

Abstract
The electrophysiological properties and functional role of GABAergic signal transmission from neurons to the gap junction-coupled astrocytic network are still unclear. GABA-induced astrocytic Cl− flux has been hypothesized to affect the driving force for GABAergic transmission by modulating [Cl−]o. Thus, revealing the properties of GABA-mediated astrocytic responses will deepen our understanding of GABAergic signal transmission. Here, we analysed the Cl− dynamics of neurons and astrocytes in CA1 hippocampal GABAergic tripartite synapses, using Cl− imaging during GABA application, and whole cell recordings from interneuron–astrocyte pairs in the stratum lacunosum-moleculare. Astrocytic [Cl−]i was adjusted to physiological conditions (40 mm). Although GABA application evoked bidirectional Cl− flux via GABAA receptors and mouse GABA transporter 4 (mGAT4) in CA1 astrocytes, a train of interneuron firing induced only GABAA receptor-mediated inward currents in an adjacent astrocyte. A GAT1 inhibitor increased the interneuron firing-induced currents and induced bicuculline-insensitive, mGAT4 inhibitor-sensitive currents, suggesting that synaptic spillover of GABA predominantly induced the astrocytic Cl− efflux because GABAA receptors are localized near the synaptic clefts. This GABA-induced Cl− efflux was accompanied by Cl− siphoning via the gap junctions of the astrocytic network because gap junction inhibitors significantly reduced the interneuron firing-induced currents. Thus, Cl− efflux from astrocytes is homeostatically maintained within astrocytic networks. A gap junction inhibitor enhanced the activity-dependent depolarizing shifts of reversal potential of neuronal IPSCs evoked by repetitive stimulation to GABAergic synapses. These results suggest that Cl− conductance within the astrocytic network may contribute to maintaining GABAergic synaptic transmission by regulating [Cl−]o.
Key points
Astrocytes encapsulate GABAergic synapses and express GABAA receptors and GABA transporters. They are tightly coupled by gap junctions, and are referred to as the gap junction-coupled astrocytic network.
With higher [Cl−]i, GABA application can mediate bidirectional Cl− fluxes in astrocytes, Cl− efflux via GABAA receptors, and Cl− influx along with GABA uptake via GABA transporters.
We focused on the Cl− dynamics of the astrocytic network under GABAergic synapse transmission. Spillover of GABA predominantly induced Cl− efflux via GABAA receptors, presumably because they are localized more closely to the synaptic cleft.
GABAA receptor-mediated currents were propagated via gap junctions within the astrocytic network. These results indicate that Cl− efflux from astrocytes mediated by GABAergic transmission is homeostatically maintained within gap junction-coupled astrocytic networks.
Blockage of gap junctional coupling by octanol promoted the collapse of the driving force for neuronal inhibitory transmission during intense activation of GABAergic synapses. Thus, the astrocytic network may play a role in maintaining GABAergic transmission by regulating [Cl−]o.
Introduction
Astrocytic processes encapsulate synapses tightly and express receptors (Verkhratsky & Steinhauser, 2000) and transporters (Eulenburg & Gomeza, 2010) for a variety of neurotransmitters. This enables astrocytes to participate in information processing of the central nervous system and to modulate neuronal signal transmission. The expression of GABAA receptors in astrocytes has been demonstrated in cell culture (Kettenmann et al. 1984b; Backus et al. 1988) and in situ in various brain regions (MacVicar et al. 1989; Muller et al. 1994). In contrast to neurons, their activation causes Cl− efflux, which results in astrocytic membrane depolarization, in cell culture (Kettenmann et al. 1987; Backus et al. 1988) and in situ (MacVicar et al. 1989; Bekar & Walz, 2002) throughout postnatal development. This depolarization stems from the high [Cl−]i maintained by the activity of the Na+/K+/2Cl− cotransporter (NKCC1) (Yan et al. 2001), but the physiological significance of astrocytic GABAA receptor activation remains to be elucidated. GABAA receptor-mediated depolarization induces morphological changes (Matsutani & Yamamoto, 1997) and a rise in cytosolic [Ca2+]i (Bernstein et al. 1996; Meier et al. 2008), implying a regulatory role in the physiological functions of astrocytes.
Kettenmann et al. (1987) hypothesized that Cl− efflux from astrocytes could buffer the [Cl−]o of the encapsulating synapse and maintain GABAergic neuronal transmission. This hypothesis has been afforded greater importance by cumulative evidence illustrating the dynamics of the driving force for neuronal GABAergic transmission during intense GABAA receptor activation (Staley et al. 1995; Kaila et al. 1997; Staley & Proctor, 1999). Synaptically activated Cl− accumulation via GABAA receptors causes collapse of the neuronal [Cl−]o/[Cl−]i gradient, inducing transient GABA-mediated depolarization (Isomura et al. 2003). This depolarization might be moderated by Cl− efflux via astrocytic GABAA receptors activated by spillover of GABA.
To estimate astrocytic participation in synaptic Cl− homeodynamics, the interactions among presynaptic GABAergic neurons, postsynaptic neurons and encapsulating astrocytes should be revealed. Astrocytic GABAA receptors may act as a siphon that counterbalance the [Cl−]o regulation of postsynaptic GABAA receptors and presynaptic and astrocytic GABA transporters (GATs), the latter co-transporting Cl− along with GABA (Kanner & Schuldiner, 1987). In addition, gap junctional coupling that equalizes the ion concentration within the astrocytic network (Rose & Ransom, 1997) may contribute to the buffering of [Cl−]o.
The properties of GABAergic neuron-to-astrocyte signal processing are still unclear because few studies have investigated the astrocytic responses induced by presynaptic GABAergic activation. Electrical stimulation of presynaptic fibres evokes concomitant K+ currents in astrocytes (Bergles & Jahr, 1997; Kinney & Spain, 2002), which hinder the precise evaluation of kinetically slow astrocytic GABAergic responses. To overcome this, we directly evaluated single GABAergic neuron–astrocyte signal transmission in the mature CA1 hippocampus by performing dual whole cell patch clamp recordings on each component. In comparison with the results of GABA application, we illustrate that GABA spillover activates astrocytic GABAA receptors localized near the synaptic clefts, and that their signals propagate to neighbouring astrocytes via gap junctions. Furthermore, we show data suggesting that such homeostatic dynamics of Cl− within the astrocytic network might contribute to maintain intense neuronal GABAergic transmission by regulating [Cl−]o.
Methods
Ethical approval
All experiments conformed to the guidelines issued by Hamamatsu University School of Medicine on the ethical use of animals for experimentation.
Slice preparation
Experiments were carried out on acute hippocampal slices prepared from 40 male, 19–30-day-old C57BL/6 heterozygous GAD67-green fluorescent protein (GFP) knock-in (GAD67+/GFP) mice (Tamamaki et al. 2003). Animals were killed by decapitation under deep anaesthesia using halothane, and hippocampal slices (350 μm thick) were cut on a microslicer (VT-1000S; Leica Microsystems, Wetzlar, Germany) in ice-cold modified artificial cerebrospinal fluid (ACSF) containing (in mm): 220 sucrose, 2.5 KCl, 1.25 NaH2PO4, 12.0 Mg2SO4, 0.5 CaCl2, 26.0 NaHCO3 and 30.0 glucose, pH 7.4 when gassed with 95% O2/5% CO2. Following sectioning, the slices were kept at room temperature for >1 h before they were used for experiments in standard ACSF solution consisting of (in mm) 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2.0 MgSO4, 2.0 CaCl2, 26.0 NaHCO3 and 20.0 glucose, pH 7.4 when gassed with 95% O2/5% CO2. For patch clamp recording experiments in astrocytes, slices were incubated in ACSF that contained 100 nm sulforhodamine 101 (SR101) for 30–40 min at room temperature to identify astrocytes (Nimmerjahn et al. 2004).
Electrophysiology
A slice was placed on the base of a recording chamber located on the stage of a microscope (ECLIPSE, Nikon Tokyo, Japan, for experiments with photo-uncaging systems, or BX61, Olympus Tokyo, Japan, for other experiments) and continuously perfused with oxygenated ACSF at a flow rate of 2 ml min−1 at 30°C. The GABAB receptor blocker, CGP55845 (3 μm), was routinely applied in the extracellular solution. The cell images were viewed on a monitor via a 40× water immersion objective lens with an infrared differential interference contrast filter and a charge coupled device (CCD) camera (ORCA-ER C4742-95 or EMCCD #C9100-02; Hamamatsu Photonics, Hamamatsu, Japan). Patch pipettes were fabricated from borosilicate capillary tubing of 1.5 mm diameter (GD-1.5; Narishige, Tokyo, Japan) with a Flaming-Brown-type horizontal puller P-97 (Sutter Instruments, Novato, CA, USA). The electrode resistance ranged from 4.5 to 6.5 MΩ.
Astrocytes in the CA1 area were identified by SR101 fluorescence excited at 540 nm and emission detected with a high-pass filter (>570 nm). Whole cell recordings were made from SR101-positive cells with the pipette solution consisting of (in mm) 104 potassium methane sulphate, 36 KCl, 2 MgCl2, 10 Hepes-NaOH, 0.2 EGTA-KOH, 2.5 Na2ATP, 0.5 Na2GTP, pH 7.4, unless otherwise stated. The Cl− concentration of the pipette solution was 40 mm, which was adjusted to previously reported physiological concentrations evaluated by gramicidin-perforated patch clamp (29 ± 3.2 mm (Bekar & Walz, 2002) or by radioactive Cl− extrusion assay (30–50 mm (Kimelberg, 1981; Kettenmann et al. 1987). Astrocytic recordings were performed with a voltage clamp configuration held at –80 mV after evaluating the stabilized electrophysiological properties in a current clamp configuration. The typical classical astrocytes displayed a highly negative resting membrane potential (−85.4 ± 0.5 mV, n= 115), a low input resistance (31.6 ± 1.7 MΩ, n= 115, evaluated by current injection of −200 pA for 1 s), and a lack of voltage-gated sodium currents.
For GABA pressure application, GABA (1 mm or 50 μm) was added to the perfusing solution, and applied through a multi-barrelled pipette placed above a slice with a horizontal distance of 30–60 μm from the recording cell. The applied pressure was set at 20–40 kPa. To study GABAA receptor antagonist-insensitive currents, recordings were made in the presence of 200 μm picrotoxin (PTX). To maintain the slice conditions under the continuous perfusion with PTX, which can induce seizure-like hyperexcitability, synaptic activity and neuronal firing were blocked with 1 μm tetrodotoxin and a Ca2+-free extracellular solution consisting of (in mm) 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 12.0 MgSO4, 26.0 NaHCO3 and 20.0 glucose, pH 7.4 when gassed with 95% O2/5% CO2. When voltage steps were applied to the recording astrocytes, 2 mm Ba2+ was added to the Ca2+-free extracellular solution to suppress the effects of voltage-gated K+ outward currents (Bekar & Walz, 2002).
In experiments with dual whole cell patch clamp recordings between an interneuron–astrocyte pair, recordings were performed in the stratum lacunosum-moleculare (SLM) or at the stratum radiatum (SR)–SLM border. In this region, a large proportion of interneurons make synapses within the SLM (Somogyi & Klausberger, 2005), including neuroglioform neurons whose axons branch close to the soma and produces an extremely dense axonal cloud (Price et al. 2005). An interneuron with a small, round soma identified by GFP fluorescence (excited at 470 nm with emission detected above 510 nm) and an astrocyte, usually 10–20 μm from the interneuron, were patch clamped with a current clamp (Ihold= 0 pA) and a voltage clamp configuration, respectively. The internal solution for the interneurons consisted of (in mm) 135 potassium methane sulphate, 5 KCl, 2 MgCl2, 10 Hepes-NaOH, 0.2 EGTA-KOH, 2.5 Na2ATP, 0.5 Na2GTP, pH 7.4. A train of action potentials was evoked by repetitive current injection of +500 pA at 50 Hz for 2 s, and the single interneuron firing-induced currents were recorded from an adjacent astrocyte. The astrocytic current amplitude was evaluated as the difference between an average peak current for 50 ms around the visually identified peak and an average baseline for 1 s to reduce the influence of current noise.
Spontaneous IPSCs were recorded in a CA1 pyramidal neuron under the voltage clamp configuration held at −60 mV. In this recording, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 20 μm), d-(–)-2-amino-5-phosphonopentanoic acid (d-AP5, 50 μm) and CGP55845 (3 μm) were applied in the ACSF to block synaptic currents other than GABAA receptor-mediated currents. The patch pipette solution consisted of (in mm): 130 CsCl, 1 CaCl2, 2 MgCl2, 10 Hepes-NaOH, 0.2 EGTA-KOH, 2.5 Na2ATP and 0.5 Na2GTP.
To analyse activity-dependent shifts in the reversal potential of neuronal IPSCs (EIPSC), whole cell voltage clamp recordings were made from a CA1 pyramidal neuron with the internal solution consisting of (in mm) 110 potassium methane sulphate, 2 KCl, 1 MgCl2, 10 Hepes-NaOH, 0.2 EGTA-KOH, 4 magnesium ATP, 20 K2-creatine phosphatase, 5 QX314 with 50 units of creatine phosphokinase, pH 7.4. In the presence of CNQX (20 μm), d-AP5 (50 μm) and CGP55845 (3 μm), tetanus stimulation (100 times at 200–400 pA, 200 μs, 50 Hz) at 1 min intervals was delivered at each holding potential, varying from −30 to −80 mV (10 mV decrements) through a monopolar glass pipette (filled with ACSF), which was placed in the SLM. The current–voltage (I–V) relationship after each stimulus was plotted to calculate the EIPSC. Current amplitudes were determined by the average of the EIPSC recorded 150–180 ms after each stimulus.
The series resistance was usually below 25 MΩ and was compensated by 60–70% in most recordings. In astrocytes, recordings were rejected when base currents changed by more than 200 pA during the experiment. The series resistance of the pyramidal neurons was monitored throughout the experiments with voltage steps, and recordings were eliminated when series resistance changed by more than 20%. Reported voltage values were corrected by liquid junction potentials of 14.5 mV for astrocytes and 20.2 mV for pyramidal neurons. Tetrodotoxin and octanol were purchased from Wako, Tokyo, Japan; CNQX, d-AP5, CGP55845, SNAP5114, carbenoxolone and SKF89976a were bought from Tocris, Bristol, UK; and PTX, bicuculline methiodide (BIC) and NO711 were obtained from Sigma-Aldrich, St. Louis, MO, USA.
Cl− imaging
Astrocytic Cl− imaging with 6-methoxy-N-ethylquinolinium iodide (MEQ; Molecular Probes, Eugene, OR, USA) in combination with whole cell patch clamp recording was conducted as previously described (Isomura et al. 2003). MEQ (0.5 mm) was dissolved in the internal solution and delivered to the astrocytes through a patch pipette. MEQ fluorescence was excited between 340 and 380 nm, and its emissions, filtered at 435–485 nm, were captured by a CCD camera (Fukuda et al. 1998). Changes in fluorescence were recorded by placing regions of interest over part of the patch clamped cell soma (5.6 × 5.6 μm), using the image acquisition system AquaCosmos 2.6 (Hamamatsu Photonics). For analysis, background fluorescence was subtracted and photobleaching of the indicator was corrected linearly according to the slope for 2 s before GABA application. The index ΔF/F was used to estimate the relative change in [Cl−]i; F is the averaged fluorescence intensity obtained for 2 s before GABA application, and ΔF is the increase in fluorescence intensity excited at a given time. Quenching of the MEQ fluorescence, corresponding to a Cl− increase, is expressed as a negative value in this index. Data were fitted to a two-dimensional local regression model using Kyplot software 4.0 (KyensLab, Inc., Tokyo, Japan). An increase in fluorescence intensity over baseline fluorescence (ΔF/F) indicated a relative decrease in [Cl−]i.
Local GABA application using photo-uncaging systems
Laser photolysis of α-carboxy-2-nitrobenzyl-caged GABA (Invitrogen, Grand Island, NY, USA) was performed using a Micropoint laser system (Photonic Instruments, St. Charles, IL, USA). In the presence of CGP55845 (3 μm), whole cell voltage clamp recordings were made in a SLM astrocyte with an internal solution containing 0.2% Lucifer yellow. Caged GABA (2.5 mm) was dissolved in phosphate-buffered saline and delivered at a flow rate of 1 μl min−1 close to the patch clamped cell using a microdialysis syringe pump (Univentor, Zejtun, Malta) and a syringe with an inner tip diameter of 100 μm. A single pulsed nitrogen laser beam (365 nm wavelength, 600 ps pulse duration; KEN-3010; Ushio, Tokyo, Japan) was delivered to the slice via a quartz optical fibre through a 40× water-immersion objective (Fluor, 40×, NA 0.8; Nikon) to trigger local photolysis of caged GABA. The UV beam formed an uncaging spot approximately 5 μm in diameter at half-maximal intensity, which was focused on to the edge of the somatic region of a patch clamped astrocyte, or the edge of the soma of a surrounding astrocyte, with or without dye coupling. Currents induced by local GABA photolysis to each area were evaluated to analyse the spatial distribution of the GABA responding region in SLM astrocytes.
Local GABA photolysis-induced currents under the suppression of K+ channels was evaluated by means of dual whole cell patch clamp recordings from a pair of astrocytes approximately 50 μm apart. BaCl2 (2 mm) was added to low Cl− ACSF consisting of (in mm) 126 C2H5NaO4S (sodium isethionate), 2.5 KCl, 1.25 NaH2PO4, 5.0 MgSO4, 26.0 NaHCO3 and 20.0 glucose, pH 7.4 when gassed with 95% O2/5% CO2. In these recordings, the voltage was clamped near the resting membrane potentials (−30 mV), which were depolarized by extracellular Ba2+ (−35 to −25 mV). To increase the driving force for Cl−, we used low Cl− ACSF. The patch pipette solution consisted of (in mm): 130 CsCl, 1 CaCl2, 2 MgCl2, 10 Hepes-NaOH, 0.2 EGTA-KOH, 2.5 Na2ATP and 0.5 Na2GTP. Electrical coupling was confirmed by evaluating a coupling coefficient for current injection as the ratio of voltage deflection in the non-injected cell (referred to as the receiving cell) to that in the current injected cell (referred to as targeted cell) under the current clamp configuration. Then, voltage was clamped at −30 mV in both cells to record the currents induced by GABA photolysis to the edge of the targeted cell.
Data acquisition and analysis
Membrane currents or membrane potentials were recorded using an Axopatch 200B or MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA), and signals were low-pass filtered at 2 kHz and digitized at 5–10 kHz by means of a Digidata 1332A data acquisition system (Molecular Devices). Pulse generation and data collection were performed using pCLAMP 9 or 10 software (Molecular Devices) and analysed offline using Clampfit 9 or 10 (Molecular Devices). The amplitudes of the GABAergic neuron firing-induced astrocytic currents were calculated by averaging over 50 ms around the peak. The time courses of these currents were calculated after low-pass filtering at 300 Hz. For clarity, recordings with peak current amplitudes below 6.0 pA were rejected for time course analysis.
All data are presented as the means ±s.e.m. One-way ANOVA followed by Dunnett's post hoc test or two-tailed Student's t test was used for statistical analysis. Two-way repeated measures ANOVA followed by Bonferroni's post-test was applied to analyse the EIPSC increment after every five tetanic stimulations.
Results
GABA application-induced currents are mediated by both GABAA receptors and mGAT4 in hippocampal CA1 astrocytes
To clarify the constituents of GABA-induced currents and Cl− flux in CA1 astrocytes, we first examined the response to GABA application using whole cell voltage clamp recordings. Under physiological conditions (holding potential of −80 mV, 40 mm[Cl−]i), pressure application of GABA (1 mm, 1 s) evoked inward currents of 81.9 ± 8.1 pA (n= 30) in CA1 astrocytes. Bath application of PTX (200 μm) and BIC (20 μm) reduced the inward currents to a similar extent (PTX, 68.8 ± 2.9% reduction, n= 24; BIC, 70.2 ± 4.0% reduction, n= 6), but residual currents remained in all conditions (Fig. 1A and B). The same results were obtained with a lower GABA concentration (50 μm; from 16.4 ± 3.1 pA to 4.1 ± 0.4 pA, with a 73.3 ± 2.1% reduction by BIC, n= 5; Fig. 1C), indicating that incomplete antagonism did not affect residual currents. As the amplitude of GABA-evoked currents and PTX-insensitive component showed no significant differences from the amplitudes of recordings obtained from the SR and SLM (data not shown), data from both layers were pooled throughout the GABA application experiments. The I–V relationship of the PTX-insensitive, GABA-evoked currents did not reverse to positive membrane potentials at a calculated equilibrium potential for Cl− (ECl) of −31 mV (Fig. 1D).
Figure 1. GABA application induces GABAA receptor-mediated and mGAT4-mediated currents in CA1 astrocytes.
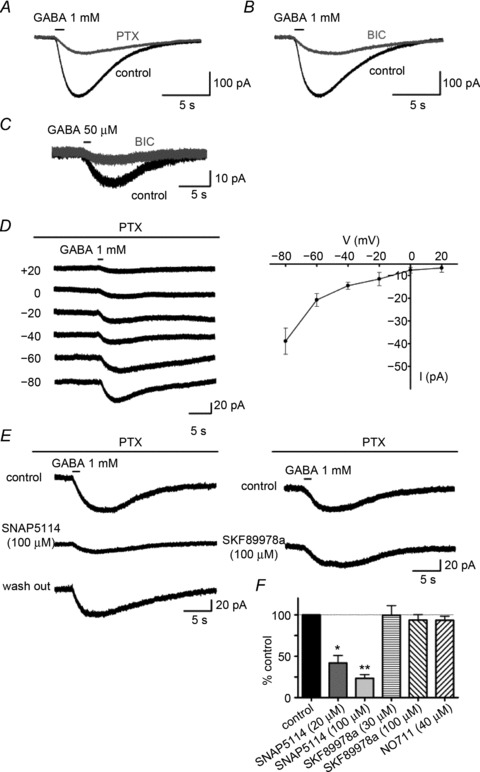
A–C, GABA-induced currents in control CA1 astrocytes (black traces) and in the presence of the GABAA receptor antagonist (grey trace). GABA (1 mm)-evoked currents were reduced but not completely blocked by 200 μm PTX (A) or 20 μm BIC (B). BIC (20 μm) had a similar effect on the inward currents as that evoked by a lower concentration of GABA (50 μm) (C). D, voltage dependence of PTX-insensitive GABA currents. The left panel shows representative PTX-insensitive currents recorded at different holding potentials in the presence of 200 μm PTX and 2 mm Ba2+. The right panel shows an I–V plot constructed from five independent experiments. The plot never crossed over to positive current amplitudes above the calculated ECl (−31 mV). E, response of PTX-insensitive GABA currents to GAT inhibitors. SNAP5114, a non-competitive mGAT4 inhibitor, reversibly reduced the PTX-insensitive currents in a dose-dependent manner, *P < 0.05, **P < 0.01 (left panel). In contrast, SKF89978a, a non-competitive GAT1 inhibitor, did not alter the PTX-insensitive currents (right panel). F, the bar graph summarizes the effects of GAT inhibitors on the PTX-insensitive currents. BIC, bicuculline methiodide; PTX, picrotoxin.
We tested the effects of subtype-specific GAT inhibitors on the PTX-insensitive GABA currents (Barakat & Bordey, 2002; Kinney & Spain, 2002). SNAP5114, a non-substrate mouse GAT4 (mGAT4; Slc6a11) selective inhibitor, significantly and reversibly reduced the PTX-insensitive GABA currents in a dose-dependent manner (58.2 ± 9.1% reduction with 20 μm SNAP5114, n= 6, P < 0.05; 76.7 ± 4.5% reduction with 100 μm SNAP5114, n= 5, P < 0.01; Fig. 1E). In contrast, non-substrate GAT1 (Slc6a1)-selective inhibitors (SKF89976a or NO711) did not significantly reduce the PTX-insensitive GABA currents (6.4 ± 6.6% reduction by 100 μm SKF89976a, n= 5; 6.6 ± 4.9% reduction by 40 μm NO711, n= 4; Fig. 1E). These results demonstrate that GABA-induced currents are mediated by GABAA receptors and mGAT4 in CA1 astrocytes.
GABA application induces bidirectional Cl− flux via GABAA receptors and mGAT4 in CA1 astrocytes
Activation of GABAA receptors produces Cl− efflux in CA1 astrocytes. It is possible that mGAT4 transfers Cl− in the opposite, inward direction, because GATs take up GABA with two Na+ and one Cl− (Kanner & Schuldiner, 1987). However, Cl− influx via GATs has not been clearly illustrated in situ. Therefore, we analysed GABA-induced [Cl−]i alterations using Cl− imaging with the Cl− indicator MEQ in combination with simultaneous whole cell voltage clamp recording. In the absence of PTX, GABA increased MEQ fluorescence (corresponding to a [Cl−]i decrease) in association with the inward currents. In contrast, MEQ fluorescence decreased (i.e. [Cl−]i increased) after PTX perfusion, accompanying the residual mGAT4 currents (Fig. 2A and B). The fluorescence signal in the same cell from SR101, which labels astrocytes, was not changed by GABA application (n= 7; Fig. 2C); therefore, the decrease in MEQ fluorescence was not affected by unrelated events such as cell swelling. These data signified that the [Cl−]i increases in astrocytes were mediated by Cl− co-transport with GABA via mGAT4. Contrastingly, in pyramidal neurons, GABA did not decrease MEQ fluorescence in the presence of PTX under similar activation protocols (Supplementary Fig. 1). Previous immunohistochemistry data showed that neuronal GAT1 is predominantly located in presynaptic GABAergic terminals (Radian et al. 1990; Heja et al. 2009). Taken together, GABA application predominantly induces Cl− efflux via GABAA receptors in CA1 astrocytes under physiological conditions, but is accompanied by a non-negligible Cl− influx via mGAT4.
Figure 2. Optical imaging of Cl− alterations induced by GABA application to CA1 astrocytes.
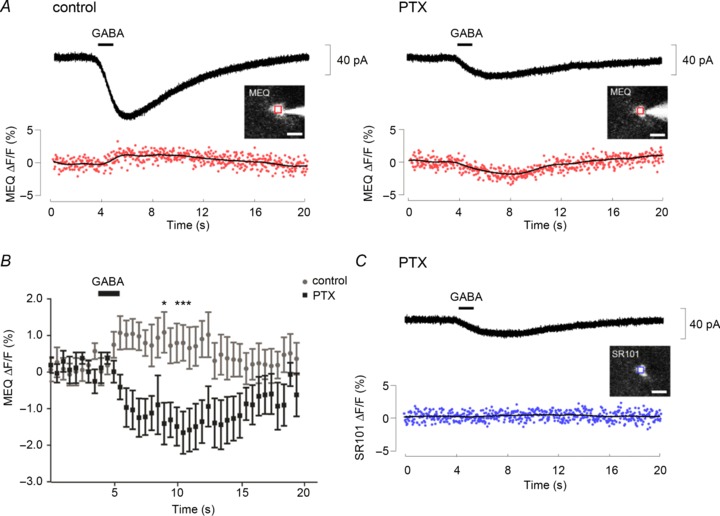
A, simultaneous recordings of currents (top) and MEQ fluorescence changes (bottom) evoked by 1 mm GABA application in controls (left panel) or in the presence of 200 μm PTX (right panel). The insets are fluorescent images of a recorded astrocyte to which MEQ was delivered through the patch electrode. The region of interest placed on the cell soma is indicated by a red square. The GABA-mediated increase in MEQ fluorescence became inverted after perfusion with PTX; both changes were synchronous with the GABA-evoked inward currents. B, average time course of MEQ fluorescence changes before (grey) and after (black) PTX perfusion (n= 8). Data at each time point were calculated by averaging for 500 ms on each recording. *P < 0.05 by two-way repeated measure ANOVA. Quenching of the MEQ fluorescence, corresponding to a Cl− increase, is expressed as a negative value. C, SR101 fluorescence of the cell shown in (A) was not altered by GABA application upon perfusion with PTX. The inset shows a fluorescent image of the cell preloaded with SR101. The region of interest placed on the cell soma is indicated by a blue square. Scale bars: 10 μm. MEQ, 6-methoxy-N-ethylquinolinium iodide; PTX, picrotoxin.
A train of single interneuron firing induces GABAA receptor-mediated currents in an adjacent astrocyte in the stratum lacunosum-moleculare layer
Astrocytic GABA receptors respond to spillover of GABA from the synaptic cleft; thus, the properties of synaptically activated responses would be different from the responses to GABA application. To examine the dynamics of perisynaptic astrocytes in GABAergic tripartite synapses, we employed dual whole cell patch clamp recordings from interneuron–astrocyte pairs using GAD67-GFP knock-in mice (Tamamaki et al. 2003).
In the SLM or at the SR-SLM border, trains of interneuron firing (50 Hz, 2 s) evoked by repetitive current injection of 500 pA induced small but obvious inward currents in an adjacent astrocyte (10–20 μm away) in 30 of 34 pairs (Fig. 3A). The mean current amplitude (6.0 ± 0.4 pA, n= 30) was not altered when the astrocytes were recorded with a Cs+-based intracellular solution (5.5 ± 1.4 pA, n= 8). The time course of these currents was characterized by a gradual increase during presynaptic firing (10–90% rise time: 1.23 ± 0.06 s, n= 17) and a relatively faster decrease after the firing (90–10% decay time: 1.14 ± 0.13 s). These single interneuron firing-evoked currents were completely and reversibly blocked by 200 μm PTX or 20 μm BIC (from 6.6 ± 1.2 to 0.3 ± 0.1 pA, n= 7, P < 0.01, using pooled data from PTX (n= 3) and BIC (n= 4); Fig. 3B, top panel). In addition, the currents were similarly blocked by a low 2 μm concentration of BIC (from 7.2 ± 1.7 to 0.4 ± 0.1 pA, n= 4, P < 0.05; Fig. 3B, bottom panel), a concentration that could only partially block the spontaneous IPSCs in CA1 pyramidal neurons (Supplementary Fig. 2). BIC antagonism of the GABA response is competitive, indicating that the single interneuron firing-evoked currents were mediated by astrocytic GABAA receptors that responded to the low concentration of GABA spilt over from the synaptic clefts.
Figure 3. Trains of interneuron firing induce GABAA receptor-mediated currents in an adjacent astrocyte in the SLM.
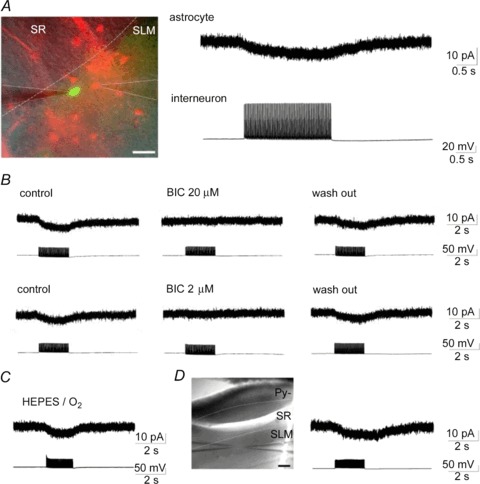
A, representative dual whole-cell patch clamp recording from SLM (dotted line) interneuron–astrocyte pair using GAD67-GFP knock-in mice. The left panel shows an overlay image of GFP (green) and SR101 (red) fluorescence and an infrared differential interference contrast image at the border of the SR and the SLM. The two patch electrodes in the SLM, emphasized with continuous lines, were placed on a close interneuron–astrocyte pair identified by GFP and SR101 fluorescence, respectively. Scale bar: 20 μm. Trains of interneuron firing initiated by repetitive current injection (right bottom) induced inward currents in the astrocyte, which we labelled as inhibitory synapse-driven astrocytic currents, Iinh-astro (right top); the arrangement is the same in subsequent figures. B, effects of BIC on Iinh-astro. At 20 μm (top panel) or at much lower concentrations (2 μm; bottom panel), BIC completely and reversibly blocked the inward currents. C, Iinh-astro recorded in Hepes-buffered extracellular solution was similar to that in HCO3−-buffered solution (A and B). (D) Iinh-astro recorded in isolated SLM. Dendrites of pyramidal neurons inserting into the SLM were isolated from the cell soma by transversely cutting a slice in the SR (left panel). Astrocytic currents were similarly induced by trains of interneuron firing in this slice (right panel). Scale bar: 100 μm. BIC, bicuculline methiodide; Py., pyramidal neurons; SLM, stratum lacunosum-moleculare; SR, stratum radiatum.
Postsynaptic GABAA receptor-mediated HCO3− conductance was previously shown to elevate [K+]o (Voipio & Kaila, 2000), which might cause the BIC-sensitive astrocytic inward currents. However, a train of interneuron firing similarly induced the astrocytic currents in HCO3−-free, Hepes-buffered extracellular solution (5.2 ± 0.6 pA, n= 3; Fig. 3C). Moreover, the astrocytic currents could be recorded in the isolated SLM, by cutting a slice in the SR (5.8 ± 2.0 pA, n= 4; Fig. 3D). As isolated dendrites of CA1 pyramidal neurons were shown to lack IPSCs (Masukawa & Prince, 1984), these findings indicate that the postsynaptic component did not mediate the interneuron firing-induced currents in astrocytes. Thus, we termed these currents inhibitory synapse-driven astrocytic currents (Iinh-astro). Iinh-astro were also recorded in wild-type mice and their amplitudes (5.5 ± 1.0 pA, n= 5) were comparable to that of GAD67-GFP knock-in mice. These data indicate that the heterozygous mutation of the GAD67 gene does not compromise signal transmission between inhibitory neurons to astrocytes, consistent with the previous report showing unaltered brain GABA content in GAD67-GFP knock-in mice (Tamamaki et al. 2003).
GABAA receptors are localized closer to the synaptic clefts than are mGAT4
In contrast to the response to GABA application, Iinh-astro did not contain mGAT4 currents. Although the Iinh-astro amplitude was much smaller than that of the 1 mm GABA-induced currents, the difference could not be explained by differences in their pharmacological affinity for GABA, because the proportion of BIC-insensitive currents induced by a lower concentration of GABA (50 μm) was equal to those induced by 1 mm GABA application (approximately a quarter, Fig. 1A). Therefore, the difference may be attributable to the distinct localization of GABAA receptors and mGAT4.
To investigate this, we analysed Iinh-astro in slices preincubated with NO711 (20 μm), which inhibits neuronal GABA uptake. The peak current amplitude (11.9 ± 2.7 pA, n= 6) and the 90–10% decay time (1.90 ± 0.27 s, n= 6) of the Iinh-astro under GAT1 blockage were significantly greater than those of controls (current amplitude, 5.5 ± 0.3 pA, n= 10, P < 0.01; decay time, 1.08 ± 0.13 s, n= 7, P < 0.05; Fig. 4A and B). These data indicated that the released GABA was largely taken up by neuronal GAT1 and only GABA spillover could activate astrocytic GABAA receptors. In the presence of NO711 (40 μm), BIC (20 μm)-insensitive currents were recorded in the Iinh-astro. Most of the residual currents were mGAT4 currents because they were significantly reduced by additional application of 20 μm SNAP5114, from 4.9 ± 1.1 to 1.7 ± 0.4 pA (n= 6, P < 0.05, Fig. 4C). Thus, mGAT4 currents evoked by single interneuron firing were recorded only when neuronal GABA uptake was inhibited, indicating that the spillover GABA was predominantly taken up by neuronal GAT1. We could not rule out a possibility of a difference in affinity for GABA between GABAA receptor and mGAT4. However, there are no reports showing the different densities between GABAA receptors and mGAT4 in astrocytic processes, and neuronal GAT1 expression is predominantly localized to the axon terminals (Radian et al. 1990). The above results suggest that GABAA receptors on astrocytic processes could be located closer to the synaptic cleft than the mGAT4 proteins.
Figure 4. Blocking neuronal GAT1 reveals firing-induced astrocytic mGAT4 currents.
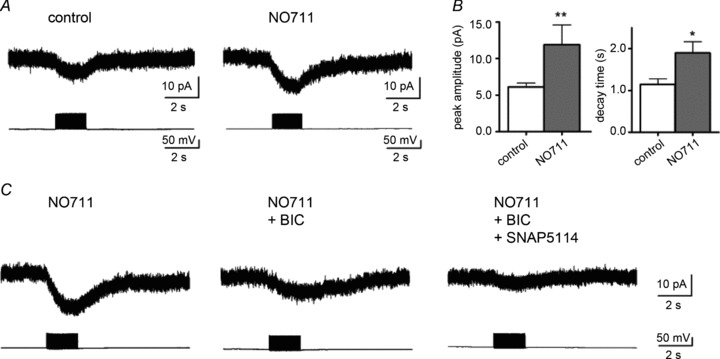
A, representative trace of Iinh-astro in a slice preincubated with 20 μm of the GAT1 inhibitor, NO711 (right panel) and in a control slice (left panel). B, the peak current amplitude and decay time were significantly increased in NO711-treated slices, *P < 0.05; **P < 0.01. C, Iinh-astro enhanced by NO711 (left panel) was reduced but not completely blocked by 20 μm BIC (middle panel). The residual currents were sensitive to additional application of the mGAT4 inhibitor, SNAP5114 (right panel). BIC, bicuculline methiodide.
Gap junction conductance helps maintain interneuron firing-induced GABAA receptor currents in stratum lacunosum-moleculare astrocytes
Iinh-astro was recorded in 89% (30 of 34) of close interneuron–astrocyte pairs (10–20 μm apart). For more distant pairs (40–60 μm apart), Iinh-astro could still be obtained in six of 15 pairs (40%), but their peak current amplitudes were significantly smaller than those of close pairs (2.5 ± 0.2 pA, n= 6 vs. 6.0 ± 0.4 pA, n= 30, P < 0.05). The distance-related differences in current amplitude may suggest the propagation of GABA-mediated signals within the gap junction-coupled network. Therefore, we examined the involvement of gap junction communication in Iinh-astro by using the gap junction inhibitors, carbenoxolone (500 μm) or octanol (1 mm) (Supplementary Fig. 3, Juszczak & Swiergiel, 2009). Bath application of carbenoxolone (500 μm) or octanol (1 mm) significantly increased the input resistance of astrocytes (carbenoxolone, from 34.6 ± 4.5 to 54.9 ± 9.3 MΩ, n= 7, P < 0.05; octanol, from 39.8 ± 4.8 to 81.0 ± 13.3 MΩ, n= 7, P < 0.05), whereas the resting membrane potential was not affected by these agents (carbenoxolone, from −88.0 ± 2.3 to −84.1 ± 2.2 mV, n= 7, P= 0.24; octanol, from −84.0 ± 2.4 to −83.0 ± 2.9 mV, n= 7, P= 0.55) in line with previous uncoupling experiments (Wallraff et al. 2006). In this condition, Iinh-astro in close pairs was significantly reduced by carbenoxolone (from 5.4 ± 0.4 to 1.6 ± 0.4 pA, n= 6, P < 0.01) or octanol (from 6.5 ± 0.8 to 2.3 ± 0.3 pA, n= 7, P < 0.001) (Fig. 5A and C). The average reduction was approximately 65%, but this varied from pair to pair (33–98% reduction). The efficacy of gap junction inhibition correlated with the distance, because the smaller Iinh-astro in distant pairs was almost completely abolished by carbenoxolone (from 2.7 ± 0.3 to 0.1 ± 0.1 pA, n= 3, P < 0.01; Fig. 5B and C).
Figure 5. Gap junction inhibitors reduce Iinh-astro.
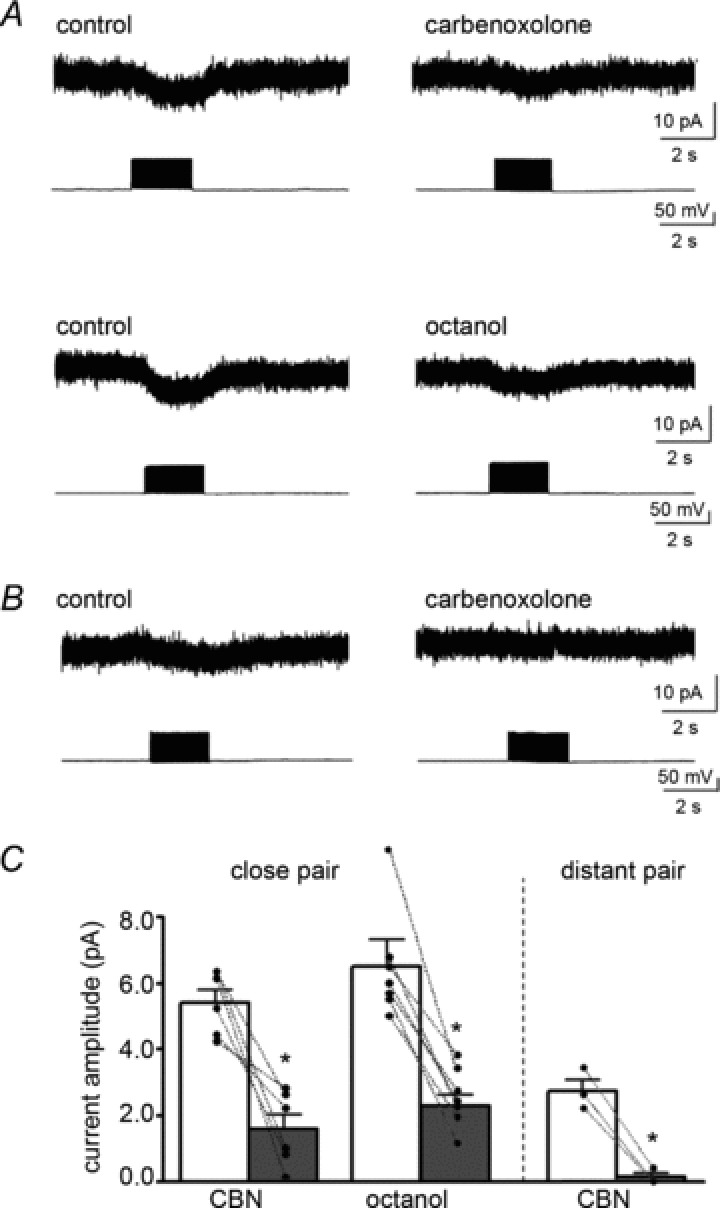
A, the effects of gap junction inhibitors on Iinh-astro in a close interneuron–astrocyte pair (10–20 μm apart). Iinh-astro was reduced by the application of CBN (500 μm, upper panels) or octanol (1 mm, lower panels). B, the effects of CBN (500 μm) on Iinh-astro in a distant interneuron–astrocyte pair (40–60 μm apart). The currents were smaller than those of close pairs and were completely blocked by CBN (500 μm). C, the effects of gap junction inhibitors on Iinh-astro in close or distant pairs. *P < 0.05. CBN, carbenoxolone.
While interneurons in the SLM are also electrically coupled (Bushong et al. 2002; Zsiros & Maccaferri, 2005), it is unlikely that they contribute to the gap junction inhibitor-sensitive components of Iinh-astro. Neuronal electrical synapses display marked low-pass filter properties, so that action potential trains barely propagate to postsynaptic interneurons (Zsiros & Maccaferri, 2005). Indeed, we did not observe action potential propagation during dual whole cell patch clamp recordings from electrically coupled interneuron pairs (Supplementary Fig. 4). These findings indicate that the observed astrocytic Iinh-astro contains current conductance via gap junctions from electrically coupled astrocytes.
To clarify the gap junction conductance of astrocytic GABAA receptor-mediated currents, we evaluated the spatial profile of local GABA application to an electrically coupled astrocyte using a UV laser photo-uncaging system (Supplementary Fig. 5). Whole cell voltage clamp recordings were obtained in a SLM astrocyte, with the internal solution containing 0.2% Lucifer yellow to visualize dye-coupled astrocytes. One to three dye-coupled astrocytes were observed in all four slices in the absence of gap junction inhibitors. Local photolysis of caged GABA focused on to a dye-coupled astrocyte or a patch-clamped astrocyte induced inward currents in three of three slices (Fig. 6A). The ratio of current amplitude induced by photolysis on the dye-coupled astrocytes to that on the patch clamped astrocytes was 0.68 ± 0.12 (n= 4). Smaller inward currents were observed when photolysis was focused on to more distant astrocytes without dye coupling. As astrocytes occupy their space exclusively, and their processes avoid extensive interdigitation (Bushong et al. 2002), spatially distant GABA-responding regions indicate the existence of gap junctional communication. As expected, the GABA-responding region was restricted to the site of uncaging in slices incubated with octanol (n= 3; Fig. 6B).
Figure 6. Local GABA application-induced currents propagate to electrically coupled astrocytes.
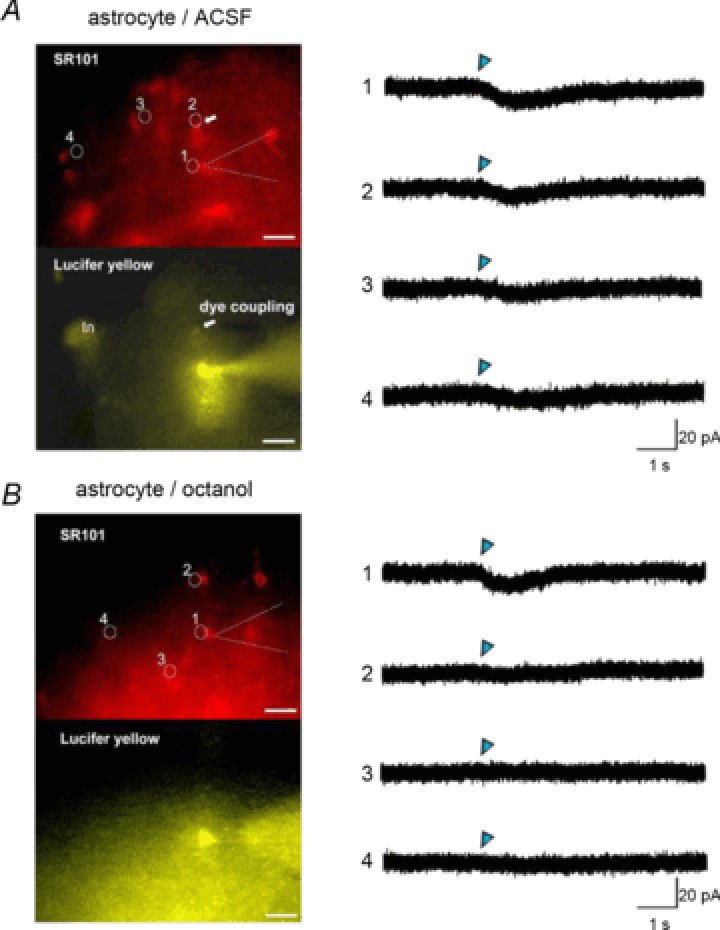
A, astrocytic responses to local GABA photolysis in the absence of gap junction inhibitors. The left upper panel is a SR101 fluorescence image. Numbered circles here and in (B) indicate uncaging spots, which correspond to the traces of uncaged GABA-induced currents in the right panel. Blue triangles indicate the time-points of a 0.9 ns uncaging flush. The patch pipette is indicated by a dashed line. The arrow indicates a dye-coupled astrocyte identified by Lucifer yellow fluorescence (left bottom panel) delivered through a patch pipette. (In) indicates an interneuron labelled by green fluorescent protein. Uncaged GABA-induced currents were recorded by photolysis, focusing on the edge of the patch clamped cell soma (1) or the edge of the dye-coupled cell soma (2). Smaller inward currents were recorded by photolysis on the distant cell somas without dye coupling (3, 4). B, these panels are the same as in (A), but in a slice preincubated with octanol (1 mm). Dye coupling was not observed. The GABA-responsive region was restricted to the vicinity of the recorded astrocyte. Scale bars: 20 μm. SR101, sulforhodamine 101.
GABA-induced depolarization might be conducted via gap junctions by an electrochemical gradient of other ions such as K+. Because of an overlap of excitation wavelength for caged GABA and MEQ, it was difficult to illustrate GABA-induced Cl− flux via gap junctions by Cl− imaging using MEQ. Thus, we examined whether the GABA-induced current could be conducted via gap junctions between a pair of astrocytes with equal holding potential and isometric ionic concentration under the suppression of astrocytic K+ currents. Dual whole cell voltage clamp recordings were performed between a pair of electrically coupled astrocytes in the SR approximately 50 μm apart in the presence of extracellular Ba2+ (2 mm). Recording astrocytes were loaded with CsCl (130 mm) via a patch pipette, and extracellular NaCl was replaced by sodium isethionate (calculated ECl= 54.3 mV). In these conditions, the resting membrane potential was depolarized to −35.4 ± 4.5 mV (n= 6), caused by strong suppression of K+ channels, and the coupling coefficient evaluated by current injection to the targeted cells was 0.025 ± 0.003. Local GABA photolysis to the vicinity of the targeted cell soma induced inward currents not only in the target cell but also in the other cell (referred to as the receiving cell) with the voltage held at −30 mV (Fig. 7A, n= 3 pairs). The currents induced by indirect photolysis were not observed in a pair at the same distance in the presence of octanol (Fig. 7B, n= 2 pairs). These findings indicate that gap junctions conduct Cl−; thus, the [Cl−]i is stably maintained in electrically coupled astrocytes. Therefore, extruded Cl− via astrocytic GABAA receptors is concomitantly complemented by an influx from other astrocytes via gap junctions, which might comprise the gap junction inhibitor-sensitive component of the inward Iinh-astro.
Figure 7. Local GABA application-induced currents conduct gap junction under the suppression of K+ channels.

A, representative voltage clamp recordings from a pair of stratum radiatum astrocytes 50 μm apart. K+ channels were blocked by extracellular Ba2+ and intracellular Cs+. Electrical coupling was confirmed by a voltage deflection in cell 2 (receiving cell) corresponding to a current injection (400 pA) to cell 1 (targeted cell) under the current clamp configuration (inset). Currents induced by local GABA photolysis (indicated by triangle) focused on the vicinity of soma of cell 1 were recorded not only in cell 1 but also in cell 2. B, traces are as in (A), but in a slice preincubated with octanol (1 mm). Pair of astrocytes electrically uncoupled as shown in the inset. GABA-photolysis-induced currents were observed only in cell 1.
Gap junction inhibitors augment the activity-dependent depolarizing shifts in reversal potential for neuronal IPSCs
Homeostatic Cl− conductance within the astrocytic network might play a vital role in the regulation of [Cl−]o in GABAergic synapses by compensating for the Cl− efflux via astrocytic GABAA receptors. To test this hypothesis, we analysed the effects of gap junction inhibitors on EIPSC. Whole cell voltage clamp recordings were made from CA1 pyramidal neurons in the presence of CNQX (20 μm), d-AP5 (50 μm) and CGP55845 (3 μm). IPSCs evoked by tetanic stimulation (50 Hz, 2 s) to the SLM were recorded with various holding potentials from −30 to −80 mV with a 1 min interval, and the EIPSC of GABA-mediated synaptic activation was evaluated.
In controls, the EIPSC of the 90th stimulus was significantly more positive than that of the second stimulus (−56.1 ± 1.7 vs. −74.9 ± 0.9 mV, n= 12, P < 0.0001). This activity-dependent depolarizing shift (ΔEIPSC=+18.6 ± 1.8 mV) resulted from a collapse of the Cl− gradient because of intracellular Cl− accumulation via GABAA receptors (Staley et al. 1995; Staley & Proctor, 1999; Isomura et al. 2003). Subsequent application of octanol (1 mm) significantly enhanced ΔEIPSC in the later phase of tetanus (on the 90th stimulus: from +19.2 ± 2.6 to +23.3 ± 3.3 mV, n= 6, P < 0.05; Fig. 8A and C). EIPSC on the 90th stimulus (from −55.1 ± 2.0 to −47.4 ± 2.3 mV, n= 6, P < 0.01), but not on the second stimulus (from −72.1 ± 2.1 to −74.5 ± 1.8 mV, P= 0.12), shifted toward depolarization (Fig. 8A and B). The peak current amplitude and decay time of the IPSCs with single or paired pulse stimulation were unaffected by octanol (single pulse stimulation-evoked IPSCs at holding potentials of −40 mV, current amplitude: 96.8 ± 10.8 vs. 93.7 ± 10.2 pA, n= 7, P= 0.69; decay time: 163.1 ± 14.2 vs. 184.9 ± 24.6 ms, n= 7, P= 0.28). Further, octanol did not affect the input resistance of neurons (from 333.0 ± 38.4 MΩ to 368.3 ± 54.5 MΩ, P= 0.14) in contrast to that of astrocytes. Taken together with the result that EIPSC in the early phase was not affected by octanol, these findings indicate that the effect of octanol on ΔEIPSC is not caused by neuronal GABAA receptor activation, which has been previously reported in vitro (Dildy-Mayfield et al. 1996).
Figure 8. A gap junction inhibitor enhances activity-dependent depolarizing shifts in the EIPSC.
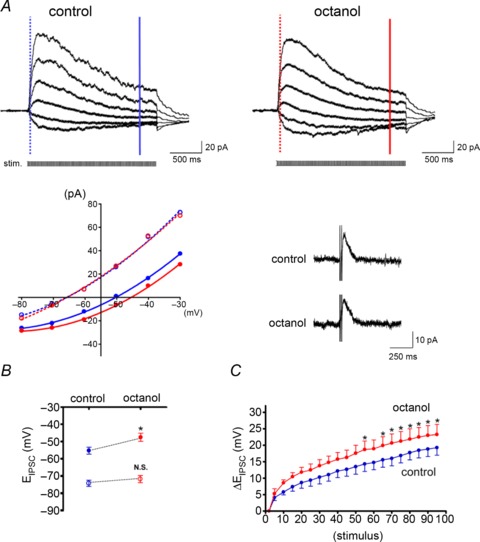
A, recordings of GABAA receptor-mediated postsynaptic currents in a CA1 pyramidal neuron evoked by tetanus stimulation (50 Hz, 2 s) to the SLM before and after perfusion of octanol (1 mm). Traces were low-pass filtered at 40 Hz to reduce stimulus artefacts. Traces were recorded with various holding potentials from −30 to −80 mV (10 mV decrement) with a 1 min interval and the baselines were superimposed. The I–V relationship after the second stimulus (early phase; indicated by the dashed lines and open circles) and the 90th stimulus (late phase; indicated by the continuous lines and filled circles) are plotted from each trace. Blue and red lines indicate before and after octanol perfusion, respectively. Note that octanol shifted the EIPSC in the late phase of tetanus but not in the early phase. The inset shows IPSCs evoked by paired stimuli in the same cell, which were not altered by octanol. B, means ±s.e.m. of the EIPSC in the early (○) and late phase (•). C, the change in EIPSC from that of the second stimulus (ΔEIPSC) plotted after every five stimulations. Octanol significantly enhanced the ΔEIPSC in the later phase of tetanus stimulation. *P < 0.05; N.S., not significant.
We could not evaluate the effects of carbenoxolone because application of carbenoxolone (100 or 500 μm) significantly reduced evoked IPSCs (Supplementary Fig. 6), presumably due to its secondary effect of elevating action potential thresholds (Rouach et al. 2000). Other gap junction inhibitors, niflumic acid or meclofenamic acid, also significantly modulated neuronal IPSC (Supplementary Fig. 6) in accordance with previous reports (Sinkkonen et al. 2003; Coyne et al. 2007). Thus, we could analyse the effect of gap junction inhibitors on the intense inhibitory synapse transmission only by octanol. Nevertheless, these results suggest that the astrocytic network may play a significant physiological role in maintaining inhibitory synaptic transmission by spatially buffering the [Cl−]o of the synaptic cleft to moderate a collapse of the Cl− gradient in postsynaptic neurons (Supplementary Fig. 7).
Discussion
The present study investigated functional properties of the astrocytic network on GABAergic tripartite synapses in the CA1 hippocampus, focusing on Cl− homeodynamics in the neuron–astrocyte network. Our main findings are as follows (and illustrated in Supplementary Fig. 7): (1) with physiologically high [Cl−]i, GABA application induced bidirectional Cl− flux in CA1 astrocytes, with Cl− efflux via GABAA receptors and Cl− influx via mGAT4; (2) synaptic spillover of GABA predominantly induced Cl− efflux from astrocytes presumably due to the localization of GABAA receptors near the synaptic clefts; and (3) gap junctions between astrocytes conducted GABA-induced currents. Blockage of gap junctions enhanced the activity-dependent depolarizing shift in EIPSC, suggesting that net Cl− extrusion from the gap junction-coupled astrocytic network might spatially buffer [Cl−]o during intense activation of GABAergic synapses.
Previous immunochemical studies have shown that GAT expression is not cell-type specific (Rattray & Priestley, 1993; Minelli et al. 1995), or specific to the hippocampus (Ribak et al. 1996), e.g. GAT1-mediated currents were recorded in glial cells in rat cortex (Kinney & Spain, 2002) and cerebellum (Barakat & Bordey, 2002). In the present study, however, most functional GATs were mGAT4 in the CA1 hippocampal astrocytes. Similarly, a recent study showed that GAT1 and GAT3 (mGAT4) immunoreactivities were specifically localized on presynaptic neurons and astrocytes, respectively (Heja et al. 2009).
GAT currents are generated by net charge translocation attributable to ions co-transported with GABA, the stoichiometry of which is generally assumed to be 1 GABA:2 Na+:1 Cl− (Kanner & Schuldiner, 1987). Although it is controversial whether Cl− is transferred by GATs (Loo et al. 2000; Karakossian et al. 2005), in this study, the increases in [Cl−]i were coincident with GABA-induced, PTX-insensitive currents and were demonstrated by Cl− imaging using MEQ. These findings indicate that Cl− was actually co-transported by mGAT4 in situ, as proven by tracer uptake measurements of GAT1 (Krause & Schwarz, 2005). The [Cl−]i decrease without PTX was a consequence of Cl− efflux via the GABAA receptor overwhelming Cl− influx via mGAT4.
mGAT4 currents were induced by single interneuron firing only when GAT1 was inhibited by NO711, indicating that the ambient GABA level was highly regulated by neuronal GAT1 when GABA release was incrementally increased by repetitive firing (Isaacson et al. 1993). This functional difference between neuronal GAT1 and astrocytic mGAT4 might be caused by their different expression intensity and/or different driving force because of the higher [Cl−]i in astrocytes. In contrast to the synaptically released GABA-induced currents, the current induced by low-dose GABA application contained a BIC-insensitive component in the absence of NO711. The above results support our hypothesis that astrocytic GABAA receptors could be localized more closely to the synaptic cleft than mGAT4. Therefore, Cl− uptake via mGAT4 could play a complementary role in the astrocyte-mediated Cl− homeostasis of GABAergic synapses. As we specifically evaluated astrocytic responses to single interneuron firing, the functional properties of mGAT4 under conditions of excessive neuronal network activity (Heja et al. 2009) remain to be elucidated.
Little is known about the functional properties of GABA spillover-induced electrical signalling in glial cells. Synaptically activated depolarization via GABAA receptors has been demonstrated in hippocampal oligodendrocyte precursor cells (Lin & Bergles, 2004) and in stellate glial cells of the pituitary (Mudrickdonnon et al. 1993). However, depolarization was attributed to direct release of GABA on to glial cells rather than spillover from synaptic clefts. Focal fibre stimulation elevated [K+]o, which makes it difficult to evaluate the slow kinetic responses to GABA spillover in astrocytes with their low input resistance. In this study, by stimulating single GABAergic neurons, signal transmission from the GABAergic neuron to astrocytes (Iinh-astro) could be directly evaluated without the contaminating effect of [K+]o uptake currents.
Several lines of evidence illustrated that Iinh-astro was evoked by GABA spillover. First, Iinh-astro displayed much slower kinetics than the evoked IPSCs stimulated by the same configuration (50 Hz, 2 s; Fig. 8). Second, Iinh-astro was completely antagonized by a low dose of BIC, which did not abolish spontaneous IPSCs. Spillover of GABA was predominantly taken up by neuronal GAT1, not by glial mGAT4. This is in contrast to the modulation of neuronal tonic currents in the rat cortex, which requires inhibition of GAT3 as well as GAT1 (Keros & Hablitz, 2005). Because GAT1 expression clusters at presynaptic boutons (Chiu et al. 2002), our electrophysiological data indicated that the distribution of GABAA receptors in astrocytic processes was peri- rather than extra-synaptic, as shown by electron microscopy of Bergmann glia (Riquelme et al. 2002). Thus, Cl− efflux via astrocytic GABAA receptors can contribute to the regulation of [Cl−]o during GABAergic transmission (Kettenmann et al. 1984a; MacVicar et al. 1989).
Strong gap junction coupling between astrocytes probably mediates the coordinated action of coupled cells and equalizes their intracellular ion concentration (Rose & Ransom, 1997). In agreement with this hypothesis, spillover GABA-induced astrocytic signalling was propagated within the astrocytic network through gap junctions. Approximately two-thirds of Iinh-astro were blocked by two different gap junction inhibitors, although the gap junction inhibitor-sensitive currents might not be equivalent to Cl− conductance via gap junctions. Because it is difficult to clamp the voltage of coupled astrocytes completely because of space clamp restrictions, these currents might be concomitant with the K+ currents that accompany GABA-induced depolarization. GABAA receptor activation has been shown to inactivate A-type outward K+ currents (IA) in depolarized astrocytes (Bekar & Walz, 2002), which might comprise part of the gap junction inhibitor-sensitive Iinh-astro. However, this is unlikely because the amplitude of Iinh-astro is not altered by intracellular Cs+ replacement, which blocks outward-rectifier K+ channels including IA (Banks & Pearce, 2000).
Alternatively, a slight GABA-mediated depolarization could be driven by voltage gradients across gap junctions with the carriage of K+. Further, GABA-mediated depolarization was counterbalanced by K+ efflux through passive K+ conductance, which sets the glial resting membrane potential close to the K+ equilibrium. In this view, the Cl− conductance component via gap junctions in Iinh-astro might be underestimated. We illustrated that GABA-evoked currents were transmitted via gap junctions between equally voltage clamped astrocytes under the suppression of K+ channels and using 130 mm CsCl pipette solution in both cells. In this condition, a driving force for GABA-induced gap junctional conductance could be predominantly produced by the chemical gradient of Cl− brought about by Cl− efflux in the GABA-activated cell. Although Cl− imaging is required to quantify a Cl− conductance via gap junctions, these results may attest to the presence of a gap junctional Cl− conductance induced by GABAA receptor activation.
Astrocytes and interneurons are connected by gap junctions (Tamas et al. 2000; Zsiros & Maccaferri, 2005). Application of the gap junction inhibitor, octanol, augmented the activity-dependent depolarizing shifts in EIPSC. Because an electrical synapse in interneurons can regulate their synchronous firing (Tamas et al. 2000), octanol might decrease the total activation of interneurons in response to tetanus stimulation. In this case, the activity-dependent depolarizing shift should be decreased rather than increased. Therefore, octanol would have only a limited effect on properties of interneuron firings in our recording protocol. Octanol can alter GABAA receptor permeability (Dildy-Mayfield et al. 1996). However, neither properties of IPSCs evoked by single pulse stimulation nor EIPSC at the second stimulus of tetanic stimulation were affected by octanol. Thus, it is unlikely that octanol significantly modulated the properties of postsynaptic GABAA receptors in this recording. Taken together, our results suggest that the astrocytic network may moderate the collapse of the inhibitory driving force for GABAergic synapses during intense GABAergic neuron firing.
Cl− conductance via gap junctions could complement the [Cl−]i decrease in astrocytes responding to GABA spillover by the GABAA receptor, which would maintain Cl− homeostasis by a siphon effect. As the GABAA receptors of postsynaptic neurons and presynaptic GATs could take up Cl− from the synaptic clefts, this astrocytic Cl− conductance might spatially buffer the change in [Cl−]o and maintain GABAergic synapse transmission. This idea is supported by a recent in vivo study using Cl−-sensitive electrodes that showed activity-dependent changes in [Cl−]o (Kroeger et al. 2010).
As the initial [Cl−]o is much higher than [Cl−]i, [Cl−]o will have a minor effect on ECl changes, determined by the log of [Cl−]o/[Cl−]i. In our study, the ΔEIPSC increment by gap junction closure was about 5 mV. In addition, because an increase in [K+]o enhances the activity-dependent depolarizing shifts in GABAergic transmission by modulating the thermodynamics of the K+–Cl− cotransporter (Staley & Proctor, 1999; DeFazio et al. 2000), blockage of glial spatial buffering of extracellular K+ by octanol could also be involved in the mechanism of depolarized EIPSC. Nevertheless, our present study suggests that [Cl−]o in the nano-domain of synaptic clefts is much more dynamic than previously assumed (Kroeger et al. 2010). In this regard, astrocytes may act as a storage for Cl−, and could help maintain [Cl−]o of the synaptic cleft. A limitation of this study is that we could analyse the functional significance of gap junction-coupled astrocytic network only by using one of the gap junction inhibitors because other drugs have shown to modulate neuronal GABAergic signalling (Supplementary Fig. 6). Further non-pharmacological analysis (e.g. genetic manipulation for astrocytic GABAA receptors and/or gap junctions) is required to elucidate the molecular basis for the role of the astrocytic network in GABAergic transmission.
Glossary
- BIC
bicuculline methiodide
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- d-AP5
d-(–)-2-amino-5-phosphonopentanoic acid
- ECl
equilibrium potential for Cl−
- EIPSC
reversal potential of neuronal IPSCs
- GAT
GABA transporter
- GFP
green fluorescent protein
- IA
A-type outward K+ current
- Iinh-astro
inhibitory synapse-driven astrocytic currents
- MEQ
6-methoxy-N-ethylquinolinium iodide
- mGAT4
mouse GABA transporter 4
- NKCC1
Na+/K+/2Cl− cotransporter
- PTX
picrotoxin
- SLM
stratum lacunosum-moleculare
- SR
stratum radiatum
- SR101
sulforhodamine 101
Additional information
Competing interests
None.
Author contributions
K.E. and A.F. contributed to the conception and design of experiments. K.E., T.F. and J.Y. contributed to the collection, analysis and interpretation of experiments. Y.Y. generated the GAD67-GFP knock-in mouse line. K.E. wrote the initial draft of the manuscript and A.F. contributed to writing and revising the manuscript. The experiments were carried out at Hamamatsu University School of Medicine. All authors approved the final version of the manuscript.
Funding
This work was supported by Grants-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology, Japan grant no. 16047213 (to A.F.), Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science grant no. 16390058, 19390058 (to A.F.), and by a Research Grant (16A-3 and 19A-6) for Nervous and Mental disorders from the Ministry of Health, Labour and Welfare, Japan (to A.F.).
Supplementary material
Supplementary Figs. S1-S7
References
- Backus KH, Kettenmann H, Schachner M. Effect of benzodiazepines and pentobarbital on the GABA-induced depolarization in cultured astrocytes. Glia. 1988;1:132–140. doi: 10.1002/glia.440010205. [DOI] [PubMed] [Google Scholar]
- Banks MI, Pearce RA. Kinetic differences between synaptic and extrasynaptic GABA(A) receptors in CA1 pyramidal cells. J Neurosci. 2000;20:937–948. doi: 10.1523/JNEUROSCI.20-03-00937.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat L, Bordey A. GAT-1 and reversible GABA transport in Bergmann glia in slices. J Neurophysiol. 2002;88:1407–1419. doi: 10.1152/jn.2002.88.3.1407. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–1308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Bekar LK, Walz W. Intracellular chloride modulates A-type potassium currents in astrocytes. Glia. 2002;39:207–216. doi: 10.1002/glia.10096. [DOI] [PubMed] [Google Scholar]
- Bernstein M, Lyons SA, Moller T, Kettenmann H. Receptor-mediated calcium signalling in glial cells from mouse corpus callosum slices. J Neurosci Res. 1996;46:152–163. doi: 10.1002/(SICI)1097-4547(19961015)46:2<152::AID-JNR3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CS, Jensen K, Sokolova I, Wang D, Li M, Deshpande P, Davidson N, Mody I, Quick MW, Quake SR, Lester HA. Number, density, and surface/cytoplasmic distribution of GABA transporters at presynaptic structures of knock-in mice carrying GABA transporter subtype 1-green fluorescent protein fusions. J Neurosci. 2002;22:10251–10266. doi: 10.1523/JNEUROSCI.22-23-10251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne L, Su J, Patten D, Halliwell RF. Characterization of the interaction between fenamates and hippocampal neuron GABAA receptors. Neurochem Int. 2007;51:440–446. doi: 10.1016/j.neuint.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFazio RA, Keros S, Quick MW, Hablitz JJ. Potassium-coupled chloride cotransport controls intracellular chloride in rat neocortical pyramidal neurons. J Neurosci. 2000;20:8069–8076. doi: 10.1523/JNEUROSCI.20-21-08069.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dildy-Mayfield JE, Mihic SJ, Liu Y, Deitrich RA, Harris RA. Actions of long chain alcohols on GABAA and glutamate receptors: relation to in vivo effects. Br J Pharmacol. 1996;118:378–384. doi: 10.1111/j.1476-5381.1996.tb15413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulenburg V, Gomeza J. Neurotransmitter transporters expressed in glial cells as regulators of synapse function. Brain Res Rev. 2010;63:103–112. doi: 10.1016/j.brainresrev.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Fukuda A, Tanaka M, Yamada Y, Muramatsu K, Shimano Y, Nishino H. Simultaneous optical imaging of intracellular Cl− in neurons in different layers of rat neocortical slices: advantages and limitations. Neurosci Res. 1998;32:363–371. doi: 10.1016/s0168-0102(98)00099-6. [DOI] [PubMed] [Google Scholar]
- Heja L, Barabas P, Nyitrai G, Kekesi KA, Lasztoczi B, Toke O, Tarkanyi G, Madsen K, Schousboe A, Dobolyi A, Palkovits M, Kardos J. Glutamate uptake triggers transporter-mediated GABA release from astrocytes. PLoS One. 2009;4:e7153. doi: 10.1371/journal.pone.0007153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA inthe hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Isomura Y, Sugimoto M, Fujiwara-Tsukamoto Y, Yamamoto-Muraki S, Yamada J, Fukuda A. Synaptically activated Cl− accumulation responsible for depolarizing GABAergic responses in mature hippocampal neurons. J Neurophysiol. 2003;90:2752–2756. doi: 10.1152/jn.00142.2003. [DOI] [PubMed] [Google Scholar]
- Juszczak GR, Swiergiel AH. Properties of gap junction blockers and their behavioural, cognitive and electrophysiological effects: animal and human studies. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:181–198. doi: 10.1016/j.pnpbp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner BI, Schuldiner S. Mechanism of transport and storage of neurotransmitters. CRC Crit Rev Biochem. 1987;22:1–38. doi: 10.3109/10409238709082546. [DOI] [PubMed] [Google Scholar]
- Karakossian MH, Spencer SR, Gomez AQ, Padilla OR, Sacher A, Loo DDF, Nelson N, Eskandari S. Novel properties of a mouse gamma-aminobutyric acid transporter (GAT4) J Membr Biol. 2005;203:65–82. doi: 10.1007/s00232-004-0732-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keros S, Hablitz JJ. Subtype-specific GABA transporter antagonists synergistically modulate phasic and tonic GABAA conductances in rat neocortex. J Neurophysiol. 2005;94:2073–2085. doi: 10.1152/jn.00520.2005. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Backus KH, Schachner M. Aspartate, glutamate and gamma-aminobutyric acid depolarize cultured astrocytes. Neurosci Lett. 1984a;52:25–29. doi: 10.1016/0304-3940(84)90345-8. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Gilbert P, Schachner M. Depolarization of cultured oligodendrocytes by glutamate and GABA. Neurosci Lett. 1984b;47:271–276. doi: 10.1016/0304-3940(84)90525-1. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Backus KH, Schachner M. Gamma-aminobutyric acid opens Cl− channels in cultured astrocytes. Brain Res. 1987;404:1–9. doi: 10.1016/0006-8993(87)91349-7. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK. Active accumulation and exchange transport of chloride in astroglial cells in culture. Biochim Biophys Acta. 1981;646:179–184. doi: 10.1016/0005-2736(81)90285-6. [DOI] [PubMed] [Google Scholar]
- Kinney GA, Spain WJ. Synaptically evoked GABA transporter currents in neocortical glia. J Neurophysiol. 2002;88:2899–2908. doi: 10.1152/jn.00037.2002. [DOI] [PubMed] [Google Scholar]
- Krause S, Schwarz W. Identification and selective inhibition of the channel mode of the neuronal GABA transporter 1. Mol Pharmacol. 2005;68:1728–1735. doi: 10.1124/mol.105.013870. [DOI] [PubMed] [Google Scholar]
- Kroeger D, Tamburri A, Amzica F, Sik A. Activity dependent layer-specific changes in the extracellular chloride concentration and chloride driving force in the rat hippocampus. J Neurophysiol. 2010;103:1905–1914. doi: 10.1152/jn.00497.2009. [DOI] [PubMed] [Google Scholar]
- Lin SC, Bergles DE. Synaptic signaling between GABAergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nat Neurosci. 2004;7:24–32. doi: 10.1038/nn1162. [DOI] [PubMed] [Google Scholar]
- Loo DD, Eskandari S, Boorer KJ, Sarkar HK, Wright EM. Role of Cl− in electrogenic Na+-coupled cotransporters GAT1 and SGLT1. J Biol Chem. 2000;275:37414–37422. doi: 10.1074/jbc.M007241200. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Tse FW, Crichton SA, Kettenmann H. GABA-activated Cl− channels in astrocytes of hippocampal slices. J Neurosci. 1989;9:3577–3583. doi: 10.1523/JNEUROSCI.09-10-03577.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masukawa LM, Prince DA. Synaptic control of excitability in isolated dendrites of hippocampal neurons. J Neurosci. 1984;4:217–227. doi: 10.1523/JNEUROSCI.04-01-00217.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsutani S, Yamamoto N. Neuronal regulation of astrocyte morphology in vitro is mediated by GABAergic signaling. Glia. 1997;20:1–9. doi: 10.1002/(sici)1098-1136(199705)20:1<1::aid-glia1>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Meier SD, Kafitz KW, Rose CR. Developmental profile and mechanisms of GABA-induced calcium signaling in hippocampal astrocytes. Glia. 2008;56:1127–1137. doi: 10.1002/glia.20684. [DOI] [PubMed] [Google Scholar]
- Minelli A, Brecha NC, Karschin C, DeBiasi S, Conti F. GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J Neurosci. 1995;15:7734–7746. doi: 10.1523/JNEUROSCI.15-11-07734.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudrickdonnon LA, Williams PJ, Pittman QJ, Macvicar BA. Postsynaptic potentials mediated by GABA and dopamine evoked in stellate glial-cells of the pituitary pars-intermedia. J Neurosci. 1993;13:4660–4668. doi: 10.1523/JNEUROSCI.13-11-04660.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller T, Fritschy JM, Grosche J, Pratt GD, Mohler H, Kettenmann H. Developmental regulation of voltage-gated K+ channel and GABAA receptor expression in Bergmann glial cells. J Neurosci. 1994;14:2503–2514. doi: 10.1523/JNEUROSCI.14-05-02503.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods. 2004;1:31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- Price CJ, Cauli B, Kovacs ER, Kulik A, Lambolez B, Shigemoto R, Capogna M. Neurogliaform neurons form a novel inhibitory network in the hippocampal CA1 area. J Neurosci. 2005;25:6775–6786. doi: 10.1523/JNEUROSCI.1135-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radian R, Ottersen OP, Storm-Mathisen J, Castel M, Kanner BI. Immunocytochemical localization of the GABA transporter in rat brain. J Neurosci. 1990;10:1319–1330. doi: 10.1523/JNEUROSCI.10-04-01319.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray M, Priestley JV. Differential expression of GABA transporter-1 messenger RNA in subpopulations of GABA neurones. Neurosci Lett. 1993;156:163–166. doi: 10.1016/0304-3940(93)90463-u. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Tong WMY, Brecha NC. GABA plasma membrane transporters, GAT-1 and GAT-3, display different distributions in the rat hippocampus. J Comp Neurol. 1996;367:595–606. doi: 10.1002/(SICI)1096-9861(19960415)367:4<595::AID-CNE9>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Riquelme R, Miralles CP, De Blas AL. Bergmann glia GABAA receptors concentrate on the glial processes that wrap inhibitory synapses. J Neurosci. 2002;22:10720–10730. doi: 10.1523/JNEUROSCI.22-24-10720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Ransom BR. Gap junctions equalize intracellular Na+ concentration in astrocytes. Glia. 1997;20:299–307. doi: 10.1002/(sici)1098-1136(199708)20:4<299::aid-glia3>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Rouach N, Glowinski J, Giaume C. Activity-dependent neuronal control of gap-junctional communication in astrocytes. J Cell Biol. 2000;149:1513–1526. doi: 10.1083/jcb.149.7.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkkonen ST, Mansikkamäki S, Möykkynen T, Lüddens H, Uusi-Oukari M, Korpi ER. Receptor subtype-dependent positive and negative modulation of GABAA receptor function by niflumic acid, a nonsteroidal anti-inflammatory drug. Mol Pharmacol. 2003;64:753–763. doi: 10.1124/mol.64.3.753. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol. 2005;562:9–26. doi: 10.1113/jphysiol.2004.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Proctor WR. Modulation of mammalian dendritic GABAA receptor function by the kinetics of Cl− and HCO3− transport. J Physiol. 1999;519(Pt 3):693–712. doi: 10.1111/j.1469-7793.1999.0693n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Tamas G, Buhl EH, Lorincz A, Somogyi P. Proximally targeted GABAergic synapses and gap junctions synchronize cortical interneurons. Nat Neurosci. 2000;3:366–371. doi: 10.1038/73936. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Steinhauser C. Ion channels in glial cells. Brain Res Brain Res Rev. 2000;32:380–412. doi: 10.1016/s0165-0173(99)00093-4. [DOI] [PubMed] [Google Scholar]
- Voipio J, Kaila K. GABAergic excitation and K+-mediated volume transmission in the hippocampus. Prog Brain Res. 2000;125:329–338. doi: 10.1016/S0079-6123(00)25022-X. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Dempsey RJ, Sun D. Expression of Na+-K+-Cl− cotransporter in rat brain during development and its localization in mature astrocytes. Brain Res. 2001;911:43–55. doi: 10.1016/s0006-8993(01)02649-x. [DOI] [PubMed] [Google Scholar]
- Zsiros V, Maccaferri G. Electrical coupling between interneurons with different excitable properties in the stratum lacunosum-moleculare of the juvenile CA1 rat hippocampus. J Neurosci. 2005;25:8686–8695. doi: 10.1523/JNEUROSCI.2810-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.