

Abstract
Histone deacetylase (HDAC) inhibitors either alone or in combination with hypomethylating agents have limited clinical effect in acute myeloid leukemia (AML). Previously we demonstrated that AML patients with higher miR-29b expression had better response to the hypomethylating agent decitabine. Therefore, an increase in miR-29b expression preceding decitabine treatment may provide a therapeutic advantage. We previously showed that miR-29b expression is suppressed by a repressor complex that includes HDACs. Thus, HDAC inhibition may increase miR-29b expression. We hypothesized that priming AML cells with the novel HDAC inhibitor (HDACI) AR-42 would result in increased response to decitabine treatment via upregulation of miR-29b. Here we show that AR-42 is a potent HDACI in AML, increasing miR-29b levels and leading to downregulation of known miR-29b targets (i.e., SP1, DNMT1, DNMT3A, and DNMT3B). We then demonstrated that the sequential administration of AR-42 followed by decitabine resulted in a stronger anti-leukemic activity in vitro and in vivo than decitabine followed by AR-42 or either drug alone. These preclinical results with AR-42 priming before decitabine administration represents a promising, novel treatment approach and a paradigm shift with regard to the combination of epigenetic-targeting compounds in AML, where decitabine has been traditionally given before HDAC inhibitors.
Keywords: acute myeloid leukemia, HDACI, AR-42, decitabine, miR-29b
INTRODUCTION
The prognosis for the majority of patients with acute myeloid leukemia (AML) receiving standard chemotherapy is poor, novel treatment strategies are needed.1–3 Aberrant promoter DNA hypermethylation and histone deacetylation are reversible processes implicated in myeloid leukemogenesis, and each are targetable by hypomethylating agents (e.g. decitabine) and histone deacetylase (HDAC) inhibitors (HDACIs), respectively.4–7 Different from other hematologic malignancies such as cutaneous T-cell lymphoma, HDACIs as single agents have resulted in limited clinical activity in AML and combination therapy with hypomethylating agents has not consistently led to a significantly improved response.4,8–11 Possible explanations for this may be related to differences in pharmacologic potency, rapid metabolism, and/or off-target activity (e.g. acetylation of non-histone substrates) of available HDACIs.12–14
Structural aspects of HDACIs that allow for access to the Zn2+ cation in the catalytic pocket of the HDAC enzyme are determinants of inhibitor activity. Researchers at The Ohio State University (OSU) synthesized a new class of HDACIs that are structurally optimized to improve access to the catalytic pocket. These new compounds have been shown to inhibit enzyme activity and cancer cell proliferation even at nanomolar concentrations.14 Among these new compounds, AR-42, has been proven to be active in non-Hodgkin lymphoma (NHL) and multiple myeloma (MM) and is in Phase I clinical trials.15–17 However, AR-42 activity in AML has not yet been investigated.
Altered expression of microRNAs (miRs), small non-coding RNA molecules, has been shown to contribute to the pathogenesis of various human cancers, including AML.18,19 miR-29b, that negatively modulates the expression of genes encoding the transcriptional activator SP1 and DNA methyltransferases (DNMT1, DNMT3A and DNMT3B), is downregulated in AML.20–22 We and others showed that lower pretreatment levels of miR-29b may be associated with worse prognosis18 and inferior response to the hypomethylating agent decitabine in older (age ≥ 60 years) AML patients.7
Expression of miR-29b is partly regulated by an SP1/NFκB transcriptional complex, which binds to a miR-29b enhancer region, recruits HDACs and decreases miR-29b expression.22 Thus, here we hypothesized that the inhibition of HDAC activity could disrupt the binding of this complex, increase miR-29b expression and in turn induce an improved response to decitabine.
MATERIALS AND METHODS
Cell lines, AML patient samples and cell culture
Kasumi-1 (CRL-2724) and NB4 (ACC 207) cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) and German Collection of Microorganisms and Cell Culture (DSMZ, Braunschweig, Germany), respectively. Murine FDC-P1 cells were purchased from ATCC and retrovirally transduced to express the oncogenic D816V KIT mutation (FDC-P1-KITmut) as previously described.22 Kasumi-1, NB4, and FDC-P1-KITmut cells were cultured as previously described.20–22 These cell lines were chosen because of a low baseline expression of miR-29b.20,22 Primary AML blasts from apheresis samples collected from 10 patients with de novo disease were obtained from the OSU Leukemia Tissue Bank. Cytogenetic analysis was available for nine of the ten patients (Supplemental Table 1). Samples from the patients 1–2 and 6–10 were used to conduct HDAC inhibition studies, while the patient samples 3–10 were used for miR expression and patients 3–5 were used for gene and protein expression studies. All patients provided written informed consent according to the Declaration of Helsinki to store and use their tissue for discovery studies according to the OSU institutional guidelines under protocols approved by the OSU Institutional Review Board. The patient samples were cultured as previously described.22
HDAC activity inhibition assay
Nuclear extracts of Kasumi-1 and NB4 cell lines and patient AML blasts were prepared using the Nuclear Extract Kit following the manufacturer’s protocol (Active Motif, Carlsbad, CA). The effects of 100 nM and 1 μM of AR-42 on HDAC activity were investigated using the Histone Deacetylase Assay Kit according to the manufacturer’s protocol (Upstate, Lake Placid, NY). Doses of AR-42 were chosen based on previous studies in hematological malignancies.15,17
Gene and microRNA expression
RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA) and quantified using the NanoDrop 2000 Spectrophotometer (Thermo Scientific, Walthan, MA). For both primary miR and messenger RNA (mRNA) gene expression, RNA was reverse transcribed into complementary-DNA using SuperScript III First Strand Synthesis (Invitrogen) according to the manufacturer’s recommendations. Real-Time PCR was performed using a 7900HT Fast Real-Time System (Applied Biosystems, Carlsbad, CA). Primer pairs and probes used were human DNMT1, DNMT3A, DNMT3B, SP1, 18S, and primary-miR-29b-1, as well as mouse Dnmt1, Dnmt3a, Dnmt3b, Sp1, and 18S purchased from Applied Biosystems. Taqman Universal PCR master Mix was purchased from Applied Biosystems. The expression of human and murine 18S were used as internal controls for both mRNA and miR expression.
Western blotting
Whole cell lysates were run on SDS-PAGE Ready Gel Precast Gels (Bio-rad, Hercules, CA) and transferred to nitrocellulose membrane as previously described.22 Immunoblotting was performed with rabbit anti-acetylated Histone H3 (06–599, Upstate), rabbit anti-acetylated Histone H4 (06–866, Upstate), rabbit anti-DNMT3A (sc-20703, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-SP1 (sc-59, Santa Cruz), goat anti-β-actin (sc-1616, Santa Cruz), rabbit anti-DNMT1 (ab87656, Abcam, Cambridge, MA), and mouse anti-DNMT3B (ab16304, Abcam).
Cell proliferation assay
Kasumi-1, NB4 and FDC-P1-KITmut cells were seeded in 96-well plates and were treated for 72 hours with vehicle, AR-42 (0.3 μM) alone, AR-42 (0.3 μM) followed by decitabine (0.5 μM) after 24 hours [AR-42→decitabine], decitabine (0.5 μM) followed by AR-42 (0.3 μM) after 24 hours [decitabine→AR-42] or decitabine (0.5 μM) alone. After 72 hours, MTS reagent [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)-2H-tetrazolium, inner salt; Promega, Madison, WI] was added to each well. Patient primary blasts were seeded in 96-well plates and were treated for 48 hours with vehicle, AR-42 (3 nM, 10 nM, 30 nM, 100 nM, 300 nM or 1 μM) or valproic acid (VPA); 600 μM or 2400 μM). After 48 hours, MTS reagent was added to each well. Plates were incubated according to the manufacturer’s protocol. The absorbance at 495 nM was measured in a Multiskan Spectrum plate reader (Thermo Electron Corporation, Vantaa, Finland). After adjustment for background interference by accounting for wavelength variation secondary to media, data in triplicate from three independent experiments were normalized to the readings from untreated cells.
Leukemogenesis in NOD/SCID mice
Four to six-week-old NOD/SCID mice (The Jackson Laboratory, Bar Harbor, ME) were intravenously injected through a tail vein with 5×106 cells of FDC-P1 cells harboring D816V KITmut. After engraftment, cell-injected mice were treated with either vehicle alone, decitabine intraperitoneally (0.4 mg/kg/d for 4 days in weeks 1 and 3), AR-42 by oral gavage (75 mg/kg/d for 2 days in weeks 1 and 3), decitabine for 4 days followed by AR-42 for 2 days on weeks 1 and 3 (doses for both drugs the same as above) or AR-42 for 2 days followed by decitabine for 4 days for weeks 1 and 3 (doses for both drugs the same as above). Spleen samples were harvested from the treated mice and processed for RNA and cDNA. Real-Time PCR was performed on all samples with murine primer/probes used for all genes and primary-miR-29b-1 as described above. These studies were performed in accordance with OSU institutional guidelines for animal care and under protocols approved by the OSU Institutional Animal Care and Use Committee.
Statistical Methods
Data were represented as mean ± standard deviations (SD) of at least three independent experiments and analyzed by the 2-tailed Student’s t-test. The means and SD were calculated and displayed in bar graphs as the height and the corresponding error bar, respectively. Mouse survival was calculated using the Kaplan-Meier method, and survival curves were compared by log-rank test. A P<0.05 was considered statistically significant.
RESULTS
AR-42 inhibits HDAC activity in AML
We first assessed the HDAC inhibitory activity of AR-42 on AML cells. Kasumi-1 and NB4 cells were treated with 100 nM and 1 μM AR-42, and HDAC enzymatic activity was measured after 24 hours. HDAC activity was reduced 82% (±1.8% SD; P<0.01) and 90% (±0.4%; P<0.01) in Kasumi-1 cells and 85% (±6.9%; P<0.01) and 90% (±3.2%; P<0.01) in NB4 cells following exposure to 100 nM and 1 μM AR-42, respectively (Figure 1a). The doses of AR-42 were chosen based on previous studies in hematological malignancies.15,17 The degree of inhibition of HDAC activity caused by AR-42 treatment was comparable to the degree of inhibition achieved by treatment with the hydroxamate analog of AR-42, Trichostatin A (TSA), a known potent HDACI often used as a control for HDACI inhibition assays.23 In Kasumi-1, concentrations of 100 nM of TSA reduced the HDAC activity by 83%, and 1 μM of TSA reduced the HDAC activity by 88%. In NB4, similar results were demonstrated (Figure 1a). The decrease in HDAC enzymatic activity in cells treated with AR-42 also led to an increase in histone acetylation. Concentrations of AR-42 as low as 30 nM induced histone H3 and H4 acetylation in both the Kasumi-1 and NB4 cell lines at 48 hours (Figure 1b).
Figure 1.

AR-42 treatment inhibits HDAC activity in AML. (a) HDAC activity in Kasumi-1 and NB4 cells at 24 hours after treatment with vehicle, AR-42 or TSA. (b) Increased histone acetylation in Kasumi-1 and NB4 and cells 48 hours after AR-42 treatment. (c) HDAC activity in primary AML patient blasts 24 hours after treatment with AR-42 (n=2; patients no.1 and no.2). (d) Increased histone acetylation in primary patient blasts 48 hours after AR-42 treatment (patients no.1 and no.2 as indicated).
The HDAC inhibition activity of AR-42 was also demonstrated in AML patient blasts. In these primary cells, we observed a dose dependent effect of HDAC activity inhibition following AR-42 treatment for 24 hours (Figure 1c). Treatment with 1 μM AR-42 reduced the total HDAC activity by 78% (±11%; P<0.01) compared to vehicle, similar to the degree of inhibition (76% ±5.6%) observed with 1 μM TSA. H3 and H4 histone acetylation was also observed in primary blasts following AR-42 treatment at 300 nM and 1 μM concentrations (Figure 1d).
We also tested 600 μM and 2400 μM VPA,24 a known HDACI, 24 as a control in both AML cell lines and additional primary patient samples (n=5) and showed that acetylation of H3 and H4 histone increased in a similar dose dependent fashion with both compounds (Supplemental Figure 1). Interestingly, similar to Stapnes et al24 we observed an anti-proliferative effect of the HDACIs, at higher concentrations (i.e., >100 nM AR-42 and 2400 μM VPA), and a more heterogenous response at lower concentrations (i.e., <100 nM AR-42 and 600 μM VPA; Supplemental Figure 2).
AR-42 upregulates miR-29b expression
We next treated Kasumi-1, NB4 and the murine FDC-P1-KITmut cell lines with AR-42 and determined the effect on miR-29b expression. These cell lines were chosen because of their low levels of endogenous miR-29b.20,22 Compared with vehicle-treated control cells, miR-29b expression was found to be upregulated 4-fold (±0.93; P<0.01) in Kasumi-1 cells, 5-fold (±1.18; P<0.05) in NB4 cells and 14-fold (±1.63; P<0.01) in FDC-P1-KITmut cells (Figure 2a) after 24 hours treatment with 1 μM of AR-42. These results were confirmed in leukemic blasts from eight primary AML patients. Treatment with 1 μM AR-42 increased miR-29b expression 6.5-fold (±2.4; P<0.05) at 24 hours compared with vehicle-treated control blasts (Figure 2b). miR-29b upregulation was also observed with 2400 μM VPA both in Kasumi-1 and NB4 cell lines as well as in primary patient blasts. While in the cell lines, VPA-induced increased in miR-29b was similar to that observed with AR-42 (Supplemental Figure 3), in primary blasts the VPA-induced increased in miR-29b was 2.5-fold (±0.17) seemingly lower than that induced by AR-42 [6.5-fold (±2.4); Figure 2b].
Figure 2.
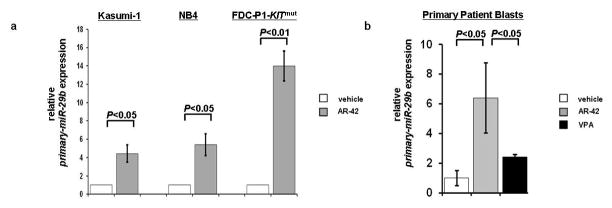
miR-29b expression increases following AR-42 treatment. (a) in Kasumi-1, NB4, and FDC-P1-KITmut cells, 24 hours after treatment with 1 μM AR-42. (b) in eight primary AML patient samples (patients nos. 3–10) at 24 hours after treatment with 1 μM AR-42 and 2400 μM VPA.
AR-42 downregulates the miR-29b targets DNMT1, DNMT3A, DNMT3B and SP1
It is known that miR-29b downregulates DNMT3A, DNMT3B and SP1 directly, and reduces expression of DNMT1 indirectly by targeting its transcription factor SP1.21 Thus, we assessed the RNA expression levels of DNMT1, DNMT3A, DNMT3B, and SP1 following AR-42 treatment. We compared the Kasumi-1 and NB4 cells treated with 1 μM AR-42 with vehicle-treated controls and observed, respectively, a reduction of DNMT1 by 80% (±7.2%; P<0.01) and 96% (±1.1%; P<0.01), DNMT3A by 95% (±0.6%; P<0.01) and 94% (±0.5%; P<0.01), DNMT3B by 78% (±10.7%; P<0.05) and 90% (±3%; P<0.05), and SP1 by 53% (±0.3%; P<0.05) and 82% (±2%; P<0.05) after 24 hours (Figures 3a and 3b). Similar results were also obtained using the murine FDC-P1-KITmut cell line treated with 1 μM AR-42, which resulted in a reduction of Dnmt1 by 63% (±0.5%; P<0.05), Dnmt3a by 40% (±2% P<0.2), Dnmt3b by 61% (±1.9%; P<0.05) and Sp1 by 73% (±0.2%; P<0.05; Figure 3b). These results were also confirmed at the protein level and although Dnmt3a by RNA was not statistically significant, western blotting confirms the downregulation. DNMT1, DNMT3A, DNMT3B, and SP1 proteins were downregulated at 24 hours following 1 μM AR-42 treatment in Kasumi-1, NB4 and FDC-P1-KITmut cell lines compared with vehicle-treated controls (Figure 3b). These findings were then validated in primary patient blasts (n=3). Twenty-four hours after treatment with 1 μM AR-42, mRNA levels were found to have a reduction of 81% (±5.8%; P<0.01) in DNMT1, 80% (±13%; P<0.01) in DNMT3A, 75% (±7.6%; P<0.01) in DNMT3B and 50% (±13%; P<0.05) in SP1 when compared with vehicle-treated controls (Figure 3c). Likewise, the DNMT1, DNMT3A, DNMT3B, and SP1 proteins were downregulated in all three patient samples following 1 μM AR-42 treatment compared with vehicle-treated controls (Figure 3d).
Figure 3.

Expression of the miR-29b targets DNMT1, DNMT3A, DNMT3B and SP1 decreases following AR-42 treatment. (a,b) Kasumi-1, NB4, and FDC-P1-KITmut cells treated with 1 μM AR-42 for 24 hours DNMT1, DNMT3A, DNMT3B and SP1, decreases on both mRNA and protein level. (c,d) Primary patient blasts were treated with 1 μM AR-42 for 24 hours with decrease of DNMT1, DNMT3A, DNMT3B and SP1 on both mRNA (n=3) and protein level (patients as indicated).
Increased anti-leukemic activity: AR-42 followed by decitabine
We have previously shown that AML patients with higher expression of miR-29b had better clinical response to decitabine.7 Therefore, we hypothesized that an AR-42-induced increase in miR-29b expression might result in increased anti-leukemic activity of decitabine. We compared the anti-leukemic activity of AR-42 followed by decitabine with that of both AR-42 and decitabine as single agents, and decitabine followed by AR-42 in Kasumi-1, NB4 and FDC-P1-KITmut cells. The cells were treated for 72 hours with vehicle, AR-42 (0.3 μM) alone, AR-42 (0.3 μM) followed by decitabine (0.5 μM) after 24 hours [AR-42→decitabine], decitabine (0.5 μM) followed by AR-42 (0.3 μM) after 24 hours [decitabine→AR-42] or decitabine (0.5 μM) alone. The lowest cell viability was observed in AR-42→decitabine group in all three cell lines (Figure 4a). Kasumi-1 cells treated with AR-42→decitabine were significantly less viable than those treated with decitabine alone (17% vs 91%; P<0.01), AR-42 alone (17% vs 40%; P<0.01), and decitabine→AR-42 (17% vs 34%; P<0.01; Figure 4a). Similar observations were made for NB4 cells [AR-42→decitabine vs decitabine alone: 59% vs 99% (P<0.01); AR-42→decitabine vs AR-42 alone: 59% vs 87% (P<0.05); and AR-42→decitabine vs decitabine→AR-42: 59% vs 75% (P<0.05)]. We also found similar changes in the FDC-P1-KITmut cells [AR-42→decitabine vs decitabine alone: 52% vs 100% (P<0.01); AR-42→decitabine vs AR-42 alone: 52% vs 90% (P<0.05); and AR-42→decitabine vs decitabine→AR-42: 51% vs 90% (P<0.05)].
Figure 4.

AR-42 followed by decitabine has the strongest activity on cell viability. Kasumi-1, NB4, and FDC-P1-KITmut cells treated with vehicle, decitabine 0.5 μM for 72 hours, decitabine 0.5 μM for 72 hours with AR-42 0.3 μM added at 24 hours, AR-42 0.3 μM for 72 hours, or AR-42 0.3 μM for 72 hours with decitabine 0.5 μM added at 24 hours. Cells treated with AR-42 followed by decitabine showed lowest cell viability.
Next we validated our in vitro findings in an in vivo AML mouse model. NOD/SCID mice engrafted with FDC-P1-KITmut cells developed AML-like disease22 and then were treated with either vehicle (n=7), decitabine alone at 0.4 mg/kg/d intraperitoneally for 4 days in weeks 1 and 3 (n=7), AR-42 alone at 75 mg/kg/d by oral gavage for 2 days in weeks 1 and 3 (n=10),15 decitabine for 4 days followed by AR-42 for 2 days at aforementioned doses (n=10), or AR-42 for 2 days followed by decitabine for 4 days at aforementioned doses for both drugs (n=17). To evaluate whether AR-42 increased miR-29b expression and downregulated miR-29b targets in vivo, five mice from each group were sacrificed 12 hours after two doses of AR-42 and compared with vehicle-treated control. RNA was extracted from spleen cells and miR-29b as well as Dnmt1, Dnmt3a, Dnmt3b, and Sp1 expression was analyzed. We found that miR-29b expression was upregulated 20-fold (±5.4) in the mice treated with AR-42 as compared with those treated with vehicle (P<0.01; Figure 5a). Dnmt1 (72.7%, ±7.4%; P<0.01), Dnmt3a (65.6%, ±8.8%; P<0.05), Dnmt3b (93.5% ±0.8%; P<0.01), and Sp1 (74.6%, ±6%; P<0.05) were found to be reduced in the AR-42 treated mice compared with vehicle-treated controls (Figure 5b). We also observed a significantly longer survival for mice treated with AR-42→decitabine, compared to vehicle treatment (P<0.001), decitabine alone (P<0.001), AR-42 alone (P<0.01) or decitabine→AR-42 (P<0.001; Figure 5a). Indeed by day 60, all of the mice died of disease with the exception of those treated with AR-42→decitabine, among which, 59% (10 of 17) were still alive at this time point (Figure 5c).
Figure 5.

Priming with AR-42 upregulates miR-29b and increases survival in murine models. (a) miR-29b levels were upregulated 20-fold in AR-42 treatment versus vehicle treatment group. (b) Dnmt1, Dnmt3a, Dnmt3b, and Sp1 were downregulated in AR-42 versus vehicle treatment group. (c) Overall survival. FDC-P1-KITmut cells injected into NOD/SCID mice showed 10 of 17 mice with survival at 60 days in AR-42 followed by decitabine group compared with no survival in mouse groups with decitabine treatment alone, decitabine followed by AR-42, or AR-42 treatment alone.
DISCUSSION
We previously showed that miR-29b has tumor suppressor activity in AML by targeting a variety of genes, including regulators of DNA methylation.20–22 Furthermore, we reported that AML patients with higher pretreatment levels of miR-29b had a better response to decitabine therapy.7 Thus, here we sought to demonstrate an increase in anti-leukemic activity of decitabine by first increasing miR-29b expression. As miR-29b is repressed by an SP1/NFκB/HDAC silencing complex in AML,22 we hypothesized that treatment with a HDAC inhibitor would increase the expression of this miR in AML cells. Indeed, Sampath et al24 recently found an upregulation of miR-29b following HDAC inhibition in chronic lymphocytic leukemia, suggesting that the expression of this miR is targetable pharmacologically and that epigenetic deregulation of miR-29b may also occur in malignancies other than AML.
To prove that HDAC inhibition resulted in miR-29b upregulation in AML, we elected to test AR-42, a novel HDAC inhibitor developed at our institution. The advantage of using AR-42 was its significantly higher HDAC inhibitory potency relative to other HDAC inhibitors’ in vitro and in vivo cancer models.26 Furthermore, AR-42 has also been well-tolerated in phase I clinical trials in patients with MM and NHL, with thrombocytopenia and fatigue as the most common adverse events. These side effects are similar to those observed with other HDACIs, which have been also associated with neurologic, gastrointestinal and cardiac adverse effects.27,28 We first demonstrated that AR-42 inhibits HDAC enzyme activity and induced histone acetylation in AML cells at concentrations in the nanomolar to micromolar ranges. We also tested the antileukemia activity of the compound. Patient samples seemingly exhibited different susceptibility to AR-42 similar to results previously reported by Stapnes, et al24 for VPA despite the different assays and the time-point of analysis utilized in the two studies. AML patients represent a very heterogeneous population with different susceptibilities to HDACIs. Studies of cytogenetic, molecular and in-vitro growth characteristics of primary blasts associated with HDACI response may provide insight into how to best select AML patients that are more likely to be responsive to this class of compounds.24,29
Next we showed that miR-29b expression increased upon treatment with AR-42 in AML cell lines and AML patient primary blasts. Upregulation of miR-29b induced by AR-42 treatment also resulted in a concurrent downregulation of the known miR-29b targets DNMT1, DNMT3A and DNMT3B and SP1. Others have also reported on the effect of HDAC inhibition on DNA methyltransferase expression.30,31 Xiong et al.30 showed that HDAC inhibition decreased DNMT3B mRNA stability in human endometrial cells, and Zhou et al.31 showed that HDAC inhibition decreased DNMT1 expression in breast cancer cells, but neither of these reports included a clear proposed mechanism for their results.
Both DNA hypermethylation and histone acetylation have been shown to contribute to tumor suppressor gene silencing in AML. The combination of HDAC inhibitors with decitabine has resulted in synergistic effects on apoptosis, DNA hypomethylation and gene re-expression in vitro32 and this combination therapy has been performed to achieve synergism. However, it has been recommended that HDACIs be administered before or concurrently with decitabine due to their ability to induce expression of p21 and other inhibitors of the cell cycle.33 Interfering with the cell cycle may decrease the activity of decitabine, as it is necessary for the active metabolite decitabine-trisphosphate to be incorporated into the nascent DNA in order to inhibit DNMT activity, induce DNA hypomethylation and gene re-expression.34 To date, treatment with HDACIs given following or concurrently with decitabine has demonstrated anti-leukemic activity in AML, but with a relatively low range of clinical response.4,8–11 In a phase I clinical study conducted by our group, we also did not observe additional clinical benefit following the addition of VPA to decitabine (concurrent) although dose escalation of VPA was limited due to the development of neurologic toxicity.11 In a phase I clinical trial of the HDACI vorinostat, administered either concomitantly or following decitabine in patients with AML or myelodysplastic syndrome (MDS), the overall response rate (ORR) was 41% with the concomitant schedule and 21% with the sequential schedule.35 Thus the synergism of post- or concurrent administration of HDACIs with DNA hypomethylating agents demonstrated in preclinical models, could not be fully recapitulated in vivo.
In a phase II clinical trial reported by our group, we recently reported the results of low dose (20 mg/m2/day × 10 days) decitabine as a single agent in untreated elderly AML.7 We showed relatively low toxicity, a complete remission rate of 47%, an ORR of 64%, and a median overall survival duration of approximately one year. The median pretreatment miR-29b expression level in responders was 2.3 times higher (i.e. more than double) than the median baseline miR-29b levels in nonresponders, suggesting relevance for this miR as a predictive marker for response to decitabine treatment. In contrast to our data, Yang et al. reported a lack of an association of miR-29b levels with clinical response in patients treated with the azanucleoside 5-azacitidine.36 However, only 10% of 5-azacitidine is reduced to decitabine and incorporated into DNA for hypomethylating activity, while the remaining 90% is incorporated into the RNA. Furthermore, we recently showed that 5-azacitidine limits its own conversion to decitabine by downregulating ribonucleotide reductase.37 Therefore, these two compounds, although both members of the same class of drugs (azanucleosides), may impact leukemia through different mechanisms, and high miR-29b levels may improve response to decitabine but not to 5-azacitidine in AML.
We demonstrated here that an increase of miR-29b expression by AR-42 improved the anti-leukemia activity of decitabine. We showed that sequential treatment of AR-42 followed by decitabine decreased cell viability significantly more than each agent alone or the previously recommended sequence of decitabine followed by AR-42. This was validated in a murine AML model where mice treated with AR-42 followed by decitabine survived significantly longer than those treated with single agent therapy or decitabine followed by AR-42. One possible explanation for the better activity of AR-42 followed by decitabine may relate to miR-29b targeting DNMT expression. With decreased amounts of DNMT enzyme present, decitabine may more effectively inhibit the activity of the remaining DNMTs resulting in improved treatment response. Indeed, we previously reported that patients with DNMT3A mutations had improved response to decitabine.38 Thus it is possible that a clinical benefit from treatment with decitabine may be derived for AML patients from low DNMT3A activity, either due to loss-of-function mutations or due to low gene expression. Another possibility may be that the lower levels of SP1 induced by increased miR-29b expression result in decreased transcription of genes known to contribute to AML leukemogenesis such as mutated and/or upregulated receptor tyrosine kinases (i.e., FLT3 and KIT) as we have previously demonstrated.22,39
Collectively our data support the notion that AR-42 is a potent HDACI that is able to increase miR-29b expression and improve clinical response to decitabine in in vivo preclinical models. Based on these preclinical findings, clinical trials utilizing AR-42 as a priming agent for decitabine treatment in patients with AML are under development.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Supported in part by CA102031, CA140158, The Coleman Leukemia Research Foundation (SS), The Harry Mangurian foundation, NIH T32 grant 60032104 (AM), K12CA133250 (AW). AW is a Paul Calabresi Clinical Scholar and a scholar of the American Society of Hematology-Amos Medical Faculty Development Program. We thank Donna Bucci and the Leukemia Tissue Bank for assistance with the primary AML samples. We would like to thank Samuel Kulp, D.V.M, Ph.D for assistance with AR-42.
Footnotes
Presented in part at the 103rd Annual Meeting of the American Association for Cancer Research, Chicago, IL, April 2012, and published in abstract form.
AUTHOR CONTRIBUTION
A.M., J.S., R.S., A.D., C.W., P.H., X.H., S.T., and S.S. performed experiments. A.M., H.W., X.H., K.K.C., D.P., C.S.C., R.G., S.S., and G.M. designed experiments and analyzed data. A.M., A.W., L.J.L., S.J., K.M., C.D.B., S.S. and G.M., wrote the manuscript. All the authors approved the manuscript. A.W., R.B.K., R.G., W.B., M.C., J.C.B., and G.M. were involved directly or indirectly in care of patients or sample procurement.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary information is available at Leukemia’s website.
References
- 1.Estey E, Döhner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 2.Estey EH. Treatment of acute myeloid leukemia. Haematologica. 2009;94:10–16. doi: 10.3324/haematol.2008.001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487–494. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- 4.Bruserud Ø, Stapnes C, Tronstad KJ, Ryningen A, Ånensen N, Gjertsen BT. Protein lysine acetylation in normal and leukaemic haematopoiesis: HDACs as possible therapeutic targets in adult AML. Expert Opin Ther Targets. 2006;10:51–68. doi: 10.1517/14728222.10.1.51. [DOI] [PubMed] [Google Scholar]
- 5.Tickenbrock L, Klein H-U, Trento C, Hascher A, Göllner S, Bäumer N, et al. Increased HDAC1 deposition at hematopoietic promoters in AML and its association with patient survival. Leuk Res. 2011;35:620–625. doi: 10.1016/j.leukres.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Cashen AF, Schiller GJ, O’Donnell MR, DiPersio JF. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol. 2009;28:556–561. doi: 10.1200/JCO.2009.23.9178. [DOI] [PubMed] [Google Scholar]
- 7.Blum W, Garzon R, Klisovic RB, Schwind S, Walker A, Geyer S, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci U S A. 2010;107:7473–7478. doi: 10.1073/pnas.1002650107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105:959–967. doi: 10.1182/blood-2004-05-1693. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Versotovsek S, et al. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108:3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111:1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 11.Blum W, Klisovic RB, Hackanson B, Liu Z, Liu S, Devine H, et al. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J Clin Oncol. 2007;25:3884–3891. doi: 10.1200/JCO.2006.09.4169. [DOI] [PubMed] [Google Scholar]
- 12.Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Natl Acad Sci U S A. 2001;98:87–92. doi: 10.1073/pnas.011405598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krämer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003;22:3411–3420. doi: 10.1093/emboj/cdg315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Q, Yang Y-T, Chen C-S, Davis M, Byrd JC, Etherton MR, et al. Zn2+-chelating motif-tethered short-chain fatty acids as a novel class of histone deacetylase inhibitors. J Med Chem. 2004;47:467–474. doi: 10.1021/jm0303655. [DOI] [PubMed] [Google Scholar]
- 15.Lucas DM, Alinari L, West DA, Davis ME, Edwards RB, Johnson AJ, et al. The novel deacetylase inhibitor AR-42 demonstrates pre-clinical activity in B-cell malignancies in vitro and in vivo. PLoS One. 2010;5:e10941. doi: 10.1371/journal.pone.0010941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang S, Suvannasankha A, Crean CD, White VL, Chen C-S, Farag SS. The novel histone deacetylase inhibitor, AR-42, inhibits gp130/Stat3 pathway and induces apoptosis and cell cycle arrest in multiple myeloma cells. Int J Cancer. 2011;129:204–213. doi: 10.1002/ijc.25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zimmerman B, Sargeant A, Landes K, Fernandez SA, Chen C-S, Lairmore MD. Efficacy of novel histone deacetylase inhibitor, AR-42, in a mouse model of, human T-lymphotropic virus type 1 adult T cell lymphoma. Leuk Res. 2011;35:1491–1497. doi: 10.1016/j.leukres.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Li Z, He C, Wang D, Yuan X, Chen J, et al. MicroRNAs expression signatures are associated with lineage and survival in acute leukemias. Blood Cells Mol Dis. 2010;44:191–197. doi: 10.1016/j.bcmd.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcucci G, Mrózek K, Radmacher MD, Garzon R, Bloomfield CD. The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood. 2011;117:1121–1129. doi: 10.1182/blood-2010-09-191312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garzon R, Heaphy CEA, Havelange V, Fabbri M, Volinia S, Tsao T, et al. MicroRNA 29b functions in acute myeloid leukemia. Blood. 2009;114:5331–5341. doi: 10.1182/blood-2009-03-211938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CEA, Callegari E, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–6418. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu S, Wu L-C, Pang J, Santhanam R, Schwind S, Wu Y-Z, et al. Sp1/NFκB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell. 2010;17:333–347. doi: 10.1016/j.ccr.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park JH, Jung Y, Kim TY, Kim SG, Jong HS, Lee JW, et al. Class I Histone Deacetylase-Selective Novel Synthetic Inhibitors Potently Inhibit Human Tumor Proliferation. Clin Cancer Res. 2004;10:5271–5281. doi: 10.1158/1078-0432.CCR-03-0709. [DOI] [PubMed] [Google Scholar]
- 24.Stapnes C, Ryningen A, Hatfield K, Øyan AM, Eide GE, Corbascio M, et al. Functional characteristics and gene expression profiles of primary acute myeloid leukaemia cells identify patient subgroups that differ in susceptibility to histone deacetylase inhibitors. Int J Oncol. 2007;31:1529–1538. [PubMed] [Google Scholar]
- 25.Sampath D, Liu C, Vasan K, Sulda M, Puduvalli VK, Wierda WG, et al. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood. 2011;119:1162–1172. doi: 10.1182/blood-2011-05-351510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kulp SK, Chen CS, Wang DS, Chen CY, Chen CS. Antitumor effects of a novel phenylbutyrate-based histone deacetylase inhibitor, (S)-HDAC-42, in prostate cancer. Clin Cancer Res. 2006;12:5199–5206. doi: 10.1158/1078-0432.CCR-06-0429. [DOI] [PubMed] [Google Scholar]
- 27.Bruserud Ø, Stapnes C, Ersvaer E, Gjertsen BT, Ryningen A. Histone deacetylase inhibitors in cancer treatment: a review of the clinical toxicity and the modulation of gene expression in cancer cell. Curr Pharm Biotechnol. 2007;8:388–400. doi: 10.2174/138920107783018417. [DOI] [PubMed] [Google Scholar]
- 28.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 29.Bruserud Ø, Gjertsen BT, Foss B, Huang TS. New strategies in the treatment of acute myelogenous leukemia (AML): in vitro culture of aml cells--the present use in experimental studies and the possible importance for future therapeutic approaches. Stem Cells. 2001;19:1–11. doi: 10.1634/stemcells.19-1-1. [DOI] [PubMed] [Google Scholar]
- 30.Xiong Y, Dowdy SC, Podratz KC, Jin F, Attewell JR, Eberhardt NL, et al. Histone deacetylase inhibitors decrease DNA methyltransferase-3B messenger RNA stability and down-regulate de novo DNA methyltransferase activity in human endometrial cells. Cancer Res. 2005;65:2684–2689. doi: 10.1158/0008-5472.CAN-04-2843. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Q, Agoston AT, Atadja P, Nelson WG, Davidson NE. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol Cancer Res. 2008;6:873–883. doi: 10.1158/1541-7786.MCR-07-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalac M, Scotto L, Marchi E, Amenqual J, Seshan VE, Bhagat G, et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood. 2011;118:5506–16. doi: 10.1182/blood-2011-02-336891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leone G, D’Aló F, Zardo G, Voso MT, Nervi C. Epigenetic treatment of myelodysplastic syndromes and acute myeloid leukemias. Curr Med Chem. 2008;15:1274–1287. doi: 10.2174/092986708784534947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirschbaum M, Gojo I, Goldberg SL, Kujawski L, Atallah E, Marks P, et al. Vorinostat in combination with decitabine for the treatment of relapsed or newly diagnosed AML (AML) or myelodysplastic syndrome (MDS): a phase I, dose- escalation study. Blood. 2009;114:2089. [Google Scholar]
- 36.Yang H, Fang Z, Wei Y, Hu Y, Calin GA, Kantarjian HM, et al. Levels of miR-29b do not predict for response in patients with acute myelogenous leukemia treated with the combination of 5-azacytidine, valproic acid, and ATRA. Am J Hematol. 2011;86:237–238. doi: 10.1002/ajh.21937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aimiuwu J, Wang H, Chen P, Xie Z, Wang J, Liu S, et al. RNA-dependent inhibition of ribonucleotide reductase is a major pathway for 5-azacytidine activity in acute myeloid leukemia. Blood. 2012;119:5229–5238. doi: 10.1182/blood-2011-11-382226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Metzeler KH, Walker A, Geyer S, Garzon R, Klisovic RB, Bloomfield CD, et al. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26:1106–7. doi: 10.1038/leu.2011.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blum W, Schwind S, Tarighat SS, Geyer S, Eisfeld AK, Whitman S, et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood. 2012 doi: 10.1182/blood-2012-03-413898. e-pub ahead of print 7 May 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.