

Background: Somatostatin regulates gut function via neuronal somatostatin receptors.
Results: Somatostatin susceptibility to degradation by endosomal endothelin-converting enzyme 1 (ECE-1) defines receptor function.
Conclusion: ECE-1 regulates the duration of somatostatin receptor signaling and trafficking.
Significance: Therapeutic somatostatin analogs are ECE-1-resistant, which underlies their prolonged actions.
Keywords: Arrestin, Endocytosis, Neurons, Neuropeptide, Receptor Recycling, Endothelin-converting Enzyme, Somatostatin
Abstract
Somatostatin (SST) 14 and SST 28 activate somatostatin 2A receptors (SSTR2A) on enteric neurons to control gut functions. SST analogs are treatments of neuroendocrine and bleeding disorders, cancer, and diarrhea, with gastrointestinal side effects of constipation, abdominal pain, and nausea. How endogenous agonists and drugs differentially regulate neuronal SSTR2A is unexplored. We evaluated SSTR2A trafficking in murine myenteric neurons and neuroendocrine AtT-20 cells by microscopy and determined whether agonist degradation by endosomal endothelin-converting enzyme 1 (ECE-1) controls SSTR2A trafficking and association with β-arrestins, key regulators of receptors. SST-14, SST-28, and peptide analogs (octreotide, lanreotide, and vapreotide) stimulated clathrin- and dynamin-mediated internalization of SSTR2A, which colocalized with ECE-1 in endosomes and the Golgi. After incubation with SST-14, SSTR2A recycled to the plasma membrane, which required active ECE-1 and an intact Golgi. SSTR2A activated by SST-28, octreotide, lanreotide, or vapreotide was retained within the Golgi and did not recycle. Although ECE-1 rapidly degraded SST-14, SST-28 was resistant to degradation, and ECE-1 did not degrade SST analogs. SST-14 and SST-28 induced transient interactions between SSTR2A and β-arrestins that were stabilized by an ECE-1 inhibitor. Octreotide induced sustained SSTR2A/β-arrestin interactions that were not regulated by ECE-1. Thus, when activated by SST-14, SSTR2A internalizes and recycles via the Golgi, which requires ECE-1 degradation of SST-14 and receptor dissociation from β-arrestins. After activation by ECE-1-resistant SST-28 and analogs, SSTR2A remains in endosomes because of sustained β-arrestin interactions. Therapeutic SST analogs are ECE-1-resistant and retain SSTR2A in endosomes, which may explain their long-lasting actions.
Introduction
G protein-coupled receptors (GPCRs)4 for peptide hormones and neurotransmitters control diverse pathophysiological processes and are a major therapeutic target. Diverse endogenous agonists and drugs can induce distinct conformations of the same receptor molecule, leading to divergent signaling and physiological outcomes. This “agonist-biased” signaling has been mostly examined in model systems treated with exogenous agonists. The ability of diverse endogenous peptides to differentially regulate receptors in the nervous system is largely unexplored.
GPCR signaling must be tightly controlled because dysregulation can cause disease. Signaling by bioactive peptides is controlled by mechanisms that regulate the activity of peptides and their receptors. Cell surface endopeptidases (e.g. neprilysin) degrade and inactivate peptides in the extracellular fluid and, thereby, attenuate their effects (1). β-arrestins interact with agonist-occupied receptors that uncouple receptors from heterotrimeric G proteins and terminate plasma membrane signaling. By recruiting receptors and signaling partners to endosomal signalosomes, β-arrestins also transmit signals from internalized receptors (2). Endosomal peptidases (e.g. endothelin-converting enzyme 1, ECE-1) degrade neuropeptides in endosomes, which disassembles signalosomes, terminates endosomal signaling, and promotes receptor recycling and resensitization of plasma membrane signaling (3). These processes are key regulators of peptide signaling. However, the capacity of these mechanisms to differentially regulate signaling by different endogenous or exogenous agonists of the same GPCR has not been examined.
We evaluated how endogenous and exogenous agonists differentially regulate somatostatin (SST) receptor 2A (SSTR2A). The cyclic peptides SST-14 and N-terminally extended SST-28 are endogenous agonists of SSTR2A that are produced by enteric neurons and enteroendocrine cells (4, 5). SSTR2A is a major mediator of the pan-inhibitory actions of SST in the gastrointestinal tract. SSTR2A is prominently localized to enteric neurons (5, 6), where activation stimulates K+ channels, leading to hyperpolarization and suppression of transit and secretion (7, 8). SSTR2A is also an important therapeutic target. Metabolically stable SST analogs (e.g. octreotide) are effective treatments for acromegaly, diarrhea, bleeding disorders, and cancer, and radiolabeled agonists can be used to detect neuroendocrine tumors (4). However, these drugs have side effects related to their actions on the gastrointestinal tract, including constipation, abdominal cramps, and nausea (4).
SST-14 induces SSTR2A endocytosis, which depends on sustained interactions with β-arrestins, followed by recycling, which requires ECE-1-dependent SST-14 degradation in endosomes (9). However, the regulation of SSTR2A by different endogenous peptides and therapeutic agonists in functionally relevant cells has not been studied. By examining SSTR2A trafficking in primary enteric neurons and model cell lines, we report that endogenous and exogenous agonists, including clinically used analogs, differentially regulate SSTR2A. Differential regulation depends on agonist susceptibility to degradation by the endosomal peptidase ECE-1, which associates with SSTR2A and governs the duration of SSTR2A and β-arrestin interactions. Thus, although resistance to extracellular proteolysis is important for development of SST analogs, resistance to intracellular proteolysis is a key determinant of their long-lasting therapeutic and side effects.
EXPERIMENTAL PROCEDURES
SSTR2A Trafficking in Myenteric Neurons
Experimental procedures on animals were approved by institutional animal care and use committees. C57BL/6 mice (male and female, 20–25 g) and Sprague-Dawley rats (6 weeks, 200–250 g) (Charles River Laboratories, Hollister, CA) were killed with sodium pentobarbital (200 mg/kg intraperitoneally) and bilateral thoracotomy. Organotypic preparations of whole-thickness cecum or colon were stabilized for 1 h in modified physiological saline solution as described (10). Tissues were incubated with the following SSTR2A agonists (1 h, 4 °C): endogenous peptides (SST-14 and SST-28), peptide analogs (octreotide, lanreotide, and vapreotide), and a non-peptide agonist (L-054,264) (1–1000 nm). Tissues were washed and incubated without agonists (0–120 min, 37 °C) to allow receptor trafficking to proceed. In some experiments, tissues were treated with hypertonic sucrose (0.45 m, blocks clathrin), dynasore (80 μm, blocks dynamin GTPase), SM-19712 (ECE-1 inhibitor, 10 μm), bafilomycin A1 (vacuolar H+-ATPase inhibitor, 1 μm), brefeldin A (disrupts Golgi, 10 μm), or vehicle (1–2 h preincubation, inclusion in all solutions). At defined times, tissues were fixed (4% paraformaldehyde, 100 mm PBS (pH 7.4), overnight, 4 °C), washed in PBS, and then wholemounts of the longitudinal muscle-myenteric plexus were prepared (10). Wholemounts were incubated with the following primary antibodies (overnight, 4 °C): rabbit anti-SSTR2A (9431 (6), 1:2000), goat anti-nitric oxide synthase (Novus Biologicals, Littleton, CO, 1:500), goat anti-ECE-1 (R&D Systems, Minneapolis, MN, 1:100), and sheep anti-TGN38 (Novus Biologicals, 1:100). Preparations were washed; incubated with donkey anti-rabbit, anti-goat, or anti-sheep IgG conjugated to FITC or rhodamine red X (Jackson ImmunoResearch Laboratories, Westgrove, PA, 1:200, 2 h, room temperature); and mounted in Prolong Gold (Invitrogen).
SSTR2A Trafficking in Neuroendocrine Cells
AtT-20 cells, a neuroendocrine tumor cell line from mouse pituitary, endogenously express SSTR2A (11). AtT-20 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin (50 units/ml), and streptomycin (50 μg/ml) (37 °C, 5% CO2). Cells were transfected with the GFP-tagged ECE-1a–d isoforms (3). Endogenous SSTR2A was detected by indirect immunofluorescence. Receptor trafficking in response to SSTR2A agonists was studied as described (9).
Image Analysis
Specimens were examined using a Zeiss LSM510 META confocal microscope (10). Plasma membrane and intracellular SSTR2A immunoreactivity (IR or immunoreactive) was quantified using ImageJ (10).
Peptide Degradation
SST-14, SST-28, lanreotide, or vapreotide (250 μm) was incubated with human ECE-1 (R&D Systems, 200 nm) in 50 mm MES-KOH (pH 5.5), or 50 mm Tris-HCl (pH 7.4) (0–240 min, 37 °C). Products were separated by high-pressure liquid chromatography (3). To determine kinetics of hydrolysis at specific sites, we used proteinase activity labeling employing 18O-enriched water (12, 13). SST-14 or SST-28 (100 μm) were incubated with ECE-1 (100 nm) in buffers containing 1:1 (v/v) H218O (Sigma-Aldrich, St. Louis, MO) (0–48 h, room temperature). Reaction products were sampled repeatedly by spotting aliquots onto a MALDI target plate with α-cyano-4-hydroxycinnamic acid (5 mg/ml in 50% acetonitrile, 0.1% trifluoroacetic acid, 1:1 (v/v) ratio). Disulfide bonds were reduced by incubating samples with dithiothreitol (50 mm final) in 25 mm NH4HCO3 (pH 8.0) (1 h, room temperature, dark). Samples were analyzed using a MALDI-TOF/TOF mass spectrometer (4800 proteomics analyzer, Applied Biosystems, Bedford, MA). Peptide identities were confirmed by MS/MS sequencing, and 18O-incorporation ratios were determined as described previously (12).
Bioluminescence Resonance Energy Transfer (BRET) SSTR2A/β-Arrestin2 and SSTR2A/ECE-1 Interaction Assays
cDNAs encoding human SSTR2A flanked by 5′ BamHI and 3′ NotI restriction sites were generated by PCR using Phusion high-fidelity PCR (New England Biosciences, Ipswich, MA) with the following primers: SSTR2A, CGCGGATCCATGGACATGGCGGATGAGC (sense) and ATAGTTTAGCGGCCGCAGGATACTGGTTTGGAGGTC (antisense). PCR products were digested with BamHI and NotI (New England Biosciences) and ligated into pcDNA3-RLuc8 using the BamHI and NotI sites. Human β-arrestin1 or 2 with C-terminal YFP constructs have been described (14). HEK-293 cells, grown as described for AtT-20 cells, were transiently transfected with human SSTR2A-RLuc8 (1 μg) and β-arrestin-YFP or pcDNA3 (control) (4 μg) and plated in a poly-D-lysine-coated 96-well plate. After 48 h, cells were incubated in Hanks' balanced salt solution with the luciferase substrate coelenterazine H (Promega, Madison, WI, 5 μm final) (5 min, 37 °C). Luminescence from RLuc8 (480 nm) and fluorescence from YFP (530 nm) were measured (LUMIstar Omega, BMG Labtech, Mornington, Australia) for a 2-min basal period and at various times after stimulation with SST-14, SST-28, or octreotide (10−12-10−6 m). In some experiments, cells were treated with ECE-1 inhibitor (SM19712, 10 μm, 1 h preincubation and inclusion throughout) or vehicle (control). The interaction between SSTR2A and the ECE-1a/d isoforms was measured as described above using the SSTR2A-RLuc8 construct cotransfected with ECE-1a/d N-terminally tagged with YFP (YFP-ECE-1a/d).
SSTR2A Agonists and Drugs
SST-14, octreotide, lanreotide, and vapreotide (Bachem, Torrance, CA); SST-28 and (1R,1′S,3′R/1R,1′R,3′S)-L-054,264 (L-054,264 (15)) (Tocris Bioscience, Minneapolis, MN); SM-19712, brefeldin A, and dynasore (Sigma-Aldrich); and bafilomycin A1 (AG Scientific, San Diego, CA) were used.
Statistical Analysis
Data are expressed as mean ± S.E. and were analyzed using Student's t test or one-way analysis of variance and Dunnett's multiple comparison test. p < 0.05 was considered significant.
RESULTS
Agonists Variably Stimulate Clathrin- and Dynamin-dependent Endocytosis of SSTR2A in Myenteric Neurons
We examined the effects of endogenous agonists (SST-14 and SST-28), octapeptide analogs (octreotide, lanreotide, and vapreotide), and a non-peptide agonist (L-054,264) on the subcellular distribution of SSTR2A in myenteric neurons. Tissues were incubated with agonists (60 min, 4 °C) to allow binding and then washed and incubated in agonist-free buffer (0–120 min, 37 °C) to enable receptor trafficking. In preparations treated with vehicle (not shown) or incubated with agonists at 4 °C, SSTR2A-IR was localized to the plasma membrane of the soma and neurites of nitric oxide synthase-IR neurons (Fig. 1A). Quantitation of the subcellular distribution of SSTR2A in the soma indicated that 87 ± 2% of SSTR2A-IR was at the plasma membrane and 14 ± 2% was internalized. After incubation with SST-14 (100 nm) and warming to 37 °C, SSTR2A-IR internalized within 10 min (not shown) and was most apparent after 30 min, when SSTR2A-IR was detected in juxtanuclear organelles resembling the Golgi apparatus (Fig. 5) and in endosomes (Fig. 4) of lamellar dendrites and neurites (Figs. 1A and 2A). SST-14 (1–1000 nm) stimulated a concentration-dependent loss of SSTR2A-IR from the plasma membrane, which mirrored its intracellular accumulation (Fig. 1, A and B). Although SST-28, octreotide, lanreotide, vapreotide, and L-054,264 also stimulated SSTR2A endocytosis, quantitation revealed differences in the extent of internalization after incubation with an equivalent agonist concentration (100 nm) (Figs. 1C and 2, A–E). The proportion of total cellular SSTR2A-IR remaining at the plasma membrane after 30 min was similar for SST-14 (56 ± 3%), SST-28 (59 ± 2%), and L-054,264 (59 ± 3%) but was significantly more than for octreotide (37 ± 3%), lanreotide (36 ± 2%), and vapreotide (36 ± 2%) (all p < 0.0001 to SST-14). Thus, peptide analogs are more effective at stimulating SSTR2A endocytosis than SST-14/28 or a non-peptide agonist.
FIGURE 1.

SSTR2A endocytosis is concentration-dependent in myenteric neurons. A, SSTR2A-IR was localized to the plasma membrane of the soma and neurites (arrowheads) in untreated nitric oxide synthase-IR neurons (0 min). SSTR2A-postive pixels above threshold are shown in the right column. SST-14 (1–1000 nm, 60 min, 4 °C, wash, 30 min recovery, 37 °C) induced a concentration-dependent internalization of SSTR2A-IR (arrows). Scale bars = 10 μm. B, quantitative image analysis of intracellular SSTR2A-IR (INT) in response to graded concentrations of SST-14. C, comparison of relative intracellular SSTR2A following internalization to different SSTR2A agonists (all 100 nm). Receptor distribution at 30 and 120 min recovery times are shown. *, p < 0.1; ***, p < 0.001 to 0 min control.
FIGURE 5.

SSTR2A traffics to the TGN and recycles in myenteric neurons. A, octreotide induced SSTR2A-IR trafficking from the plasma membrane (arrowheads, 0 min) to TGN38-IR structures (arrows, 30 min). B, brefeldin A disrupted the TGN and caused a redistribution of internalized SSTR2A-IR to multiple vesicles. C, brefeldin A did not affect SST-14-stimulated endocytosis of SSTR2A-IR (arrow, 30 min) but caused intracellular retention of SSTR2A-IR up to 120 min (arrows). Scale bars = 10 μm. NOS, nitric oxide synthase. D, column graph showing quantitative analysis of receptor distribution. PM, plasma membrane-associated; INT, internalized; BFA, brefeldin A. ***, p < 0.001 to vehicle control.
FIGURE 4.

ECE-1 regulates SSTR2A recycling in myenteric neurons. A, in neurons treated with the ECE-1 inhibitor SM-19712 (10 μm), SST-14 caused internalization of SSTR2A-IR, which was retained even after 120-min recovery (arrows). In vehicle-treated neurons, SSTR2A-IR was fully recycled after 120-min recovery (arrowheads). NOS, nitric oxide synthase. B, column graph showing quantitative analysis of receptor distribution. PM, plasma membrane-associated; INT, internalized; SM, SM-19712. ***, p < 0.001 to vehicle control. C, bafilomycin A1 similarly inhibited SSTR2A-IR recycling at 120 min (arrow). D, ECE-1-IR was present in SSTR2A-IR myenteric neurons of the rat distal colon. In untreated preparations (0 min), there was no colocalization of ECE-1-IR and SSTR2A-IR (arrowheads). SST-28 induced trafficking of SSTR2A-IR to ECE-1-IR intracellular structures within the soma (arrows). Scale bars = 10 μm.
FIGURE 2.

Agonists induce endocytosis of SSTR2A in myenteric neurons. Photomicrographs show the localization of SSTR2A-IR in nitric oxide synthase-IR myenteric neurons. The threshold image shows SSTR2A-positive staining in white. Arrowheads show SSTR2A-IR at the plasma membrane, and arrows show internalized SSTR2A-IR. Column graphs show the proportion of SSTR2A-IR in the soma at the plasma membrane (PM) and internalized (INT). Whole-mounts were incubated with agonists, washed, and recovered for the indicated times at 37 °C. A, SST-14-induced trafficking of SSTR2A-IR. In untreated neurons (0 min), SSTR2A-IR was localized to the plasma membrane of the soma and neurites. After incubation with SST-14, washing, and recovery, SSTR2A-IR internalized (30 min), and then recycled (60–120 min). NOS, nitric oxide synthase. B, SST-28 also induced SSTR2A-IR endocytosis, but the receptor did not fully recycle after 120 min. C, L-054,264 stimulated transient SSTR2A-IR internalization (30 min) followed by a complete recycling within 60 min. D and E, octreotide and vapreotide induced sustained internalization with no detectable recycling. Scale bars = 10 μm. ***, p < 0.001 versus 0 min.
To evaluate the mechanism of endocytosis, we treated neurons with hypertonic sucrose or dynasore, which, respectively, block clathrin- and dynamin-dependent endocytosis (16). Both treatments inhibited SSTR2A endocytosis stimulated by SST-14 (not shown) and octreotide (Fig. 3, A and B). Sucrose or dynasore had no effect on the subcellular localization of SSTR2A in unstimulated neurons (not shown). Thus, clathrin and dynamin mediate agonist-stimulated endocytosis of SSTR2A in myenteric neurons.
FIGURE 3.
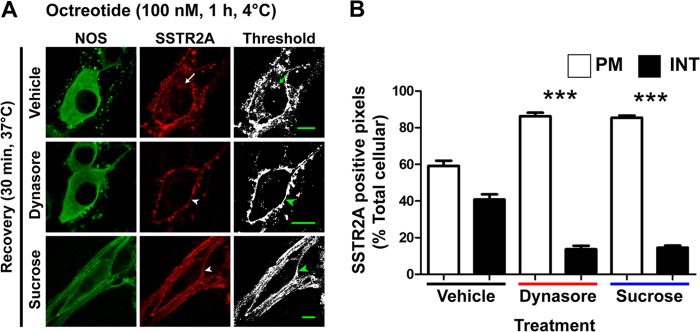
Agonists induce dynamin- and clathrin-dependent endocytosis of SSTR2A in myenteric neurons. A, octreotide stimulated marked SSTR2A-IR endocytosis after 30 min (arrow, top row), which was prevented by dynasore and sucrose (arrowheads). Scale bars = 10 μm. NOS, nitric oxide synthase. B, column graph indicating that dynasore and sucrose significantly inhibit octreotide-stimulated endocytosis of SSSTR2A-IR. PM, plasma membrane; INT, internalized. ***, p < 0.001 to vehicle control.
Agonists Stimulate Divergent Intracellular Trafficking of SSTR2A in Myenteric Neurons
Within 60 min of exposure of neurons to SST-14 or L-054,264, SSTR2A-IR was no longer detected in intracellular locations, and after 120 min, there was complete recovery of plasma membrane levels of the receptor (Fig. 2, A and C; percent SSTR2A-IR at plasma membrane at 120 min, SST-14 75 ± 4%, L-054,264 86 ± 3%). Conversely, the return of SSTR2A-IR to the plasma membrane was diminished in SST-28-treated neurons (Fig. 2B, 60 ± 3% at 120 min, p < 0.0001 to SST-14). In neurons treated with octreotide, lanreotide, or vapreotide, there was no detectable recovery of SSTR2A at the plasma membrane after 120 min of recovery (Figs. 2, D–E, and 1C; at 120 min, octreotide 42 ± 3%, lanreotide 26 ± 2%, vapreotide 37 ± 2%, p < 0.0001 to SST-14). In neurons treated with SST-28 or the peptide analogs, SSTR2A-IR was retained in Golgi-like structures in the soma and in endosomes of lamellar dendrites. Thus, although SSTR2A efficiently recycles in neurons treated with SST-14 and a non-peptide agonist, recycling is delayed after stimulation with SST-28 and prevented in neurons treated with SST analogs.
ECE-1 Controls SSTR2A Recycling in Myenteric Neurons
GPCRs that exhibit sustained interactions with β-arrestins are retained in endosomes until they dissociate from β-arrestins and recycle (2). By degrading neuropeptides, such as SST-14, in acidified endosomes, ECE-1 disrupts the peptide/receptor/β-arrestin complex and promotes receptor recycling (3). To determine whether this mechanism controls SSTR2A recycling in myenteric neurons, we treated neurons with the ECE-1-selective inhibitor SM-19712 or vehicle (control). SM-19712 had no effect on basal SSTR2A-IR localization or on SST-14-induced endocytosis after 30 min (Fig. 4, A and B). However, SM-19712 prevented SSTR2A recycling at 60 and 120 min after SST-14 treatment relative to controls, causing receptor retention in juxtanuclear structures in the soma and endosomes in dendrites (Fig. 4, A and B; percent SSTR2A-IR at plasma membrane at 120 min, SM-19712 36 ± 3%, vehicle 75 ± 4%, p < 0.0001). Bafilomycin A1, which inhibits vacuolar H+-ATPase and neutralizes acidic vesicles (17), also prevented recycling in SST-14-treated neurons, consistent with the requirement for acidification for optimal ECE-1 activity (3) (Fig. 4C; percent SSTR2A-IR at plasma membrane at 120 min, bafilomycin 59 ± 2%, vehicle 75 ± 4%, p < 0.0001). Conversely, SM-19712 did not affect SSTR2A-IR recycling in neurons stimulated with L-054,264, a non-peptide that is not an ECE-1 substrate (percent SSTR2A-IR at plasma membrane at 120 min, SM-19712 85 ± 1%, vehicle 86 ± 3%, data not shown). We studied SSTR2A trafficking to ECE-1-containing organelles in rat colon, where ECE-1-IR is readily detectable in endosomes (10, 13). In unstimulated preparations, SSTR2A-IR was localized to the plasma membrane of myenteric neurons, and ECE-1-IR was intracellular (Fig. 4D). At 30 min after incubation with SST-28, SSTR2A-IR colocalized with ECE-1-IR. Thus, activated SSTR2A traffics to ECE-1-containing organelles. SSTR2A recycling requires ECE-1 activity, an ECE-1 cleavable ligand, and endosomal acidification.
SSTR2A Recycling Requires an Intact Golgi Apparatus in Myenteric Neurons
Although SSTR2A is endocytosed to the trans-Golgi network (TGN) in cell lines and hippocampal neurons (18–20), the importance of Golgi trafficking in enteric neurons is not known. After stimulation with SST-14, SST-28 (not shown), or octreotide (Fig. 5A), SSTR2A-IR trafficked to juxtanuclear structures that labeled with an antibody to the TGN (TGN-38). In cells treated with brefeldin A, the TGN was disrupted, and internalized SSTR2A-IR was not colocalized with TGN-38 (Fig. 5B). Brefeldin A did not affect SST-14-stimulated endocytosis of SSTR2A-IR but prevented recycling of SSTR2A-IR (Fig. 5, C and D; percent SSTR2A-IR at plasma membrane at 120 min, brefeldin A 52 ± 2%, vehicle 75 ± 4%, p < 0.0001). Thus, activated SSTR2A traffics to the TGN, and an intact TGN is necessary for receptor recycling in myenteric neurons.
SSTR2A Colocalizes with ECE-1 Isoforms in Neuroendocrine Cells
There are four isoforms of ECE-1 that differ in their subcellular distribution. All isoforms can be detected in endosomes, but ECE-1b and ECE-1d are predominantly endosomal, whereas ECE-1a and ECE-1c are mainly at the plasma membrane (13). ECE-1b has also been detected in the TGN (21). Although myenteric neurons express all ECE-1 isoforms (10), the lack of isoform-selective antibodies precluded investigation of SSTR2A trafficking to individual isoforms in neurons. To determine whether agonists stimulate trafficking of SSTR2A to individual ECE-1 isoforms, we expressed ECE-1a–d with GFP tags in AtT-20 neuroendocrine cells that endogenously express SSTR2A but not ECE-1 (22). Cells were incubated with agonists, endogenous SSTR2A was detected by immunofluorescence, and ECE-1 isoforms were localized using GFP. In unstimulated cells, SSTR2A-IR was confined to the plasma membrane, ECE-1b and ECE-1d were predominantly in endosomes, and ECE-1a and ECE-1c were mainly at the plasma membrane (Fig. 6A for ECE-1d; data not shown for ECE-1a, ECE-1b, and ECE-1c). Agonists stimulated trafficking of SSTR2A-IR to endosomes and juxtanuclear structures that predominantly contained ECE-1d-GFP (Fig. 6A for SST-28) and ECE-1b (data not shown). ECE-1d-GFP partially colocalized with Golgin97 in AtT-20 cells, suggesting that SSTR2A traffics to ECE-1d-GFP in endosomes and the Golgi apparatus (Fig. 6B). SST-14, SST-28, L-054,264, vapreotide (Fig. 6C), lanreotide, and octreotide (data not shown) all stimulated endocytosis of endogenous SSTR2A-IR in AtT-20 cells. After 120 min, SSTR2A-IR was fully recycled in cells treated with L-054,264, partially recycled after SST-14 and SST-28, and had not recycled after exposure to vapreotide (Fig. 6C), lanreotide, and octreotide (data not shown). Thus, activated SSTR2A predominantly traffics to endosomes and the Golgi apparatus containing ECE-1d.
FIGURE 6.

SSTR2A trafficking in At-T20 cells. A, under basal conditions (0 min), SSTR2A-IR was at the plasma membrane (red, arrowhead), and ECE-1d-GFP was intracellular. SST-28 stimulated trafficking of SSTR2A-IR to intracellular locations that colocalized with ECE-1d-GFP (30–120 min, arrows). B, ECE-1d-GFP partially colocalized with Golgin97 in the Golgi apparatus (arrows). C, SST-14, SST-28 and L-054,264 and vapreotide stimulated internalization of SSTR2A-IR (30 min). SSTR2A-IR was recycled 60 min after L-054,264, 120 min after SST-14 and SST-28, but not at all after vapreotide. Scale bars = 5 μm.
SSTR2A Interacts with ECE-1 at the Cell Surface and in Endosomes
The interaction between SSTR2A and the ECE-1a or ECE-1d isoforms was investigated using BRET. The SSTR2A-RLuc8 construct was coexpressed with YFP-tagged ECE-1a or ECE-1d in HEK293 cells, and the transfer of energy between these two proteins was measured upon addition of SST-14, SST-28, or octreotide (100 nm each). The BRET signal between ECE-1a and SSTR2A-RLuc8 decreased upon addition of all three ligands, suggesting an increase in the distance between the energy donor protein and the acceptor (Fig. 7A, p < 0.001 for 6–20 min). In contrast, the BRET signal between ECE-1d and SSTR2A-RLuc8 increased upon addition of SST-14, SST-28, and octreotide, indicating an increased proximity between SSTR2A and ECE-1d (Fig. 7B, p < 0.01 for 9–20 min). These results are in agreement with the observation that ECE-1a is primarily located at the plasma membrane. Thus, agonist-stimulated receptor internalization would be expected to reduce the SSTR2A-RLuc8 and ECE-1a-YFP BRET signal. Because ECE-1d is mainly located in endosomes, our results suggest that internalized SSTR2A traffics to endosomes, increasing the SSTR2A-RLuc8/ECE-1d-YFP BRET signal.
FIGURE 7.
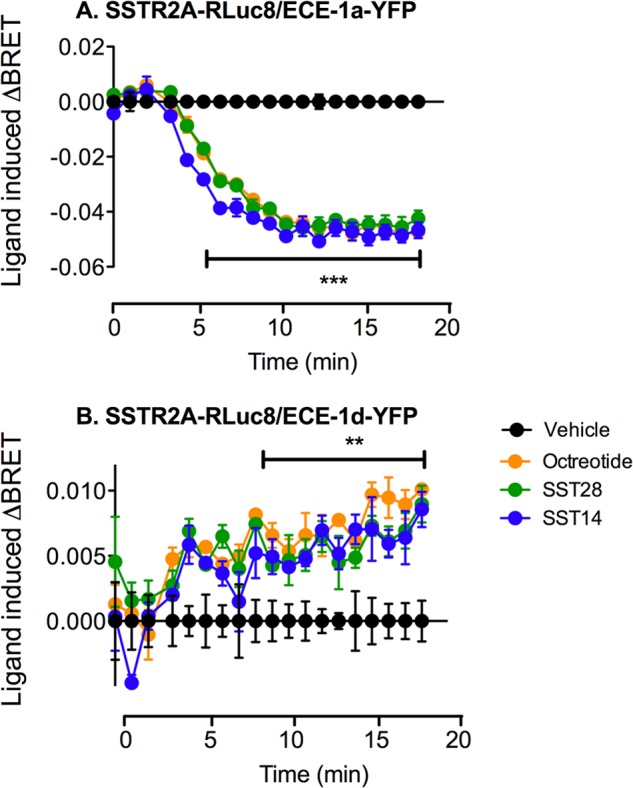
SSTR2A interacts with the ECE-1a and ECE-1d isoforms. SSTR2A-RLuc8 and ECE-1a-YFP or ECE-1d-YFP were coexpressed in HEK cells. A, stimulation with SST-14, SST-28 and octreotide (100 nm) resulted in a significant reduction in BRET signal between SSTR2A-RLuc8 and the plasma membrane-associated ECE-1a-YFP isoform, indicating a loss of interaction between these two proteins. B, in contrast, agonist stimulation gave rise to a significant increase in BRET signal between SSTR2A-RLuc8 and the endosome-associated ECE-1d-YFP isoform, indicating increased interaction. **, p < 0.01; ***, p < 0.001.
SST-28, Lanreotide, and Vapreotide Are Resistant to Degradation by ECE-1
ECE-1 degrades SST-14 but not octreotide at acidic endosomal pH. However, the kinetics and sites of hydrolysis have not been defined, and it is not known whether N-terminally extended SST-28 and peptide analogs are protected from degradation. To determine whether susceptibility to hydrolysis by ECE-1 could explain the differential effects of peptides on SSTR2A recycling, we compared peptide degradation by this enzyme. When SST-14 (250 μm) was incubated with ECE-1 (200 nm) at endosomal pH 5.5, there was complete degradation after 120 min at 37 °C (3.6 ± 0.5% intact). Under the same conditions, SST-28 was degraded more slowly (54.9 ± 3.0% intact, 120 min), and lanreotide and vapreotide were not degraded (lanreotide 100.4 ± 1.4% intact, vapreotide 102.6 ± 3.6% intact, 240 min; data not shown).
To define the kinetics and sequence of SST-14 and SST-28 hydrolysis, we monitored the appearance of products using the activity-based mass spectrometry proteinase activity labeling employing 18O-enriched water (12). Newly formed cleavage products were identified by the incorporation of 18O atoms into nascent C-terminal carboxylic groups, allowing for precise determination of the sequence of degradation. When SST-14 (100 μm) was incubated with ECE-1 (100 nm) at endosomal pH 5.5, products were formed that were consistent with initial hydrolysis at Thr10-Phe11, followed by cleavage at Phe6-Phe7 and then Asn5-Phe6 (Fig. 8, A–C). ECE-1 cleaved SST-28 at the corresponding residues Thr24-Phe25 and then Phe20-Phe21 and Asn19-Phe20 but at a much reduced rate compared with SST-14 (Fig. 8, D–G). Because all cleavage sites reside within the intramolecular disulfide bond, the cleavage products remained linked after the initial hydrolytic step, which resulted in a gain in mass of 18 Da corresponding to water incorporation. Reduction of disulfide bonds prior to analysis did not alter cleavage sites, but products were, as expected, not further linked. At extracellular pH 7.4, the same proteolytic processing of SST-14 occurred, but at a much reduced rate, and SST-28 processing at pH 7.4 was negligible (Fig. 8, B and F).
FIGURE 8.

ECE-1 degrades SST-14 at endosomal pH. SST-14 (A and B) and SST-28 (E and F) were incubated with ECE-1 at endosomal (pH 5.5) and extracellular (pH 7.4) pH. Degradation and formation of products was assessed by proteinase activity labeling employing 18O-enriched water. A–C, SST-14 (1637 Da, black) was degraded at Thr10-Phe11, and the products remained disulfide-linked, which resulted in a mass gain of 18 Da because of water incorporation (1655, blue). ECE-1 then cleaved at Phe6-Phe7, forming a second linked product (1093, orange). Degradation was far more rapid at pH 5.5 than pH 7.4. E–G, at pH 5.5, SST-28 (3147 Da, black) was degraded at Thr24-Phe25, and the products remained disulfide-linked, which resulted in a gain of mass of 18 Da because of water incorporation (3165, blue). ECE-1 then cleaved at Phe20-Phe21, forming a second linked product (2456, orange). Degradation of SST-28 was negligible at pH 7.4. D, comparison of the rate of degradation of SST-14 and SST-28 at pH 5.5.
Thus, although ECE-1 can rapidly degrade SST-14 at endosomal acidity, SST-28 is protected, and octapeptide SST analogs are completely resistant. These differences likely account for the differential effects of these agonists on SSTR2A recycling.
ECE-1 Destabilizes the SSTR2A/β-Arrestin Endosomal Complex in a Ligand-dependent Manner
The stability of GPCR/β-arrestin complexes in endosomes determines the rate of receptor recycling, which is required for resensitization and maintenance of plasma membrane signaling (2). To examine agonist- and ECE-1-dependent SSTR2A/β-arrestin interactions, we coexpressed SSTR2A-RLuc8 with β-arrestin1-YFP or β-arrestin2-YFP in HEK293 cells, which are commonly used to evaluate SSTR2A trafficking and express endogenous ECE-1. We used BRET assays to examine the kinetics of interaction between SSTR2A and β-arrestins (Fig. 9A). SST-14, SST-28, and octreotide all stimulated the rapid recruitment of β-arrestin1 and β-arrestin2 to SSTR2A-RLuc8 (Fig. 9, B and C). The highest concentration (1 μm) of all agonists stimulated detectable recruitment within 1 min, which was maximal after ∼3 min and was similar for β-arrestin1 and β-arrestin2. However, there were differences in the potency of recruitment: SSTR2A-RLuc8 and β-arrestin1-YFP (log EC50) octreotide −8.2 > SST-14 −7.9 > SST-28 −6.9 (Fig. 9B); SSTR2A-RLuc8 and β-arrestin2-YFP (log EC50) octreotide −8.4 > SST-14 −8.0 > SST-28 −6.5 (Fig. 9C).
FIGURE 9.

ECE-1 regulates SSTR2A/β-arrestin interactions. A, SSTR2A-RLuc8 and β-arrestin-YFP were coexpressed in HEK cells. Agonist stimulation induced BRET, which is indicative of SSTR2A-RLuc8 and β-arrestin-YFP interactions. B and C, effects of graded concentrations of SST-14, SST-28, and octreotide on the interaction between SSTR2A-RLuc8 and β-arrestin1-YFP (B) and β-arrestin2-YFP (C). D, time course of SSTR2A-RLuc8 and β-arrestin2-YFP interaction in the continued presence of SST-14 (left panel), SST-28 (center panel), or octreotide (right panel). In vehicle-treated cells, SST-14- and SST-28-induced BRET declined, whereas octreotide-induced BRET was sustained. SM-19712 maintained SST-14- and SST-28-induced BRET. E, SSTR2A-RLuc8 and β-arrestin2-YFP interaction measured at various times after stimulation with SST-14 (left panel), SST-28 (center panel), or octreotide (right panel). In vehicle-treated cells, SST-14- and SST-28-induced BRET declined after washing, whereas octreotide-induced BRET was sustained. SM-19712 maintained SST-14- and SST-28-induced BRET. *, p < 0.001 to vehicle control.
To assess the stability of interactions between SSTR2A and β-arrestin2, we either examined the decay in BRET signal in the continued presence of agonist over 10 min or measured the BRET signal at various times after agonist exposure and recovery of cells in agonist-free medium. In the presence of SST-14 or SST-28 (100 nm), the SSTR2A/β-arrestin2 interaction declined within 5 min (Fig. 9D, left and center panels), whereas in the presence of octreotide (100 nm), the SSTR2A/β-arrestin2 interaction was sustained for at least 10 min (right panel). The SSTR2A/β-arrestin2 interaction declined to basal levels 50 min after agonist removal (SST-14, 0.033 ± 0.004; SST-28 0.023 ± 0.002, Fig. 9E, left and center panels). However, the BRET signal was sustained in octreotide-treated cells for at least 130 min after washing (0.184 ± 0.015 at 50 min) (Fig. 9E, right panel). The ECE-1 inhibitor SM-19712 prevented the decay in BRET signal during incubation with SST-14 and SST-28 and attenuated the decline after agonist removal. SM-19712 did not affect the BRET signal during or after incubation with octreotide. Thus, by degrading internalized SST-14 and SST-28, ECE-1 controls the duration of SSTR2A and β-arrestin interactions. The sustained SSTR2A and β-arrestin interactions in octreotide-treated cells depends on the resistance of octreotide to degradation by ECE-1.
DISCUSSION
We report that diverse endogenous and therapeutic agonists differentially regulate SSTR2A in myenteric neurons. Although all agonists stimulate clathrin- and dynamin-dependent SSTR2A endocytosis and trafficking to the Golgi apparatus, they evoke distinct patterns of postendocytic receptor sorting. When activated by SST-14, SSTR2A transiently interacts with β-arrestins and rapidly recycles, but when activated by peptide analogs, SSTR2A shows sustained interactions with β-arrestins (peptide analogs) and recycles slowly (SST-28) or not at all (peptide analogs). These differences depend on agonist susceptibility to degradation by ECE-1. By degrading SST-14 in endosomes or the Golgi apparatus, ECE-1 promotes SSTR2A dissociation from β-arrestins and rapid receptor recycling. Agonists that are resistant to ECE-1 degradation promote stable interactions between SSTR2A and β-arrestins, which lead to intracellular retention of the receptor. This differential regulation of a key pan-inhibitory receptor may explain different physiological effects of endogenous agonists and could account for the long-lasting therapeutic actions and side effects of clinically used agonists.
Agonist-biased Trafficking of SSTR2A
“Biased agonism,” in which different agonists induce distinct conformations of the same receptor that lead to divergent signals and physiological outcomes, is an emerging theme of GPCR activation that has been examined extensively for synthetic agonists of receptors expressed in model cell lines. By studying the effects of six endogenous and synthetic agonists of SSTR2A, we observed agonist-biased endocytosis and recycling of a GPCR in primary neurons.
Although all agonists stimulated endocytosis of SSTR2A, octreotide, and related octapeptide derivatives induced a greater degree of endocytosis than SST-14, SST-28, and L-054,264. One possible explanation for this difference is that the octapeptides are more potent agonists, although these agonists all activate SSTR2A with similar potencies (23, 24). Alternatively, the octapeptide analogs may stabilize conformations that promote more sustained interactions of SSTR2A with β-arrestins, which couple GPCRs to clathrin and AP2 and, thereby, favor receptor endocytosis. Additional experiments are required to assess these possibilities.
There were marked differences in the postendocytic sorting of SSTR2A in neurons exposed to different agonists. Although SST-14 and l-054,264 induced transient internalization of SSTR2A and complete recycling to the plasma membrane within 60–120 min, SST-28, octreotide, lanreotide, and vapreotide caused sustained SSTR2A retention in endosomes and the Golgi apparatus. This difference can be explained by the sensitivity of the different agonists to degradation by ECE-1. We observed that ECE-1 rapidly degrades SST-14 at endosomal acidity by cleaving at three sites within the dicysteine linked region, which would inactivate the peptide. An ECE-1-selective inhibitor or suppression of endosomal acidification (and, thus, ECE-1 activity) prevented SST-14-induced recycling of SSTR2A. As expected, ECE-1 inhibition did not affect SSTR2A recycling after treatment with L-054,264, a non-peptide that would not be degraded by ECE-1. ECE-1 degraded SST-28 at corresponding sites, but degradation was delayed, which indicates that the N-terminal extension protects the bioactive peptide from proteolytic inactivation. SST-28-activated SSTR2A recycled slowly in AtT-20 cells but not at all in neurons. The octapeptide analogs were completely resistant to ECE-1 degradation, and these caused sustained retention of SSTR2A in endosomes and the Golgi apparatus. These findings are consistent with our previous observations that ECE-1 degrades substance P, calcitonin gene-related peptide, and SST-14 in endosomes to promote receptor recycling and resensitization (3, 9, 13, 25).
Our results demonstrate, for the first time, the close interaction between a GPCR and ECE-1. Moreover, we show that susceptibility of agonists to degradation by ECE-1 regulates the duration of interactions between SSTR2A and β-arrestins and that this process controls the rate of receptor recycling. The affinity of GPCR interactions with β-arrestins is a critical determinant of GPCR recycling. Most studies indicate that this interaction is an intrinsic property of the receptor that depends on the extent of phosphorylation of intracellular Ser and Thr residues by G protein-coupled receptor kinases that are required for β-arrestin binding. “Class A” GPCRs (e.g. β2 adrenergic receptor, neurokinin 3 receptor) have few phosphorylation sites, exhibit low affinity and transient interactions with β-arrestin2, and rapidly recycle (26–28). “Class B” GPCRs (e.g. neurokinin 1 receptor, neurotensin 1 receptor) are highly phosphorylated, interact with both β-arrestin1 and 2 with high affinity for prolonged periods in endosomes, and slowly recycle. We report that the susceptibility of the agonist to degradation by ECE-1 in endosomes is a second determinant of GPCR interactions with β-arrestins and the rate of receptor recycling. By using BRET to assess the duration of interaction between SSTR2A and β-arrestins, we found that although SST-14 and SST-28 cause transient interactions, metabolically stable octreotide causes a sustained interaction. The transient interaction after treatment with SST-14 and SST-28 was prolonged by an ECE-1 inhibitor, whereas the sustained interaction after octreotide was unaffected by ECE-1 inhibition. With time, the interaction between SSTR2A and β-arrestins waned even in octreotide- and ECE-1 inhibitor-treated cells, suggesting that additional mechanisms may also drive SSTR2A/β-arrestin dissociation. These may include degradation of the peptide or receptor by other proteases or endosomal acidification that promotes ligand dissociation. Endosomal ECE-1, by cleaving SST-14 and SST-28, presumably accelerates this dissociation, freeing SSTR2A from β-arrestins and promoting receptor recycling.
Functional Relevance of Agonist-biased Trafficking of SSTR2A
GPCR trafficking and signaling are inextricably linked. In addition to G protein-mediated signaling at the plasma membrane, internalized GPCRs can continue to signal by G protein-independent mechanisms (2). By uncoupling receptors from G proteins, β-arrestins terminate plasma membrane signaling. β-arrestins also recruit GPCRs and down-stream signaling partners, notably mitogen-activated protein kinases, to endosomes and, thereby, mediate sustained signals from internalized receptors. Dissociation from β-arrestins and recycling are then required for resensitization and maintenance of plasma membrane signaling. Our results show that the nature of the agonist and its susceptibility to degradation by intracellular ECE-1 determine the stability of SSTR2A/β-arrestin interactions and, thus, the rate of SSTR2A recycling, with likely consequences for SSTR2A signaling. Those agonists that promote rapid SSTR2A recycling (SST-14, L-054,264) would facilitate a rapid resensitization of plasma membrane but transient intracellular signaling. Conversely, agonists that induce sustained retention of SSTR2A in endosomes and the Golgi apparatus (SST-28, peptide analogs) would favor sustained intracellular signaling and a prolonged suppression of plasma membrane signaling. Further studies are required to define the importance of agonist-dependent SSTR2A signaling from the plasma membrane and endosomes of enteric neurons. Although many GPCRs can signal from endosomes, GPCR signaling from the Golgi apparatus has not been examined in detail.
Octreotide is used clinically to treat endocrine tumors and the associated effects of altered pituitary hormone release. It is also used to inhibit the release of peptide hormones from the gut and endocrine pancreas. Octreotide is remarkably resistant to degradation by both extracellular and intracellular peptidases (29), a contributing factor to its therapeutic effectiveness. Our observation that clinically useful SSTR2A agonists are highly resistant to ECE-1 suggests that design of SSTR2A agonists with reduced ECE-1 susceptibility may lead to the development of better SSTR2A ligands for use in diagnostic imaging or treatment of neuroendocrine tumors (30) or controlling disorders such as acromegaly (31). Alternatively, agonists that are readily degraded by ECE-1 will enable more rapid receptor recycling and resensitization. We have also demonstrated clear differences in receptor recycling following SST-14 and SST-28 treatment that are attributable to their susceptibility to degradation by ECE-1. This has functional implications because SST-28 may have more prolonged actions or bias toward endosomal signaling (32).
In conclusion, this study identifies ECE-1 as a major regulator of recycling of internalized SSTR2A in myenteric neurons. Receptor recycling is highly dependent on the susceptibility of agonists to degradation by ECE-1, and recycling correlates with the duration of interaction between SSTR2A and β-arrestins. Therapeutic SST analogs are highly resistant to degradation by ECE-1, which may explain their long-lasting actions.
This work was supported, in whole or in part, by NIDCR National Institutes of Health Grant 1R01DE019796 (to M. H. and N. W. B). This work was also supported by National Health and Medical Research Council (NHMRC) Grants 63303 and 103188 and Monash University (to N. W. B.) and by NHMRC Grant 454858 (to D. P. P.).
- GPCR
- G protein-coupled receptor
- ECE-1
- endothelin converting enzyme 1
- SST
- somatostatin
- SSTR
- somatostatin receptor
- IR
- immunoreactivity
- BRET
- bioluminescence resonance energy transfer
- TGN
- trans-Golgi network.
REFERENCES
- 1. Lu B., Figini M., Emanueli C., Geppetti P., Grady E. F., Gerard N. P., Ansell J., Payan D. G., Gerard C., Bunnett N. (1997) The control of microvascular permeability and blood pressure by neutral endopeptidase. Nat. Med. 3, 904–907 [DOI] [PubMed] [Google Scholar]
- 2. Murphy J. E., Padilla B. E., Hasdemir B., Cottrell G. S., Bunnett N. W. (2009) Endosomes. A legitimate platform for the signaling train. Proc. Natl. Acad. Sci. U.S.A. 106, 17615–17622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roosterman D., Cottrell G. S., Padilla B. E., Muller L., Eckman C. B., Bunnett N. W., Steinhoff M. (2007) Endothelin-converting enzyme 1 degrades neuropeptides in endosomes to control receptor recycling. Proc. Natl. Acad. Sci. U.S.A. 104, 11838–11843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lamberts S. W., van der Lely A. J., de Herder W. W., Hofland L. J. (1996) Octreotide. N. Engl. J. Med. 334, 246–254 [DOI] [PubMed] [Google Scholar]
- 5. Van Op den Bosch J., Adriaensen D., Van Nassauw L., Timmermans J. P. (2009) The role(s) of somatostatin, structurally related peptides and somatostatin receptors in the gastrointestinal tract. A review. Regul. Pept. 156, 1–8 [DOI] [PubMed] [Google Scholar]
- 6. Sternini C., Wong H., Wu S. V., de Giorgio R., Yang M., Reeve J., Jr., Brecha N. C., Walsh J. H. (1997) Somatostatin 2A receptor is expressed by enteric neurons, and by interstitial cells of Cajal and enterochromaffin-like cells of the gastrointestinal tract. J. Comp. Neurol. 386, 396–408 [PubMed] [Google Scholar]
- 7. Abdu F., Hicks G. A., Hennig G., Allen J. P., Grundy D. (2002) Somatostatin SST (2) receptors inhibit peristalsis in the rat and mouse jejunum. Am. J. Physiol. 282, G624–633 [DOI] [PubMed] [Google Scholar]
- 8. Grider J. R. (2003) Neurotransmitters mediating the intestinal peristaltic reflex in the mouse. J. Pharmacol. Exp. Ther. 307, 460–467 [DOI] [PubMed] [Google Scholar]
- 9. Roosterman D., Kempkes C., Cottrell G. S., Padilla B. E., Bunnett N. W., Turck C. W., Steinhoff M. (2008) Endothelin-converting enzyme-1 degrades internalized somatostatin-14. Endocrinology 149, 2200–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pelayo J. C., Poole D. P., Steinhoff M., Cottrell G. S., Bunnett N. W. (2011) Endothelin-converting enzyme-1 regulates trafficking and signalling of the neurokinin 1 receptor in endosomes of myenteric neurones. J. Physiol. 589, 5213–5230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sarret P., Nouel D., Dal Farra C., Vincent J. P., Beaudet A., Mazella J. (1999) Receptor-mediated internalization is critical for the inhibition of the expression of growth hormone by somatostatin in the pituitary cell line AtT-20. J. Biol. Chem. 274, 19294–19300 [DOI] [PubMed] [Google Scholar]
- 12. Robinson S., Niles R. K., Witkowska H. E., Rittenbach K. J., Nichols R. J., Sargent J. A., Dixon S. E., Prakobphol A., Hall S. C., Fisher S. J., Hardt M. (2008) A mass spectrometry-based strategy for detecting and characterizing endogenous proteinase activities in complex biological samples. Proteomics 8, 435–445 [DOI] [PubMed] [Google Scholar]
- 13. Cottrell G. S., Padilla B. E., Amadesi S., Poole D. P., Murphy J. E., Hardt M., Roosterman D., Steinhoff M., Bunnett N. W. (2009) Endosomal endothelin-converting enzyme-1. A regulator of beta-arrestin-dependent ERK signaling. J. Biol. Chem. 284, 22411–22425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kocan M., Dalrymple M. B., Seeber R. M., Feldman B. J., Pfleger K. D. (2010) Enhanced BRET technology for the monitoring of agonist-induced and agonist-independent interactions between GPCRs and β-arrestins. Front. Endocrinol. 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang L., Guo L., Pasternak A., Mosley R., Rohrer S., Birzin E., Foor F., Cheng K., Schaeffer J., Patchett A. A. (1998) Spiro[1H-indene-1,4′-piperidine] derivatives as potent and selective non-peptide human somatostatin receptor subtype 2 (sst2) agonists. J. Med. Chem. 41, 2175–2179 [DOI] [PubMed] [Google Scholar]
- 16. Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., Kirchhausen T. (2006) Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10, 839–850 [DOI] [PubMed] [Google Scholar]
- 17. Johnson L. S., Dunn K. W., Pytowski B., McGraw T. E. (1993) Endosome acidification and receptor trafficking. Bafilomycin A1 slows receptor externalization by a mechanism involving the receptor's internalization motif. Mol. Biol. Cell 4, 1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sarret P., Esdaile M. J., McPherson P. S., Schonbrunn A., Kreienkamp H. J., Beaudet A. (2004) Role of amphiphysin II in somatostatin receptor trafficking in neuroendocrine cells. J. Biol. Chem. 279, 8029–8037 [DOI] [PubMed] [Google Scholar]
- 19. Csaba Z., Lelouvier B., Viollet C., El Ghouzzi V., Toyama K., Videau C., Bernard V., Dournaud P. (2007) Activated somatostatin type 2 receptors traffic in vivo in central neurons from dendrites to the trans Golgi before recycling. Traffic 8, 820–834 [DOI] [PubMed] [Google Scholar]
- 20. Lelouvier B., Tamagno G., Kaindl A. M., Roland A., Lelievre V., Le Verche V., Loudes C., Gressens P., Faivre-Baumann A., Lenkei Z., Dournaud P. (2008) Dynamics of somatostatin type 2A receptor cargoes in living hippocampal neurons. J. Neurosci. 28, 4336–4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schweizer A., Valdenaire O., Nelböck P., Deuschle U., Dumas Milne Edwards J. B., Stumpf J. G., Löffler B. M. (1997) Human endothelin-converting enzyme (ECE-1). Three isoforms with distinct subcellular localizations. Biochem. J. 328, 871–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Muller L., Barret A., Etienne E., Meidan R., Valdenaire O., Corvol P., Tougard C. (2003) Heterodimerization of endothelin-converting enzyme-1 isoforms regulates the subcellular distribution of this metalloprotease. J. Biol. Chem. 278, 545–555 [DOI] [PubMed] [Google Scholar]
- 23. Nunn C., Cervia D., Langenegger D., Tenaillon L., Bouhelal R., Hoyer D. (2004) Comparison of functional profiles at human recombinant somatostatin sst2 receptor. Simultaneous determination of intracellular Ca2+ and luciferase expression in CHO-K1 cells. Br. J. Pharmacol. 142, 150–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Q., Cescato R., Dewi D. A., Rivier J., Reubi J. C., Schonbrunn A. (2005) Receptor signaling and endocytosis are differentially regulated by somatostatin analogs. Mol. Pharmacol. 68, 90–101 [DOI] [PubMed] [Google Scholar]
- 25. Padilla B. E., Cottrell G. S., Roosterman D., Pikios S., Muller L., Steinhoff M., Bunnett N. W. (2007) Endothelin-converting enzyme-1 regulates endosomal sorting of calcitonin receptor-like receptor and β-arrestins. J. Cell Biol. 179, 981–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oakley R. H., Laporte S. A., Holt J. A., Caron M. G., Barak L. S. (2000) Differential affinities of visual arrestin, β arrestin1, and β arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275, 17201–17210 [DOI] [PubMed] [Google Scholar]
- 27. Oakley R. H., Laporte S. A., Holt J. A., Barak L. S., Caron M. G. (2001) Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-β-arrestin complexes after receptor endocytosis. J. Biol. Chem. 276, 19452–19460 [DOI] [PubMed] [Google Scholar]
- 28. Schmidlin F., Déry O., Bunnett N. W., Grady E. F. (2002) Heterologous regulation of trafficking and signaling of G protein-coupled receptors. β-Arrestin-dependent interactions between neurokinin receptors. Proc. Natl. Acad. Sci. U.S.A. 99, 3324–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hofland L. J., van Koetsveld P. M., Waaijers M., Zuyderwijk J., Breeman W. A., Lamberts S. W. (1995) Internalization of the radioiodinated somatostatin analog [125I-Tyr3]octreotide by mouse and human pituitary tumor cells. Increase by unlabeled octreotide. Endocrinology 136, 3698–3706 [DOI] [PubMed] [Google Scholar]
- 30. Oberg K. E., Reubi J. C., Kwekkeboom D. J., Krenning E. P. (2010) Role of somatostatins in gastroenteropancreatic neuroendocrine tumor development and therapy. Gastroenterology 139, 742–753 [DOI] [PubMed] [Google Scholar]
- 31. Gilroy J. J., James R. A. (2002) Optimizing somatostatin analog therapy in acromegaly. Long-acting formulations. Treat. Endocrinol. 1, 149–154 [DOI] [PubMed] [Google Scholar]
- 32. Whalen E. J., Rajagopal S., Lefkowitz R. J. (2011) Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. Med. 17, 126–139 [DOI] [PMC free article] [PubMed] [Google Scholar]