

Abstract
Bipolar cells (BCs), the second order neurons in the vertebrate retina, receive two types of GABAergic feedback inhibition at their axon terminal: reciprocal and lateral inhibition. It has been suggested that two types of inhibition may be mediated by different pathways. However, how each inhibition is controlled by excitatory BC output remains to be clarified. Here, we applied single/dual whole cell recording techniques to the axon terminal of electrically coupled BCs in slice preparation of the goldfish retina, and found that each inhibition was regulated independently. Activation voltage of each inhibition was different: strong output from a single BC activated reciprocal inhibition, but could not activate lateral inhibition. Outputs from multiple BCs were essential for activation of lateral inhibition. Pharmacological examinations revealed that composition of transmitter receptors and localization of Na+ channels were different between two inhibitory pathways, suggesting that different amacrine cells may mediate each inhibition. Depending on visual inputs, each inhibition could be driven independently. Model simulation showed that reciprocal and lateral inhibition cooperatively reduced BC outputs as well as background noise, thereby preserving high signal-to-noise ratio. Therefore, we conclude that excitatory BC output is efficiently regulated by the dual operating mechanisms of feedback inhibition without deteriorating the quality of visual signals.
Key points
Two different forms of feedback inhibition, reciprocal and lateral inhibition, are ubiquitously observed throughout the nervous system.
In the retina, the axon terminal of bipolar cells receives reciprocal and lateral GABAergic inhibitory inputs from amacrine cells, but how a variety of visual inputs activate each inhibition remains largely unexplored.
Here we show that each inhibition is independently controlled by different types of bipolar cell outputs; reciprocal inhibition is driven by strong output from each bipolar cell, whereas lateral inhibition is driven by outputs from multiple bipolar cells even when each output is weak.
Composition of transmitter receptors and localization of Na+ channels were different between two inhibitory pathways, suggesting that different amacrine cells may mediate each inhibition.
The dual feedback inhibition can cooperatively reduce bipolar cell outputs in response to various visual inputs without deteriorating the quality of visual signals, thereby contributing to efficient signal transmission in the visual pathway.
Introduction
Diverse connectivity of inhibitory neurons renders neural circuits capable of various computations. In the vertebrate retina, two inhibitory neurons form characteristic microcircuits to modulate the visual signal, which is processed successively by photoreceptors, bipolar cells (BCs) and ganglion cells (GCs). One is the horizontal cell (HC), which regulates signal transmission between photoreceptors and BCs in the outer retina. The other is the amacrine cell (AC), which controls the activity of BCs, GCs and other ACs in the inner retina. HCs contribute to the formation of the centre-surround antagonism of BCs and GCs (Hirasawa et al. 2012; Thoreson & Mangel, 2012). On the other hand, ACs are involved in a variety of computations, such as edge extraction (Roska & Werblin, 2001), detection of relative motion (Ölveczky et al. 2003) and direction selectivity (Lee et al. 2010). The source of these elaborate computations, however, is often elusive, partly because of the complexity of microcircuits in the inner retina. Actually, the operating condition of feedback inhibition from ACs to BCs, which should affect all the subsequent computations in the inner retina, remains largely unexplored.
Feedback inhibition from ACs to BCs can take various forms (Eggers & Lukasiewicz, 2011), but it is usually dichotomized into reciprocal and lateral inhibition (Grimes, 2012). Ultrastructural studies have found their probable morphological correlates: reciprocal and non-reciprocal synapses (Marc & Liu, 2000). Pharmacological studies have characterized their different synaptic compositions (Vigh & von Gersdorff, 2005; Chávez et al. 2006, 2010; Vigh et al. 2011). Underpinned by these pathways, feedback inhibition plays important roles in signal transmission from BCs to postsynaptic neurons: enhancing temporal contrast (Dong & Werblin, 1998), limiting spill-over (Matsui et al. 2001; Sagdullaev et al. 2006) and promoting correlated release (Freed et al. 2003). Nevertheless, the contribution of reciprocal and lateral inhibition to such roles has been often ambiguous, and their dynamic behaviour in response to a variety of visual inputs remains to be addressed.
Here, we investigate the mechanisms controlling each inhibition by using various methods to inhibit/activate parts of the entangled retinal circuits: physical severance of BC axons, local puff application of drugs, Ca2+ uncaging in a single BC terminal and confined light stimulation. Results show that reciprocal inhibition is driven by strong BC output, whereas lateral inhibition is driven by multiple BC outputs even when each output is weak. Model simulation based on these findings suggests that the dual feedback inhibitory circuits can reduce BC outputs with minimal deterioration of signal-to-noise ratio (SNR).
Methods
Ethical approval
Experiments were performed in accordance with “A Manual for the Conduct of Animal Experiments in The University of Tokyo” and “Guiding Principles for the Care and Use of Animals in the Field of Physiological Sciences, The Physiological Society of Japan”.
Retinal slice preparation
Retinal slices were prepared from goldfish of either sex (Carassius auratus; 6–10 cm) housed in a 12 h light/dark cycle (light: 08.00–20.00 h) at 23°C as described previously (Arai et al. 2010). Briefly, in the daytime, the retina of a dark-adapted goldfish was isolated under dim red light, cut into 150–200 μm slices (ST-20-S; Narishige Scientific Instrument Lab., Tokyo, Japan), and used for experiments at room temperature (∼23°C). The retina was sometimes sliced on a tilted (∼10°) board to efficiently obtain axotomized terminals.
Electrophysiology
Whole cell recordings were performed from axotomized/ intact terminals of Mb1 BC (ON-type, which evokes a depolarizing response to illumination of the receptive-field center; Tachibana, 1999) under visualization with IR-DIC optics (Eclipse E600FN; Nikon Corp., Tokyo, Japan), an IR-CCD camera (C2400-79H; Hamamatsu Photonics, Hamamatsu, Shizuoka, Japan), and a TV monitor (TM-920; JVC KENWOOD, Yokohama, Kanagawa, Japan). The recorded terminals were identified as axotomized or intact by their characteristic membrane properties and morphology.
In voltage clamp experiments without light stimulation, the bath solution contained (in mm) 117 NaCl, 2.6 KCl, 20 Hepes, 10 d-glucose, 2.5 CaCl2 and 1 MgCl2 (pH 7.4 with NaOH). Otherwise, the bath solution contained (in mm) 104 NaCl, 2.6 KCl, 28 NaHCO3, 10 d-glucose, 2.5 CaCl2, 1 MgCl2 and 2 mg l−1 phenol red, which was equilibrated with 95% O2/5% CO2. In the Ca2+-free solution, 2.5 mm CaCl2 in the bath solution containing Hepes was replaced with 2.5 mm MgCl2. In voltage clamp experiments, the pipette solution contained (in mm) 118 CsMeSO3, 10 TEA-Cl, 10 Hepes, 0.5 EGTA, 0.05 CaCl2, 2 MgCl2, 5 ATP-Na2, 0.5 GTP-Na3 and 0.08% Lucifer yellow-2K (pH 7.4 with CsOH). In current clamp experiments, the pipette solution contained (in mm) 128 potassium gluconate, 10 KCl, 10 Hepes, 0.5 EGTA, 0.05 CaCl2, 2 MgCl2, 5 ATP-Na2, 0.5 GTP-Na3 and 0.08% Lucifer yellow-2K (pH 7.4 with KOH). ECl was ∼−55 mV if not specified. In experiments in which ECl was equated to ∼−70 mV, TEA-Cl or KCl was lowered to 3 mm and the osmolarity was adjusted with CsMeSO3 or potassium gluconate, respectively. In some experiments with light stimulation, 5 mm phosphocreatine-2Na was added to the pipette solution in place of 7 mm potassium gluconate. The membrane potential was corrected for a junction potential, which was measured for each combination of pipette and bath solutions. The bath solution was supplied at a rate of ∼1.7 ml min−1.
For pharmacological experiments, drugs were dissolved in the bath solution and supplied at a rate of 2.5 ml min−1 for > 3 min before examination of their effects. Mefloquine was bath applied for >2 h. l-2-amino-4-phosphonobutyric acid (l-AP4) was dissolved in the bath solution on the day for use. The bath solution contained < 0.05% (v/v) dimethyl sulphoxide as a solvent for mefloquine, picrotoxin and 2,3-dioxo-6-nitro-1, 2, 3, 4-tetrahydrobenzo[f]quinoxaline-7-sulphonamide (NBQX). Drugs were purchased as follows: picrotoxin, strychnine, philanthotoxin-433 (PhTX) and mefloquine from Sigma-Aldrich Corp. (St. Louis, MO, USA); bicuculline (BIC), NBQX, tetrodotoxin (TTX), l-AP4 and CdCl2 from Wako Pure Chemical Industries, Ltd. (Osaka, Japan), (1,2,5,6-tetrahydropyridin-4-yl) methylphosphinic acid (TPMPA) and d-(–)-2-amino-5-phosphonopentanoic acid (d-AP5) from Tocris Bioscience (Bristol, UK); and DM-nitrophen from Calbiochem (La Jolla, CA, USA).
Recordings were performed with EPC 9/2 (HEKA Elektronik, Lambrecht/Pfalz, Germany) controlled by patchmaster (v2.52, HEKA Elektronik). Recorded currents were low-pass filtered at 2.9 kHz and sampled at 10 kHz. Membrane capacitance was measured by sine + DC method (sinusoidal voltage of 30 mV in peak-to-peak amplitude at 1 kHz) to axotomized terminals voltage clamped at −70 mV. A borosilicate glass (CNC 1.5; Ken Enterprise, Atsugi, Kanagawa, Japan) was pulled with a horizontal puller (P97; Sutter Instrument co., Navoto, CA, USA) and used for a recording pipette or a puff pipette (the resistance was 7–12 MΩ and 8–9 MΩ, respectively). Recording pipettes for the ΔCm measurement were coated by wax (Apiezon Wax W; M & I Materials, Manchester, UK).
Recordings were discontinued when the series resistance was high (>60 MΩ) or the leak current was large (>50 pA for axotomized terminals and >150 pA for intact terminals at −70 mV). The reciprocal IPSC was recorded with a ∼60 s interval to avoid possible synaptic depression (Li et al. 2007). Before application of the depolarizing pulse to a terminal, 10 hyperpolarizing pulses from −70 to −90 mV were applied for leak subtraction and calculation of input resistance. The data were discarded when the peak ICa evoked by depolarization of axotomized terminal to −10 mV was small (<100 pA) or when large fluctuations of current or voltage were spontaneously evoked immediately before stimulation. The data with oscillatory spontaneous IPSCs were also excluded. In current clamp experiments, constant current was injected to hold the membrane potential (Vm) to a desired value. In experiments with light stimulation, we discarded data when the maximal peak of light responses was <1 mV or when run-down of light responses was observed (>50%) during the recording. Axotomized terminals usually underwent run-down of glutamate release in a few minutes. Thus, we used only up to the first six evoked responses for analysis, and pharmacological effects on the reciprocal IPSC were assessed by between-cell comparisons. Similarly, for assessment of the voltage dependence of ΔCm (Fig. 1B), an axotomized terminal was depolarized at most twice to minimize the run-down of exocytosis. For comparison of the effects of puff-applied drug on the reciprocal and lateral IPSCs (Fig. 4D–I), both IPSCs were recorded under the same condition: the holding potential (−10 mV), the time window for calculating QIPSCs (200 ms), and the method of assessing the pharmacological effects (within cell). To evaluate the pharmacological effects on the reciprocal IPSC within-cell responses, the depolarizing pulse was alternately applied every ∼1 min with and without drugs three times for each, and the order of drug application was counter-balanced.
Figure 1. Properties of reciprocal inhibition.
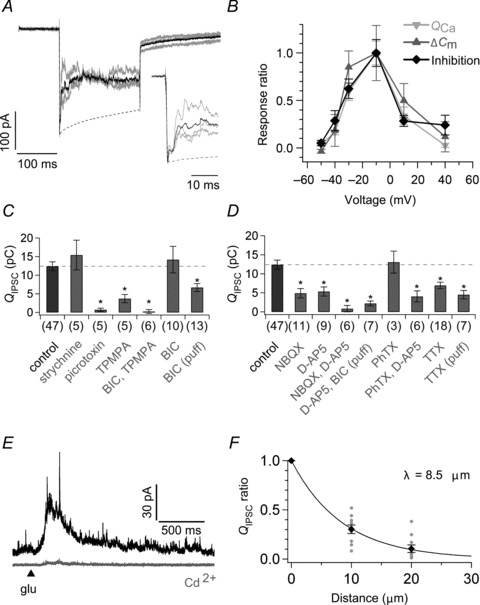
A, quantification of reciprocal inhibition. An axotomized terminal was depolarized from −70 to −10 mV for 200 ms. The charge of reciprocal IPSC (reciprocal QIPSC) was calculated by integrating the difference between the averaged current trace (black) of three responses (grey) and the peak normalized ICa template (broken line; see also Fig. S1A). The ICa template was obtained as the average of responses recorded from 11 axotomized terminals which were depolarized from −70 to −10 mV for 200 ms in the presence of bath-applied 200 μm picrotoxin and 10 μm strychnine. Twenty mm Hepes was included in the bath solution to suppress the proton feedback on ICa (Palmer et al. 2003). The equilibrium potential for Cl− (ECl) was −55 mV. The inset shows the expanded traces. B, voltage dependence of QCa (light grey; n= 4–11), ΔCm (dark grey; n= 4–7), and reciprocal inhibition (black; n= 4–11). Axotomized terminals were depolarized from −70 mV to various membrane potentials (Vm) for 200 ms (Fig. S1B). QCa was the charge of the current response during the 200 ms pulse in the presence of puff-applied BIC and TPMPA. Capacitance jump ΔCm was determined as the difference between the averaged capacitance before the stimulus onset (from −220 to −20 ms) and that after the stimulus offset (from 500 to 700 ms). The amount of reciprocal inhibition was quantified as QIPSC/(Vm−ECl), where QIPSC is the charge of the reciprocal IPSC obtained as the difference between the current responses before and during puff application of 100 μm BIC and 800 μm TPMPA. Data are shown as the ratio to the value at −10 mV. ECl was −70 mV. C and D, effects of various pharmacological blockers on the reciprocal QIPSC obtained as in A. The asterisk indicates significant difference between the control and each blocker (+) condition by Welch's t test with Bonferroni correction. E, glutamate (5 mm) was puff applied for 10 ms (arrowhead) to an axotomized terminal voltage clamped at −10 mV in the absence (black) or presence of bath-applied 200 μm Cd2+ (grey). F, the glutamate-evoked QIPSC/800 ms (see Methods) became smaller as the puff pipette was laterally shifted from the recorded terminal (n= 10). Data were fitted to a single exponential function and the length constant (λ) was 8.5 μm. BIC, bicuculline; d-AP5, d-(–)-2-amino-5-phosphonopentanoic acid; NBQX, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulphonamide; PhTX, philanthotoxin-433; TPMPA, (1,2,5,6-tetrahydropyridin-4-yl) methylphosphinic acid; TTX, tetrodotoxin.
Figure 4. Effects of pharmacological blockers on the reciprocal and lateral IPSCs.
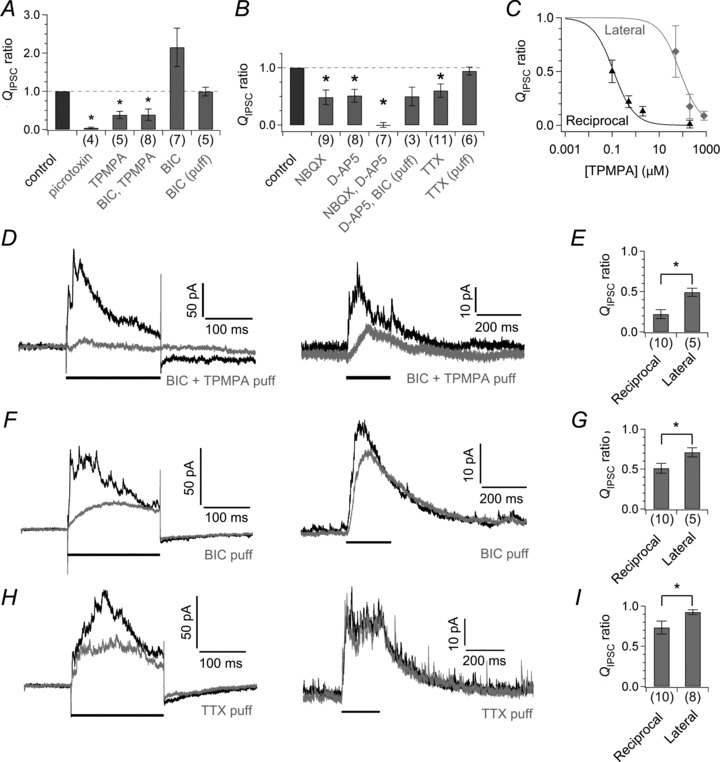
A and B, effects of various blockers on the lateral QIPSC/400 ms measured from axotomized terminals voltage clamped at −90 mV. Values are normalized to control (before the application of blockers). The asterisk indicates significant difference between before and during the application of blockers. C, dose (TPMPA)-dependent suppression of the reciprocal QIPSC (black triangle, n= 6–9) and the lateral QIPSC/200 ms measured from axotomized terminals voltage clamped at −90 mV (grey diamond, n= 3–5 pairs) in the presence of 100 μm BIC. Data were fitted to Hill functions. ID50 for the reciprocal QIPSC and lateral QIPSC is 0.12 μm and 76.02 μm, respectively. D and E, effects of a puff-applied mixture of BIC (100 μm) and TPMPA (20 μm) on the reciprocal IPSC (above) and the lateral IPSC (below) obtained under the same condition (D, see Methods). The reciprocal IPSC is shown as the difference between the averaged current response and the peak normalized ICa template for clarity. The horizontal bar indicates the period of the depolarizing pulse that elicited the reciprocal or lateral IPSC. Abscissa shows the ratio of QIPSC/200 ms under the puff application to that under control condition (E). The asterisk indicates significant interaction between the type of IPSC and the effect of puff-applied drug. F–I, effects of puff-applied 100 μm BIC (F and G) and puff-applied 1 μm TTX (H and I) on the reciprocal and lateral IPSCs as in D and E. BIC, bicuculline; d-AP5, d-(–)-2-amino-5-phosphonopentanoic acid; NBQX, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulphonamide; TPMPA, (1,2,5,6-tetrahydropyridin-4-yl) methylphosphinic acid; TTX, tetrodotoxin.
Puff application of drugs
For puff application of drugs, the pipette tip was located within a few μm from the recorded terminal. To examine the pharmacological effects on lateral inhibition, drugs were puff-applied to an axotomized terminal. The pressure of the puff application was set to ∼5–20 kPa and the stability of the pressure was carefully checked with the DIC image on the TV monitor for each application. The duration of puff application was determined by preliminary experiments in which the localized effect of drugs and its recovery was confirmed. Glutamate was dissolved in the bath solution containing Hepes and puff applied for 10 ms. Puff application of BIC (400 ms in duration) started 600 ms before the stimulus onset. Puff application of TTX or a mixture of BIC and TPMPA (600 ms in duration) started 650 ms before the stimulus onset. Puff application of a mixture of NBQX and d-AP5 (200 ms in duration) started 250 ms before the stimulus onset. When the pharmacological effect of glutamate-evoked reciprocal IPSC was examined, the puff-applied solution included both glutamate and drugs. These careful procedures of puff application confirmed the localized effects of puff-applied drugs: lateral shift (∼20 μm) of a puff pipette from the target terminal caused a steep reduction in the drug effect (see Fig. 1F).
Light stimulation
Retinal slice preparation was illuminated by a white light bar projected from a computer-controlled LCD monitor (LCT-V6MF0; Toei Electronics co., Ltd., Tokyo, Japan) through a condenser lens of the upright microscope (Eclipse E600FN). The light bar was centred on the recorded terminal and its orientation was perpendicular to the retinal layers. At the focal plane, the resolution was 9.8 μm per pixel. The light intensity of the arbitrary unit (a.u.) of 100 (corresponding to 1.95 × 104 photon μm−2 s−1) was used if not specified. In experiments with light stimulation, the bicarbonate buffer solution was used because the Hepes buffer solution tended to reduce spontaneous/evoked IPSCs.
Ca2+ uncaging
In Ca2+ uncaging experiments, the recording pipette solution contained 5 mm DM-nitrophen and 4 mm CaCl2, and Lucifer yellow was excluded. The osmolarity was adjusted with CsMeSO3. The intact terminal filled with DM-nitrophen was stimulated through a 60 × objective lens by a 365 nm flash (N2 pulsed laser through a laser dye BPBD 365; MicroPoint PIJ-3010; Andor Technology plc., Belfast, UK).
Analysis
For figures and quantitative analyses, three or more responses were averaged, if not specified. Amplitude of the peak response was measured from the mean resting potential for 200 ms just before the stimulus onset to the peak potential appeared in the first 200 ms during stimulation. QIPSC was calculated by integrating the evoked current for an arbitrary time window (200–800 ms from the stimulus onset; shown as [QIPSC/200 ms, …, QIPSC/800 ms]) relative to the mode of the resting current (for 200 or 500 ms just before the stimulus onset). Our conclusions were not affected by changing the time window in this range.
Cross-correlogram C(τ) of 5 s current traces (I1 and I2) was calculated as follows:
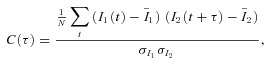 |
where τ is the time shift (<±600 ms; 1 ms bin width), the bar on a variable represents the sample mean of the variable, N is the sample number of the current traces and σ is the standard deviation of each current trace.
Statistical analysis was performed using paired two-tailed t test if not specified. Welch's t test was used in Figs 1C and D and 3F, where P < 0.05 was further corrected by the Bonferroni method. The two-way ANOVA was used in Figs 4D–I and 6A–F. In Fig. 6, multiple comparisons were performed with Ryan's method. A significance level of P < 0.05 was accepted throughout. Results were expressed as means ±s.e.m., if not specified. Error bars denote s.e.m. and parenthesized numbers represent the number of recordings. Correlation was assessed by Pearson's r.
Figure 6. Pharmacological separation of reciprocal and lateral inhibition.
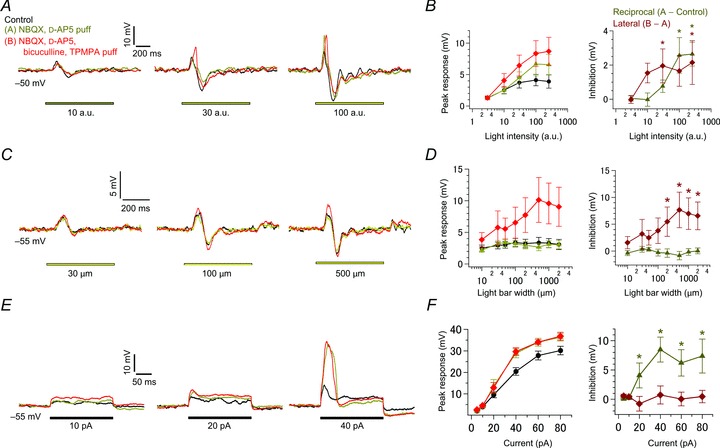
A, effects of light intensity. Voltage responses in an intact terminal (Vm: ∼−50 mV; current-clamped condition) to various intensities of light (yellow bar; 500 μm in width) were recorded in control (black), in the presence of puff-applied 10 μm NBQX and 20 μm d-AP5 (yellow; selective suppression of reciprocal inhibition), and in the presence of puff-applied 100 μm BIC and 800 μm TPMPA in addition to NBQX and d-AP5 (red; suppression of reciprocal and lateral inhibition). ECl was −70 mV. B, the relationship between the peak response amplitude and light intensity in each condition as shown in A (left; n= 9). Activation of reciprocal (dark yellow) and lateral inhibition (dark red) was estimated as the difference between responses in each condition (right). C and D, effects of light bar width as in A and B (n= 10–12 in D). Voltage responses in an intact terminal (Vm: ∼−55 mV; current-clamped condition) to various widths of light (yellow bar; intensity of 100 a.u.) were recorded in each condition as shown in A (C). ECl was −70 mV. E and F, effects of current injection as in A and B (n= 8–11 in F). Voltage responses to current injection (black bar) were recorded from an intact terminal (Vm: ∼−55 mV; current-clamped condition)in each condition as shown in A (E). ECl was −70 mV. BIC, bicuculline; d-AP5, d-(–)-2-amino-5-phosphonopentanoic acid; NBQX, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulphonamide; TPMPA, (1,2,5,6-tetrahydropyridin-4-yl) methylphosphinic acid.
Model simulation
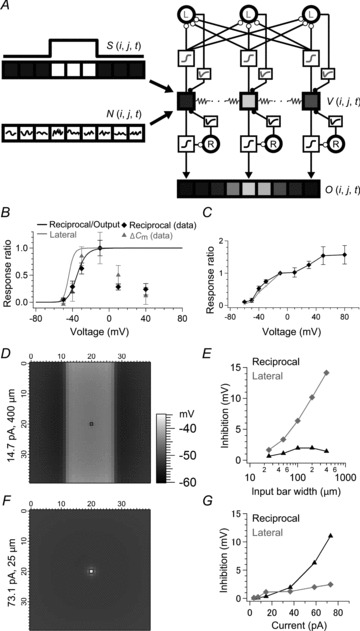
Using Igor Pro 6 (WaveMetrics, Inc., Lake Oswego, OR, USA), we constructed the model of an array of 40 × 40 BC units (25 μm/pixel) with reciprocal and lateral inhibition, where each BC unit was electrically coupled to the neighbouring four BC units. In this model, an intact Mb1 BC with dendrites, soma, axon and axon terminal was regarded as an equipotential unit (BC unit) because using a realistic Mb1 BC array model (Arai et al. 2010) we confirmed that the voltage change in an Mb1 BC terminal propagates to its dendrites with little decay. The membrane potential of each BC unit V(i, j, t) was initially set to −50 mV, which was dynamically changed by input I(i, j, t) and input through gap junctions G(i, j, t) with the time constant of 5 ms (τm) and 10 ms (τg), respectively:
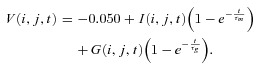 |
I(i, j, t) consisted of the visual signal S(i, j, t), intrinsic noise N(i, j, t) (used in Fig. 8), reciprocal inhibitory input R(i, j, t) and lateral inhibitory input L(i, j, t):
Figure 8. Simulated Mb1 bipolar cell outputs in response to various inputs.
A–D, model output (A) in response to a square input (14.7 pA, 400 × 400 μm) at the time indicated by the grey vertical line in C and D. The output profile of the horizontal grey line in A (B), mean of the outputs in the inner edge region (magenta in A) (μout; C), and SNR of the outputs (SNRout; D) to the square input are shown for each model: the model with both inhibition (red), that without reciprocal inhibition (dark red), that without lateral inhibition (yellow), and that without any inhibition (black). SNR of the outputs was calculated as (μi−μo)/σo, where μi and μo is mean of the outputs in the inner edge region (magenta in A) and the outer edge region (green in A), respectively, and σo is standard deviation of the outputs in the outer edge region. The yellow bar indicates the position (B) or the period (C and D) of the applied square input. The horizontal black bar in C and D (last 250 ms of the square input) indicates the period, in which mean and SNR of the outputs are calculated in E and F. E and F, mean of the outputs in the inner edge region (E) and SNR of the outputs (F) as a function of the intensity and side length of the applied square input (averaged values for 250 ms period shown as the black bar in C and D). Values were normalized to that in the model w/o inh (black traces in B–D). The colour and symbols for each model correspond to those in B–D. SNR, signal-to-noise ratio; w/o inh, without any inhibition.
G(i, j, t) was calculated as follows:
where g was 5.0 to satisfy the voltage ratio of ∼0.3 between neighbouring BC units (Arai et al. 2010). The output from each BC unit O(i,j,t) was described as a Hill function of V(i, j, t):
to satisfy the voltage-ΔCm plot in Fig. 1B (see Fig. 7B). R(i, j, t) was calculated as O(i, j, t) convolved with an alpha function αR(t):
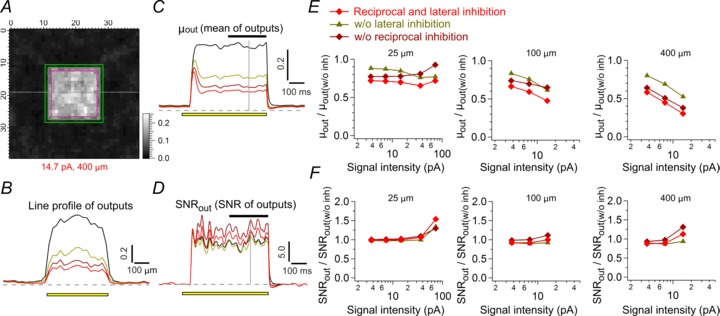
Figure 7. A model of an electrically coupled array of Mb1 bipolar cell (BC) terminals.
A, simplified diagram of the model (see Methods). Only three or nine BC units in the model array are illustrated for clarity. The visual signal S(i, j, t) and intrinsic noise N(i, j, t) charges the membrane potential of electrically coupled BC units V(i, j, t), which drives reciprocal (R) and lateral (L) inhibition through a Hill function and an alpha function for each to regulate V(i, j, t). The BC output O(i, j, t) is determined by a Hill function of V(i, j, t). In the lateral inhibitory pathway, the supralinear integration of weak inputs and the requirement of population inputs are implemented by a low voltage-dependent Hill function (grey) and by spatial summation of exponentially weighted BC inputs (λ = 100 μm), respectively. B, Hill functions for reciprocal (black) and lateral inhibition (grey) used in the model. The Hill function for reciprocal inhibition and BC output was determined to satisfy the voltage dependence of reciprocal inhibition (black diamond) and ΔCm (grey triangle) shown in Fig. 1B. The Hill function for lateral inhibition was determined to satisfy the voltage dependence of lateral inhibition as shown in C. C, the voltage dependence of lateral inhibition of the model (grey). Two neighbouring BC units at the central region of the model array were voltage clamped. Depolarization of one BC unit from −70 to −10 mV for 200 ms evoked lateral IPSP in the other voltage clamped at −90 mV. The lateral IPSP was integrated for 400 ms from the pulse onset and plotted against the membrane potentials (grey), which satisfied the experimental data in Fig. 3C (black diamond). The IPSP was normalized to the value at −10 mV. D, the output voltage profile of BC units in response to a bar input (14.7 pA in intensity, 400 μm in width) at 250 ms after the input onset. A hyperpolarizing current was injected into the BC unit at the black square (Vm: ∼−55 mV) as in Fig. 6C and D. E, effects of input bar width. Inputs with various bar widths were applied to the model as in D (14.7 pA in intensity). Reciprocal IPSP (black) and lateral IPSP (grey) in the black square region shown in D are shown. F, the output voltage profile of BC units in response to a local input (73.1 pA in intensity, 25 μm in width) applied to a single BC unit (black) at 250 ms after the input onset. A hyperpolarizing current was injected to the BC unit (Vm: ∼−55 mV) as in Fig. 6E and F. G, effects of local input intensity. Local input of various intensities was applied as in F. Reciprocal IPSP (black) and lateral IPSP (grey) in the black square region shown in F are shown. In this figure, the intrinsic noise is excluded for clarity.
where
in which AR was 3.0 × 10−4 and τR was 20 ms. L(i,j,t) was calculated as the exponentially weighted sum of OL(i, j, t) with a length constant (λL) of 100 μm, which was then convolved with an alpha function αL(t):
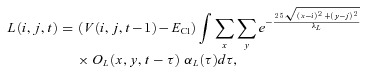 |
where
in which AL was 3.0 × 10−6 and τL was 20 ms. OL(i, j, t) was described as a Hill function of V(i, j, t):
to satisfy the voltage-lateral QIPSC plot in Fig. 3C (see Fig. 7B and C).
Figure 3. Properties of lateral inhibition.
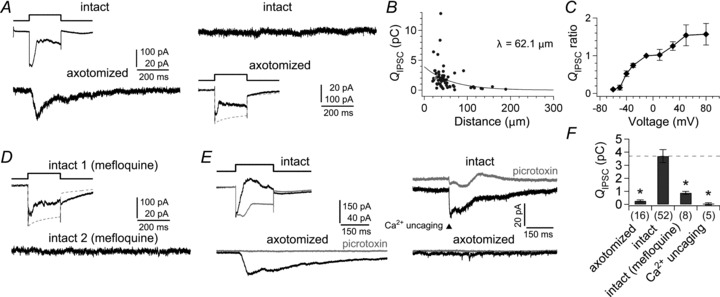
A, depolarization of an intact terminal from −70 to −10 mV for 200 ms evoked the lateral IPSC in a neighbouring (26.0 μm apart) axotomized terminal voltage clamped at −90 mV (left). Depolarization of the axotomized terminal elicited the reciprocal IPSC in itself, but failed to induce a detectable response in the intact terminal voltage clamped at −90 mV (right). ECl was −55 mV. B, the relationship between the lateral QIPSC/200 ms at −90 mV and the inter-terminal distance (circles; n= 64 pairs). Data were fitted to a single exponential function (λ = 62.1 μm). The grey circle indicates the lateral QIPSC shown in A. C, voltage dependence of lateral inhibition (n= 4–8 pairs). An intact terminal was depolarized from −70 mV to various membrane potentials for 200 ms, and the lateral QIPSC/400 ms was obtained from a nearby (<60 μm) axotomized terminal voltage clamped at −90 mV. The lateral QIPSC is normalized to the value at −10 mV in each recording. D, after bath application of 10 μm mefloquine for 2 h, depolarization of an intact terminal (intact 1) from −70 to −10 mV for 200 ms evoked the reciprocal IPSC, but failed to elicit the lateral IPSC in a nearby intact terminal (intact 2; 34.0 μm apart) voltage clamped at −90 mV. E, Ca2+ uncaging experiment. Paired recordings were performed from an intact terminal filled with 5 mm DM-nitrophen and a nearby (57.8 μm apart) axotomized terminal voltage clamped at −90 mV. Depolarization of the intact terminal from −70 to −10 mV for 200 ms elicited the lateral IPSC in the axotomized terminal (left). UV laser flash applied to the intact terminal voltage clamped at −90 mV evoked the reciprocal IPSC in the terminal but failed to evoke the lateral IPSC in the axotomized terminal (right). The bath solution contained 100 μm l-2-amino-4-phosphonobutyric acid (l-AP4) to reduce artefacts of the UV flash. IPSC was obtained as the difference between the current responses before and during bath application of 200 μm picrotoxin (grey traces). The reduction of the resting current by picrotoxin was not included for calculation of the IPSCs. F, summary of QIPSC/400 ms in unstimulated terminals in paired recordings (<60 μm apart). Stimulated terminals were 16 axotomized terminals (P < 0.001), 52 intact terminals, eight mefloquine-treated intact terminals (P < 0.001), and five Ca2+ uncaged intact terminals (P < 0.001). P-values are calculated by Welch's t test with Bonferroni correction.
N(i, j, t) was Gaussian noise (low-pass filtered at 20 Hz) and scaled such that the resulting V(i, j, t) in the model satisfies experimental data (standard deviation =∼1 mV). Input intensity of S(i, j, t) in Figs 7 and 8 was presented as V/RN, where V was the voltage response to a local input of the intensity in the model without any inhibition, and RN was 300 MΩ, the typical input resistance of intact Mb1 BCs. Analyses were confined to the central region (500 × 500 μm) of the array to exclude the artefacts originated from the edge region of the array. Inputs were limited to the range where O(i, j, t) in the model without any inhibition did not saturate (<0.9). Simulation was performed with the time step of 1 ms.
Results
Quantitative analysis of reciprocal inhibition
To characterize the properties of inhibitory inputs to BC terminals, we used a goldfish retinal slice preparation and performed whole cell recordings from the axon terminal of axotomized Mb1 BCs, which receives almost exclusively GABAergic inhibitory inputs from ACs (Vigh & von Gersdorff, 2005; Palmer, 2006). Depolarization of an axotomized terminal from −70 to −10 mV elicited L-type inward calcium current (ICa) that was immediately truncated by an outward IPSC, reflecting reciprocal inhibition from ACs (Fig. 1A). First, we examined the activation range of reciprocal inhibition by depolarizing an axotomized terminal to various membrane potentials (Fig. 1B). The amount of reciprocal inhibition was closely correlated with the charge of ICa (QCa; r= 0.76, n= 38, P < 0.001) and the capacitance jump associated with exocytosis (ΔCm; r= 0.75, n= 34, P < 0.001). These three values peaked at −10 mV, and declined at >−10 mV, where the driving force for Ca2+ decreased.
Next, we applied various pharmacological blockers to identify the transmitter receptors and ion channels involved in reciprocal inhibition (Fig. 1C and D). The reciprocal IPSC (see Fig. 1A; QIPSC, the charge of IPSC: 12.39 ± 1.22 pC, n= 47) was not reduced by glycine receptor blocker (1 μm strychnine; QIPSC: 125 ± 32% of control, n= 5, P= 0.665), whereas it was abolished by GABA receptor (GABAR) blocker (200 μm picrotoxin; 6 ± 3%, n= 5, P < 0.001) or by a mixture of GABAA receptor (GABAAR) blocker (100 μm BIC) and GABAC receptor (GABACR) blocker (200 μm TPMPA) (2 ± 4%, n= 6, P < 0.001). TPMPA alone was not sufficient to abolish the reciprocal IPSC (30 ± 9%, n= 5, P= 0.001), indicating that both GABAARs and GABACRs mediate reciprocal inhibition (Vigh & von Gersdorff, 2005). Nevertheless, BIC alone did not reduce the reciprocal IPSC (115 ± 29%, n= 10, P= 0.848), which may be ascribed to suppression of the serial inhibition among ACs (Watanabe et al. 2000; Eggers & Lukasiewicz, 2009) and/or of the presynaptic inhibition at other BC terminals. To circumvent the possible effect of disinhibition, BIC was puff applied locally to the recorded axotomized terminal, which resulted in reduction of the reciprocal IPSC by ∼46% (54 ± 9%, n= 13, P= 0.014). Thus, bath-applied BIC would have disinhibited the GABACR component of the reciprocal IPSC by ∼113%.
Reciprocal inhibition is mediated by activation of glutamate receptors in ACs. Actually, a mixture of AMPA/KA receptor (AMPA/KAR) blocker (20 μm NBQX) and NMDA receptor (NMDAR) blocker (50 μm d-AP5) abolished the reciprocal IPSC (Fig. 1D; QIPSC: 7 ± 7% of control, n= 6, P < 0.001). The reciprocal IPSC was partially reduced either by NBQX alone (39 ± 10%, n= 11, P= 0.002) or by d-AP5 alone (43 ± 10%, n= 9, P= 0.010). Puff application of BIC in the presence of bath-applied d-AP5 strongly suppressed the reciprocal IPSC (18 ± 5%, n= 7, P < 0.001), which confirmed the previous report showing that the GABAAR component of reciprocal inhibition is driven by AMPAR-mediated activation of ACs (Vigh & von Gersdorff, 2005).
It has been shown that reciprocal inhibition at rod BC terminals in the rat retina is evoked by Ca2+ influx through Ca2+-permeable AMPARs in A17 ACs (Chávez et al. 2006). In goldfish Mb1 BC terminals, however, the reciprocal IPSC was not reduced by Ca2+-permeable AMPAR blocker (1 μm PhTX; QIPSC: 106 ± 23% of control, n= 3, P= 0.925). Moreover, a mixture of PhTX and d-AP5 reduced but did not abolish the reciprocal IPSC (33 ± 12%, n= 6, P= 0.008). These results indicate that neither Ca2+ influx through Ca2+-permeable AMPARs nor NMDARs may be essential for reciprocal inhibition, though we cannot exclude the contribution of PhTX-insensitive Ca2+-permeable AMPARs (Bowie, 2012). It is known that Cd2+ blocks voltage-gated Ca2+ (CaV) channels (Chávez et al. 2010) but does not block either Ca2+-permeable AMPARs (Rossi et al. 2008) or NMDARs (Chen et al. 2000). Thus, we examined whether CaV channels in ACs are required for reciprocal inhibition. To evoke reciprocal IPSC by directly stimulating ACs without activating L-type Ca2+ channels in BC terminals, glutamate was puff applied to an axotomized terminal. The evoked IPSC (Fig. 1E; QIPSC/800 ms: 26.74 ± 7.90 pC, n= 10) could be eliminated by picrotoxin (QIPSC/800 ms: 5 ± 3% of control, n= 3, P < 0.001). Puff-applied glutamate was spatially confined to the vicinity of the recorded terminal; the glutamate-evoked IPSC decreased when the puff pipette was laterally shifted > ∼10 μm from the terminal (Fig. 1F). The glutamate-evoked reciprocal IPSC was abolished by bath application of 200 μm Cd2+ (Fig. 1E; 4 ± 1%, n= 5, P < 0.001) or in the Ca2+-free solution (QIPSC: 5 ± 3% of control, n= 3, P < 0.001). Therefore, Ca2+ influx through CaV channels in ACs seems to be essential for reciprocal inhibition.
The reciprocal IPSC was reduced by bath application of voltage-gated Na+ (NaV) channel blocker (1 μm TTX; QIPSC: 56 ± 7% of control, n= 18, P= 0.006), suggesting that NaV channels in ACs would contribute to boosting of reciprocal inhibition (Fig. 1D). Interestingly, puff-applied TTX also reduced the reciprocal IPSC (QIPSC: 36 ± 9% of control, n= 7, P < 0.001), indicating that the NaV channels would be expressed near the GABA-releasing sites of ACs in the reciprocal inhibitory pathway. Larger effectiveness of puff-applied TTX than bath-applied TTX may suggest that bath-applied TTX had disinhibited the reciprocal IPSC by ∼56%.
Narrow spread of inhibition evoked by activation of a single axotomized bipolar cell terminal
Puff-applied glutamate elicited IPSC in an axotomized terminal only when the puff pipette was positioned close to the terminal (Fig. 1F). However, it has been shown that BC terminals receive not only reciprocal inhibitory inputs but also lateral inhibitory inputs from ACs (Chávez et al. 2010; Vigh et al. 2011). One may wonder why puff-applied glutamate at remote sites failed to evoke lateral inhibition.
To examine the range of lateral spread of inhibition, we recorded simultaneously from a pair of axotomized terminals. Depolarization of an axotomized terminal to −10 mV, which elicited maximal ICa, evoked the reciprocal IPSC in itself but did not induce a detectable response in the counterpart of the pair (Fig. 2A). Similar results were obtained in the bicarbonate buffer solution (data not shown). The QIPSC recorded from the unstimulated counterparts during the 200 ms pulse (0.18 ± 0.05 pC, n= 8 pairs, <40 μm apart; see also Fig. 3F) was almost indistinguishable from the charge of spontaneous IPSCs (QIPSC/200 ms: 0.12 ± 0.03 pC, n= 26, P= 0.279, Welch's t test). Although the QIPSC in the unstimulated counterparts showed a significant negative correlation with the inter-terminal distance of the pairs (Fig. 2B; r=−0.42, P= 0.037), the lateral spread of IPSC was limited only to neighbouring terminals (<30 μm).
Figure 2. Paired recordings from axotomized Mb1 bipolar cell terminals.
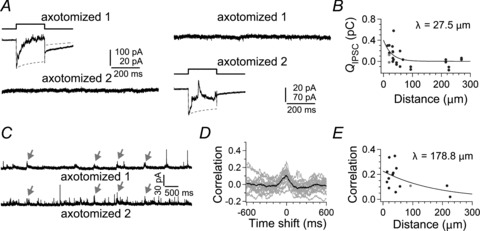
A, depolarization of an axotomized terminal (axotomized 1) from −70 to −10 mV for 200 ms elicited the reciprocal IPSC in itself, but no response was observed in a neighbouring axotomized terminal (axotomized 2; 19.2 μm apart) voltage clamped at −90 mV (left). Depolarization of the axotomized terminal 2 elicited the reciprocal IPSC in itself but failed to evoke detectable response in the axotomized terminal 1 (right). ECl was −55 mV. B, the relationship between the QIPSC/200 ms in the unstimulated terminal voltage clamped at −90 mV and the inter-terminal distance of the pairs as in A (circles; n= 16 pairs). The grey circle indicates the value calculated from the pair shown in A. Data were fitted to a single exponential function (λ= 27.5 μm). C, spontaneous IPSCs recorded from a pair of axotomized terminals voltage clamped at −10 mV (axotomized 1 and 2; 92.5 μm apart). Arrows indicate nearly synchronized IPSCs. D, cross-correlograms calculated from eighteen 5 s segments of current traces simultaneously recorded from the pair shown in C (grey). The peak of the averaged cross-correlogram (black) was at the time shift of ∼0 ms, reflecting the synchronized spontaneous IPSCs. See also Fig. S2. E, the relationship between the peak value of cross-correlograms and the inter-terminal distance of pairs as in C (n= 14). Data were fitted to a single exponential function (λ = 178.8 μm). The grey circle indicates the value calculated from the pair shown in C.
Nevertheless, spontaneous IPSCs recorded simultaneously from a pair of distant axotomized terminals sometimes occurred in synchrony (Fig. 2C). Cross-correlation analysis revealed a peak at 0 time shift (Fig. 2D). Shuffled traces recorded from the pair showed no correlation (Fig. S2A). The peak correlation–distance plot revealed a long range lateral interaction (>150 μm) (Fig. 2E; see also Fig. S2B–D), suggesting that distant Mb1 BC terminals may receive common inhibitory inputs from ACs, presumably through the lateral inhibitory pathway.
Requirement of multiple Mb1 bipolar cell activation for lateral inhibition
Spontaneous activity of retinal neurons often occurs in synchrony (Neuenschwander et al. 1999; Trong & Rieke, 2008). Thus, synchronous activation of multiple Mb1 BCs may be involved in activation of the lateral inhibitory pathway in Fig. 2C. It has been demonstrated that Mb1 BCs are electrically coupled through gap junctions at their dendrites in the outer retina (Arai et al. 2010), and thus, depolarization of an intact Mb1 BC can activate multiple Mb1 BCs.
We simultaneously recorded pairs of an intact terminal and an axotomized terminal. Strikingly, depolarization of the intact terminal to −10 mV elicited a conspicuous inward current in the axotomized terminal voltage clamped at −90 mV (Fig. 3A). This laterally evoked current was GABAergic IPSC because it reversed polarity at ECl (∼−55 mV) and it was abolished by picrotoxin (n= 4). Depolarization of the axotomized terminal did not induce a detectable response in the intact terminal, similar to the data shown in Fig. 2A. The lateral IPSC could be evoked in all the nearby pairs examined (the lateral QIPSC/200 ms: 2.61 ± 0.15 pC, n= 30 pairs; <40 μm apart), indicating that the synapses and processes responsible for lateral inhibition were sufficiently preserved in our slice preparation. The lateral QIPSC became smaller as the inter-terminal distance of the pair was increased (Fig. 3B; r=−0.31, n= 30 pairs, P= 0.016) with the length constant of ∼60 μm (Fig. 3B).
Depolarization of an intact terminal to various voltages further yielded interesting results (Fig. 3C). First, the lateral IPSC could be evoked by smaller depolarization than the reciprocal IPSC (the lateral QIPSC at −40 mV/the lateral QIPSC at −10 mV = 0.52 ± 0.08, n= 5 pairs vs. the reciprocal QIPSC at −40 mV/the reciprocal QIPSC at −10 mV = 0.29 ± 0.11, n= 11, P= 0.003, Welch's t test; the half activation voltage of a Hill function fitted to the data of ≤−10 mV was at −39.9 mV and −33.9 mV for the lateral and reciprocal QIPSCs, respectively), suggesting that some mechanisms could boost weak BC outputs in the lateral inhibitory pathway (see also Fig. 6A and B). Second, the lateral IPSC was increased as an intact terminal was depolarized over −10 mV, where the driving force for Ca2+ was reduced (see Fig. 1B). This result indicates that activation of electrically coupled neighbouring Mb1 BCs may contribute to the lateral IPSC.
Bath application of gap junction (connexin 36/50) blocker (10 μm mefloquine) for 2 h severely reduced the gap junction conductance between simultaneously recorded nearby intact terminals (Fig. S3C; < 40 μm apart; control: 1225.8 ± 139.7 pS, n= 23 pairs; mefloquine: 51.8 ± 12.0 pS, n= 4 pairs, P < 0.001, Welch's t test). Under this condition, depolarization of an intact terminal hardly evoked the lateral IPSC in the unstimulated counterpart (Fig. 3D and F; QIPSC/400 ms: 0.88 ± 0.12 pC, n= 4 pairs, <40 μm apart, P < 0.001). The reciprocal IPSC observed in the depolarized terminal (QIPSC: 7.20 ± 3.02 pC, n= 8) was not significantly different from that under the control condition (QIPSC: 12.39 ± 1.22 pC, n= 47, P= 0.101, Welch's t test), suggesting that the non-specific effect of mefloquine would be negligible. Therefore, depolarization of an intact Mb1 BC terminal elicited the lateral IPSC through outputs from electrically coupled multiple Mb1 BCs.
Further evidence was obtained by Ca2+ uncaging experiments (Fig. 3E). Ca2+ uncaging in an intact terminal induced the reciprocal IPSC in itself (QIPSC/400 ms: 7.42 ± 3.14 pC; n= 5), but failed to evoke a detectable response in a nearby axotomized terminal (Fig. 3E and F; QIPSC/400 ms: 0.05 ± 0.08 pC; n= 5 pairs, <60 μm apart). These results consistently support the hypothesis that activation of multiple Mb1 BCs is required for generation of lateral inhibition.
It is known that a tonic GABAergic IPSC is spontaneously induced in Mb1 BCs through various unknown sources (Jones & Palmer, 2009). In axotomized terminals, the total conductance of the tonic GABAergic current was 189.3 ± 42.5 pS (Fig. S3A; n= 8). Interestingly, hyperpolarization of an intact terminal from −70 to −90 mV reduced the tonic GABAergic conductance in a nearby axotomized terminal by 24.3 ± 6.3 pS (Fig. S3B, n= 15 pairs, <40 μm apart). Therefore, at least 12.8% of the tonic GABAergic current in Mb1 BC terminals seems to be ascribed to continuously driven lateral inhibition, which would account for synchronized spontaneous IPSCs observed in pairs of axotomized terminals (Fig. 2C). This is consistent with the boosting mechanism in the lateral inhibitory pathway, which allows integration of spontaneously induced weak Mb1 BC outputs.
Different pathways for reciprocal and lateral inhibition
Lateral and reciprocal inhibition may be mediated by different pathways because the lateral IPSC was induced by weaker depolarization than the reciprocal IPSC (Fig. 3C). To identify the transmitter receptors and ion channels that mediate lateral inhibition, we applied various pharmacological blockers (Fig. 4A and B). The lateral IPSC was eliminated by picrotoxin (QIPSC: 4 ± 2% of control, n= 4, P < 0.001) and partially suppressed by TPMPA (200 μm; 38 ± 9%, n= 5, P= 0.004) (Fig. 4A), suggesting involvement of both GABAARs and GABACRs. However, the lateral IPSC was not abolished by a mixture of 100 μm BIC and 200 μm TPMPA (39 ± 15%, n= 8, P= 0.027). When TPMPA was bath applied together with BIC (Fig. 4C), 800 μm TPMPA was required to sufficiently block the lateral IPSC (9 ± 4%, n= 3, P= 0.040), whereas 2 μm TPMPA was enough to suppress the reciprocal IPSC (2 ± 4%, n= 6, P < 0.001). Similarly, puff application of a mixture of 20 μm TPMPA and 100 μm BIC suppressed the reciprocal IPSC more strongly than the lateral IPSC (Fig. 4D and E; reciprocal QIPSC: 22 ± 6%, n= 10; lateral QIPSC: 49 ± 5%, n= 5 pairs, P= 0.003) under the same experimental condition (see Methods). The resistance to TPMPA can be ascribed to the expression of TPMPA-resistant GABACR subunits such as ρ1B and ρ2B (Pan et al. 2005) and/or to the release of a large amount of GABA into the synaptic cleft, which could reduce the potency of a competitive blocker such as TPMPA (Ragozzino et al. 1996). Importantly, in either case, the GABACR-mediated synapses in the lateral inhibitory pathway may be distinct from those in the reciprocal inhibitory pathway.
Similar to the reciprocal IPSC (Fig. 1C), the lateral IPSC was not reduced by bath-applied BIC (Fig. 4A; QIPSC: 215 ± 50% of control, n= 7, P= 0.055), presumably because of disinhibition. However, puff-applied BIC, which would circumvent disinhibition, hardly reduced the lateral IPSC (99 ± 11%, n= 5, P= 0.797). Actually, the reciprocal IPSC was more susceptible to puff-applied BIC than the lateral IPSC (Fig. 4F and G; reciprocal QIPSC: 51 ± 6%, n= 10; lateral QIPSC: 71 ± 6%, n= 5 pairs, P= 0.020), indicating that the GABAAR component is larger in the reciprocal IPSC than in the lateral IPSC. We did not examine the dose dependence of BIC on IPSCs because isolation of the GABAAR component of the lateral IPSC required a high concentration of TPMPA, which would also affect GABAARs (Ragozzino et al. 1996).
The lateral IPSC was completely abolished by a mixture of NBQX and d-AP5 (Fig. 4B; QIPSC: 0 ± 4% of control, n= 7, P < 0.001). The lateral IPSC was partially reduced either by NBQX (48 ± 13%, n= 9, P= 0.039) or by d-AP5 (51 ± 11%, n= 8, P= 0.045), indicating the contribution of both AMPA/KARs and NMDARs. Puff application of BIC in the presence of bath-applied d-AP5 could not eliminate the lateral IPSC (50 ± 16%, n= 3, P= 0.236), suggesting that AMPA/KARs in ACs are sufficient to activate GABACRs in the lateral inhibitory pathway different from the reciprocal inhibitory pathway.
The lateral IPSC was partially reduced by bath-applied TTX (QIPSC: 60 ± 12% of control, n= 11, P= 0.008), but it was not reduced by puff-applied TTX (94 ± 7%, n= 6, P= 0.203) (Fig. 4B). These results suggest that NaV channels in ACs mediating lateral inhibition may be expressed at remote sites from synapses on to Mb1 BC terminals, such as the soma or proximal dendrites. The reciprocal IPSC was more susceptible to puff-applied TTX than the lateral IPSC (Fig. 4H and I; reciprocal QIPSC: 73 ± 8%, n= 10; lateral QIPSC 92 ± 3%, n= 8 pairs, P= 0.009). Therefore, NaV channels expressed in reciprocal and lateral inhibitory pathway are likely to distribute at different sites, presumably in different ACs.
Supralinear integration of light signals in the lateral inhibitory pathway
Depolarization of an intact terminal could drive lateral inhibition through activation of multiple Mb1 BC outputs (see Fig. 3F). We further examined how lateral inhibition is driven by light stimulation. Illumination of the retinal slice by a light bar (500 μm in width) evoked an outward current in an axotomized terminal voltage clamped at −10 mV (Fig. 5A; QIPSC/500 ms: 5.64 ± 0.94 pC, n= 10). The current was GABAergic IPSC because it reversed polarity at ECl (∼−55 mV) and it was abolished by picrotoxin (QIPSC/500 ms: 5 ± 3%, n= 3, P= 0.005). As axotomized terminals receive inputs only from ACs, the IPSC should be the lateral IPSC from ACs activated by other BCs. We sometimes observed IPSCs evoked at the light offset (Vigh et al. 2011; Vickers et al. 2012), but we did not explore this further because of its instability under our experimental condition. In the mammalian retina, the contribution of glutamate transporters to lateral inhibition has been reported (Veruki et al. 2006; Ichinose & Lukasiewicz, 2012). However, the contribution seems to be small at Mb1 BC terminals, because lateral inhibition evoked either by depolarization of an intact terminal or by light stimulation was totally abolished by picrotoxin (Figs 3E left, and 5A).
Figure 5. Properties of light-evoked lateral inhibition.
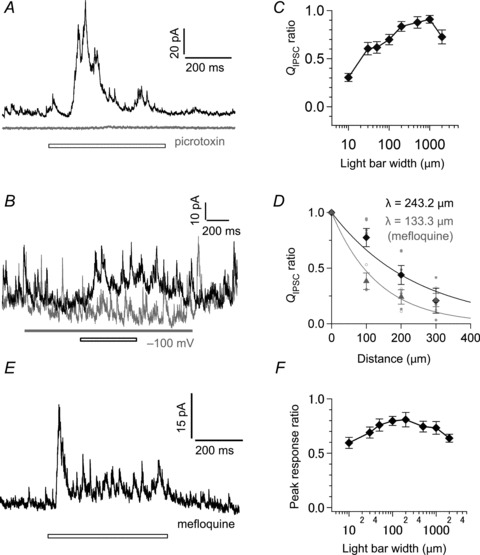
A, light-evoked lateral IPSC in an axotomized terminal voltage clamped at −10 mV in the absence (black) and presence of bath-applied 200 μm picrotoxin (grey). The light bar (white bar) was 500 μm in width. ECl was −55 mV. B, light-evoked lateral IPSC in an axotomized terminal voltage clamped at −10 mV with a neighbouring (19.5 μm) intact terminal current-clamped at 0 pA (black, Vm: ∼−50 mV) or voltage clamped at −100 mV (grey; the grey bar indicates the period). The intact terminal was located at the centre of the light bar (30 μm in width; white bar). C, the relationship between the light-evoked lateral QIPSC/500 ms at −10 mV and the light bar width (n= 10). The QIPSC was normalized to the maximum response in each recording. D, the light-evoked lateral QIPSC/500 ms at −10 mV became smaller as the light bar (50 μm in width) was laterally shifted from the recorded terminal in control (individual data: closed circle; mean: black diamond, n= 3–6) and in the presence of bath-applied 10 μm mefloquine (individual data: open circle; mean: grey triangle, n= 3). The QIPSC was normalized to the response evoked by central illumination (at 0 μm). Data were fitted to a single exponential function (λ= 243.2 μm and 133.3 μm under control and mefloquine, respectively). E, light-evoked lateral IPSC recorded from an axotomized terminal voltage clamped at −10 mV in the presence of bath-applied 10 μm mefloquine. The light bar (white bar) was 500 μm in width. F, the relationship between the peak depolarization of an intact terminal (Vm: ∼−55 mV; current-clamped condition) and the light bar width (n= 13). The peak response was normalized to the maximum response in each recording. ECl was −70 mV.
The light-evoked lateral IPSC was severely suppressed by group III metabotropic glutamate receptor agonist (100 μm l-AP4; QIPSC/500 ms: 0.3 ± 9.6%, n= 3, P= 0.009), which blocks light responses of ON-type BCs, including Mb1 BCs. Moreover, the light-evoked lateral IPSC was greatly reduced when a neighbouring intact terminal was voltage clamped at −100 mV (Fig. 5B; QIPSC/500 ms: 17 ± 8%, n= 3; P= 0.008). These results suggest that the light-evoked lateral IPSC in Mb1 BCs derives largely from activation of Mb1 BCs.
The light-evoked lateral IPSC became larger as the light bar width was expanded up to 1000 μm (Fig. 5C), indicating that activation of multiple Mb1 BCs efficiently evokes lateral inhibition. Note, however, that further expansion of the light bar width reduced the light-evoked lateral IPSC, presumably by inhibition from HCs and/or from serially connected ACs. The light-evoked lateral IPSC spread as far as ∼245 μm when a light bar (50 μm in width) was laterally shifted from the recorded axotomized terminal (Fig. 5D, black).
After bath application of mefloquine for 2 h, light illumination of large area still evoked the lateral IPSC (Fig. 5E, n= 5). This result suggests that lateral inhibition can be evoked by light-induced depolarization of multiple Mb1 BCs without gap junctions, and that the integration of multiple Mb1 BC outputs may be mediated by a wide-field AC rather than electrically coupled narrow-field ACs. We also realized that the length constant of the light-evoked lateral IPSC was reduced from 243.2 μm (control) to 133.3 μm (mefloquine) (Fig. 5D, grey). This result indicates that the long range propagation of the lateral IPSC would be partly ascribed to the lateral spread of light signals through gap junctions among photoreceptors, Mb1 BCs and/or ACs.
We next assessed how large depolarization of Mb1 BCs was required for driving the light-evoked lateral inhibition. The light-evoked depolarization of an intact terminal was often as small as a few mV in peak amplitude (see Fig. 6A–D, black) and decreased as the light bar width was expanded >200 μm (Fig. 5F). Therefore, small outputs from Mb1 BCs in a wide area seem to be supralinearly integrated in the lateral inhibitory pathway.
Independent activation of reciprocal and lateral inhibition
Lateral inhibition could be driven by small depolarization of multiple Mb1 BCs through the pathway different from reciprocal inhibition, suggesting independent operation of each inhibition in visual signal processing. To examine whether reciprocal and lateral inhibition could be differently driven by light stimulation, we pharmacologically separated each inhibition. The reciprocal IPSC could be selectively suppressed by puff application of NBQX and d-AP5 (Fig. S4A; reciprocal QIPSC was reduced to −9 ± 8% of control, n= 8, P < 0.001, Welch's t test; lateral QIPSC/200 ms at −10 mV was reduced to 93 ± 4%, n= 3 pairs, P= 0.259). On the other hand, both reciprocal and lateral IPSCs could be strongly suppressed by puff application of BIC and TPMPA in addition to NBQX and d-AP5 (Fig. S4B; reciprocal QIPSC: 11 ± 8%, n= 6, P < 0.001, Welch's t test; lateral QIPSC/200 ms at −10 mV: 12 ± 21%, n= 3 pairs, P= 0.054).
We first examined the activation range of each inhibition. As the intensity of a light bar (500 μm in width) was increased, light-evoked depolarization of an intact terminal became larger in amplitude (Fig. 6A and B, black). Selective blockage of reciprocal inhibition increased the light-evoked depolarization especially for strong light intensities (yellow), indicating that strong light efficiently drives reciprocal inhibition. In contrast, additional blockage of lateral inhibition revealed that weak light was sufficient to drive lateral inhibition (red), which supports the idea that the lateral inhibitory pathway can supralinearly integrate multiple weak inputs.
The different operating range of reciprocal and lateral inhibition suggests that they may be independently driven under certain conditions. For instance, light illumination of a wide area would be suitable for driving lateral inhibition (see Fig. 5C), but it would not drive reciprocal inhibition without sufficient depolarization of Mb1 BCs. Actually, when a hyperpolarizing constant current was injected into an intact terminal to prevent large depolarization, light illumination of a wide area activated lateral inhibition but failed to activate reciprocal inhibition (Fig. 6C and D). On the other hand, local injection of a depolarizing current pulse into an intact terminal activated only reciprocal inhibition (Fig. 6E and F), showing that local depolarization of Mb1 BCs is not suitable for driving lateral inhibition. One may argue that this result contradicts the previous finding that lateral inhibition could be evoked by depolarization of an intact terminal (Fig. 3A). It should be noted, however, that the current-clamped intact terminal with the K+-based pipette solution was depolarized to at most ∼−20 mV only for ∼50 ms during 200 ms current injection in Fig. 6E and F, which would have been insufficient to elicit enough depolarization in neighbouring Mb1 BCs. We conclude that different features of visual inputs may drive reciprocal and lateral inhibition independently.
Contribution of reciprocal and lateral inhibition to signal transmission
The above results highlighted contrasting operating mechanisms of reciprocal and lateral inhibition, which could allow their independent activation. To examine how visual inputs dynamically drive each inhibition to regulate Mb1 BC outputs, we constructed a model of an electrically coupled array of BC units (simplified Mb1 BCs), in which reciprocal and lateral inhibition based on the experimental results was implemented (Fig. 7A–C; see Methods). The model satisfied the independent activation of each inhibition (Fig. 7D–G).
When various square inputs were applied to the model, activation of reciprocal and lateral inhibition suppressed BC outputs to ∼0.3–0.7 (Fig. 8A–C and E, red). Removal of reciprocal inhibition (dark yellow) or lateral inhibition (dark red) from the model increased the outputs, suggesting cooperative suppression of BC outputs by reciprocal and lateral inhibition. Lateral inhibition contributed more to the suppression, especially when inputs were applied to a wide area, whereas reciprocal inhibition contributed more to the suppression when a strong input was locally applied. Similar results were obtained when inputs of different patterns were applied to the model (Fig. S5A–C), showing that reciprocal and lateral inhibition can cooperatively suppress BC outputs in response to a variety of visual inputs.
Suppression of BC outputs, however, might disturb signal transmission from BCs to postsynaptic neurons. To assess the ‘visibility’ of the contour of a square input, SNR of the outputs in the edge region of the square input was calculated as a measure of the quality of signal transmission. Strikingly, in response to various square inputs, SNR was little degraded in most cases, and was even improved, especially when the input was strong (Fig. 8D and F, red). The preservation of SNR could be ascribed to the reduction of background noise by feedback inhibition (Fig. S5D, red; see also Supplemental movie). Removal of lateral inhibition from the model degraded SNR (Fig. 8F, dark yellow), suggesting the critical role of lateral inhibition in improving SNR. Indeed, lateral inhibition alone could considerably reduce background noise (Fig. S5D, dark red), resulting in improvement of SNR in most cases (Fig. 8F, dark red). The supralinear integration of weak inputs in the lateral inhibitory pathway allows reduction of background noise without driving massive reciprocal inhibition, which could disturb signal transmission. Therefore, the dual organization of feedback inhibition seems to be suitable for reducing presynaptic outputs with minimal degradation of the quality of signal transmission to postsynaptic neurons.
Discussion
BC outputs are essential for elaborate retinal computations such as non-linear spatial summation (Schwartz et al. 2012), and encoding of luminance and contrast (Oesch & Diamond, 2011; Odermatt et al. 2012). We show here that Mb1 BC output is independently regulated by reciprocal and lateral inhibition through distinct pathways. Each Mb1 BC output drives reciprocal inhibition when the output is strong enough. Multiple Mb1 BC outputs drive lateral inhibition even when each output is weak. We also show that reciprocal and lateral inhibition is driven differently by a variety of visual inputs. Furthermore, model simulation showed that reciprocal and lateral inhibition cooperatively reduced BC outputs as well as background noise, thereby preserving high SNR. Therefore, we conclude that excitatory BC output is efficiently regulated by the dual operating mechanisms of feedback inhibition without deteriorating the quality of visual signals.
Synaptic pathways for reciprocal and lateral inhibition
Recent studies suggest that reciprocal and lateral inhibition may be mediated by different types of ACs (Chávez et al. 2010; Vigh et al. 2011). In accord with these reports, our pharmacological examination showed different composition of each inhibitory pathway. Especially, puff application of TTX suggested that NaV channels located different sites in the reciprocal and lateral inhibitory pathways: close to and away from the GABA-releasing sites, respectively (Fig. 4H and I). This result strongly suggests that each inhibition is mediated by different ACs, which could account for independent activation of reciprocal and lateral inhibitory pathways (Fig. 6C–F).
Depolarization of a single Mb1 BC evoked reciprocal inhibition, but could not elicit detectable IPSCs in neighbouring Mb1 BCs even when the depolarization activated ICa maximally (Fig. 2A). In other words, GABAergic synapses that mediate reciprocal inhibition in an Mb1 BC terminal could be activated only by its output. This may be enabled by narrow-field ACs that release GABA mainly on to a single Mb1 BC terminal (Masland, 2001). Alternatively, this can result from electrotonically isolated GABA-releasing sites in ACs such as varicosities of A17 ACs in the mammalian retina (Grimes et al. 2010). Curiously, reciprocal inhibition required stronger Mb1 BC output than lateral inhibition regardless of the expression of NaV channels near the GABA-releasing sites in the reciprocal inhibitory pathway. Therefore, in the reciprocal inhibitory pathway, release of GABA may be prevented by some mechanisms such as BK channels in A17 ACs (Grimes et al. 2009).
The light-evoked lateral inhibition in an Mb1 BC was maximally activated when a light bar of 1000 μm in width was applied (Fig. 5C), indicating that ACs mediating lateral inhibition could integrate inputs from a wide area. The propagation distance of the lateral inhibition evoked by a light bar of 50 μm in width was ∼245 μm (Fig. 5D, black), suggesting that the dendritic field diameter of the ACs would be ∼490 μm, or perhaps much longer because long processes of ACs might be severed in our slice preparation of ∼200 μm thickness (see Cook et al. 1998). Note, however, that light stimulation also elicits lateral spread of depolarization through an electrically coupled Mb1 BC network in the outer retina. Indeed, in the presence of mefloquine, the propagation distance of lateral inhibition was reduced to ∼135 μm (Fig. 5D, grey). Therefore, it is possible that the dendritic field diameter of the ACs is ∼270 μm. None the less, the preservation of lateral inhibition in the presence of mefloquine (Fig. 5E) indicates that these ACs can integrate weak inputs from multiple Mb1 BCs only by their own dendrites. NaV channels in the soma or proximal dendrites may help the ACs to integrate these weak inputs and to promote signal propagation to distant GABA-releasing sites (Azuma et al. 2004). However, as bath-applied TTX did not abolish lateral inhibition (Fig. 4B), other mechanisms such as Ca2+ spikes or NMDA spikes (Schiller et al. 2000) may also contribute to enhancement of weak inputs.
The relevance of feedback inhibition in visual signal processing
Reciprocal and lateral inhibition could reduce Mb1 BC outputs in response to a variety of visual inputs (Figs 8E and S5C, red). The reduction of BC outputs may contribute to prevention of presynaptic depletion (Sagdullaev et al. 2011) or expansion of the dynamic range of postsynaptic responses (Sagdullaev et al. 2006). Moreover, the reduction of BC outputs may spare unnecessary energy consumption in the visual pathway. Lateral inhibition was continuously activated to induce IPSCs in Mb1 BC terminals (Figs 2C and S3B), and thus, the spontaneous spike discharges in postsynaptic GCs may be reduced. Because maintenance of spike discharges requires a high energy cost, reduction of the spontaneous and light-evoked discharges in GCs by presynaptic inhibition may be relevant for efficient transmission of visual information from the retina, one of the most energy-consuming tissues (Niven & Laughlin, 2008).
Model simulation demonstrated that reduction of Mb1 BC outputs was accompanied by noise reduction (Fig. S5D, red), which could account for the minimal degradation of SNR (Fig. 8F, red). Our results also showed that lateral inhibition was largely responsible for the noise reduction (Fig. S5D, dark red). Under scotopic condition, noise generated in the retinal circuitry is problematic for the detection of photon signals reliably (Field et al. 2005). Therefore, the low threshold lateral inhibition may be advantageous for the detection of photon signals from such noise or for the retention of high acuity under dim light. Note, however, that feedback inhibition slightly decreased SNR especially when a weak signal was applied to a wide area, which might partly account for the loss of sensitivity in the retina (see Borghuis et al. 2009). Reciprocal inhibition decreased SNR in most cases (Fig. 8F, dark yellow), but reciprocal inhibition contributed to noise reduction (Fig. S5D, dark yellow) when strong noise was sparsely applied (arrows), suggesting that reciprocal inhibition can improve SNR under some conditions, such that spontaneous Ca2+ spikes are sparsely evoked (Protti et al. 2000).
Lateral inhibition can further play an important role in the retina: formation of the centre-surround antagonism in BCs and GCs. Our results showed that light-evoked depolarization of Mb1 BCs was decreased by lateral inhibitory inputs from ACs (Fig. 6C and D). Note, however, that the centre-surround antagonism was still observed in Mb1 BCs even when lateral inhibition from ACs was suppressed (Fig. 6D left, red). Therefore, both ACs and HCs would contribute to the receptive field organization of Mb1 BCs. Lateral inhibition from ACs could be driven by dim light, suggesting that ACs and HCs may operate at a different range of light intensity (Ichinose & Lukasiewicz, 2005).
Dual feedback inhibition in the nervous system
We found that the composition of inhibitory pathways to goldfish Mb1 BCs was different from that to mammalian rod BCs: the requirement of Ca2+-permeable AMPARs for reciprocal inhibition and NaV channels for lateral inhibition (Chávez et al. 2006, 2010). Nevertheless, both BCs seem to share some functionally relevant properties. First, output from rod BCs induces independent activation of each varicosity in A17 ACs, resulting in local reciprocal inhibition (Grimes et al. 2010). Second, reciprocal and lateral inhibition in rod BCs seems to be mediated by distinct subtypes of ACs (Chávez et al. 2010). These similarities suggest that independent control of reciprocal and lateral inhibition may be a common feature of feedback inhibition in the inner retina.
In our model simulation, parameters were determined based on experimental results. However, modification of some parameters (e.g. summation area of lateral inhibition, strength of inhibition or time course of inhibition) yielded similar results under the conditions where core properties were preserved: the requirement of a strong output for reciprocal inhibition and the supralinear integration of weak multiple outputs for lateral inhibition. Therefore, such organization of reciprocal and lateral inhibition could play similar roles in a variety of neural circuits. Dual organization of reciprocal and lateral inhibition is widely observed throughout nervous systems (Windhorst, 1996; Isaacson & Strowbridge, 1998; Cohen & Yarom, 2000). Moreover, some of their organizations show intriguing parallelism with feedback inhibition in Mb1 BCs: low threshold for lateral inhibition (Cohen & Yarom, 2000) and requirement of population activity for lateral inhibition (Arevian et al. 2008; Kapfer et al. 2007). At Mb1 BC terminals, lateral inhibition contributed to reduction of presynaptic outputs without degrading the quality of signal transmission to postsynaptic neurons. A variety of microcircuits in nervous systems may sometimes share common connectivity required for general computations.
Acknowledgments
We thank T. Sakaba for helpful comments.
Glossary
- AC
amacrine cell
- AMPAR
AMPA receptor
- AMPA/KAR
AMPA/KA receptor
- BC
bipolar cell
- BIC
bicuculline
- CaV channel
voltage-gated Ca2+ channel
- d-AP5
d-(–)-2-amino-5-phosphonopentanoic acid
- GABAAR
GABAA receptor
- GABACR
GABAC receptor
- GABAR
GABA receptor
- GC
ganglion cell
- HC
horizontal cell
- l-AP4
l-2-amino-4-phosphonobutyric acid
- NaV channel
voltage-gated Na+ channel
- NBQX
2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide
- NMDAR
NMDA receptor
- PhTX
philanthotoxin-433
- SNR
signal-to-noise ratio
- TPMPA
(1,2,5,6-tetrahydropyridin-4-yl) methylphosphinic acid
- TTX
tetrodotoxin
Additional information
Competing interests
None.
Author contributions
The studies were undertaken at the University of Tokyo, Tokyo, Japan. Conception and design of the experiments: M. Tanaka and M. Tachibana. Collection, analysis and interpretation of data: M. Tanaka and M. Tachibana. Drafting and revising the article for important intellectual content: M. Tanaka and M. Tachibana. All authors approved the final version of the manuscript.
Funding
This work was supported by KAKENHI21300148 and Japan Science and Technology Agency, CREST to M. Tachibana and Grant-in-Aid for JSPS Fellows to M. Tanaka.
Supplementary material
Supplemental Materials
Supplemental Movie
References
- Arai I, Tanaka M, Tachibana M. Active roles of electrically coupled bipolar cell network in the adult retina. J Neurosci. 2010;30:9260–9270. doi: 10.1523/JNEUROSCI.1590-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arevian AC, Kapoor V, Urban NN. Activity-dependent gating of lateral inhibition in the mouse olfactory bulb. Nat Neurosci. 2008;11:80–87. doi: 10.1038/nn2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma T, Enoki R, Iwamuro K, Kaneko A, Koizumi A. Multiple spatiotemporal patterns of dendritic Ca2+ signals in goldfish retinal amacrine cells. Brain Res. 2004;1023:64–73. doi: 10.1016/j.brainres.2004.07.025. [DOI] [PubMed] [Google Scholar]
- Borghuis BG, Sterling P, Smith RG. Loss of sensitivity in an analog neural circuit. J Neurosci. 2009;29:3045–3058. doi: 10.1523/JNEUROSCI.5071-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D. Redefining the classification of AMPA-selective ionotropic glutamate receptors. J Physiol. 2012;590:49–61. doi: 10.1113/jphysiol.2011.221689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez AE, Grimes WN, Diamond JS. Mechanisms underlying lateral GABAergic feedback onto rod bipolar cells in rat retina. J Neurosci. 2010;30:2330–2339. doi: 10.1523/JNEUROSCI.5574-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez AE, Singer JH, Diamond JS. Fast neurotransmitter release triggered by Ca influx through AMPA-type glutamate receptors. Nature. 2006;443:705–708. doi: 10.1038/nature05123. [DOI] [PubMed] [Google Scholar]
- Chen WR, Xiong W, Shepherd GM. Analysis of relations between NMDA receptors and GABA release at olfactory bulb reciprocal synapses. Neuron. 2000;25:625–633. doi: 10.1016/s0896-6273(00)81065-x. [DOI] [PubMed] [Google Scholar]
- Cohen D, Yarom Y. Cerebellar on-beam and lateral inhibition: two functionally distinct circuits. J Neurophysiol. 2000;83:1932–1940. doi: 10.1152/jn.2000.83.4.1932. [DOI] [PubMed] [Google Scholar]
- Cook PB, Lukasiewicz PD, McReynolds JS. Action potentials are required for the lateral transmission of glycinergic transient inhibition in the amphibian retina. J Neurosci. 1998;18:2301–2308. doi: 10.1523/JNEUROSCI.18-06-02301.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong CJ, Werblin FS. Temporal contrast enhancement via GABAC feedback at bipolar terminals in the tiger salamander retina. J Neurophysiol. 1998;79:2171–2180. doi: 10.1152/jn.1998.79.4.2171. [DOI] [PubMed] [Google Scholar]
- Eggers ED, Lukasiewicz PD. Interneuron circuits tune inhibition in retinal bipolar cells. J Neurophysiol. 2009;103:25–37. doi: 10.1152/jn.00458.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers ED, Lukasiewicz PD. Multiple pathways of inhibition shape bipolar cell responses in the retina. Vis Neurosci. 2011;28:95–108. doi: 10.1017/S0952523810000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field GD, Sampath AP, Rieke F. Retinal processing near absolute threshold: from behavior to mechanism. Annu Rev Physiol. 2005;67:491–514. doi: 10.1146/annurev.physiol.67.031103.151256. [DOI] [PubMed] [Google Scholar]
- Freed MA, Smith RG, Sterling P. Timing of quantal release from the retinal bipolar terminal is regulated by a feedback circuit. Neuron. 2003;38:89–101. doi: 10.1016/s0896-6273(03)00166-1. [DOI] [PubMed] [Google Scholar]
- Grimes WN. Amacrine cell-mediated input to bipolar cells: variations on a common mechanistic theme. Vis Neurosci. 2012;29:41–49. doi: 10.1017/S0952523811000241. [DOI] [PubMed] [Google Scholar]
- Grimes WN, Li W, Chávez AE, Diamond JS. BK channels modulate pre- and postsynaptic signaling at reciprocal synapses in retina. Nat Neurosci. 2009;12:585–592. doi: 10.1038/nn.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes WN, Zhang J, Graydon CW, Kachar B, Diamond JS. Retinal parallel processors: more than 100 independent microcircuits operate within a single interneuron. Neuron. 2010;65:873–885. doi: 10.1016/j.neuron.2010.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa H, Yamada M, Kaneko A. Acidification of the synaptic cleft of cone photoreceptor terminal controls the amount of transmitter release, thereby forming the receptive field surround in the vertebrate retina. J Physiol Sci. 2012;62:359–375. doi: 10.1007/s12576-012-0220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose T, Lukasiewicz PD. Inner and outer retinal pathways both contribute to surround inhibition of salamander ganglion cells. J Physiol. 2005;565:517–535. doi: 10.1113/jphysiol.2005.083436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose T, Lukasiewicz PD. The mode of retinal presynaptic inhibition switches with light intensity. J Neurosci. 2012;32:4360–4371. doi: 10.1523/JNEUROSCI.5645-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Strowbridge BW. Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron. 1998;20:749–761. doi: 10.1016/s0896-6273(00)81013-2. [DOI] [PubMed] [Google Scholar]
- Jones SM, Palmer MJ. Activation of the tonic GABAC receptor current in retinal bipolar cell terminals by nonvesicular GABA release. J Neurophysiol. 2009;102:691–699. doi: 10.1152/jn.00285.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapfer C, Glickfeld LL, Atallah BV, Scanziani M. Supralinear increase of recurrent inhibition during sparse activity in the somatosensory cortex. Nat Neurosci. 2007;10:743–753. doi: 10.1038/nn1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim K, Zhou ZJ. Role of ACh-GABA cotransmission in detecting image motion and motion direction. Neuron. 2010;68:1159–1172. doi: 10.1016/j.neuron.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GL, Vigh J, von Gersdorff H. Short-term depression at the reciprocal synapses between a retinal bipolar cell terminal and amacrine cells. J Neurosci. 2007;27:7377–7385. doi: 10.1523/JNEUROSCI.0410-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marc RE, Liu W. Fundamental GABAergic amacrine cell circuitries in the retina: nested feedback, concatenated inhibition, and axosomatic synapses. J Comp Neurol. 2000;425:560–582. doi: 10.1002/1096-9861(20001002)425:4<560::aid-cne7>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Masland RH. The fundamental plan of the retina. Nat Neurosci. 2001;4:877–886. doi: 10.1038/nn0901-877. [DOI] [PubMed] [Google Scholar]
- Matsui K, Hasegawa J, Tachibana M. Modulation of excitatory synaptic transmission by GABAC receptor-mediated feedback in the mouse inner retina. J Neurophysiol. 2001;86:2285–2298. doi: 10.1152/jn.2001.86.5.2285. [DOI] [PubMed] [Google Scholar]
- Neuenschwander S, Castelo-Branco M, Singer W. Synchronous oscillations in the cat retina. Vision Res. 1999;39:2485–2497. doi: 10.1016/s0042-6989(99)00042-5. [DOI] [PubMed] [Google Scholar]
- Niven JE, Laughlin SB. Energy limitation as a selective pressure on the evolution of sensory systems. J Exp Biol. 2008;211:1792–1804. doi: 10.1242/jeb.017574. [DOI] [PubMed] [Google Scholar]
- Odermatt B, Nikolaev A, Lagnado L. Encoding of luminance and contrast by linear and nonlinear synapses in the retina. Neuron. 2012;73:758–773. doi: 10.1016/j.neuron.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesch NW, Diamond JS. Ribbon synapses compute temporal contrast and encode luminance in retinal rod bipolar cells. Nat Neurosci. 2011;14:1555–1561. doi: 10.1038/nn.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ölveczky BP, Baccus SA, Meister M. Segregation of object and background motion in the retina. Nature. 2003;423:401–408. doi: 10.1038/nature01652. [DOI] [PubMed] [Google Scholar]
- Palmer MJ. Functional segregation of synaptic GABAA and GABAC receptors in goldfish bipolar cell terminals. J Physiol. 2006;577:45–53. doi: 10.1113/jphysiol.2006.119560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Hull C, Vigh J, von Gersdorff H. Synaptic cleft acidification and modulation of short-term depression by exocytosed protons in retinal bipolar cells. J Neurosci. 2003;23:11332–11341. doi: 10.1523/JNEUROSCI.23-36-11332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Khalili P, Ripps H, Qian H. Pharmacology of GABAC receptors: responses to agonists and antagonists distinguish A- and B-subtypes of homomericρ receptors expressed in Xenopus oocytes. Neurosci Lett. 2005;376:60–65. doi: 10.1016/j.neulet.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Protti DA, Flores-Herr N, von Gersdorff H. Light evokes Ca2+ spikes in the axon terminal of a retinal bipolar cell. Neuron. 2000;25:215–227. doi: 10.1016/s0896-6273(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Ragozzino D, Woodward RM, Murata Y, Eusebi F, Overman LE, Miledi R. Design and in vitro pharmacology of a selectiveγ-aminobutyric acidC receptor antagonist. Mol Pharmacol. 1996;50:1024–1030. [PubMed] [Google Scholar]
- Roska B, Werblin F. Vertical interactions across ten parallel, stacked representations in the mammalian retina. Nature. 2001;410:583–587. doi: 10.1038/35069068. [DOI] [PubMed] [Google Scholar]
- Rossi B, Maton G, Collin T. Calcium-permeable presynaptic AMPA receptors in cerebellar molecular layer interneurones. J Physiol. 2008;586:5129–5145. doi: 10.1113/jphysiol.2008.159921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagdullaev BT, Eggers ED, Purgert R, Lukasiewicz PD. Nonlinear interactions between excitatory and inhibitory retinal synapses control visual output. J Neurosci. 2011;31:15102–15112. doi: 10.1523/JNEUROSCI.1801-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagdullaev BT, McCall MA, Lukasiewicz PD. Presynaptic inhibition modulates spillover, creating distinct dynamic response ranges of sensory output. Neuron. 2006;50:923–935. doi: 10.1016/j.neuron.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Schiller J, Major G, Koester HJ, Schiller Y. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000;404:285–289. doi: 10.1038/35005094. [DOI] [PubMed] [Google Scholar]
- Schwartz GW, Okawa H, Dunn FA, Morgan JL, Kerschensteiner D, Wong RO, Rieke F. The spatial structure of a nonlinear receptive field. Nat Neurosci. 2012;15:1572–1580. doi: 10.1038/nn.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M. Regulation of transmitter release from retinal bipolar cells. Prog Biophys Mol Biol. 1999;72:109–133. doi: 10.1016/s0079-6107(99)00003-6. [DOI] [PubMed] [Google Scholar]
- Thoreson WB, Mangel SC. Lateral interactions in the outer retina. Prog Retin Eye Res. 2012;31:407–441. doi: 10.1016/j.preteyeres.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trong PK, Rieke F. Origin of correlated activity between parasol retinal ganglion cells. Nat Neurosci. 2008;11:1343–1351. doi: 10.1038/nn.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veruki ML, Mørkve SH, Hartveit E. Activation of a presynaptic glutamate transporter regulates synaptic transmission through electrical signaling. Nat Neurosci. 2006;9:1388–1396. doi: 10.1038/nn1793. [DOI] [PubMed] [Google Scholar]
- Vickers E, Kim M-H, Vigh J, von Gersdorff H. Paired-pulse plasticity in the strength and latency of light-evoked lateral inhibition to retinal bipolar cell terminals. J Neurosci. 2012;32:11688–11699. doi: 10.1523/JNEUROSCI.0547-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigh J, Vickers E, von Gersdorff H. Light-evoked lateral GABAergic inhibition at single bipolar cell synaptic terminals is driven by distinct retinal microcircuits. J Neurosci. 2011;31:15884–15893. doi: 10.1523/JNEUROSCI.2959-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigh J, von Gersdorff H. Prolonged reciprocal signaling via NMDA and GABA receptors at a retinal ribbon synapse. J Neurosci. 2005;25:11412–11423. doi: 10.1523/JNEUROSCI.2203-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Koizumi A, Matsunaga S, Stocker JW, Kaneko A. GABA-mediated inhibition between amacrine cells in the goldfish retina. J Neurophysiol. 2000;84:1826–1834. doi: 10.1152/jn.2000.84.4.1826. [DOI] [PubMed] [Google Scholar]
- Windhorst U. On the role of recurrent inhibitory feedback in motor control. Prog Neurobiol. 1996;49:517–587. doi: 10.1016/0301-0082(96)00023-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.