b. The BAC 52 transgene contains even less of this transcription unit, as it is missing exons 1a, 1b, and 2. As described in the accompanying paper, this large transcription unit encodes a novel member of the basic–helix-loop-helix (bHLH)–PAS domain family of transcription factors (King et al., 1997b). Therefore, on the basis of in vivo complementation, we conclude that this candidate is the Clock gene.
Transgene Expression and Correlation with Rescue
Based on copy number and phenotypic analyses, transgene-positive animals from rescuing lines were expected to express higher levels of Clock mRNA. A Northern blot of RNA isolated from hypothalamic tissue of Clock/+ transgenic and non-transgenic animals from different lines was probed with a Clock-specific cDNA, yz50 (Figure 6A), and a bendless probe (Muralidhar and Thomas, 1993) (Figure 6B). The ~8 and ~11 kb Clock -transcripts were present in all RNA samples; however, transgenic animals from rescuing lines (TG14, TG36, and TG55) showed significantly higher levels of Clock mRNA than non-transgenic littermates, and these levels roughly correlated with transgene copy number. A small difference was seen in line TG60 (other experiments, data not shown), which is consistent with the low transgene copy number (1–2 copies) in this line. An additional transcript of 2.4 kb was present in line TG55; this could represent a truncated fragment at the transgene integration site (see transgene integrity results above). Alternatively, this additional transcript could be revealing a change in RNA processing, as the 3-to 5-fold overexpression level of the 8 and 11 kb transcripts does not appear to match the 8-to 10-fold transgene copy number. Clock mRNA expression levels were not different between transgenic and non-transgenic mice in the non- rescuing lines TG97 and TG121. Finally, all animals (transgenic and non-transgenic from rescuing and non-rescuing lines) showed the same level of expression of bendless 1.3 and 4.0 kb transcripts. Thus, rescue of the Clock phenotype directly correlates with the higher expression level of only one transcription unit in the transgenes: Clock.
Figure 6.
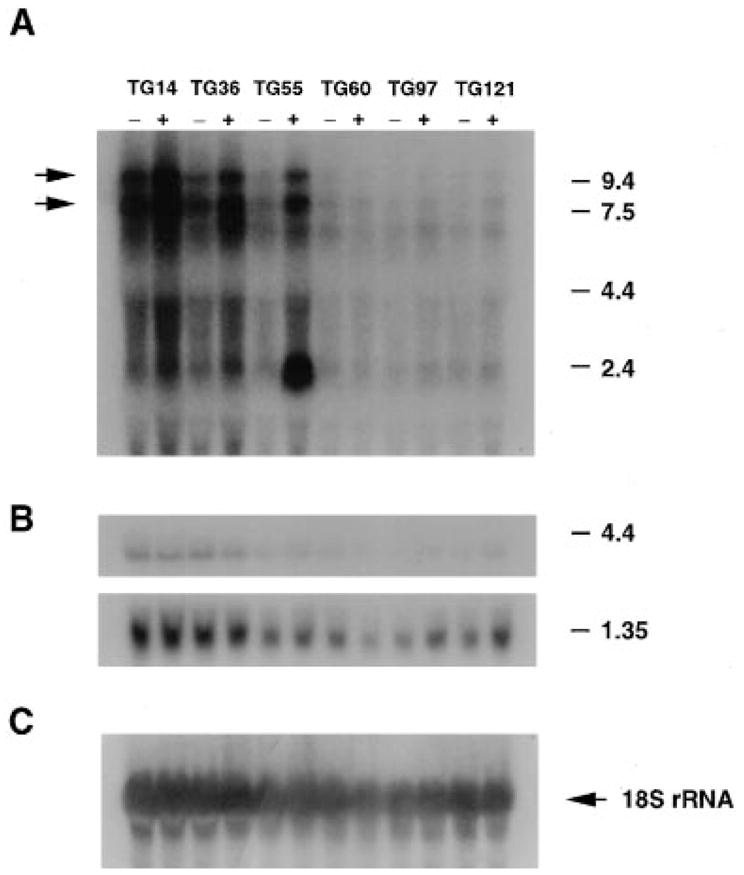
Clock mRNA Expression in Rescuing and Non-rescuing Lines
Total RNA was isolated from the hypothalamic tissue of different BAC transgenic and control mice for Northern blot hybridization. Total RNA (25 μg) was hybridized with 32P-labeled 51-base oligonucleotide complementary to 18S ribosomal RNA (C). RNA loading differs across the lines, as can be seen with the 18S ribosomal probe; however, the critical pairwise comparisons between non-transgenic (−) and transgenic (+) mice within each line can still be made. The BAC 54 transgenic lines TG14, TG36, and TG55 showed increased levels of Clock mRNA expression in transgene-positive mice, whereas the 100 kb BAC 54 line (TG97) and the BAC 52 line (TG121) showed similar levels of Clock mRNA between non-transgenic and transgenic mice. In this experiment, increased expression of Clock mRNA was not apparent in the BAC 54 line TG60 due to the low copy number of the transgene. Transgenic mice from the TG55 line also expressed a novel 2.4 kb transcript.
To determine the expression pattern of the BAC 54 transgene in a rescuing line, we performed in situ hybridization with brain sections from line TG36 Clock/Clock mice using riboprobes from the yz50cDNAclone (Figure 7). The expression of Clock mRNA was lower in Clock homozygous mice, as seen with Northern blot analysis in the accompanying paper (King et al., 1997b). Hybridization signal was found in SCN and pyriform cortex (Figure 7) as well as supraoptic nucleus, thalamic and hypothalamic paraventricular nuclei, medial habenula, and hippocampus (data not shown). Both transgenic and non-transgenic animals had the same overall hybridization pattern; however, mice carrying the transgene uniformly showed a stronger hybridization signal in the SCN region than non-transgenic littermates. These results suggest that the presence of the BAC 54 transgene leads to an increase in Clock mRNA expression in the SCN that correlates with the functional rescue of the circadian behavioral phenotype in these mice.
Figure 7.
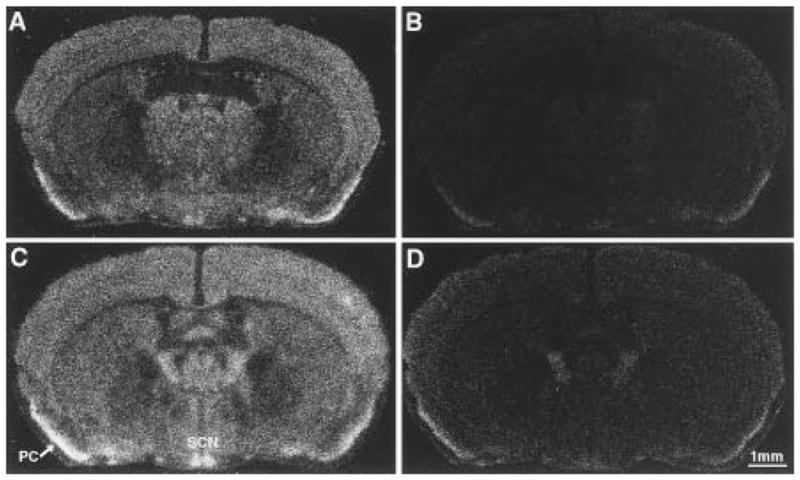
In Situ Hybridization Analysis of Clock mRNA in Line TG36
Coronal brain sections (20 μm) from TG36 Clock/Clock littermates, either lacking the transgene (A and B) or positive for the transgene (C and D), are shown. (A) and (C) are autoradiograms using antisense yz50probe, while (B) and (D) show the sense probe controls. Expression of Clock mRNA is widely distributed in the brains of both non-transgenic and transgenic animals. Highest levels of Clock mRNA were found in the suprachiasmatic nucleus (SCN) and pyriform cortex (PC). The transgene-positive animal (C) shows a selectively stronger signal in SCN as compared to Clock/Clock control animal (A). These representative sections are from the midportion of the SCN.
Discussion
Circadian rhythmicity represents a complex behavioral and physiological phenotype. Clock genes have been identified in other organisms, most notably the period (per) and timeless (tim) genes in Drosophila and the frequency (frq) gene in Neurospora. Interestingly, both per and frq were cloned by rescue (Bargiello et al., 1984; Zehring et al., 1984; McClung et al., 1989). Indeed, per is the first example of any gene to be cloned by p-element transformation in Drosophila. In mammals, no clock genes have been previously cloned and shown to have a functional role in the circadian system. Using a deliberate ENU mutagenesis screen, we identified a single gene, Clock, that is essential for circadian rhythmicity (Vitaterna et al., 1994). In the studies reported here, we used transgenic rescue as an approach to identify the Clock gene at the molecular level. We have shown that a large transcription unit with 24 exons spanning ~100,000 base pairs of genomic sequence is the only candidate gene consistent with the transgenic rescue experiments. Taken together with the results of positional cloning reported in the accompanying paper (King et al., 1997b), which define the molecular nature of the Clock mutation, we can definitively conclude that a novel member of the bHLH–PAS domain family of transcription factors functions as the Clock locus.
The use of transgenic technology to rescue a mutant phenotype provides the most powerful means available to prove that a cloned candidate gene (or a large DNA segment identified from positional cloning) is indeed the locus responsible for a particular mutant phenotype. The strategy of positional cloning ultimately relies upon a correlation between a sequence alteration identified in a candidate gene and a mutant phenotype. This association is strengthened if sequence alterations are identified for each of the multiple mutant alleles of a gene, but it nevertheless remains correlative in nature. However, such corroboration is not possible in cases of genes for which only one mutation has been identified, such as Clock. In contrast, rescue of a mutant phenotype by transgenic insertion of a candidate gene sequence allows for direct functional assay of the candidate and does not require identification of multiple alleles. Thus, functional rescue represents the strongest evidence that a candidate gene identified by position is in fact the gene responsible for the mutant phenotype.
Recently, substantial progress has been made in generating transgenic mice by insertion of large DNA segments, mainly YACs (reviewed in Peterson et al., 1997). The insertion of large (several hundred kb) fragments of genomic DNA into the mouse germline allows one to study the phenotypic expression of an entire gene, multigene loci, or even distant regulatory sequences in their native sequence environment. The large size of YAC clones, while advantageous for mapping purposes, makes them very difficult to handle. BACs, while significantly smaller in size and thus amenable to manipulation, can still represent a large genomic segment. BACs are circular DNA molecules that can be handled as conventional plasmids, allowing cesium chloride preparations that result in the highly pure samples necessary for microinjections. Therefore, we chose BACs as an alternative to YACs both for cloning and for microinjection. The success rate for creating transgenics with BAC DNA was comparable with that for YAC transgenesis: about 10%.
We generated transgenic mice using a set of overlapping BAC clones and then tested these mice phenotypically for Clock mutation complementation. The phenotypic rescue obtained in these experiments was complete, quantitative, and unambiguous. Clock was originally identified on the basis of an altered circadian period: heterozygotes have free-running circadian periods approximately one hour longer than normal, while homozygotes have periods approximately four hours longer than normal and eventually lose circadian rhythmicity with time in constant darkness (Vitaterna et al., 1994). In heterozygous mutant animals, the BAC 54 transgene shortened the free-running period length of the activity rhythm to the wild-type range. In homozygous mutant animals, the BAC 54 transgene both shortened the free-running period to the wild-type range and prevented the loss of circadian rhythmicity. Neither BAC 52 nor the linear100 kb BAC54 DNA transgenes affected the circadian period of the mutant mice. The efficiency of transgenesis and the complete rescue obtained with the circular 140 kb BAC 54 DNA injections are striking and could be due to a number of factors: first, the use of circular BAC DNA facilitates one’s ability to obtain extremely pure DNA samples for microinjection; second, the BAC vector sequence is small (~5%) relative to the genomic DNA; and third, this genomic segment contains the entire Clock transcription unit and appears to contain all necessary regulatory elements of the Clock locus.
The functional rescue of the Clock phenotype is in complete agreement with the transcription unit identification results of King et al. (1997b) in which a novel gene, Clock, was identified as a primary candidate for the mutant phenotype. Other transcription units in the region included pFT27 (Akagi et al., 1988) and a homolog of the Drosophila gene bendless (Figure 1). BAC 54 does not contain the full pFT27 gene, while its complete sequence is found in BAC 52, a clone that did not rescue the mutation in either of two lines tested. The bendless sequence homolog is contained within all three BAC constructs tested; therefore, the lack of rescue in the BAC 52 and 100 kb BAC 54 lines excluded this candidate. In addition, bendless appears to be a processed pseudogene: genomic sequence analysis shows that the bendless homolog lacks introns and is flanked by direct repeats (Yamaguchi et al., 1996). Furthermore, its expression is not increased in the rescuing transgenic lines (Figure 6). Thus, Clock is the only transcription unit completely included in this genomic interval that rescues the mutant phenotype. Taken together with the identification of a sequence alteration in the Clock mutant (King et al., 1997b), we can conclude that the gene contained in full-length BAC 54 is indeed responsible for Clock function.
The rescue of the Clock mutation in mice contrasts with the phenotypic rescue of circadian clock genes in Drosophila in several ways. In per, only partial rescue by p-element-mediated transformation was accomplished (Bargiello et al., 1984; Zehring et al., 1984), whereas we observed full rescue of both of Clock’s major phenotypic effects with the BAC 54 transgene. Rescue of per0 and per− null alleles was attempted, whereas we have demonstrated rescue of a semi-dominant allele with Clock. With transgenic rescue of per, strong positional effects were detected (Bargiello et al., 1984; Zehring et al., 1984; Hamblen et al., 1986; Baylies et al., 1987), whereas we see no evidence of positional effects among the BAC 54 lines. In Drosophila, partial per rescue can be accomplished without a promoter region (Frisch et al., 1994), whereas in mice the 100 kb BAC 54 linear fragment, which represents a Clock promoterless construct, fails to rescue the mutant phenotype. Finally, in mice, one or two copies of the transgene (TG60) is sufficient to produce phenotypic rescue. Because multiple lines carrying the same transgenic construct have not been examined in all Clock genotypes, it is not possible to evaluate quantitatively the effects of the transgene copy number at this time. It will be of interest to test whether a period shortening–copy number relationship holds for Clock as has been described for per (Baylies et al., 1987).
To that end, we previously have examined the effects of varying gene dosage by comparison of different Clock mutant, wild-type, and deletion (W19H) allelic combinations (King et al., 1997a). From such an analysis, we concluded that Clock is an anti-morphic mutation; that is, it has a phenotypic effect that is antagonistic with the normal allele (Muller, 1932). This interpretation of the Clock mutation is based primarily on the finding that a single copy of the mutant allele (Clock/W19H) produces a more severely affected phenotype (i.e., longer circadian period) than one mutant allele and one wild-type allele (Clock/+), but not as severely affected as the phenotype produced by two mutant alleles (Clock/Clock). If Clock is indeed an anti-morphic allele, this leads one to predict that transgenic overexpression of wild-type Clock should have the hypermorphic effect of shortening circadian period. The rescue results are entirely consistent with this; in Clock wild-type mice, the presence of multiple copies of the BAC 54 140 kb transgene results in period shortening beyond the normal wild-type values. Similar results have been seen with the Drosophila per gene, where overexpression by additional copies of wild-type per (Smith and Konopka, 1981; Baylies et al., 1987) or an overactive allele (Rutila et al., 1992) can shorten circadian period. Although constitutive overexpression of per abolishes rhythmicity (Zeng et al., 1994), a general relationship between expression level and period is observed (Coté and Brody, 1986). Our results also demonstrate a positive correlation between Clock expression level and circadian period, as if Clock were rate limiting. In fact, the shortened period in wild-type transgenic mice suggests the existence of a regulatory apparatus in which the level of the Clock gene product (CLOCK) in wild-type non-transgenic mice is sub-saturating. King et al. (1997b) have predicted that the Clock mutant gene product is a transcription factor that appears capable of dimerization but is defective in activation. This type of dominant-negative interaction is common among other transcription factors such as p53 (Levine, 1990; Vogelstein and Kinzler, 1992). In Clock heterozygous and homozygous mutant mice, the presence of sufficient wild-type monomers from the BAC 54 transgene could overcome the effect of the mutant gene product. It will be of interest to determine the exact nature of CLOCK’s interactions with other proteins and the mechanism by which the Clock mutation affects the mutant phenotype.
In conclusion, we suggest that transgenic rescue of mutations in mice using BAC clones may become the method of choice in many positional cloning projects. First, the creation of BAC transgenic lines is efficient and much easier than YAC transgenics because circular DNA preparations can be used. Second, in cases where no candidate genes are initially found or where close recombinants are absent, a series of overlapping BAC clones can cover a large genomic interval (Hamilton et al., 1997). In the case of Clock, transgenic rescue definitively reduced the critical interval to 140 kb. The value of such functional corroboration during a positional cloning effort cannot be overestimated and can accelerate cloning efforts immensely. Third, the pattern of rescue in the initial overlapping set of BAC transgenics can reduce the number of candidate genes substantially. Finally, in the future, we anticipate that BAC clones will become the optimal clone source for sequencing of human and mouse genomes and that DNA sequence analysis will be the primary method for transcription unit identification. Thus, one can foresee the following scenario for a future positional cloning project in the mouse. One would map a mutation with high resolution to a set of physically ordered BAC clones. This set of BAC clones then would be used for transgenic rescue of the mutant phenotype. Once a rescuing BAC was obtained, the genomic sequence would be analyzed for candidate genes. Finally, conventional methods would be used for mutation identification. This type of scenario is already a reality in Caenorhabditis elegans; we expect the mouse should soon follow.
Experimental Procedures
BAC DNA Purification
Mouse BAC clones were isolated and described in the accompanying paper (King et al., 1997a). The BAC library was obtained from Research Genetics. Genomic DNA originated from the CJ7 cell line from the 129/Sv strain (Shizuya et al., 1992) and is cloned into the pBeloBAC11 vector at the HindIII cloning site (Kim et al., 1996). BAC 54 and 52 circular DNA were isolated using an alkaline lysis and cesium chloride gradient ultracentrifugation protocol described for cosmid DNA purification (Favello et al., 1995) with some modifications. After the last ethanol precipitation step, DNA was dissolved in injection buffer (10 mM Tris–HCl [pH 7.5], 0.1 mM EDTA, 100 mM NaCl, 30 μM spermine, 70 μM spermidine) and quantitated using a TKO 100 fluorometer (Hoefer Scientific Instruments). A small aliquot was digested with NotI (New England Biolabs), and restriction fragments were separated using pulsed-field gel electrophoresis (PFGE) for 18 hr on 1% SeaKem LE agarose (FMC) gel using the following conditions: included angle 120°, 6 V/cm, switch time ramping from 0.1 s to 12 s (CHEF Mapper II, BioRad). After ethidium bromide staining, two fragments were identified for both BACs used: 100 kb and 40 kb for BAC 54; 110 and 50 kb for BAC 52.
The 100 kb linear NotI fragment of BAC 54 was purified as described in Smith et al. (1995). After the i-agarase digestion step, DNA was dialyzed on a floating 0.05 μm Millipore membrane filter against the injection buffer and, if necessary, concentrated using a Millipore Ultrafree MC unit (100,000 NMWL).
Generation of BAC Transgenic Mice
Isolated BAC DNA (either circular plasmid or linear fragment) was injected at a concentration of 1 ng/μl into fertilized mouse oocytes isolated from crosses between either wild-type CD1 females and (BALB/cJ x C57BL/6J) F2 Clock/Clock males or wild-type CD1 females and wild-type CD1 males as described (Hogan et al., 1994). Microinjections were performed using Leitz micromanipulators attached to a Leitz DM IRB microscope. Oocytes that survived injection were transferred later the same day to both oviducts of pseudopregnant CD1 foster mothers. Transgenic mice were identified both by PCR and Southern blot analysis of genomic DNA prepared from tail biopsies as described below (Hogan et al., 1994).
Detection of Transgene by PCR Analysis
To identify transgenic animals the following sets of primers were used: 5′ ATACGACTCACTATAGGGCGAATTCG and 5′ TTAGCCTGT TCTGTGCTACATCG (amplified a 290 bp fragment from the T7-end of the BAC 54 vector–insert junction) and 5′ GACCATGATTACGCCA AGCTATTTAG and 5′ GCTGTATTTCCACATAAGCAGGG (amplified a 220 bp fragment from the Sp6-end of the full-length BAC 54 and the 100 kb NotI fragment vector–insert junction); 5′ GACCATGATTA CGCCAAGCTATTTAG and 5′ AGATAACACTTTTTTCTGTGCCCC (amplified a 290 bp fragment from the Sp6 end of BAC 52). PCR assays were performed in a final volume of 20 μl buffer consisting of 50 mM potassium chloride, 10 mM Tris–HCl (pH 8.3), 1.5 mM MgCl 2,1 μM each primer, 0.2 mM dNTPs, 0.5 μg genomic DNA, and 0.2 μl AmpliTaq polymerase (5 U/μl, Perkin Elmer). PCR conditions were: 3 min at 94°C; followed by 35 cycles of 1 min at 94°C, 1min at 55°C, and 1 min at 72°C. The reaction products were analyzed on a 1.2% agarose gel.
Transgene Examination by Southern Blot Analysis
For transgene analysis, 10 μg of genomic DNA were digested with BamHI, EcoRI, or BglII and size fractionated on a 1% agarose gel in 1x Tris–acetate–EDTA. Gels were blotted onto nylon membranes (GeneScreen Plus, NEN Research Products) and hybridized according to standard procedures (Sambrook et al., 1989). A pBeloBAC11 vector fragment (nucleotides 1607–2900) and clone L4 (a cDNA clone from the 3′ untranslated region of the Clock transcript that was present in all transgene constructs) were used as hybridization probes. For copy number estimation, 5 μg of genomic DNA from F1 transgenic animals and non-transgenic littermates was digested with BglII or BamHI. BAC 54 DNA equivalent to 1, 3, and 5 copies per genome was digested with the same enzyme and mixed with digested control genomic DNA. All samples were size fractionated on the same gel and then hybridized with L4 (BglII digests) or pBeloBAC11 (BamHI digests) probes. Autoradiograms were exposed to X-ray film at −70°C with an intensifying screen and analyzed using the Quantity One (Version 3.0) densitometry program (PDI, Inc., Huntington Station, NY). For transgene integrity assessment, 10 μg of genomic DNA and 0.5 ng of BAC 54 DNA were digested with BamHI, EcoRI, or BglII and hybridized with probes corresponding to different regions of BAC 52 and BAC 54. These probes were generated from M13 shotgun sequencing library clones from BAC 52 (clones 052A2GH10, 052A2FB08, and 052A3AA01, the last of which aligns with the STS yREB14 [Brunkow et al., 1995]) by PCR amplification using M13 vector primers. All probes were 32P labeled to a specific activity of about 1–2 × 109 cpm/μg DNA using random priming (Ready-To-Go Kit, Pharmacia).
Clock Genotyping
All founder animals and F1 progeny were genotyped for the Clock locus using simple sequence length polymorphisms (SSLPs) (Dietrich et al., 1992), with some modifications as described (King et al., 1997a). SSLP primer pairs were obtained from Research Genetics. One SSLP proximal (D5Mit201) and one distal (D5Mit309) to the Clock locus were used to assign the animal’s Clock genotype. Transgenic animals recombinant for the Clock locus were excluded from the analysis. Recombinant animals that did not carry the transgene were behaviorally tested for circadian locomotor activity, and the presumed Clock genotype was assigned by phenotype.
Total RNA Isolation and Northern Blot Analysis
Total RNA was prepared by the method described in Chomczynski and Sacchi (1987). Mice were sacrificed at ZT6 (Zeitgeber time; 6 hours after lights on) after being kept in a 12 hours light:12 hours dark (LD 12:12) light cycle for at least 2 weeks. RNA isolated from hypothalamic tissue was separated on 1% agarose denaturing formaldehyde gels, transferred to nylon membranes (Duralon-UV, Stratagene), and cross-linked to the membrane by UV irradiation using a UV Stratalinker 1800 (Stratagene). Blots were pre-hybridized for at least 1 hour at 42°C in 50% formamide, 0.1% Blotto, 6x SSPE, 0.5% SDS, and 0.5 mg/ml salmon sperm DNA. Following hybridization with yz50 probe or bendless probe (see Results), blots were washed once with 2x SSC, 0.5% SDS at room temperature and then twice with 0.3x SSC, 0.5% SDS at 65°C. Blots were normalized for the RNA loading by hybridization with a 51-mer oligonucleotide complementary to 18S ribosomal RNA: 5′GATGTGGTAGCCGTTTCTCAGG CTCCCTCTCCGGAATCGAACCCTGATTCC 3′. The probe was labeled with [′-32P]ATP using T4 polynucleotide kinase and purified by polyacrylamide gel electrophoresis. Hybridization was performed as described in King et al., (1997b).
In Situ Hybridization
Brains were taken from littermate sibling pairs of Clock homozygous mice, with and without the transgene, from the TG36 transgenic line. Mice were sacrificed at ZT6 after at least 2 weeks in LD 12:12. Brains were rapidly dissected and frozen on dry ice. Coronal sections at 20 μm were collected. In situ hybridization was performed as described by Suhr et al. (1989). 35S-labeled riboprobes were synthesized from linearized plasmid templates from the coding region of the Clock gene (clone yz50) using SacI digestion and NotI digestion for linearization of antisense and sense templates, respectively.
Locomotor Activity Rhythm Monitoring and Analysis
All mice were bred and raised in the Northwestern University Center for Experimental Animal Resources (CEAR). Mice were housed in LD 12:12 (lights on at 5 AM Central Standard Time). Pups were weaned at 3–4 weeks of age and housed with same-sex littermates until 8 weeks of age, when they were behaviorally tested. Locomotor activity was monitored essentially as described in Vitaterna et al. (1994). Mice were individually housed in cages equipped with a running wheel in the same LD 12:12 cycle as previously for one week, and then transferred to constant darkness for at least 3 weeks. Activity was monitored with an on-line PC computer system (Stanford Software Systems, Chronobiology Kit).
Period calculations were performed for a 20-day interval in constant darkness (days 1–20) by X2 periodogram analysis (Sokolove and Bushel, 1978, Chronobiology Kit). Periods at 1-min increments ranging from 20 to 30 hr were tested. It should be noted that this estimation of free-running period differs from those previously reported for the Clock mutation (Vitaterna et al., 1994), i.e., the first 20 days in constant darkness was used rather than the first, the second, or a steady-state 10-day interval. The 20-day interval was chosen so that the same time period could be analyzed for all three Clock genotypes. Many Clock heterozygotes express a circadian period less than 24 hr, comparable to wild-type mice, when initially placed in constant darkness, but the period lengthens over time. However, many Clock homozygotes lose circadian periodicity by the second 10 days in constant darkness. Estimation of steady-state period tended to include a different interval dependent upon the Clock genotype. By using a 20-day interval, period estimates represent an average across an interval during which period (at least for Clock heterozygotes and homozygotes) was changing in many mice. Hence, the period estimates for Clock mutants reported here are often shorter than those obtained from an interval during which the period was at steady state. Out of a total of 266 mice that were behaviorally tested, 21 were excluded from periodogram analysis due to detection of multiple periodicities, a lack of significant period, or missing data.
Fast Fourier analysis was performed for two 10-day intervals in constant darkness (days 1–10 and days 11–20). Frequencies ranging from 0 to 6 cycles per day were tested with a 6-min step size. The analysis involves first calculating the Hartley transform and then converting the result into the Fourier transformation (Bracewell, 1986). The amplitude of the power spectral density (PSD) of the circadian peak (peak closest to 1 cycle/day) was measured. For statistical analysis, the logarithm of the PSD amplitude was used to maintain homogeneity of variance. Low levels of activity could result in artifactual lowering of the PSD. For this reason, 16 of 266 mice that were behaviorally tested were excluded from PSD analysis.
Both the free-running period and the log PSD were analyzed by Generalized Linear Models (GLM) Analysis of Variance (ANOVA), using Number Cruncher Statistical Systems (NCSS ver. 6.0; Kaysville, UT). First, a 3-way ANOVA was performed to test for the main effects: Clock genotype, presence/absence of the transgene, and transgenic construct. For the log PSD, a repeated measures design for the two 10-day intervals was used, so that interval number added a dimension to the ANOVA. Then, within each transgenic construct, the effects of Clock genotype, presence/absence of the transgene, and transgenic line were tested. A Tukey’s test was used for all post hoc pairwise comparisons.
Acknowledgments
We thank William F. Dove for his encouragement concerning transgenic rescue; Fred W. Turek and our colleagues in the NSF Center for Biological Timing for advice and support; Yelena Khenkina for technical assistance; Gunther Schutz, Lluis Montoliu, Edward M. Rubin, Desmond Smith, J. Doug Engel, Jorg Bungert, Kenneth Lieuw, and Jie Fan for advice on transgenic mouse production; and Bruce Hamilton and E. M. Rubin for unpublished information on transgenic rescue of vibrator.
Research was supported by the NSF Center for Biological Timing, an Unrestricted Grant in Neuroscience from Bristol-Myers Squibb (NIMH grant R37 MH39592), and Northwestern University (J. S. T.), MSTP fellowship T32 GM08152 (L. D. W.), and NIH grant P30 HD28048 (F. W. Turek).
References
- Akagi JM, Nomiyama H, Setoyama C, Akagi M. Messenger RNA expressed in mouse teratocarcinoma stem cells and down-regulated by a tumor-promoting phorbol ester codes for a novel transmembrane protein. Biochem Biophys Res Commun. 1988;157:548–557. doi: 10.1016/s0006-291x(88)80284-5. [DOI] [PubMed] [Google Scholar]
- Al-Ubaidi MR, Pittler SJ, Champagne MS, Triantafyllos JT, McGinnis JF, Baehr W. Mouse opsin: gene structure and molecular basis of multiple transcripts. J Biol Chem. 1990;265:20563–20569. [PubMed] [Google Scholar]
- Aronson BD, Johnson KA, Loros JJ, Dunlap JC. Negative feedback defining a circadian clock: autoregulation of the clock gene frequency. Science. 1994;263:1578–1584. doi: 10.1126/science.8128244. [DOI] [PubMed] [Google Scholar]
- Bargiello TA, Jackson FR, Young MW. Restoration of circadian behavioural rhythms by gene transfer in Drosophila. Nature. 1984;312:752–754. doi: 10.1038/312752a0. [DOI] [PubMed] [Google Scholar]
- Baylies MK, Bargiello TA, Jackson FR, Young MW. Changes in abundance or structure of the per gene product can alter periodicity of the Drosophila clock. Nature. 1987;326:390–392. doi: 10.1038/326390a0. [DOI] [PubMed] [Google Scholar]
- Bird A. The essentials of DNA methylation. Cell. 1992;70:5–8. doi: 10.1016/0092-8674(92)90526-i. [DOI] [PubMed] [Google Scholar]
- Block GD, Khalsa SBS, McMahon DG, Michel S, Geusz M. Biological clocks in the retina: cellular mechanisms of biological timekeeping. Int Rev Cytol. 1993;146:83–144. doi: 10.1016/s0074-7696(08)60381-2. [DOI] [PubMed] [Google Scholar]
- Bracewell RN. The Hartley Transform. New York: Oxford University Press; 1986. [Google Scholar]
- Brunkow ME, Nagle DL, Bernstein A, Bucan M. A 1.8Mb YAC contig spanning three members of the receptor tyrosine kinase gene family (Pdgfra, Kit, and Flk1) on mouse chromosome 5. Genomics. 1995;5:421–432. doi: 10.1016/0888-7543(95)80042-k. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Coté GG, Brody S. Circadian rhythms in Drosophila melanogaster: analysis of period as a function of gene dosage at the per (Period) locus. J Theor Biol. 1986;121:487–503. doi: 10.1016/s0022-5193(86)80104-7. [DOI] [PubMed] [Google Scholar]
- Dietrich W, Katz H, Lincoln SE, Shin HS, Friedman J, Dracopoli NC, Lander ES. A genetic map of the mouse suitable for typing intraspecific crosses. Genetics. 1992;131:423–447. doi: 10.1093/genetics/131.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove WF. Molecular genetics of Mus musculus: point mutagenesis and milliMorgans. Genetics. 1987;116:5–8. doi: 10.1093/genetics/116.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap JC. Genetic analysis of circadian clocks. Annu Rev Physiol. 1993;55:683–728. doi: 10.1146/annurev.ph.55.030193.003343. [DOI] [PubMed] [Google Scholar]
- Dunlap JC. Genetic and molecular analysis of circadian rhythms. Annu Rev Genet. 1996;30:579–601. doi: 10.1146/annurev.genet.30.1.579. [DOI] [PubMed] [Google Scholar]
- Favello A, Hillier L, Wilson R. Genomic DNA sequencing methods. Methods in Cell Biology. 1995:551–569. doi: 10.1016/s0091-679x(08)61403-x. [DOI] [PubMed] [Google Scholar]
- Frisch B, Hardin PE, Hamblen-Coyle MJ, Rosbash M, Hall JC. A promoterless period gene mediates behavioral rhythmicity and cyclical per expression in a restricted subset of the Drosophila nervous system. Neuron. 1994;12:555–570. doi: 10.1016/0896-6273(94)90212-7. [DOI] [PubMed] [Google Scholar]
- Gibson S, Somerville C. Isolating plant genes. Trends Biochem Sci. 1993;21:93–111. doi: 10.1016/0167-7799(93)90019-6. [DOI] [PubMed] [Google Scholar]
- Hall JC. Tripping along the trail to the molecular mechanisms of biological clocks. Trends Neurosci. 1995;18:230–240. doi: 10.1016/0166-2236(95)93908-g. [DOI] [PubMed] [Google Scholar]
- Hamblen M, Zehring WA, Kyriacou CP, Reddy P, Yu Q, Wheeler DA, Zweibel LJ, Konopka RJ, Rosbash M, Hall JC. Germ-line transformation involving DNA from the period locus in Drosophila melanogaster: over-lapping genomic fragments that restore circadian and ultradian rhythmicity to per0 and per mutants. J Neurogenet. 1986;3:249–291. doi: 10.3109/01677068609106855. [DOI] [PubMed] [Google Scholar]
- Hamilton BA, Smith DJ, Mueller KL, Kerrebrock AW, Bronson RT, van Berkel V, Daly MJ, Kruglyak L, Reeve MP, Nemhauser JL, et al. The vibrator mutation causes neurodegeneration via reduced expression of PITPa: positional complementation cloning and extragenic suppression. Neuron. 1997 doi: 10.1016/s0896-6273(00)80312-8. in press. [DOI] [PubMed] [Google Scholar]
- Hardin PE, Hall JC, Rosbash M. Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature. 1990;343:536–540. doi: 10.1038/343536a0. [DOI] [PubMed] [Google Scholar]
- Hardin PE, Hall JC, Rosbash M. Circadian oscillations in period gene mRNA levels are transcriptionally regulated. Proc Natl Acad Sci USA. 1992;89:11711–11715. doi: 10.1073/pnas.89.24.11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Constantini F, Lacey E. Manipulating the Mouse Embryo, A Laboratory Manual. 2. Plainview, New York: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Kim UJ, Birren BW, Slepak T, Mancino V, Boysen C, Kang HL, Simon MI, Shizuya H. Construction and characterization of a human bacterial artificial chromosome library. Genomics. 1996;34:213–218. doi: 10.1006/geno.1996.0268. [DOI] [PubMed] [Google Scholar]
- King DP, Vitaterna MH, Chang AM, Dove WF, Pinto LH, Turek FW, Takahashi JS. The mouse Clock mutation behaves as an antimorph and maps within the W19H deletion, distal of Kit. Genetics. 1997a doi: 10.1093/genetics/146.3.1049. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DP, Zhao Y, Sangoram AM, Wilsbacher LD, Tanaka M, Antoch MP, Steeves TDL, Vitaterna MH, Kornhauser JM, Lowrey PL, Turek FW, Takahashi JS. Positional cloning of the mouse circadian Clock gene. Cell. 1997b doi: 10.1016/s0092-8674(00)80245-7. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DC, Moore RY, Reppert SM. Suprachiasmatic Nucleus—The Mind’s Clock. New York: Oxford University Press; 1991. [Google Scholar]
- Levine AJ. Tumor suppressor genes. Bioessays. 1990;12:60–66. doi: 10.1002/bies.950120203. [DOI] [PubMed] [Google Scholar]
- McClung CR, Fox BA, Dunlap JC. Frequency, a clock gene in Neurospora, shares a sequence element with the Drosophila clock gene period. Nature. 1989;339:558–562. doi: 10.1038/339558a0. [DOI] [PubMed] [Google Scholar]
- Meijer JH, Rietveld WJ. Neurophysiology of the suprachiasmatic circadian pacemaker in rodents. Physiol Rev. 1989;69:671–707. doi: 10.1152/physrev.1989.69.3.671. [DOI] [PubMed] [Google Scholar]
- Mello C, Fire A. DNA transformation. In: Epstein HF, Shakes DC, editors. Methods in Cell Biology. Caenorhabditis elegans: Modern Biological Analysis of an Organism. New York: Academic Press; 1995. pp. 452–482. [Google Scholar]
- Moore RY. Organization of the mammalian circadian system. In: Chadwick DJ, Ackrill K, editors. Ciba Foundation Symposium on Circadian Clocks and Their Adjustment. West Sussex, England: John Wiley and Sons; 1995. pp. 88–106. [PubMed] [Google Scholar]
- Moore RY. Hypothalamic Integration of Circadian Rhythms. Amsterdam: Elsevier; 1996. Entrainment pathways and the functional organization of the circadian system; pp. 103–119. [DOI] [PubMed] [Google Scholar]
- Muller HJ. Sixth International Congress of Genetics. Ithaca, New York: Brooklyn Botanic Gardens; 1932. Further studies on the nature and causes of gene mutations. [Google Scholar]
- Muralidhar MG, Thomas JB. The Drosophila bendless gene encodes a neural protein related to a ubiquitin-conjugating enzyme. Neuron. 1993;11:253–266. doi: 10.1016/0896-6273(93)90182-q. [DOI] [PubMed] [Google Scholar]
- Peterson KR, Clegg CH, Li Q, Stamatoyannopoulos G. Production of transgenic mice with yeast artificial chromosomes. Trends Genet. 1997;13:61–66. doi: 10.1016/s0168-9525(97)01003-2. [DOI] [PubMed] [Google Scholar]
- Pittendrigh CS. Temporal organization: reflections of a Darwinian clock-watcher. Annu Rev Physiol. 1993;55:17–54. doi: 10.1146/annurev.ph.55.030193.000313. [DOI] [PubMed] [Google Scholar]
- Ralph MR, Foster RG, Davis FC, Menaker M. Transplanted suprachiasmatic nucleus determines circadian period. Science. 1990;247:975–978. doi: 10.1126/science.2305266. [DOI] [PubMed] [Google Scholar]
- Rosbash M, Allada R, Dembinska M, Guo WQ, Le M, Marrus S, Quian Z, Rutila J, Yaglom J, Zeng H. A Drosophila circadian clock. Cold Spring Harbor Symp Quant Biol. 1996 in press. [PubMed] [Google Scholar]
- Rutila JE, Edery I, Hall JC, Rosbash M. The analysis of new short-period circadian rhythm mutants suggests features of D. melanogaster period gene function. J Neurogenet. 1992;8:101–113. doi: 10.3109/01677069209084155. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schedl A, Montoliu L, Kelsey G, Schütz G. A yeast artificial chromosome covering the tyrosinase gene confers copy number–dependent expression in transgenic mice. Nature. 1993;362:258–261. doi: 10.1038/362258a0. [DOI] [PubMed] [Google Scholar]
- Sehgal A, Rothenfluh-Hilfiker M, Hunter-Ensor M, Chen Y, Myers MP, Young MW. Rhythmic expression of timeless: a basis for promoting circadian cycles in period gene autoregulation. Science. 1995;270:808–810. doi: 10.1126/science.270.5237.808. [DOI] [PubMed] [Google Scholar]
- Shizuya H, Birren B, Kim UJ, Mancino V, Slepak T, Tachiiri Y, Simon M. Cloning and stable maintenance of 300kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc Natl Acad Sci USA. 1992;89:8794–8797. doi: 10.1073/pnas.89.18.8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Zhu Y, Zhang J, Cheng JF, Rubin EM. Construction of a panel of transgenic mice containing a contiguous 2-Mb set of YAC/P1 clones from human chromosome 21q22.2. Genomics. 1995;27:425–434. doi: 10.1006/geno.1995.1073. [DOI] [PubMed] [Google Scholar]
- Smith RF, Konopka RJ. Circadian clock phenotypes of chromosome aberrations with a breakpoint at the per locus. Mol Gen Genet. 1981;183:243–251. doi: 10.1007/BF00270625. [DOI] [PubMed] [Google Scholar]
- Sokolove PG, Bushel WN. The Chi square periodogram: its utility for analysis of circadian rhythms. Federation Proc. 1978;38:2589–2595. [Google Scholar]
- Takahashi JS. Molecular neurobiology and genetics of circadian rhythms in mammals. Annu Rev Neurosci. 1995;18:531–553. doi: 10.1146/annurev.ne.18.030195.002531. [DOI] [PubMed] [Google Scholar]
- Takahashi JS, Kornhauser JM, Koumenis C, Eskin A. Molecular approaches to understanding circadian oscillations. Annu Rev Physiol. 1993;55:729–753. doi: 10.1146/annurev.ph.55.030193.003501. [DOI] [PubMed] [Google Scholar]
- Takahashi JS, Murakami N, Nikaido SS, Pratt BL, Robertson LM. The avian pineal, a vertebrate model system of the circadian oscillator: cellular regulation of circadian rhythms by light, second messengers, and macromolecular synthesis. Rec Prog Hormone Res. 1989;45:279–352. doi: 10.1016/b978-0-12-571145-6.50010-8. [DOI] [PubMed] [Google Scholar]
- Tosini G, Menaker M. Circadian rhythms in cultured mammalian retina. Science. 1996;272:419–421. doi: 10.1126/science.272.5260.419. [DOI] [PubMed] [Google Scholar]
- Turek FW. Circadian rhythms. In: Bardin W, editor. Recent Progress in Hormone Research. New York: Academic Press; 1994. pp. 43–90. [DOI] [PubMed] [Google Scholar]
- Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, Takahashi JS. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264:719–725. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. p53 function and dysfunction. Cell. 1992;70:523–526. doi: 10.1016/0092-8674(92)90421-8. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Kim NS, Sekine S, Seino H, Osaka F, Yamao F, Kato S. Cloning and expression of cDNA encoding a human ubiquitin-conjugating enzyme similar to the Drosophila bendless gene product. J Biochem. 1996;120:494–497. doi: 10.1093/oxfordjournals.jbchem.a021440. [DOI] [PubMed] [Google Scholar]
- Zehring WA, Wheeler DA, Reddy P, Konopka RJ, Kyriacou CP, Rosbash M, Hall JC. P-element transformation with period locus DNA restores rhythmicity to mutant, arrhythmic Drosophila melanogaster. Cell. 1984;39:369–376. doi: 10.1016/0092-8674(84)90015-1. [DOI] [PubMed] [Google Scholar]
- Zeng H, Hardin PE, Rosbash M. Constitutive overexpression of the Drosophila period protein inhibits period mRNA cycling. EMBO J. 1994;13:3590–3598. doi: 10.1002/j.1460-2075.1994.tb06666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]