

Abstract
Hypomelanosis of the skin is a frequently encountered problem in childhood, being totally innocent or representing the first sign of a multisystem disorder. Medical history, clinical examination, Wood’s light investigation, histological analysis of the skin and a multidisciplinary consultation can contribute to a correct and early diagnosis of the different types of hypopigmentations. In the present paper, we present a systematic clinical approach to the differential diagnosis of those skin disorders.
Keywords: Depigmentation, hypomelanosis, hypopigmentation, review, vitiligo, Wood’s light
INTRODUCTION
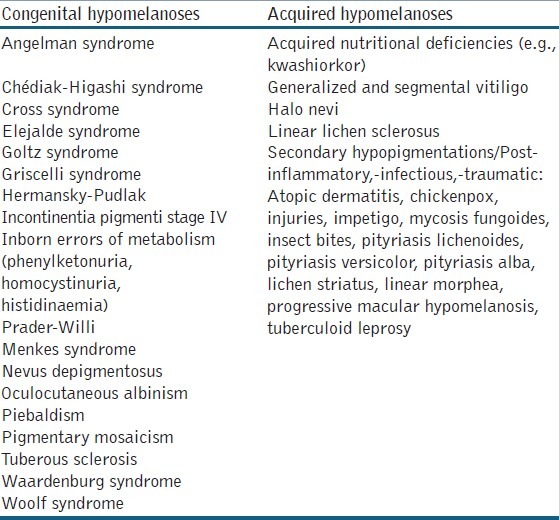
Cutaneous hypopigmentions enclose a wide range of disorders, which can be mainly differentiated based on the age of onset (congenital or acquired) [Table 1], the extent of involvement (diffuse or localized) and the underlying aetiology. Diffuse or generalized hypopigmentation in early childhood often have a genetic origin, in contrast to the acquired localized form at a later age. An adequate medical history and a thorough clinical evaluation are very useful tools to make a presumptive diagnosis, which can be followed by further diagnostic investigations. In routine clinical practice, the clinician can ask several key questions concerning age of onset (congenital or acquired), evolution of the lesions (stable or progressive), family history and associated disorders (e.g., autoimmune disorders, loss of sight or hearing, developmental disorders, neurological disorders, skeletal anomalies). In addition, other clinical features have been proven to be crucial in the differentiation of hypomelanoses: Subtype of pigment disorder (hypopigmentation, depigmentation or hyperpigmentation), distribution pattern and shape of the lesions (body location, unilateral or bilateral, solitary or multiple lesions, linear pattern, Blaschkoid distribution or leaf-like pattern) or other distinct clinical findings (anomalies of hair, eyes, teeth, nervous system or skeleton).[1] In this paper, we describe the differential diagnosis of hypomelanoses in children (ranging from birth to end of puberty).
Table 1.
Most common congenital and acquired hypomelanoses
WOOD’S LIGHT EXAMINATION
In diagnosing pigmentary disorders, Wood’s light examination plays an important role, distinguishing hypo- and depigmentation. While a decreased pigment production is reported as hypopigmentation, depigmentation has been defined as loss of pigment. In a similar way, partial lack of melanin is known as hypomelanosis while amelanosis is the total absence of melanin. In case of doubt, supplemental histological and electron microscopic investigations can be helpful.[2] Wood’s lamp inspection is particularly useful in lighter skin phototypes (I-II), and neonates due to a decreased pigment contrast under visible light.
DIFFUSE HYPOMELANOSES
Diffuse hypomelanoses of the total body surface in children often have an underlying genetic cause [Figure 1]. Although the number of melanocytes can be normal in this group of conditions, there is a reduced melanin production or a reduced transport and/or transfer of melanosomes to the keratinocytes. During Wood’s light examination, a hypopigmentation rather than a complete depigmentation is observed. Multiple possible mechanisms have been proposed to explain the reduced melanogenesis: Absence of tyrosinase activity (oculocutaneous albinism [OCA] 1A), reduced tyrosinase activity (OCA 1B), or impaired tyrosinase function caused by underlying metabolic disorders or nutritional deficiencies (copper, selenium or phenylalanine hydroxylase).[1]
Figure 1.
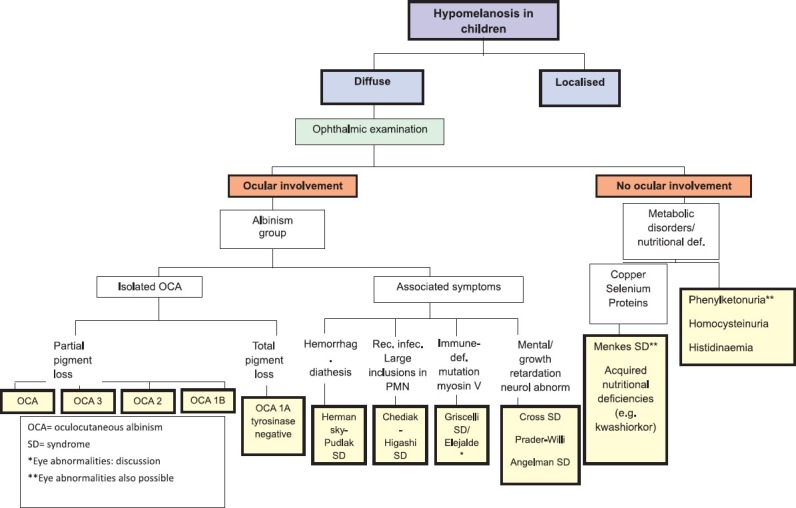
diffuse hypomelanoses in children. OCA= oculocutaneous albinism, SD= syndrome, *Eye abnormalities: discussion, **Eye abnormalities also possible
Albinism
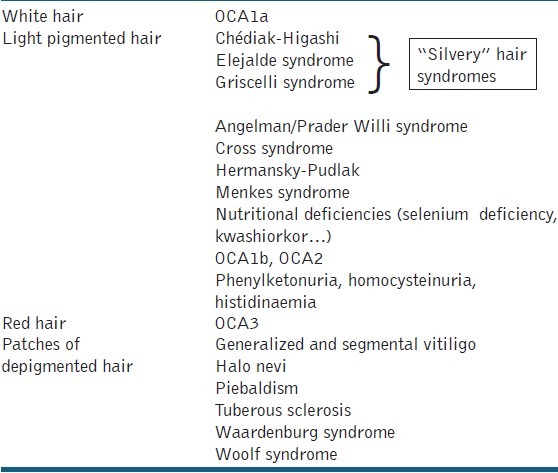
Two different types of albinism have been identified: Ocular (OA) and OCA albinism.[3] In OCA, the ocular manifestations (including nystagmus, photophobia and iris translucency) are often the first clue leading to the diagnosis. This clinical entity is divided into four subtypes with a wide variation in color of hair and skin, ranging from a pigmentless condition (OCA 1A) to subtle pigmentary dilution (OCA 1B-4). Genetic counseling to specify the subtype of OCA is recommended to determine the prognosis and evolution as well as the consequences in case of possible pregnancy. Besides a stringent ophthalmologic follow up, dermatological care should focus on the development of (pre) malignant lesions, sun protection and cosmetic advice (camouflage). In the differential diagnosis of OCA other more rare syndromes of albinism, requiring an internal and pediatric work-up, should be kept in mind.[4] These syndromes are characterized by defects in packaging of melanin and other cellular proteins, rather than by defects in melanin production seen in OCA. The Hermansky-Pudlak syndrome can be suspected if there are signs of an associated platelet dysfunction: repeated episodes of epistaxis, abnormal development of bruise. The lysosomal ceroid storage defect, which is another finding in the Hermansky-Pudlak syndrome, can cause pulmonary fibrosis, inflammatory bowel disease and kidney disease.[5] On the other hand, patients with the Chédiak-Higashi syndrome typically exhibit a silvery hair color, prolonged bleeding times, recurrent infections and large intracytoplasmic granules in the circulating polymorphonuclear cells [Table 2]. This syndrome should be differentiated from the Griscelli syndrome, which is a rare and often life-threatening autosomal recessive disorder characterized by hypopigmentation, immunological and neurological deficiencies.[6] Children with the Elejalde syndrome have a similar clinical presentation, with silvery hair and neurological impairment, but lack immunological problems.[7] Other genetic disorders with associated pigmentary dilution of the skin are Cross syndrome, Woolf syndrome, Prader-Willi and Angelman syndrome. Cross syndrome is a very rare oculocerebral syndrome defined by generalised hypopigmentation, psychomotor impairment, growth retardation and progressive neurological manifestations.[8] Woolf syndrome, also called Ziprkowski-Margolis or albinism-deafness syndrome, is an X-linked disorder (gene locus Xq26.3-q27.1). Affected boys have congenital neural deafness and a severe piebaldism-like phenotype.[9] Children with Prader-Willi syndrome present with infantile hypotonia, characteristic obesity, mental retardation, short stature and typical facial abnormalities; half of these patients have mild to moderate hypopigmentation. Hypopigmentation in Prader-Willi patients correlates with a deletion of the P gene on chromosome 15 which is also affected in OCA2. Reduced pigmentation of the skin is also a common feature in Angelman syndrome.[10]
Table 2.
Cutaneous hypomelanoses with localized or diffuse light hair
Inborn errors of metabolism and nutritional deficiencies
Certain inborn errors of metabolism can disturb melanin synthesis. Phenylketonuria is transmitted in an autosomal recessive manner and is caused by a deficiency of the enzyme phenylalanine hydroxylase, resulting in an accumulation of phenylalanine. The deficient melanin synthesis in infants with phenylketonuria results in very fair skin and hair. If the treatment (a lifelong low phenylalanine diet) is neglected, mental retardation, epilepsy and extra-pyramidal signs can arise.[11]
Selenium deficiency is associated with a diffuse loss of pigmentation of hair and skin, both returning to normal after adequate supplementation.[12] As tyrosinase is a copper-dependent enzyme in the melanin production process, hypopigmentation is a feature of copper deficiency, as observed in patients with the Menkes syndrome.[13]
Acquired nutritional deficiencies, with a lack of copper and selenium in particular, can cause a hypopigmentation of the skin, as well. In non-developing countries, these conditions can be seen in children receiving long-term parental nutrition or in case of insufficient nutrition intake (kwashiorkor). The low protein intake in kwashiorkor leads to specific clinical signs such as edema, and changes in hair and skin pigmentation. The skin lesions are initially erythematous to red-brown in color with marked desquamation. This can be followed by irregular or patchy skin discoloration (both hypomelanosis and hypermelanosis). The hypomelanosis usually begins on the face. Hypomelanotic kwashiorkor responds to dietary protein, although the skin is said to repigment slowly.
LOCALIZED HYPOMELANOSES
The differential diagnosis of localized hypomelanoses in children [Figure 2] can be based on the distinction between depigmentation and hypopigmentation, using Wood’s light examination. Furthermore, a careful medical history of the approximate time of onset can be helpful in differentiating congenital (e.g., piebaldism, Waardenburg syndrome) from acquired depigmentations (e.g., vitiligo). If a hypopigmentation is diagnosed with Wood’s light examination, clinical data about the number of lesions and their distribution will be most important to establish the final diagnosis.
Figure 2.
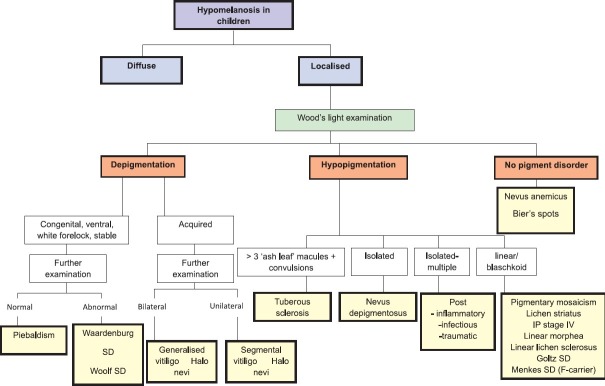
localised hypomelanoses in children. SD= syndrome
Depigmentation
Vitiligo
Vitiligo is undoubtedly one of the most familiar forms of localized hypomelanoses (amelanoses) [Figure 3a]. It is an acquired condition resulting from the progressive loss of melanocytes. The usual age of onset is often between the age of 20 years and 30 years. The estimated prevalence is 0.5–1%, without ethnic or gender preference. Depending on the pattern of skin lesions, which are typical milky white sharply demarcated maculae, vitiligo is divided into two different subtypes: generalized (non-segmental) vitiligo (bilateral maculae, often distributed in an acrofacial pattern or scattered symmetrically over the entire body) and segmental vitiligo (unilateral maculae in a segmental/band shape distribution). The segmental type of vitiligo is more often seen in children, as it starts generally earlier in life than the non-segmental one.[14] The diagnosis of vitiligo is essentially based on clinical examination because the lesions have a typical appearance. However, if the lesions are not distributed according to the classic vitiligo localizations, confusion with other hypomelanoses can arise. The presence of a family history for vitiligo, presence of Koebner phenomenon, of leukotrichia and associated autoimmune disorders such as thyroid disease are typical data by medical history that can support the diagnosis. On histological examination, a complete absence of melanocytes is reported, except in early lesions where some persistent melanocytes can be found. Current common conventional treatment options include topical steroids, topical calcineurin inhibitors or UV therapy, all of them often with variable results.
Figure 3.
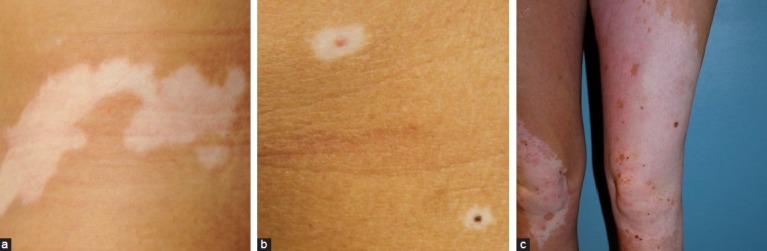
Depigmentation; (a) Vitiligo; (b) Halo nevi; (c) Piebaldism
The association between vitiligo and halo nevi is well established, although they can also be present separately. A halo nevus is a melanocytic nevus which acquires a surrounding depigmented rim, resulting ultimately in a complete regression of the nevus [Figure 3b]. Several reports have been published regarding the development of vitiligo simultaneously or shortly after the occurrence of a halo nevus. In our previous study, halo nevi were present in 31.1% of all vitiligo patients.[15]
Piebaldism
Piebaldism [Figure 3c] is a rare autosomal dominant disorder with an underlying defect of the tyrosine kinase transmembrane receptor on melanocytes, leading to an impaired embryonic migration and survival of melanocytes in the skin. It results from a mutation in the c-kit proto-oncogene, mapped to the proximal long arm of chromosome 4 (4q12) or from deletions in the SLUG gene, which is a zincfinger neural crest transcription factor. Recently reported cases of piebaldism, which are milder or more severe than genetically expected, indicate that other factors (e.g., a modifier gene of MC1R) influence skin and hair color.[16] Piebaldism is clinically characterized by congenital, extensive, symmetrically distributed depigmentations mainly on forehead, front of the thorax and extremities [Figure 3c]. The extent of the lesions is variable, ranging from only a midfrontal poliosis or white forelock (present in 80–90% of the patients) and minimal areas of depigmentation to a depigmentation of almost the entire body and hair. Piebaldism should be differentiated from the Waardenburg syndrome, which is also an autosomal dominant disorder with a similar clinical presentation. It is associated with heterochromia iridis, dystopia canthorum, congenital deafness and occasionally a congenital megacolon (Hirschsprung’s disease). The Waardenburg syndrome is an expression of a neurocristopathy, not only involving the melanocytes in the skin, but also those at the level of the eyes, hair, cochlea and meninges. Four subtypes have been identified. Another differential diagnosis is vitiligo as comparable depigmented maculae are observed. However, the predominantly ventral distribution of the lesions, congenital character, the stable course, the white forelock and the presence of hyperpigmented maculae within the areas of depigmentation are suggestive for piebaldism.[16] Nowadays surgical grafting techniques offer a good therapeutic option for stable depigmentations as in piebaldism or segmental vitiligo. Surprisingly good results (with a success rate > 90%) have been reported with autologous non-cultured epidermal cell transplantation [Figure 4a–b]. With this technique, only a small patch of the patient’s normally pigmented skin is required to treat large surfaces of depigmentation.[17,18]
Figure 4.
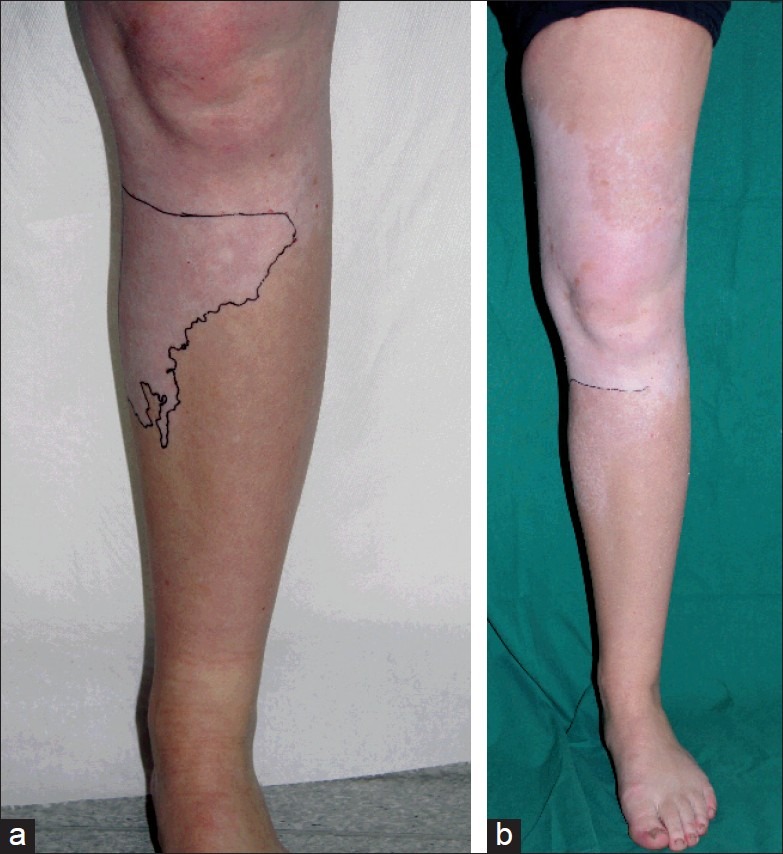
Piebaldism; (a) Before and (b) After treatment with non-cultured epidermal cell transplantation
Hypopigmentation
Nevoid hypomelanoses and nevus depigmentosus
A common localized hypomelanosis seen in children is the nevus depigmentosus, which becomes visible from birth or in the 1st year of life and does not change in shape [Figure 5a]. Especially in the 1st year of life, some confusion is possible with segmental or focal vitiligo. Skin inspection with Wood’s light examination is an essential part in the diagnosis. In contrast to what its name suggests, the nevus depigmentosus presents itself as a hypopigmentation rather than a depigmentation. Other factors discriminating the nevus depigmentosus from vitiligo are the absence of a marked sensitivity to sun exposure and the potential of slightly tanning. According to some authors, there are three subtypes within the group of the nevi depigmentosi: an isolated form (solitary and well defined lesions), a segmental form (unilateral, band-shaped lesions, sometimes Blaschkoid distribution) and a systematized from (extensive whorls and streaks of hypopigmentation, following the lines of Blaschko [hypomelanosis of Ito or pigmentary mosaicism]).[19] The theory of Happle postulates that all linear hypo- and depigmented maculae following the lines of Blaschko are a consequence of mosaicism, even though a chromosomal mosaicism could not always be demonstrated.[20] Whenever this blaschkoid pattern is seen, neurological and musculo-skeletal abnormalities should be suspected. More recently it has been suggested that pigmentary mosaicism (of the ‘hypomelanosis of Ito’ type) can be regarded as a continuum of different subtypes, amongst which the pigmentary disorder can be present with and without associated neurological anomalies. This approach is contrary to the ancient view, in which the associated conditions were used as a condition sine qua non to diagnose ‘hypomelanosis of Ito’. If present, the neurological anomalies usually manifest themselves within the 1st years of life. A recent study demonstrated that in 92.3% of patients with a linear hypo-or hyperpigmentation following the lines of Blaschko, the associated neurological disorder was detected before the age of two.[21] Developmental disorders and epilepsy were the most reported problems. According to this study, neurological defects were more frequently observed in linear hyperpigmentations compared to linear hypopigmentations. There is no consensus in literature about the prevalence of neurological symptoms associated with ‘hypomelanosis of Ito’ as different definitions are being used. This percentage however, does appear to be higher in more disseminated forms, probably due to an earlier mutation in the embryogenesis compared to the localized nevus depigmentosus. A subsequent neurological, pediatric and genetic work-up seems to be most appropriate in children under the age of two, presenting with a linear and/or Blaschkoid pattern of hypopigmentation. In asymptomatic children older than two, watchful waiting seems to be an acceptable strategy. More recently phylloid hypomelanoses has been described by Happle et al. as another pattern hypopigmentation.[22] It is a rare neurocutaneous syndrome characterized by hypopigmented leaf-like or oblong macules reminiscent of floral ornaments. Associated extracutaneous anomalies include cerebral, Ocular, and skeletal defects. It has been suggested that this phenotype originates from mosaic partial or complete trisomy 13. Other entities to be considered in the differential diagnosis of linear hypomelanoses in children are lichen striatus, linear scleroderma, linear lichen sclerosis, post-inflammatory or traumatic changes, incontinentia pigmenti stage IV and the Goltz-syndrome. In a solitary rather round/oval lesion, distinction must be made with a nevus anaemicus, which is characterized by a normal presence of melanocytes and melanin and is based on vascular disturbances.
Figure 5.
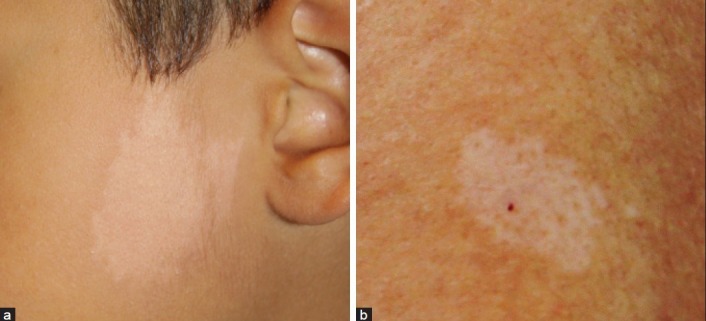
Hypopigmentation; (a) Naevus depigmentosus; (b) ‘Ash leaf’ macula in tuberous sclerosis
Tuberous sclerosis complex
Tuberous sclerosis is an autosomal dominant condition (mutation in tuberous sclerosis protein 1 gene [TSC1] and TSC2) resulting in a hyperplasia of ectodermal and mesodermal cells.[23] This rare multisystem genetic disease is characterized by the triad of seizures, mental retardation and hypopigmented oval (‘ash leaf spots’) or dot-like (‘confetti’) maculae [Figure 5b]. Studies have shown that the likelihood of tuberous sclerosis is significantly higher if the patient has more than three of these hypopigmented maculae. The associated hamartomas found in several organs (brain, eyes, heart, kidneys, lungs and skin) are also typical. In the skin, angiofibromas in the face, periungual fibromas, and presence of a ‘shagreen’ patch can be found. The conditions associated with tuberous sclerosis generally become apparent between the age of five and late puberty. The decision of screening depends largely on the number of hypopigmented maculae as well as of the presence of other traits of the tuberous sclerosis complex. Additional investigations consist of neurological and ophthalmologic examinations, cranial computerized tomography and/or nuclear magnetic resonance (to look at the presence of calcifications). A renal and cardial echography should be performed to detect renal angiomyolipomas and cardial rhabdomyomas, which are diagnosed in 90% of tuberous sclerosis patients under the age of two. Finally, molecular analysis of the two tuberous sclerosis genes plays an important role in the diagnosis. Other entities to be considered in the differential diagnosis of oval round macular hypomelanoses are pityriasis alba, vitiligo, post-inflammatory hypopigmentation (atopic dermatitis, pityriasis rosea, pityriasis lichenoides chronica), naevus anaemicus and lichen sclerosus.[24,25]
Secundairy hypopigmentations and hypopigmentations without pigment disorders
An acquired diminution or loss of melanin in the skin is detected in patients with post-inflammatory hypomelanosis, which is influenced by inter-individual differences in sensitivity. The hypopigmentation can be a consequence of an impaired transfer of melanin to the keratinocytes secondary to the inflammatory process or the result of application of potent topical steroids.
Pityriasis versicolor, caused by yeasts of the Malassezia genus, is one of the most common yeast infection associated with pigmentary changes. Well-demarcated finely scaling patches, hyper- or hypopigmentations are found on clinical examination. If the lesions are hypopigmented, the name ‘pityriasis versicolor alba’ has been used. As most Malassezia species are lipid-dependent, pytiriasis versicolor is commonly seen in adolescence, associated with a high sebaceous activity in the skin. The clinical picture is often suggestive enough to establish the diagnosis, though confirmation by microscopy can be performed. A yellow-green fluorescence may be observed on examination of affected areas with Wood’s light.[26] Pityriasis alba is another common dermatosis characterized by hypopigmentation, presenting with pale white, well to moderate defined, very slightly scaling plaques. A relationship with sun exposure, xerosis cutis and atopy has been suggested. This disease is usually reported in children between 6 years and 16 years old, with lesions typically occurring in the face and upper arms.[27] Lichen striatus is an asymptomatic linear dermatosis that occurs mostly in children and has been reported to follow the lines of Blaschko. The primary lesions (small, flat, skin-colored to pink papules) can disappear spontaneously after several months or years, often leaving a linear macular hypopigmentation (post-inflammatory).[11] Other skin infections/inflammations like impetigo and chickenpox, tuberculoid leprosy, insect bites, atopic dermatitis, physical agents such as burns or minor injuries or pytiriasis lichenoides chronica, hypopigmented mycosis fungoides and progressive macular hypomelanosis can also cause secondary or post-inflammatory hypopigmentation.[28] The latter four disorders usually manifest at later age, but can be present during puberty or adolescence.
Hypopigmentation without pigment disorder
Naevus anaemicus is a solitary hypopigmented lesion, characterized by well-defined, irregularly shaped, confluent maculae. On histological examination, a normal presence of melanocytes and melanin is seen. This lesion is present from birth and most often located on the trunk. The underlying mechanism is an aberrant, hypersensitive response to catecholamines, resulting in pallor of the skin. Hence, it has been called a pharmacological naevus. When the skin in these lesions is rubbed, no red coloring is observed as there is no vasodilatation possible.[29] A similar pale skin can be observed in Bier’s spots, which is also linked to vascular changes. They are usually found on the arms and legs of young adults as small, hypopigmented macules. The intervening skin may seem erythematous but blanches with pressure so that the pale macules disappear. This is in general a physiological vascular anomaly.
CONCLUSION
As hypomelanoses of the child can fit a wide range of pigment disorders, this review focused on the practical approach and differential diagnosis. Information about age of onset, distribution of lesions and degree of pigment loss should lead the clinician in routine clinical practice. Additional neurological, pediatric and genetic research can be important to detect associated conditions.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Tey HL. A practical classification of childhood hypopigmentation disorders. Acta Derm Venereol. 2010;90:6–11. doi: 10.2340/00015555-0794. [DOI] [PubMed] [Google Scholar]
- 2.Gupta LK, Singhi MK. Wood’s lamp. Indian J Dermatol Venereol Leprol. 2004;70:131–5. [PubMed] [Google Scholar]
- 3.Grønskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis. 2007;2:43. doi: 10.1186/1750-1172-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheinfeld NS. Syndromic albinism: A review of genetics and phenotypes. Dermatol Online J. 2003;9:5. [PubMed] [Google Scholar]
- 5.Thielen N, Huizing M, Krabbe JG, White JG, Jansen TJ, Merle PA, et al. Hermansky-Pudlak syndrome: The importance of molecular subtyping. J Thromb Haemost. 2010;8:1643–5. doi: 10.1111/j.1538-7836.2010.03898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reddy RR, Babu BM, Venkateshwaramma B, Hymavathi Ch. Silvery hair syndrome in two cousins: Chediak-Higashi syndrome vs Griscelli syndrome, with rare associations. Int J Trichology. 2011;3:107–11. doi: 10.4103/0974-7753.90825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambert J, Vancoillie G, Naeyaert JM. Elejalde syndrome revisited. Arch Dermatol. 2000;136:120–1. doi: 10.1001/archderm.136.1.120. [DOI] [PubMed] [Google Scholar]
- 8.Tezcan I, Demir E, Aþan E, Kale G, Müftüoðlu SF, Kotiloðlu E. A new case of oculocerebral hypopigmentation syndrome (Cross syndrome) with additional findings. Clin Genet. 1997;51:118–21. doi: 10.1111/j.1399-0004.1997.tb02432.x. [DOI] [PubMed] [Google Scholar]
- 9.Shiloh Y, Litvak G, Ziv Y, Lehner T, Sandkuyl L, Hildesheimer M, et al. Genetic mapping of X-linked albinism-deafness syndrome (ADFN) to Xq26.3-q27.1. Am J Hum Genet. 1990;47:20–7. [PMC free article] [PubMed] [Google Scholar]
- 10.Spritz RA, Bailin T, Nicholls RD, Lee ST, Park SK, Mascari MJ, et al. Hypopigmentation in the Prader-Willi syndrome correlates with P gene deletion but not with haplotype of the hemizygous P allele. Am J Med Genet. 1997;71:57–62. doi: 10.1002/(sici)1096-8628(19970711)71:1<57::aid-ajmg11>3.0.co;2-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruiz-Maldonado R. Hypomelanotic conditions of the newborn and infant. Dermatol Clin. 2007;25:373–82. ix. doi: 10.1016/j.det.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Vinton NE, Dahlstrom KA, Strobel CT, Ament ME. Macrocytosis and pseudoalbinism: Manifestations of selenium deficiency. J Pediatr. 1987;111:711–7. doi: 10.1016/s0022-3476(87)80247-0. [DOI] [PubMed] [Google Scholar]
- 13.Ishizaki H, Spitzer M, Wildenhain J, Anastasaki C, Zeng Z, Dolma S, et al. Combined zebrafish-yeast chemical-genetic screens reveal gene-copper-nutrition interactions that modulate melanocyte pigmentation. Dis Model Mech. 2010;3:639–51. doi: 10.1242/dmm.005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Geel N, Mollet I, Brochez L, Dutré M, De Schepper S, Verhaeghe E, et al. New insights in segmental vitiligo: Case report and review of theories. Br J Dermatol. 2012;166:240–6. doi: 10.1111/j.1365-2133.2011.10650.x. [DOI] [PubMed] [Google Scholar]
- 15.van Geel N, Vandenhaute S, Speeckaert R, Brochez L, Mollet I, De Cooman L, et al. Prognostic value and clinical significance of halo naevi regarding vitiligo. Br J Dermatol. 2011;164:743–9. doi: 10.1111/j.1365-2133.2010.10154.x. [DOI] [PubMed] [Google Scholar]
- 16.Oiso N, Fukai K, Kawada A, Suzuki T. Piebaldism. J Dermatol. 2012 doi: 10.1111/j.1346-8138.2012.01583.x. In press. [DOI] [PubMed] [Google Scholar]
- 17.van Geel N, Ongenae K, De Mil M, Haeghen YV, Vervaet C, Naeyaert JM. Double-blind placebo-controlled study of autologous transplanted epidermal cell suspensions for repigmenting vitiligo. Arch Dermatol. 2004;140:1203–8. doi: 10.1001/archderm.140.10.1203. [DOI] [PubMed] [Google Scholar]
- 18.van Geel N, Wallaeys E, Goh BK, De Mil M, Lambert J. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186–93. doi: 10.1111/j.1365-2133.2010.10014.x. [DOI] [PubMed] [Google Scholar]
- 19.Di Lernia V. Segmental nevus depigmentosus: Analysis of 20 patients. Pediatr Dermatol. 1999;16:349–53. doi: 10.1046/j.1525-1470.1999.00091.x. [DOI] [PubMed] [Google Scholar]
- 20.Happle R. Mosaicism in human skin. Understanding the patterns and mechanisms. Arch Dermatol. 1993;129:1460–70. [PubMed] [Google Scholar]
- 21.Pinheiro A, Mathew MC, Thomas M, Jacob M, Srivastava VM, Cherian R, et al. The clinical profile of children in India with pigmentary anomalies along the lines of Blaschko and central nervous system manifestations. Pediatr Dermatol. 2007;24:11–7. doi: 10.1111/j.1525-1470.2007.00325.x. [DOI] [PubMed] [Google Scholar]
- 22.Happle R. Phylloid hypomelanosis is closely related to mosaic trisomy 13. Eur J Dermatol. 2000;10:511–2. [PubMed] [Google Scholar]
- 23.Tomasoni R, Mondino A. The tuberous sclerosis complex: Balancing proliferation and survival. Biochem Soc Trans. 2011;39:466–71. doi: 10.1042/BST0390466. [DOI] [PubMed] [Google Scholar]
- 24.Thoma W, Krämer HJ, Mayser P. Pityriasis versicolor alba. J Eur Acad Dermatol Venereol. 2005;19:147–52. doi: 10.1111/j.1468-3083.2004.01085.x. [DOI] [PubMed] [Google Scholar]
- 25.Kohrman MH. Emerging treatments in the management of tuberous sclerosis complex. Pediatr Neurol. 2012;46:267–75. doi: 10.1016/j.pediatrneurol.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Hu SW, Bigby M. Pityriasis versicolor: A systematic review of interventions. Arch Dermatol. 2010;146:1132–40. doi: 10.1001/archdermatol.2010.259. [DOI] [PubMed] [Google Scholar]
- 27.Blessmann Weber M, Sponchiado de Avila LG, Albaneze R, Magalhães de Oliveira OL, Sudhaus BD, Cestari TF. Pityriasis alba: A study of pathogenic factors. J Eur Acad Dermatol Venereol. 2002;16:463–8. doi: 10.1046/j.1468-3083.2002.00494.x. [DOI] [PubMed] [Google Scholar]
- 28.Ruiz-Maldonado R, Orozco-Covarrubias ML. Postinflammatory hypopigmentation and hyperpigmentation. Semin Cutan Med Surg. 1997;16:36–43. doi: 10.1016/s1085-5629(97)80034-x. [DOI] [PubMed] [Google Scholar]
- 29.Ahkami RN, Schwartz RA. Nevus anemicus. Dermatology. 1999;198:327–9. doi: 10.1159/000018169. [DOI] [PubMed] [Google Scholar]