

Abstract
Fast inhibitory neurotransmission in the central nervous system is mediated by γ-aminobutyric acid (GABA) and glycine, which are accumulated into synaptic vesicles by a common vesicular inhibitory amino acid transporter (VIAAT) and are then co-released. However, the mechanisms that control the packaging of GABA + glycine into synaptic vesicles are not fully understood. In this study, we demonstrate the dynamic control of the GABA–glycine co-transmission by the neuronal glutamate transporter, using paired whole-cell patch recording from monosynaptically coupled cultured spinal cord neurons derived from VIAAT-Venus transgenic rats. Short step depolarization of presynaptic neurons evoked unitary (cell-to-cell) inhibitory postsynaptic currents (IPSCs). Under normal conditions, the fractional contribution of postsynaptic GABA or glycine receptors to the unitary IPSCs did not change during a 1 h recording. Intracellular loading of GABA or glycine via a patch pipette enhanced the respective components of inhibitory transmission, indicating the importance of the cytoplasmic concentration of inhibitory transmitters. Raised extracellular glutamate levels increased the amplitude of GABAergic IPSCs but reduced glycine release by enhancing glutamate uptake. Similar effects were observed when presynaptic neurons were intracellularly perfused with glutamate. Interestingly, high-frequency trains of stimulation decreased glycinergic IPSCs more than GABAergic IPSCs, and repetitive stimulation occasionally failed to evoke glycinergic but not GABAergic IPSCs. The present results suggest that the enhancement of GABA release by glutamate uptake may be advantageous for rapid vesicular refilling of the inhibitory transmitter at mixed GABA/glycinergic synapses and thus may help prevent hyperexcitability.
Key points
Inhibition mediated by GABA and glycine is essential for controlling a balance of excitation and inhibition in the spinal cord.
Although these transmitters are known to be co-released from the same synaptic vesicles, the mechanisms that control the packaging of GABA + glycine into synaptic vesicles have not been fully characterized.
In this study, using paired whole-cell recording, we found that raised extracellular glutamate levels increased the amplitude of GABAergic IPSCs by enhancing glutamate uptake but reduced glycine release.
High-frequency trains of stimulation decreased glycinergic IPSCs more than GABAergic IPSCs at GABA/glycine mixed synapses, and repetitive stimulation occasionally failed to evoke glycinergic but not GABAergic IPSCs.
The present results suggest that the use of GABA as a transmitter at GABA/glycine mixed synapses may afford protection against pathophysiological hyperexcitability associated with increased extracellular glutamate concentration.
Introduction
The two inhibitory neurotransmitters γ-aminobutyric acid (GABA) and glycine have been shown to be co-released at central synapses in the brainstem (Nabekura et al. 2004; Dufour et al. 2010), cerebellum (Dumoulin et al. 2001) and spinal cord (Jonas et al. 1998). At these inhibitory synapses, GABA and glycine are accumulated into synaptic vesicles by the activity of vesicular inhibitory amino acid transporter (VIAAT), also known as vesicular GABA transporter (VGAT). The proportions of GABA and glycine in vesicles are presumed to be determined by the relative cytoplasmic concentrations of these neurotransmitters. GABA is synthesized from glutamate by the enzyme glutamic acid decarboxylase (GAD), while inhibitory nerve terminals are also equipped with a GABA uptake system. In contrast, the intracellular glycine concentration is predominantly regulated by the neuronal plasma membrane glycine transporter type 2 (GlyT2; Rousseau et al. 2008). GAD and GlyT2 are both present in inhibitory neurons that co-release GABA and glycine (Tanaka & Ezure, 2004).
The developmental shift from one inhibitory neurotransmitter to another occurs in a variety of regions in the central nervous system. For example, immature inhibitory pathways in the auditory brainstem are predominantly GABAergic and switch to being glycinergic with time (Kotak et al. 1998; Nabekura et al. 2004), whereas GABAergic neurotransmission becomes dominant in developing collicular neurons (Meier et al. 2002). The switching of transmission phenotype may be caused by either presynaptic (Nabekura et al. 2004) or postsynaptic (Keller et al. 2001; Meier et al. 2002) mechanisms. The percentage of GABA/glycine co-release remains constant during perinatal spinal cord development (Gao et al. 2001) and their co-release persists in adults (Coull et al. 2003; Dufour et al. 2010). In addition, a shift from the glycine receptor towards GABAA-receptor-dominated transmission in adult rat spinal lamina I has also been reported in conditions characterized by hyperexcitability, such as chronic pain (Coull et al. 2003). At GABA/glycinergic co-release synapses, therefore, the proportions of synaptically activated GABAA and glycine receptors easily change in response to changes in the surrounding microenvironment. However, little is known about how the vesicular GABA/glycine content is regulated in individual GABA/glycine mixed synapses.
In the present study, we used paired whole-cell patch-clamp recordings from cultured spinal cord neurons to study the presynaptic factors regulating the GABAergic/glycinergic phenotypes. We showed that the inhibitory transmission phenotype was regulated by the cytoplasmic levels of inhibitory and excitatory transmitters. Raising the extracellular glutamate level increased GABA release but decreased glycine release at spinal GABA/glycine mixed synapses by enhancing presynaptic glutamate uptake. Interestingly, during high frequency transmission, glycinergic IPSCs decreased to a greater extent than GABAergic IPSCs; in addition, glycinergic, but not GABAergic, IPSCs were occasionally absent. Because GABAergic IPSCs had slower decay kinetics, these results illustrate a novel form of synaptic plasticity that maintains the relative balance between excitation and inhibition.
Methods
Cell culture
VIAAT (VGAT)-Venus rats, which express the fluorescent protein Venus in the inhibitory neurons (Uematsu et al. 2008), were used in this study. Spinal cord primary cultures were prepared from embryonic day 14 animals, as described previously (Kitamura et al. 2008; Ishibashi et al. 2009). In brief, embryos were obtained from pregnant rats under isoflurane anaesthesia, and their spinal cords were dissected out and incubated for 20 min at 32°C in Earle's balanced salt solution containing papain (16 U ml−1; Funakoshi, Tokyo, Japan). Single cells were isolated by trituration, seeded onto 0.2% polyethyleneimine-coated glass coverslips (2.7 × 104–5.4 × 104 cells cm−2), and maintained in Neurobasal medium containing l-glutamine (0.5 mm), B27 supplement (Invitrogen, Tokyo, Japan), penicillin (100 units ml−1) and streptomycin (100 μg ml−1) at 37°C and 5% CO2. Cell cultures were maintained by changing one-half of the medium once a week and used for experiments after 12–28 days in vitro as indicated.
Electrophysiological measurements
Cultured neurons were viewed under phase contrast on an inverted microscope (IX71; Olympus, Tokyo, Japan), and presynaptic inhibitory neurons were identified by Venus expression. Electrical measurements were performed using the conventional whole-cell patch-clamp recording technique at room temperature. The resistance between the patch pipette filled with the internal solution and the reference electrode in the normal external solution was 4–6 MΩ. The neurons were voltage-clamped at a holding potential (VH) of −70 mV except during 1 ms step depolarizations to +30 mV to evoke an action potential in unclamped axonal processes. Ionic currents were measured with a patch-clamp amplifier EPC-7plus (Heka Elektronik, Lambrecht, Germany) or Multiclamp 700B (Axon Instruments, Union City, CA, USA), and recorded with a sampling frequency of 10 kHz after low-pass filtering at 3 kHz. Currents were recorded, and voltage protocols were applied, using pCLAMP software (Clampex 10; Molecular Devices, Union City, CA, USA). With the exception of high-frequency stimulations, recordings were made following 10–20 mV hyperpolarizing step pulses delivered 2 s after the stimulation to monitor the access resistance, and recordings were discontinued if the access resistance changed markedly. Autaptic inhibitory currents were sometimes recorded from inhibitory neurons and were included in the current study.
Solutions
The standard external solution used for electrical recording contained (in mm): 150 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 10 Hepes, 10 glucose. When elevating the extracellular K+ concentration, Na+ ions were replaced with equimolar K+. For Na+-free solution, all extracellular Na+ was replaced with N-methyl-d-glucamine. The pH of the external solutions was adjusted to 7.4 with Tris-OH. The control patch pipette solution contained (in mm): 100 potassium methanesulfonate, 30 KCl, 8 NaCl, 1 MgCl2, 10 Hepes, 2 EGTA, 4 ATP-Mg. The composition of the pipette solution containing 100 mm inhibitory or excitatory amino acids was (in mm): 100 inhibitory or excitatory amino acids, 50 potassium methanesulfonate, 30 KCl, 8 NaCl, 1 MgCl2, 10 Hepes, 2 EGTA, 4 ATP-Mg. The internal solution containing 20 mm amino acids was obtained by diluting this solution with the control pipette solution. All pipette solutions were buffered to pH 7.3 with Tris-OH, and their osmolality ranged from 290 to 300 mosmol kg-1. Unless otherwise indicated, the test solutions containing drugs were applied using the Y-tube system for rapid solution exchange (Murase et al. 1989). In some experiments, standard extracellular solution containing both glycine (100 μm) and GABA (100 μm) was briefly (50 ms) puffed at 2 Hz via a glass pipette (3–6 μm diameter) using a valve-controlled pressure application system (IM300 Microinjector, Narishige, Tokyo, Japan).
Immunocytochemistry
Cultured spinal cord neurons were fixed with paraformaldehyde (4% w/v) in phosphate buffered saline (PBS) and then permeabilized with Triton X-100 (0.1%, v/v). Non-specific antibody binding sites were blocked with PBS containing normal horse serum (2%, v/v) and 1% bovine serum albumin (1%, w/v). Cells were incubated with anti-EAAT3 primary antibody (1:50; sc-7761; Santa Cruz Biotechnology, Heidelberg, Germany) overnight at 4°C, followed by Alexa Fluor-conjugated secondary antibody (Alexa Fluor 647; 1:1000; Invitrogen) for 2 h at room temperature. Coverslips were mounted in PermaFluor aqueous mounting medium (Thermo Shandon, Pittsburgh, PA, USA), and the immunofluorescent images were acquired with a confocal laser scanning microscope (Nikon A1, Plan Apo VC ×60 H, NA 1.4, Tokyo, Japan).
Drugs
The drugs used in the present study were: dl-(−)-2-amino-5-phosphonopentanoic acid (AP-5), ATP-Mg, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), EGTA, GABA, glutamic acid, glycine, N-methyl-d-glucamine, 6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid (SR95531), strychnine and (1,2,5,6-tetrahydro-pyridin-4-yl)methylphosphinic acid hydrate (TPMPA) from Sigma (St Louis, MO, USA); dl-threo-β-ben-zyloxyaspartic acid (TBOA) and (2S)- 3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenyl-methyl)phosphinic acid hydrochloride (CGP55845) from Tocris Bioscience (Bristol, UK); potassium methanesulfonate from Alfa Aesar (Ward Hill, MA, USA). All other chemicals were obtained from Wako Pure Chemical Industries (Tokyo, Japan).
Statistical analysis
The data are expressed as mean ± SEM, with the number of data (n). Statistical significance was assessed using paired and unpaired Student's t tests. One-way analysis of variance (post hoc: Dunnett's test) was used to compare l-glutamate loading and high-K+ loading. Means with two-tail P values < 0.05 were considered significantly different. The number of asterisks in the figures corresponds to level of significance: *P < 0.05, **P < 0.01 and ***P < 0.001.
Results
Whole-cell recordings were obtained from pairs of monosynaptically coupled cultured spinal cord neurons. Short depolarizing steps were applied to the presynaptic neuron at 0.1 Hz under voltage-clamp conditions at a VH of −70 mV in the presence of 10 μm CNQX and 20 μm AP-5. This stimulation generated action potentials in an unclamped axon. The action potential then propagated along the axon giving rise to a unitary (cell-to-cell) IPSC when the recorded cell was synaptically connected. It should be noted that presynaptic stimulation was performed in voltage-clamp mode, because subthreshold somatic depolarization can spread electrotonically into the axon and modulate subsequent spike-evoked transmission (Shu et al. 2006; Christie et al. 2011). In nine paired recordings, application of 3 μm SR95531, a selective GABAA receptor antagonist, inhibited the evoked unitary IPSCs by 41.1 ± 7.2% immediately after obtaining paired recording within 5 min of rupturing the patch membrane of the presynaptic neurons. Although the inhibition ratio produced by SR95531 varied among cells, the remaining current was fully inhibited by 1 μm strychnine, a glycine receptor blocker, suggesting that these presynaptic neurons had a mixed GABAergic/glycinergic phenotype (Fig. 1). In these pairs, the SR95531-induced inhibition 30 and 60 min after starting the recordings was 44.6 ± 7.0% and 45.5 ± 8.5%, respectively. These values were not significantly different from that obtained at the onset of the recordings, suggesting that the fractional contribution of each receptor did not change significantly during 1 h recording under our experimental conditions.
Figure 1. Unitary IPSC recordings from cultured rat spinal cord neurons.
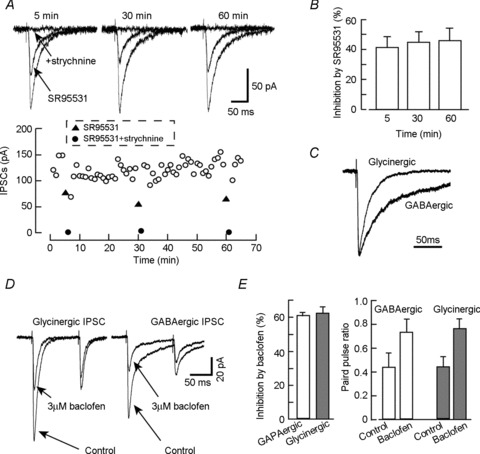
Current recordings were performed in the presence of 10 μm CNQX and 20 μm AP-5. A, effect of SR95531 on the unitary (cell-to-cell) IPSCs. The unitary IPSCs were evoked by 1 ms step depolarizations to +20 mV from a holding potential (VH) of −70 mV applied to the presynaptic neuron. Upper panel shows the superimposed current traces obtained under control, 3 μm SR95531, and SR95531 with 1 μm strychnine conditions. SR95531 was applied at 5 min, 30 min and 60 min after rupturing the patch membrane of presynaptic neurons. The currents in the presence of SR95531 were fully inhibited by applying 1 μm strychnine. Lower panel shows the time course of the recording as shown in A. Each point is the average of 3 responses. B, effect of 3 μm SR95531 on the unitary IPSC amplitude. Each column represents mean ± SEM from 9 independent experiments. C, superimposed traces of glycinergic and GABAergic unitary IPSCs; currents were normalized to the peak. D and E, effect of the GABAB receptor agonist baclofen on GABAergic and glycinergic IPSCs.
We compared the kinetics of the glycine- and GABAA-receptor-mediated unitary IPSC components. Glycinergic IPSCs were recorded in the presence of 3 μm SR95531, and GABAergic IPSC was recorded in the presence of 0.3 μm strychnine. The 20–80% rise time and decay time constants (weighted τ value) were 1.1 ± 0.1 ms and 23.4 ± 5.1 ms for glycinergic and 1.2 ± 0.2 ms and 57.5 ± 6.0 ms for GABAergic components, respectively. The glycinergic IPSC decayed significantly faster than the GABAergic IPSC (P < 0.01, n= 9) (Fig. 1C). In order to clarify that GABA and glycine would be released from the same presynaptic release sites, we further studied basic features of synaptic transmission including paired-pulse ratio and modulation by presynaptic receptors. As shown in Fig. 1D and E, an activation of presynaptic GABAB receptor inhibited both GABAergic and glycinergic components. The inhibition of the unitary IPSC evoked by 1st pulse was 60.2 ± 1.3% in glycinergic and 61.8 ± 3.1% in GABAergic transmission (n= 5). These values showed no significant difference. In addition, the GABAergic and glycinergic IPSCs showed a similar paired-pulse ratio in the absence and presence of GABAB receptor agonist baclofen. These results suggest that GABA and glycine have similar release mechanisms.
The mechanisms that regulate the vesicular phenotype of inhibitory neurons are not fully understood. Expression of GlyT2 increases intracellular glycine concentration and promotes glycine release (Roux & Supplisson, 2000: Rousseau et al. 2008), suggesting that cytoplasmic glycine concentration is important for glycine release. We previously reported that a developmental shift from GABAergic to glycinergic transmission is accompanied by a decrease in GAD expression (Nabekura et al. 2004). GAD catalyses the conversion of glutamate to GABA. Thus, not only cytoplasmic GABA but also glutamate concentration is thought to be important for GABA release. However, the effect of intracellular inhibitory and excitatory neurotransmitters on the GABA/glycinergic co-release synapse remains to be clarified. In the present study, therefore, we investigated the effects of intracellular GABA, glutamate and glycine on unitary IPSCs. Infusion of 100 mm GABA into presynaptic neurons via the patch pipette dramatically increased the SR95531-induced inhibition of the IPSC. The SR95531 (3 μm) inhibition of unitary IPSCs was 48.2 ± 8.2% and 92.4 ± 2.3% (n= 7, P < 0.01) after 5 min and 60 min perfusion, respectively (Fig. 2A and C). This result suggests that presynaptic intracellular loading of GABA enhanced the SR95531-sensitive GABAergic component of inhibitory transmission. Similar results were obtained using the presynaptic pipette solution containing l-glutamate. When 100 mm glutamate was loaded into presynaptic neurons, SR95531 inhibited the evoked IPSCs by 44.9 ± 16.9% and 78.9 ± 8.1% at 5 min and 60 min, respectively, after starting the recordings (n= 6, P < 0.05). In 2 of 6 neurons in which SR95531 produced no detectable inhibition at the beginning of paired recording, 1 h loading of glutamate produced an SR955531-sensitive component of the IPSC (Fig. 2B). In contrast, infusion of 100 mm glycine into presynaptic neurons markedly reduced the SR95531-induced inhibition from 43.5 ± 8.1% to 5.4 ± 3.5% (n= 6, P < 0.001). In all cases, IPSCs in the presence of SR95531 were fully inhibited by 0.3 μm strychnine. When internal amino acid (100 mm) was replaced with equimolar mannitol as a control, the SR95531-induced inhibition did not change during 1 h recording (33.8 ± 8.1% at 5 min, 37.6 ± 7.0% at 60 min (n= 5); data not shown). The loading of 20 mm inhibitory or excitatory amino acids into presynaptic neurons had no significant effect on the SR95531-induced inhibition of unitary IPSCs (data not shown).
Figure 2. Effects of intracellular inhibitory and excitatory neurotransmitters on unitary inhibitory synaptic transmission.

A, effect of intracellular perfusion of GABA. Representative current traces showing the effect of SR95531 and strychnine at 5 min and 60 min after rupture of the patch membrane of a presynaptic cell, which was intracellularly perfused with pipette solution containing 100 mm GABA. B, effect of intracellular infusion of glutamate. In this case, SR95531 produced no detectable inhibition on the evoked IPSCs at 5 min after starting the recording. C, effect of intracellular perfusion of GABA, glutamate and glycine. Each panel shows the inhibitory action of SR95531 on unitary IPSCs at 5 min and 60 min after starting the recording. Concentration of each transmitter was 100 mm. Values obtained by one paired recording are filled circles connected by line. Open circles show mean ± SEM.
The decay kinetics of the GABA- and glycine-mediated IPSCs were modulated after the loading of the neurotransmitters. Intracelluar infusion of 100 mm GABA reduced the decay time constant, τ, of glycinergic IPSC by 29.3 ± 2.9% but increased that of GABAergic IPSC by 23.3 ± 4.0% (n= 5). Similarly, the loading of 100 mm glutamate reduced the glycinergic decay time constant by 18.5 ± 4.2% but increased GABAergic decay by 15.1 ± 3.3% (n= 4). On the other hand, the loading of 100 mm glycine increased the glycinergic decay time constant by 19.7 ± 2.7% but reduced the GABAergic decay time constant by 14.3 ± 2.9% (n= 6). These results suggest that the vesicular phenotype of the inhibitory neuron depends on the intracellular concentration of inhibitory or excitatory amino acids.
Synaptic GABA release is dependent upon extracellular glutamate, which is taken up directly into inhibitory terminals by sodium-dependent excitatory amino acid transporters (EAATs: Mathews & Diamond, 2003), or indirectly by the astrocytic glutamate–glutamine cycle (Liang et al. 2006). However, it is unclear whether increased glutamate uptake enhances the GABAergic component at GABA/glycinergic co-release synapses. The EAAT type 3 (EAAT3) co-localizes with GAD at inhibitory nerve terminals (Conti et al. 1998; He et al. 2000). We therefore next tested whether extracellular glutamate regulates the GABA/glycinergic co-transmission. The experiments were performed in the neuronal pair whose IPSC was inhibited by SR95531 by 40–60%. We first examined the effect of extracellular glutamate on GABAergic unitary IPSCs in the presence of 0.3 μm strychnine. Glutamate was applied extracellularly for 10 min, and after washout of extracellular glutamate the GABAergic compontent was markedly increased to 192 ± 30% of the control (n= 9, P < 0.01; Fig. 3A). This increase was blocked by the EAAT inhibitor TBOA (300 μm). The efficiency of glutamate uptake is influenced by extracellular Na+ concentrations since glutamate transport is coupled with the co-transport of 3Na+. In the present study, removal of extracellular Na+ during glutamate application fully inhibited the glutamate-induced potentiation of GABAergic IPSCs. Extracellular glutamine (100 μm) produced no significant effect on the GABAergic IPSCs (Fig. 3B).
Figure 3. Effect of extracellular glutamate on the GABAergic component of evoked IPSCs.

A, effect of glutamate on the GABAergic component of unitary IPSCs. Current recordings were performed in the presence of 0.3 μm strychnine. Current traces were obtained before and after application of 100 μm glutamate (10 min). B, relative GABAergic IPSC amplitudes under control conditions, after glutamate application without and with TBOA or Na+-free solution. Glutamine showed no significant effect on the GABAergic IPSCs. *P < 0.05, compared with control. C, effect of high K+ (10 mm) with and without inhibitors, as indicated. *P < 0.05, compared with control. D, effect of TPMPA on the GABAergic IPSC with or without extracellular glutamate loading. *P < 0.05.
The concentration of transmitters in the synaptic cleft has extensively been studied at excitatory glutamatergic synapses using a rapidly dissociating AMPA receptor antagonist (Diamond & Jahr, 1997; Wadiche & Jahr, 2001). This strategy has also been applied to inhibitory synapses to evaluate the concentration of synaptic neurotransmitters (Barberis et al. 2005; Liang et al. 2006). In the present study, to evaluate further whether the increase in amplitude of GABAergic IPSCs after glutamate application was due to increased GABA release from presynaptic nerve terminals, we used 1,2,5,6-tetrahydropyridin-4-yl-methylphosphinic acid (TPMPA; 100 μm). TPMPA is a low-affinity, rapidly equilibrating antagonist at the GABAA receptor (Jones et al. 2001). We reasoned that if GABA uptake into synaptic vesicles is increased, then the neurotransmitter concentration in the synaptic cleft would also increase and the IPSC blockade by TPMPA would be attenuated. After glutamate application for 10 min, the GABAergic IPSC increased, and TPMPA blockade of IPSCs was reduced from 20.7 ± 3.5% to 12.2 ± 2.1% (P < 0.05; n= 6). Thus, the uptake of glutamate into GABA/glycinergic nerve terminals enhances the GABA concentration in the synaptic cleft, probably resulting from an increase in vesicular GABA content.
We also examined the effect of l-glutamate uptake on glycinergic IPSCs recorded in the presence of 3 μm SR95531. The amplitude of these IPSCs decreased after 10 min exposure to l-glutamate (control: 0.94 ± 0.06, n= 5; glutamate: 0.70 ± 0.06, n= 6; P < 0.05; Fig. 4). Because SR95531 can also inhibit glycine receptors, albeit with low affinity (Wang & Slaughter, 2005; Beato et al. 2007), the fraction of current blocked by high concentrations of SR95531 is thought to increase as the amount of glycine release decreases. We therefore examined the effect of high concentrations of SR95531 on glycinergic IPSCs and found that 100 μm SR95531 reduced the glycinergic IPSC amplitude by 36.7 ± 4.3% (n= 5) under control conditions. After glutamate application, the SR95531-induced inhibition was significantly potentiated (64.7 ± 4.0%; n= 5; P < 0.001). As shown in Fig. 5, EAAT3 was expressed in the cultured spinal neurons with and without Venus expression, suggesting that inhibitory neurons also express EAAT3 (52.9 ± 0.02%, n= 1770). These results suggest that uptake of glutamate increases the GABA release, but decreases glycine release in the mixed GABAergic/glycinergic inhibitory synapse.
Figure 4. Effect of extracellular glutamate on the glycinergic component of evoked IPSCs.

A, effect of glutamate on the glycinergic component of unitary IPSCs. Current recordings were performed in the presence of 3 μm SR95531. Current traces were obtained before and after application of 100 μm glutamate (10 min). B, effect of a high concentration of SR95531 (100 μm) on the glycinergic IPSC with or without glutamate.
Figure 5. Immunocytochemistry confirmed the expression of the glutamate transporter in inhibitory neurons.
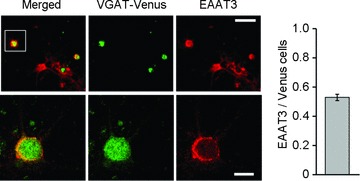
Venus-expressing (green) cultured inhibitory neurons were immunostained for EAAT3 (red). Venus-expressing neurons in the inset of upper left panel were digitally enlarged in lower panels. The histogram (right) shows the population of EAAT3-expressing cells in Venus cells. Upper scale bar, 30 μm; lower scale bar, 10 μm.
The extracellular glutamate concentration increased during excessive neuronal activity (Meldrum, 1994). Thus, enhanced neuronal activity alone may affect the GABA/glycinergic IPSCs. To test this hypothesis, we examined the effect of high K+ on the GABA/glycinergic IPSCs. The extracellular solution containing 10 mm K+ was bath perfused for 10 min. After washout of the high K+ solution, the GABAergic component increased by 68.2 ± 12.0% of the control (n= 7, P < 0.05; Fig. 3C), whereas the glycinergic component declined by 45.2 ± 10.2% (n= 7, P < 0.05; data not shown). Augmentation of GABAergic IPSCs by high K+ was inhibited by the glutamate transporter inhibitor TBOA (300 μm, n= 7), but not by NO-711, a GABA uptake blocker (10 μm, n= 7: Fig. 3C). These results suggested that glutamate uptake during neuronal circuit activity may alter the components of inhibitory synaptic transmission.
Synaptic transmission depends on filling newly formed synaptic vesicles and/or refilling endocytosed vesicles. To investigate this process in the GABA/glycine mixed synapse, we recorded the unitary GABAergic and glycinergic IPSCs occurring during prolonged high-frequency transmissions in the presence of strychnine (0.3 μm) and SR95531 (3 μm), respectively. In this series of experiments, CGP55845 (1 μm), a specific GABAB receptor antagonist, was added to block presynaptic GABAB receptors. Figure 6A and B shows the profiles of GABAergic and glycinergic unitary IPSCs in a neuronal paired recording stimulated at 2 Hz. The average time course of the IPSC amplitude during stimulation is shown in Fig. 6C. The amplitudes of GABAergic and glycinergic IPSCs gradually decreased during repetitive stimulation and reached a steady-state level of 20.9 ± 0.4% (n= 6) and 8.6 ± 1.2% (n= 5, P < 0.05) relative to the initial IPSCs, respectively. Repetitive stimulation shortens the evoked IPSC decay phase. The decay time constants under control conditions and the late phase of stimulation were 52.2 ± 4.2 and 38.6 ± 4.7 ms for GABAergic (n= 6, P < 0.001), and 24.6 ± 5.0 and 19.9 ± 5.4 for glycinergic (n= 5, P < 0.001), respectively. During the late phase of the stimulation, failure of the postsynaptic responses was occasionally observed in glycinergic, but not GABAergic, IPSCs, and the amplitude of glycinergic IPSCs had a greater coefficient of variation. On the other hand, the glycine-induced current elicited by puff application (2 Hz) of glycine (100 μm) with GABA (100 μm) in the presence of SR95531 never showed failure (n= 6, Fig. 6F). These results suggest that usage of GABA as a transmitter at co-release synapses may be advantageous for maintaining high-fidelity inhibitory transmission during neuronal hyperexcitability.
Figure 6. Unitary GABAergic and glycinergic IPSCs during high frequency stimulation.

A and B, traces of glycinergic and GABAergic IPSCs recorded at the onset (1–5 stimulation) and during steady state (901–905 stimulation) at 2 Hz. Numbers (1–5 and 901–905) are stimulus number. C, time course of the depression of GABAergic and glycinergic IPSCs. Responses evoked by 2 Hz stimulation were normalized by the average amplitude recorded with 0.1 Hz stimulation. Each point represents the average amplitude of 5 unitary IPSCs. D and E, number of failures (D) and coefficient of variation (E) during 901–1000 stimulation. F, glycine-induced currents evoked by pressure puff application (2 Hz) of glycine. Numbers shown above each trace (1–6 and 995–1000) are stimulus number.
Discussion
We have analysed the regulation of the GABAergic/glycinergic vesicular phenotypes in cultured rat spinal inhibitory neurons. Most unitary IPSCs from paired whole-cell recordings consisted of SR95531-sensitive GABAergic and strychnine-sensitive glycinergic components. These mixed transmissions are expected from immature spinal cord inhibitory neurons because of co-expression of GlyT2 and GAD during the spinal cord development (Mackie et al. 2003). Although the proportion of the GABAergic or glycinergic components varied in each cell, their fractional contributions remained constant during the 1 h recording of unitary IPSCs evoked at 0.1 Hz under control conditions without intracellular perfusion of inhibitory or excitatory amino acids. These results suggest that local presynaptic mechanisms maintain the glycinergic and GABAergic transmissions. Because synaptic vesicles are recycled locally, these two inhibitory transmitters should also be supplied locally at axon terminals.
Infusion of high concentrations of GABA or glycine into the somata of inhibitory neurons via the patch pipette modified the contribution of each transmitter to the unitary IPSCs, indicating the crucial roles of presynaptic cytosolic inhibitory amino acids in the transmission phenotype. This may be because GABA and glycine are secluded into synaptic vesicles by VIAAT (McIntire et al. 1997; Sagnéet al. 1997; Wojcik et al. 2006). Because VIAAT has a lower affinity for glycine than for GABA (McIntire et al. 1997; Sagnéet al. 1997), the glycine concentration should highly exceed the GABA concentration when the glycinergic transmission is predominant. Cytosolic GABA concentration ranges vary from 1 mm to 6 mm in mammalian neurons (Otsuka et al. 1971), whereas cytoplasmic glycine is assumed to be 10 mm in glycinergic neurons (Ottersen et al. 1990; Roux & Supplisson, 2000). In the present study, on the other hand, intracellular loading of 20 mm inhibitory transmitters had no significant effect on the transmission. This may be due to the reduced efficacy of neurotransmitter diffusion in axon (Koike & Nagata, 1979). Further study is needed to evaluate the transmitter concentration at the nerve terminals.
Intracellular GABA is synthesized from glutamate by the GAD isoforms GAD65 and GAD67. GAD67 is a cytosolic enzyme and is distributed throughout the cell. GAD65 is localized to the nerve terminal where it is reversibly bound to the membrane of synaptic vesicles (Kaufman et al. 1991; Christgau et al. 1992). In addition, GAD65 makes a functional coupling to VIAAT, increasing the efficiency of GABA synthesis from glutamate and GABA loading into the presynaptic vesicles (Jin et al. 2003). In the present study, we found that intracellular loading of glutamate increased the GABAergic component of unitary IPSCs. In two glycinergic neurons in which SR95531 produced no detectable inhibition under control conditions (Fig. 2B), infusion of glutamate into the soma triggered GABA release from the nerve terminal. This suggests that these glycinergic neurons have GAD activity but do not have sufficient glutamate uptake/synthesis to release GABA. In addition, we found that a subset of inhibitory neurons expressed EAAT3 (Fig. 5). Therefore, GABA synthesis by GAD after glutamate uptake plays an important role in the regulation of inhibitory GABA/glycinergic co-transmission.
Prolonged alterations in excitatory synaptic activity cause adaptations to inhibitory synapses to maintain a level of network activity (Turrigiano, 2007). For instance, chronic epilepsy increases GAD expression in the rat hippocampus (Esclapez & Houser, 1999), and chronic suppression of network activity in cultured rat cortex down-regulates the GABA content (Ramakers et al. 1994). Thus, the GABAergic network system is thought to contribute to homeostatic plasticity in neuronal networks. Long-term potentiation at GABAergic synapses on spinal lamina I neurons is also triggered by enhanced activity in primary afferent nerve fibres (Fenselau et al. 2011). Thus, the facilitation of neuronal circuits, including excitatory ones, could enhance the GABAergic synaptic transmission. In the present study, we found that glutamate uptake by nerve terminals that co-release GABA and glycine enhanced the amplitude of GABAergic IPSCs (Fig. 3), which was accompanied by reduced TPMPA inhibition, suggesting that glutamate uptake increased the GABA concentration in the synaptic cleft. Because neurotransmitter concentration in the cleft is dependent primarily on its concentration in synaptic vesicles, glutamate uptake is likely to increase the GABA concentration in the synaptic vesicles. In contrast, after extracellular glutamate loading, glycinergic IPSCs decreased with the increased inhibition by high concentrations of SR95531, suggesting a decline in glycine concentration in the synaptic vesicles. These observations indicate that glutamate-derived GABA is not additively taken up into the synaptic vesicles but replaces glycine. These results can be explained by saturation in the amount of inhibitory amino acids that can be stored in synaptic vesicles. The neurotransmitter content of a synaptic vesicle may depend on transmitter uptake as well as leakage of vesicular transmitter, as observed in isolated synaptic vesicle preparations (Floor et al. 1995). The modelling study estimated the intravesicular concentration of inhibitory transmitter to be approximately 100 mm (Axmacher et al. 2004).
During high frequency stimulation (2 Hz), glycinergic IPSCs at mixed synapses were more effectively inhibited than GABAergic IPSCs, and occasionally failed to occur. In addition, the amplitude of glycinergic IPSCs had a greater coefficient of variation. These differences between GABAergic and glycinergic IPSCs may be due to reduced function of presynaptic glycine release, since the current elicited by puff application of glycine did not show such insecurity. Thus the refilling of glycine into synaptic vesicles during high frequency transmission is not always enough to elicit detectable glycine receptor activation. Further studies are needed to elucidate the detail of mechanisms underlying the difference between glycine and GABA release during high-frequency transmission.
The balance between excitation and inhibition is crucial for the functioning of neuronal circuits and disruption of this equilibrium may lead to diseases such as epilepsy. The plasticity of inhibitory synaptic transmission plays an important role in balancing excitation and inhibition within a neuronal circuit. Coull et al. (2003) reported a shift from glycine-receptor- towards GABAA-receptor-dominated synaptic transmission in spinal lamina I after peripheral nerve injury. In the present study, elevated extracellular glutamate levels enhanced the GABA component at the mixed GABA/glycine synapses via neuronal glutamate uptake. In addition, high frequency stimulation leads to a greater decrease in glycinergic IPSC amplitude and occasional synaptic failures. GABAergic IPSCs have a slower decay time constant than glycinergic IPSCs, leading to prolonged inhibition in postsynaptic cells. Therefore, an increased GABAergic component would enhance target cell inhibition and become one of the rapid homeostatic mechanisms in mixed inhibitory synapses to control the excitation–inhibition balance. Thus, the use of GABA as a transmitter at GABA/glycine mixed synapses may afford protection against pathophysiological hyperexcitability associated with increased extracellular glutamate concentration. In addition, since GABA is also a trophic factor (Represa & Ben-Ari, 2005), increased GABA release may support the reorganization of neuronal circuits following changes in network activity.
Acknowledgments
VIAAT (VGAT)-Venus transgenic rats were kindly provided by Y. Kawaguchi (NIPS, Okazaki, Japan). Confocal images were acquired at the Spectrography and Bioimaging Facility, NIBB Core Research Facilities (Okazaki, Japan).
Glossary
- EAAT
excitatory amino acid transporter
- GABA
γ-aminobutyric acid
- GAD
glutamic acid decarboxylase
- GlyT2
glycine transporter type 2
- IPSC
inhibitory postsynaptic current
- VGAT
vesicular GABA transporter
- VIAAT
vesicular inhibitory amino acid transporter
Additional information
Competing interests
None.
Author contributions
H.I. and J.N. designed the study. H.I., J.Y. and Y.N. performed the experiments. H.I. and J.Y. were the primary writers of the manuscript while all authors contributed to critical interpretation and manuscript preparation and approved the final version. The experiments were carried out in National Institute for Physiological Sciences, Japan.
Funding
This study was supported by JSPS KAKENHI (grant numbers 25253017, 24590739) and JST, CREST.
References
- Axmacher N, Stemmler M, Engel D, Draguhn A, Ritz R. Transmitter metabolism as a mechanism of synaptic plasticity: a modeling study. J Neurophysiol. 2004;91:25–39. doi: 10.1152/jn.00797.2003. [DOI] [PubMed] [Google Scholar]
- Barberis A, Lu C, Vicini S, Mozrzymas JW. Developmental changes of GABA synaptic transient in cerebellar granule cells. Mol Pharmacol. 2005;67:1221–1228. doi: 10.1124/mol.104.006437. [DOI] [PubMed] [Google Scholar]
- Beato M, Burzomato V, Sivilotti LG. The kinetics of inhibition of rat recombinant heteromeric α1β glycine receptors by the low-affinity antagonist SR-95531. J Physiol. 2007;580:171–179. doi: 10.1113/jphysiol.2006.126888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christgau S, Aanstoot HJ, Schierbeck H, Begley K, Tullin S, Hejnaes K, Baekkeskov S. Membrane anchoring of the autoantigen GAD65 to microvesicles in pancreatic β-cells by palmitoylation in the NH2-terminal domain. J Cell Biol. 1992;118:309–320. doi: 10.1083/jcb.118.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Chiu DN, Jahr CE. Ca2+-dependent enhancement of release by subthreshold somatic depolarization. Nat Neurosci. 2011;14:62–68. doi: 10.1038/nn.2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti F, DeBiasi S, Minelli A, Rothstein JD, Melone M. EAAC1, a high-affinity glutamate tranporter, is localized to astrocytes and gabaergic neurons besides pyramidal cells in the rat cerebral cortex. Cereb Cortex. 1998;8:108–116. doi: 10.1093/cercor/8.2.108. [DOI] [PubMed] [Google Scholar]
- Coull JAM, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour A, Tell F, Kessler J-P, Baude A. Mixed GABA-glycine synapses delineate a specific topography in the nucleus tractus solitarii of adult rat. J Physiol. 2010;588:1097–1115. doi: 10.1113/jphysiol.2009.184838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumoulin A, Triller A, Dieudonne S. IPSC kinetics at identified GABAergic and mixed GABAergic and glycinergic synapses onto cerebellar Golgi cells. J Neurosci. 2001;21:6045–6057. doi: 10.1523/JNEUROSCI.21-16-06045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esclapez M, Houser CR. Up-regulation of GAD65 and GAD67 in remaining hippocampal GABA neurons in a model of temporal lobe epilepsy. J Comp Neurol. 1999;412:488–505. [PubMed] [Google Scholar]
- Fenselau H, Heinke B, Sandkühler J. Heterosynaptic long-term potentiation at GABAergic synapses of spinal lamina I neurons. J Neurosci. 2011;31:17383–17391. doi: 10.1523/JNEUROSCI.3076-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floor E, Leventhal PS, Wang Y, Meng L, Chen W. Dynamic storage of dopamine in rat brain synaptic vesicle preparation in vitro. J Neurochem. 1995;64:689–699. doi: 10.1046/j.1471-4159.1995.64020689.x. [DOI] [PubMed] [Google Scholar]
- Gao BX, Stricker C, Ziskind-Conhaim L. Transition from GABAergic to glycinergic synaptic transmission in newly formed spinal networks. J Neurophysiol. 2001;86:492–502. doi: 10.1152/jn.2001.86.1.492. [DOI] [PubMed] [Google Scholar]
- He Y, Janssen WG, Rothstein JD, Morrison JH. Differential synaptic localization of glutamate transporter EAAC1 and glutamate receptor subunit GluR2 in the rat hippocampus. J Comp Neurol. 2000;418:255–269. [PubMed] [Google Scholar]
- Ishibashi H, Hirao K, Yamaguchi J, Nabekura J. Inhibition of chloride outward transport by gadolinium in cultured rat spinal cord neurons. Neurotoxicology. 2009;30:155–159. doi: 10.1016/j.neuro.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Jin H, Wu H, Osterhaus G, Wei J, Davis K, Sha D, Floor E, Hsu CC, Kopke RD, Wu JY. Demonstration of functional coupling between γ-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc Natl Acad Sci U S A. 2003;100:4293–4298. doi: 10.1073/pnas.0730698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Sandkühler J. Corelease of two fast neurotransmitters at a central synapse. Science. 1998;281:419–424. doi: 10.1126/science.281.5375.419. [DOI] [PubMed] [Google Scholar]
- Jones MV, Jonas P, Sahara Y, Westbrook GL. Microscopic kinetics and energetics distinguish GABAA receptor agonists from antagonists. Biophys J. 2001;81:2660–2670. doi: 10.1016/S0006-3495(01)75909-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman DL, Houser CR, Tobin AJ. Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J Neurochem. 1991;56:720–723. doi: 10.1111/j.1471-4159.1991.tb08211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller AF, Coull JAM, Chéry N, Poisbeau P, Koninck YD. Region-specific developmental specialization of GABA-glycine cosynapses in laminas I-II of the rat spinal dorsal horn. J Neurosci. 2001;21:7871–7880. doi: 10.1523/JNEUROSCI.21-20-07871.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura A, Ishibashi H, Watanabe M, Takatsuru Y, Brodwick M, Nabekura J. Sustained depolarizing shift of the GABA reversal potential by glutamate receptor activation in hippocampal neurons. Neurosci Res. 2008;62:270–277. doi: 10.1016/j.neures.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Koike H, Nagata Y. Intra-axonal diffusion of [3H]acetylcholine and [3H]γ-aminobutyric acid in a neurone of Aplysia. J Physiol. 1979;295:397–417. doi: 10.1113/jphysiol.1979.sp012976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Korada S, Schwartz IR, Sanes DH. A developmental shift from GABAergic to glycinergic transmission in the central auditory system. J Neurosci. 1998;18:4646–4655. doi: 10.1523/JNEUROSCI.18-12-04646.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S-L, Carlson GC, Coulter DA. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J Neurosci. 2006;26:8537–8548. doi: 10.1523/JNEUROSCI.0329-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM. Identification and characterization of the vesicular GABA transporter. Nature. 1997;389:870–876. doi: 10.1038/39908. [DOI] [PubMed] [Google Scholar]
- Mackie M, Hughes DI, Maxwell DJ, NJK Tillakaratne, Todd AJ. Distribution and colocalisation of glutamate decarboxylase isoforms in the rat spinal cord. Neuroscience. 2003;119:461–472. doi: 10.1016/s0306-4522(03)00174-x. [DOI] [PubMed] [Google Scholar]
- Mathews GC, Diamond JS. Neuronal glutamate uptake contributes to GABA synthesis and inhibitory synaptic strength. J Neurosci. 2003;23:2040–2048. doi: 10.1523/JNEUROSCI.23-06-02040.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier J, Jüttner R, Kirischuk S, Grantyn R. Synaptic anchoring of glycine receptors in developing collicular neurons under control of metabotropic glutamate receptor activity. Mol Cell Neurosci. 2002;21:324–340. doi: 10.1006/mcne.2002.1161. [DOI] [PubMed] [Google Scholar]
- Meldrum BS. The role of glutamate in epilepsy and other CNS disorders. Neurology. 1994;44:S14–23. [PubMed] [Google Scholar]
- Murase K, Ryu PD, Randić M. Excitatory and inhibitory amino acids and peptide-induced responses in acutely isolated rat spinal dorsal horn neurons. Neurosci Lett. 1989;103:56–63. doi: 10.1016/0304-3940(89)90485-0. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Katsurabayashi S, Kakazu Y, Shibata S, Matsubara A, Jinno S, Mizoguchi Y, Sasaki A, Ishibashi H. Developmental switch from GABA to glycine release in single central synaptic terminals. Nat Neurosci. 2004;7:17–23. doi: 10.1038/nn1170. [DOI] [PubMed] [Google Scholar]
- Otsuka M, Obata K, Miyata Y, Tanaka Y. Measurement of γ-aminobutyric acid in isolated nerve cells of cat central nervous system. J Neurochem. 1971;18:287–295. doi: 10.1111/j.1471-4159.1971.tb00567.x. [DOI] [PubMed] [Google Scholar]
- Ottersen OP, Storm-Mathisen J, Laake JH. Cellular and subcellular localization of glycine studied by quantitative electron microscopic immunocytochemistry. In: Ottersen OP, Storm-Mathisen J, editors. Glycine Neurotransmission. Chichester, UK: Wiley; 1990. pp. 303–328. [Google Scholar]
- Ramakers GJ, van Galen H, Feenstra MG, Corner MA, Boer GJ. Activity-dependent plasticity of inhibitory and excitatory amino acid transmitter systems in cultured rat cerebral cortex. Int J Dev Neurosci. 1994;12:611–621. doi: 10.1016/0736-5748(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Represa A, Ben-Ari Y. Trophic action of GABA on neuronal development. Trends Neurosci. 2005;28:278–283. doi: 10.1016/j.tins.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Rousseau F, Aubrey KR, Supplisson S. The glycine transporter GlyT2 controls the dynamics of synaptic vesicle refilling in inhibitory spinal cord neurons. J Neurosci. 2008;28:9755–9768. doi: 10.1523/JNEUROSCI.0509-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux MJ, Supplisson S. Neuronal and glial glycine transporters have different stoichiometries. Neuron. 2000;25:373–383. doi: 10.1016/s0896-6273(00)80901-0. [DOI] [PubMed] [Google Scholar]
- Sagné C, El Mestikawy S, Isambert MF, Hamon M, Henry JP, Giros B, Gasnier B. Cloning of a functional vesicular GABA and glycine transporter by screening of genome databases. FEBS Lett. 1997;417:177–183. doi: 10.1016/s0014-5793(97)01279-9. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, Duque A, Yu Y, McCormick DA. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature. 2006;441:761–765. doi: 10.1038/nature04720. [DOI] [PubMed] [Google Scholar]
- Tanaka I, Ezure K. Overall distribution of GLYT2 mRNA-containing versus GAD67 mRNA-containing neurons and colocalization of both mRNAs in midbrain, pons, and cerebellum in rats. Neurosci Res. 2004;49:1874–1882. doi: 10.1016/j.neures.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic signalling: the positive side of negative feedback. Curr Opin Neurobiol. 2007;17:318–324. doi: 10.1016/j.conb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Uematsu M, Hirai Y, Karube F, Ebihara S, Kato M, Abe K, Obata K, Yoshida S, Hirabayashi M, Yanagawa Y, Kawaguchi Y. Quantitative chemical composition of cortical GABAergic neurons revealed in transgenic Venus-expressing rats. Cereb Cortex. 2008;18:315–330. doi: 10.1093/cercor/bhm056. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- Wang P, Slaughter MM. Effects of GABA receptor antagonists on retinal glycine receptors and on homomeric glycine receptor alpha subunits. J Neurophysiol. 2005;93:3120–3126. doi: 10.1152/jn.01228.2004. [DOI] [PubMed] [Google Scholar]
- Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N, Rhee JS. A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron. 2006;50:575–587. doi: 10.1016/j.neuron.2006.04.016. [DOI] [PubMed] [Google Scholar]