

Background: Glutathionylation is a major post-translational modification that regulates protein function.
Results: Human glutathione transferase Omega 1 (GSTO1-1) can catalyze the deglutathionylation of protein thiols in vitro and in cell culture.
Conclusion: GSTO1-1, but not GSTO2-2, catalyzes the deglutathionylation of actin and other proteins under physiological conditions.
Significance: Genetic variation in GSTO1-1-catalyzed deglutathionylation may influence the function of multiple proteins in signaling pathways.
Keywords: Enzyme Catalysis, Genetic Polymorphism, Glutathionylation, Redox Regulation, Thiol, Glutathione Transferase Omega
Abstract
The glutathionylation of intracellular protein thiols can protect against irreversible oxidation and can act as a redox switch regulating metabolic pathways. In this study we discovered that the Omega class glutathione transferase GSTO1-1 plays a significant role in the glutathionylation cycle. The catalytic activity of GSTO1-1 was determined in vitro by assaying the deglutathionylation of a synthetic peptide by tryptophan fluorescence quenching and in T47-D epithelial breast cancer cells by both immunoblotting and the direct determination of total glutathionylation. Mutating the active site cysteine residue (Cys-32) ablated the deglutathionylating activity of GSTO1-1. Furthermore, we demonstrate that the expression of GSTO1-1 in T47-D cells that are devoid of endogenous GSTO1-1 resulted in a 50% reduction in total glutathionylation levels. Mass spectrometry and immunoprecipitation identified β-actin as a protein that is specifically deglutathionylated by GSTO1-1 in T47-D cells. In contrast to the deglutathionylation activity, we also found that GSTO1-1 is associated with the rapid glutathionylation of cellular proteins when the cells are exposed to S-nitrosoglutathione. The common A140D genetic polymorphism in GSTO1 was found to have significant effects on the kinetics of both the deglutathionylation and glutathionylation reactions. Genetic variation in GSTO1-1 has been associated with a range of diseases, and the discovery that a frequent GSTO1-1 polymorphism affects glutathionylation cycle reactions reveals a common mechanism where it can act on multiple proteins and pathways.
Introduction
The irreversible oxidation of protein thiols to sulfonic acids can lead to the inactivation and proteolysis of the modified protein. However, the formation of mixed disulfides between glutathione and protein thiols (glutathionylation) during oxidative stress provides an efficient defense against irreversible oxidation as the disulfide bonds can be subsequently enzymatically reduced or “exchanged” (1). Glutathionylation is also important in other physiological roles as there is mounting evidence that even in the absence of oxidative stress the selective glutathionylation/deglutathionylation of specific protein thiols can contribute to cell signaling and the regulation of some metabolic pathways (2, 3). Glutathionylation has been shown to have an impact on several cellular processes including regulation of the cell cycle (4), apoptosis (5–7), and drug response in cancer (8) and in the progression of neurodegeneration (9). The cohort of proteins that are structurally and functionally modified by glutathionylation is collectively known as the “disulfide proteome” or “glutathionome” and have been recently reviewed (3, 10).
Studies of the enzymes involved in the glutathionylation cycle have focused significantly on the glutaredoxins and related enzymes such as thioredoxin and protein disulfide isomerase (11, 12). In addition, sulfiredoxin and glutathione transferase Pi have been shown to catalyze the deglutathionylation and glutathionylation of specific proteins, respectively (8, 13). The Omega class GSTs (GSTO1-12 and GSTO2-2) were discovered by bioinformatics (14–16), and immunohistochemical studies identified GSTO1-1 in a diverse range of tissues with varied expression levels (17). Whereas GSTs from the Alpha, Mu, Pi, Theta, and Zeta classes have a tyrosine or serine residue in the active site, the Omega class GSTs have an active site cysteine residue and catalyze several atypical reactions including dehydroascorbate reductase and S-(phenacyl)glutathione reductase activities as well as glutaredoxin activity (formerly termed thioltransferase) (18–20). Furthermore, the active site of GSTO1 is positioned in a wide crevice that can potentially accommodate large substrates such as proteins (14).
Since their first description, the Omega class GSTs have been investigated in relation to a number of biologically significant pathways and clinical disorders including drug resistance (21), Alzheimer disease (22–24), Parkinson disease (25), vascular dementia and stroke (26, 27), and amyotrophic lateral sclerosis (28), the action of anti-inflammatory drugs (29), the disposition of arsenic (30), and susceptibility to chronic obstructive pulmonary disease (31–33) and cancer (34–40). However, the mechanism by which the Omega GSTs mediate their effects in these diverse settings has not been clear. In this study we show that GSTO1-1 has significant deglutathionylation activity with a model peptide substrate. GSTO1-1-dependent deglutathionylation of cellular proteins can also be detected in cultured cell lines. In contrast, our study found that the expression of GSTO1-1 in cultured T47-D breast cancer cells promotes the rapid glutathionylation of cellular proteins when the cells are exposed to nitrosoglutathione. The discovery that GSTO1-1 participates in the glutathionylation cycle and targets specific proteins could explain the association of GSTO1 polymorphisms with a diverse range of clinical disorders and provide a novel therapeutic target.
EXPERIMENTAL PROCEDURES
Materials
Unless otherwise indicated, all chemicals were purchased from Sigma.
Protein Expression and Purification
Human (h-) GSTO1-1 allelic variants, hGSTO2-2, hGSTK1-1, and CLIC2, were expressed from cDNA constructs in the bacterial expression plasmid pHUE and purified as described previously using nickel affinity chromatography (41).
Cell Culture
Cells lines were purchased from ATCC and were maintained at 37 °C, 5% CO2 in RPMI media supplemented with 10% fetal bovine serum. T47-D cells were stably transfected with a GSTO1-encoding pCDNA3 plasmid. Stable expression was confirmed by immunoblotting with rabbit anti-human GSTO1-1 antibody. The human lymphoblastoid cell lines expressing GSTO1 variants were gifts from Dr. Juleen Cavanaugh.
Peptide Synthesis
The peptide used in this study (SQLWCLSN) was synthesized by the ACRF Biomolecular Resource Facility (BRF), John Curtin School of Medical Research, Australian National University. The synthesized peptide was dissolved in 0.1 m ammonium acetate, pH 8.0, and glutathionylated by incubation with a 10-fold molar excess of oxidized glutathione for 24 h at room temperature with constant stirring. The peptide was then diluted in sterile water (3×) before freeze-drying and purification by HPLC before use.
Tryptophan Quenching Assay
The deglutathionylation assay was based on a previously described procedure that recorded the change in tryptophan fluorescence as glutathione is removed from a model peptide (42). Fluorescence was recorded at an excitation wavelength of 280 nm and an emission wavelength of 356 nm at room temperature over a time period of 10–30 min. For the deglutathionylation reaction, the reaction mix contained McIlvaine's buffer (2 m sodium hydrogen phosphate, 1 m citric acid, pH 7.0), 1 mm GSH, 50 μΜ NADPH, 0.25 units of glutathione reductase, 1 mm EDTA, 0–20 μm peptide, 1.4 μg GSTO1-1. Glutathione reductase and GSTO1-1 were diluted in McIlvaine's buffer containing 1 μg/μl BSA. GSH affinity was similarly measured by varying GSH concentrations from 0 to 2 mm, maintaining protein concentration constant at 1.4 μg and substrate concentration at 5 μm. Deglutathionylation activities of GSTO2-2, CLIC2, and GSTK were tested up to an enzyme concentration of 6 μg. All measurements were recorded in triplicate.
Glutathionylation of the peptide was measured as described previously (42) with a modification. The reaction mix was made in McIlvaine's buffer (described above) with 5 mm GSSG, 1 mm EDTA, 5 μm peptide, 6 μg of enzyme, and 2 mm t-butylhydroxyperoxide, and 10 μg of GSTA2-2 were added to the reaction to recycle the GSH to GSSG.
Preparation of S-Nitrosoglutathione
S-Nitrosoglutathione was synthesized as described previously (17). An equimolar amount of reduced glutathione was mixed with sodium nitrite, and the pH was adjusted to 1.5 with concentrated HCl. The reaction was allowed to proceed at room temperature for 5 min followed by neutralization of the synthesized nitrosoglutathione (GSNO) with 5 m NaOH. The concentration of GSNO was estimated by measuring absorbance at 544 and 332 nm on a Cary UV spectrophotometer using extinction coefficients of 15 and 750 m−1cm−1, respectively.
Immunoblotting
SDS-PAGE and non-reducing SDS-PAGE were performed as described previously by Laemmli (43). The sample buffer was devoid of reducing agents when analyzing glutathionylated proteins. Separated proteins were transferred onto nitrocellulose membranes and immunodetected using antibodies raised against human GSTO1-1, β-actin (Abcam), and glutathione (Virogen) at a 1:1000 dilution and probed with goat anti-rabbit and goat anti mouse HRP-conjugated immunoglobulins (Dako). Chemiluminescence was detected by the ECL rapid step chemiluminescence detection system (Calbiochem, Merck).
Total Protein Glutathionylation Assay
Protein glutathionylation was determined by a previously described method (44) Cells were lysed in a non-reducing buffer (0.5 m Tris, 300 mm NaCl, 1% Nonidet P-40, 50 mm N-ethylmaleimide, 2 mm 4-(2-aminoethyl)benzenesulfonyl fluoride), and the protein concentration was determined using a BCA protein estimation kit according to manufacturer's instructions (Pierce). Cellular proteins (100 μg) were precipitated in ice-cold acetone at −20 °C for >2h. Precipitated proteins were collected by centrifugation at 20,000 × g for 15 min and resuspended in 0.1% Triton X-100 and reduced with 5 mm tris(2-carboxyethyl)phosphine (TCEP) for 30 min at room temperature. Reduced proteins were precipitated with 200 mm salicylic acid and centrifuged at 20,000 × g for 15 min. The eluted GSH in the supernatant was assayed with 2,3-naphthalenedicarboxyaldehyde and compared with a standard curve. The assay was carried out in duplicate in five independent experiments.
Glutaredoxin Thioltransferase Assay
Reactions were carried out as described previously (16). 25 μg of purified protein was added to a reaction mix containing 0.1 m Tris HCl, pH 8.0, 0.3 mm NADPH, 1 mm GSH, 1 unit glutathione reductase from Saccharomyces cerevisiae, 0.1 mg/ml bovine serum albumin, 1.5 mm EDTA, and 0.75 mm 2-hydroxyethyl disulfide. The reaction rate was measured at 340 nm at 30 °C, and the specific activity was calculated based on an extinction coefficient of 6.22 mm−1·cm−1. Background rates (sample buffer only) were subtracted from the enzyme-catalyzed reaction rates.
Dehydroascorbate Reductase Assay
Reactions were carried out as previously described (18). Dehydroascorbate was prepared fresh on the day and used within 3 h. A typical reaction mix contained 200 mm sodium phosphate, pH 6.85, 3 mm GSH, and 1.5 mm dehydroascorbate. The reaction was initiated by adding 1 μg of purified protein, and the rate of the reaction was recorded at 265 nm at 30 °C. The specific activity of the enzymes was calculated with an extinction coefficient of 14.7 mm−1·cm−1. Background rates (sample buffer only) were subtracted from the enzyme-catalyzed reaction rates.
Glutathione Transferase Assay
Reactions were carried out in 0.1 m sodium phosphate buffer, pH 6.5, with 1 mm GSH and 1 mm freshly made 1-chloro-2,4-dinitrobenzene. The conjugation reaction was initiated by adding 5 μg of enzyme and recorded at 340 nm at 30 °C for 10 min (45). Specific activity was calculated with an extinction coefficient of 9.6 mm−1cm−1.
Glutathionylation via Thiyl Radical Formation
The assay was carried out as previously described (46). The Fe(II)ADP complex was prepared by incubating 20 mm FeCl2 and 100 mm ADP for at least 1 h before use. The final reaction mix was made up in 0.1 m sodium potassium phosphate buffer and contained 0.2 mm NADPH, 0.5 mm GSH, 2 units/ml glutathione reductase, and 50 μm hydrogen peroxide. The reaction was initiated with premixed 0.5 mm FeCl2 and 2.5 mm ADP and recorded spectrophotometrically at 340 nm.
Quantification of GSSG
The assay was carried out as described previously (47). 100 μl of samples and standards were treated with 10% 2-vinylpyridine for 1 h followed by pH neutralization with 6% triethanolamine. Samples were incubated with premixed 5,5′-dithiobis(nitrobenzoic acid) (0.67 mg/ml) and glutathione reductase (250 u/ml) for 30 s to 1 min as per protocol and mixed with NADPH (0.67 mg/ml) and immediately read at 412 nm using a microplate reader.
Immunoprecipitation
T47-D cells were treated in culture with 50 mm N-ethylmaleimide for 30 min followed by lysis in non-reducing buffer. 1 mg of total cell lysate protein was used per sample as measured by BCA protein estimation. Immunoprecipitation was carried out using Pierce cross-linking immunoprecipitation kit. Cellular GSH was removed by precipitating the proteins with 5 times the volume of acetone overnight at −20 °C. Precipitated proteins were resuspended in lysis buffer as per the manufacturer's instructions. Immunoprecipitated proteins were separated on 12% SDS-PAGE and immunoblotted as above. The nitrocellulose membrane was incubated with a rabbit polyclonal anti-actin (Abcam) antibody at a 1:000 dilution for 1 h followed by incubation with a goat anti-rabbit antibody.
Mass Spectrometry
Immunoprecipitation was performed as described above. Proteins were vacuum-dried and resuspended in 7 m urea, 2 m thiourea, 20 mm Tris, 4% CHAPS. Samples were then incubated in DTT (final concentration 100 mm) for 1 h followed by iodoacetamide (final concentration 15 mm) for 1 h in the dark and run on a 12% SDS-PAGE. The gel was stained in Coomassie Blue R-250 overnight, and bands absent in the T47-D/GSTO1-1 lysates were excised from the T47-D/pCDNA3 lysate. One-dimensional nanoLC electrospray ionization MS/MS was carried out at the Australian Proteome Analysis Facility. Data generated were submitted to Mascot (Matrix Science Ltd, London, UK). Listed proteins were identified with a significance threshold of p < 0.02 with Mascot cut-off of 34.
G-actin/F-actin Assay
Cells were plated in 6-well plates and treated with 1 mm GSNO as described above. Cells were washed twice with PBS and lysed in 1% Triton X-100. The detergent-soluble supernatant (containing G-actin) was collected, and the pellet (containing F-actin) was washed in PBS and resuspended in 1× SDS sample loading buffer (48). Proteins were run on SDS-PAGE and immunoblotted. Actin was detected after incubation with anti-actin (Abcam), and the ratio of G/F actin was determined by densitometry. Glutathionylated actin was detected by immunoblotting with anti-glutathione antibody.
Phalloidin Staining
Cells were plated on coverslips (5 × 104 cells) and treated with GSNO as described. For immunostaining, the cells were washed with PBS to remove unattached cells and fixed in 3.7% paraformaldehyde (in PBS) for 15 min at room temperature. The cells were washed in PBS 3 times and permeabilized in 0.1% Triton X-100 for 10 min at room temperature. Permeabilized cells were washed in PBS and incubated with 50 μg/ml phalloidin-FITC stain for 1 h at room temperature in the dark. Samples were washed extensively in PBS and mounted on glass slides with mounting medium with DAPI (Vectashield). Fluorescence was recorded using a confocal microscope (Leica SP5).
Statistical Analysis
Data were expressed as the means ± S.E. and analyzed using Prism 4 (Graphpad software Inc.). Statistical significance was calculated by standard t tests. All experiments were performed in triplicate unless otherwise stated.
RESULTS
In Vitro Deglutathionylation by GSTO1-1
To determine if the Omega class GSTs participate in the glutathionylation cycle, a synthetic peptide incorporating a single cysteine residue adjacent to a tryptophan residue (SQLWCLSN) was glutathionylated (SG) on the cysteine residue (SQLWC−[SG]LSN) and used as a substrate (42). Deglutathionylation was measured by monitoring the change in fluorescence emitted by tryptophan as GSH was removed from the neighboring cysteine. Fig. 1 shows a significant increase in fluorescence in the presence of GSTO1-1. In contrast, the closely related GSTO2-2 isoenzyme did not catalyze deglutathionylation of the peptide. Because recombinant GSTO2-2 exhibited normal glutaredoxin activity and the expected high level of dehydroascorbate reductase activity (Table 1), we concluded it was not degraded.
FIGURE 1.
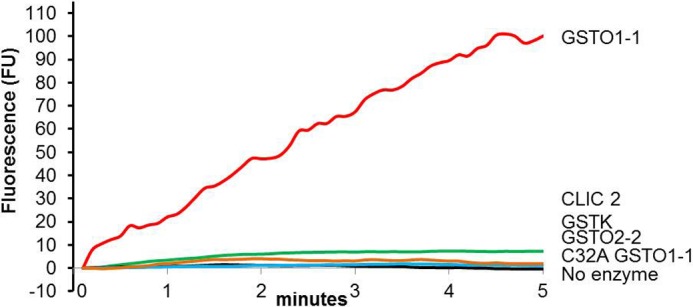
GSTO1-1 catalyzes the deglutathionylation of a peptide substrate. The increase in tryptophan fluorescence indicates the rate of peptide (SQLWC−[SG]LSN) deglutathionylation by recombinant human GSTO1-1 in the presence of GSH. Other enzymes showed no significant activity. The results are representative traces from three replicates. The reaction rate is provided in Table 1.
TABLE 1.
Genetic variants of GSTO1-1 exhibit significantly different deglutathionylation reaction kinetics
Data are based on triplicate measurements, and standard errors are shown.
| Enzyme | Km peptide | kcat peptide | kcat/Km peptide | Km GSH | Deglutathionylation-specific activity | Thiol transferase-specific activity | Dehydroascorbate reductase specific-activity |
|---|---|---|---|---|---|---|---|
| μm | s−1 | μm−1 s−1 | mm | nmol/mg of protein/min | μmol/mg of protein/min | μmol/mg of protein/min | |
| Ala-140/Glu-208/ Cys-32 | 7.9 ± 0.97 | 2.8 ± 0.28 | 0.35 | 0.4 ± 0.09 | 83.4 ± 4.4 | 1.0 ± 0.03 | 0.1 ± 0.03 |
| Asp-140/Glu-208/Cys-32 | 3.4 ± 0.5 | 1.3 ± 0.15 | 0.38 | 0.5 ± 0.12 | 48.6 ± 2.9 | 1.0 ± 0.03 | 0.2 ± 0.04 |
| Ala-140/Lys-208/Cys-32 | 2.6 ± 0.49 | 0.4 ± 0.07 | 0.15 | 0.3 ± 0.05 | 17.8 ± 2.53 | 1.0 ± 0.03 | 0.1 ± 0.04 |
| Ala-140/Glu-208/Ala-32 | NDa | ND | 0.6 ± 0.09 | ||||
| GSTO2-2 | ND | 1.2 ± 0.02 | 12.9 ± 0.88 | ||||
| GSTKb | ND | ND | ND |
a ND, activity below the limit of detection.
b GSTK conjugation activity with CDNB was measured at 25.0 ± 1.46 μmol/mg of protein/min.
We also studied two additional proteins that could potentially catalyze deglutathionylation reactions. Chloride intracellular channel 2 protein (CLIC-2) is a member of the cytosolic GST structural family and, like the Omega class GSTs, has a cysteine residue in its proposed “active site” (49, 50). Despite the presence of a potential active site cysteine residue, we did not detect any deglutathionylation activity. Previous studies of CLIC-2 have failed to identify any significant enzymatic activity. The Kappa class glutathione transferase (GSTK1-1) is found in mitochondria and is structurally and evolutionarily distinct from the cytosolic GSTs like GSTO1-1 (45). GSTK1-1 is structurally related to the prokaryotic disulfide bond-forming proteins and can also be considered as a potentially active participant in the glutathionylation cycle. However, recombinant human GSTK1-1 did not catalyze the deglutathionylation of the glutathionylated peptide. We confirmed the viability of the GSTK1-1 protein by measuring its glutathione-conjugating activity with 1-chloro-2.4-dinitrobenzene (25.0 ± 1.5 μmol/min/mg).
Deglutathionylation by GSTO1-1 Variants
Our previous studies identified several polymorphic variants of GSTO1 that could potentially vary in their substrate affinity and have an impact on the regulation of glutathionylation levels (15). The specific activity and other kinetic parameters shown in Table 1 were calculated from V/[S] plots. The Asp-140 variant had a significantly lower deglutathionylation activity (p < 0.001) than the Ala-140 isoenzyme, and this difference mainly reflected a lower kcat. We have also previously identified an E208K polymorphism that is closely linked with a deletion of Glu-155 (15). The deletion of Glu-155 causes a marked decrease in stability of GSTO1-1, and it is difficult to distinguish the separate effects of the lysine substitution at position 208. In this case we have prepared recombinant enzyme with the Lys-208 substitution and retained the glutamate at position 155. This enzyme had a significantly lower specific activity (p < 0.001) when compared with the normal enzyme with glutamate in position 208 (Table 1). To confirm that these differences were not due to the differential degradation of the recombinant enzymes during their preparation, we compared their glutaredoxin and dehydroascorbate reductase activities. These activities were found to be in agreement with our previous studies, and the values obtained for the different variants were equivalent.
Our previous studies have shown that the active site residue in GSTO1-1 is cysteine 32 and that mutation of Cys-32 to Ala-32 results in the total loss of glutaredoxin and S-phenacylglutathione reductase activity (16, 20). In this study we found a similar loss of deglutathionylation activity in the C32A mutant enzyme (Table 1), confirming the importance of cysteine 32 in the deglutathionylation reaction. Surprisingly the C32A enzyme had increased dehydroascorbate reductase activity compared with the other GSTO1-1 isoforms. This suggests that Cys-32 is not required for the reduction of dehydroascorbate. We previously reported that the C32A mutation causes an increase in the enzyme capacity to use 1-chloro-2,4-dinitrobenzene as a substrate (16).
pH Dependence of Deglutathionylation by GSTO1-1
The reaction conditions were found to be optimum at pH 7 with significantly lower rates at both pH extremes (pH 4 and pH 9 data not shown).
The Expression of GSTO1-1 Caused an Abatement of Protein Glutathionylation in T47-D Cells
In previous studies, we have shown that T47-D cells are deficient in functional GSTO1-1 (51, 52). In the present study we compared the levels of protein glutathionylation in T47-D cells transfected with either an empty pCDNA3 plasmid vector, cells transfected with pCDNA3-GSTO1C32A, or cells transfected with pCDNA3-GSTO1. The absence of GSTO1-1 expression in T47-D cells transfected with empty pCDNA3 and the level of stable GSTO1-1 expression in T47-D cells transfected with either pCDNA3-GSTO1 or pCDNA3-GSTO1C32A is shown in Fig. 2A. The expression of GSTO1-1 in T47-D cells reduced total glutathionylation levels by ∼50% in comparison cells transfected with the empty vector or pCDNA3-GSTO1C32A (Fig. 2B). Immunoblots of T47-D cell lysates probed with an anti-PSSG antibody are shown in Fig. 2C. The left-hand panel shows a marked depletion of glutathionylated protein in cells where GSTO1-1 is expressed. Although many proteins show a reduced level of glutathionylation in the presence of GSTO1-1, it is evident that the extent of deglutathionylation varies between proteins (see the arrows Fig. 2C), suggesting a degree of substrate specificity. In the right-hand panel of Fig. 2C, the lysate was treated with 100 mm DTT to reduce any glutathione mixed disulfides, and the general absence of bands on this blot confirms that the antibody is detecting specific glutathionylation. It has been previously considered that glutaredoxin was primarily responsible for deglutathionylation reactions, and the present results could be explained if the expression of GSTO1-1 resulted in an elevation in glutaredoxin expression. Western blotting showed that this was not the case as GSTO1-1 expression in T47-D cells resulted in a decrease in glutaredoxin expression (Fig. 2C). It is not clear why the level of glutaredoxin is low in GSTO1-1-expressing cells, but it is possible that the level of glutaredoxin expression may be regulated by the general level of protein glutathionylation or by the glutathionylation of a specific protein that is deglutathionylated by GSTO1-1.
FIGURE 2.

Expression of GSTO1-1 causes the deglutathionylation of proteins in T47-D breast cancer cells. A, an immunoblot shows the expression of GSTO1-1 in T47-D cells transfected with pcDNA3-empty vector, pcDNA3-GSTO1, or pcDNA3-GSTO1C32A. B, the expression of active GSTO1-1 caused a significant reduction in total protein glutathionylation (p < 0.001) compared with cells expressing GSTO1-1C32A. The values are the mean ± S.E. of five observations. C, the lower left panel shows immunoblots for glutathionylated protein comparing T47-D cells transfected with the empty pCDNA3 vector and T47-D cells expressing active GSTO1-1. Proteins showing a marked decrease in glutathionylation are indicated by arrows. The lower right panel (C) shows that treatment of cellular proteins with dithiothreitol removes all glutathionylation. The upper panel (C) shows glutaredoxin 1 (Grx1) expression in T47-D cells transfected with the empty pCDNA3 vector and lower expression in T47-D cells expressing active GSTO1-1.
Targets of GSTO1-1-catalyzed Deglutathionylation in T47-D Cells
Mass spectrometry was used to identify specific proteins (see arrows Fig. 2C) deglutathionylated by GSTO1-1 expressed in T47-D cells. Glutathionylated proteins were immunoprecipitated from T47-D cells and T47-D cells that express GSTO1-1. The immunoprecipitated proteins from each preparation were compared by SDS-PAGE and staining with Coomassie Blue. Glutathionylated proteins that were present in T47-D cells and absent from cells expressing GSTO1-1 were excised from the gel, digested with trypsin, and analyzed by mass spectrometry. This analysis revealed β-actin and three other potential targets of deglutathionylation by GSTO1-1 (Table 2). Because β-actin is known to be a target for glutathionylation (53), we specifically examined the effect of GSTO1-1 expression on the glutathionylation of β-actin. As shown in Fig. 3A, β-actin that was immunoprecipitated from T47-D cells was strongly glutathionylated, but β-actin from T47-D cells expressing GSTO1-1 was not detectably glutathionylated. The immunoblot was subsequently probed with anti β-actin to confirm that β-actin was immunoprecipitated from the GSTO1-1-expressing cells. In an additional experiment to confirm this result, glutathionylated proteins were immunoprecipitated from T47-D cells with anti-PSSG serum, and after SDS-PAGE they were immunoblotted with anti-β-actin. The results shown in Fig. 3A, lower panel, indicate that glutathionylated β-actin is only present in T47-D pCDNA3 cells that are deficient in GSTO1-1.
TABLE 2.
Proteins deglutathionylated by GSTO1-1 identified by mass spectrometry
| Protein | NCBI number | Type | Cellular localization | Molecular mass |
|---|---|---|---|---|
| kDa | ||||
| Actin-B | NP_001092.1 | Cytoskeleton | Cytoplasmic | 42 |
| Heat shock protein 70 (GRP75) | NP_004125.3 | Stress protein | Mitochondrial | 70 |
| Heat shock protein 7C (Hsp7C) | NP_006588.1 | Stress protein | Cytoplasmic | 71 |
| Prolactin-inducible protein | NP_032869.2 | Signaling, metastasis marker | Cytoplasmic | 16.5 |
FIGURE 3.

Specific deglutathionylation of β-actin by GSTO1-1. A, top panel, β-actin was immunoprecipitated (IP), fractionated by SDSPAGE, blotted (IB), and probed with anti-PSSG. Middle panel, the immune precipitation of β-actin was confirmed by reprobing with anti-β-actin. Lower panel, glutathionylated proteins were immunoprecipitated with anti-PSSG, and the blot was probed with anti-β-actin. B, globular and filamentous β-actin were separated from T47-D pCDNA3 cells, and T47-D GSTO1-1 cells (see “Experimental Procedures”), were blotted and probed with anti-β-actin. Expression of GSTO1-1 decreased the proportion of G actin and significantly altered the G/F actin ratio. C, fluorescence microscopy revealed the high levels of filamentous actin in cells expressing GSTO1-1. Treatment of cells with GSNO caused increased glutathionylation and the formation of G actin.
Deglutathionylation of Actin by GSTO1-1 Influences Its Physiological Activity
Previous studies have shown that the glutathionylation of globular actin (G-actin) can prevent its polymerization to form filaments (F-actin). To further examine the functional effects of GSTO1-1-dependent deglutathionylation of actin, the ratio of G-actin/F-actin was determined in T47-D cells and T47-D GSTO1-1-expressing cells. The relative expression of G actin was found to be decreased in the GSTO1-1-expressing cells, causing a decrease in the G/F actin ratio (Fig. 3B). Confocal fluorescence microscopy also demonstrated the increase in the proportion of filamentous actin in cells expressing GSTO1-1 (Fig. 3C).
In Vitro Glutathionylation by GSTO1-1
The glutathionylating activity of GSTO1-1, GSTO2-2, CLIC2, and GSTK1-1 was tested using the unglutathionylated form of the SQLWCLSN peptide in the presence of high GSSG concentrations. Although we recorded a low spontaneous rate of glutathionylation (minus enzyme control reaction, Fig. 4A), the rate only increased marginally in the presence of GSTO1-1, GSTO2-2, and CLIC2 (Fig. 4A). The absolute rate of glutathionylation was very low when compared with the deglutathionylating activity of GSTO1-1 (Fig. 1). These results suggest that under these conditions GSTO1-1 acts primarily as a deglutathionylating enzyme.
FIGURE 4.

The increase in cellular glutathionylation after GSNO treatment is accelerated by GSTO1-1. A, the decrease in tryptophan fluorescence indicates the very low rate of glutathionylation of the peptide SQLWCLSN by GSTO1-1, GSTO2-2, and CLIC2. The results are the average traces from three replicates. B, shows that the increase in protein glutathionylation after treatment of cells with 1 mm GSNO is significantly higher in T47-D cells expressing active GSTO1-1 than in GSTO1-1-deficient cells transfected with the empty pCDNA3 vector or the C32A mutant enzyme. The data points represent the mean ± S.E. of three replicates. C, shows an immunoblot for glutathionylated protein in untreated T47-D cells and cells treated with 1 mm GSNO. GSNO caused the glutathionylation of several proteins that were deglutathionylated in untreated cells expressing GSTO1-1. D, the levels of GSSG are significantly higher after GSNO treatment in T47-D cells expressing GSTO1-1.
GSTO1-1 Promotes Glutathionylation in Cells Treated with GSNO
Although GSNO is not a strong glutathionylating agent in vitro (54, 55), it has previously been shown to strongly induce glutathionylation in cultured cells (44, 56). In this study we examined the time course of intracellular glutathionylation after treatment with 1 mm GSNO and compared T47-D cells transfected with either an empty pCDNA3 plasmid vector, cells transfected with pCDNA3-GSTO1C32A, or cells transfected with pCDNA3-GSTO1. The level of glutathionylation rose in each cell line; however, over the first 30 min the increased level of glutathionylation that occurred in cells expressing catalytically active GSTO1-1 vastly exceeded that occurring in the control pCDNA3 and pCDNA3-GSTO1C32A-transfected T47-D cells (Fig. 4B). Subsequently, the level of glutathionylation declined in the GSTO1-1-expressing cells but continued to climb slowly in both of the control cells. This marked difference in response suggests that GSTO1-1 is associated with the glutathionylation of proteins when cells are exposed to high GSNO concentrations and subsequently catalyzes the deglutathionylation of proteins when GSNO is depleted. An immunoblot of glutathionylated proteins shows that many proteins that are poorly glutathionylated in GSTO1-1-expressing cells become glutathionylated after the cells are treated with GSNO (Fig. 4C). This result is similar to the result shown in Fig. 3C where treatment of cells with GSNO caused a decrease in filamentous actin as a result of increased glutathionylation.
To investigate the mechanism of increased glutathionylation in GSTO1-1-expressing cells after GSNO exposure, the endogenous GSSG levels were measured. After 30 min the cells expressing active GSTO1-1 had significantly higher (p < 0.05) levels of free GSSG than the GSTO1-1-deficient cells. This suggests that GSTO1-1 may promote the formation of GSSG as an intermediate in the metabolism of GSNO, and the elevated GSSG level could contribute to the increased glutathionylation of proteins in these cells (Fig. 4D). The return of GSSG levels to control levels after 1 h probably results from its reduction to GSH by glutathione reductase and its efflux from the cells by multidrug resistance-related protein (MRP1).
Glutathione Thiyl Radicals as a GSTO1-1 Substrate
Previous studies have demonstrated that glutaredoxin can react with glutathione thiyl radicals (GS·) to form a disulfide anion radical intermediate (GRX-SSG⨪) and subsequently transfer the GS· to GSH or protein thiols to form GSSG or PSSG disulfides (24). In the present study we found that GSTO1-1 can also catalyze this reaction and that the rate of formation of GSSG by GSTO1-1D140 is significantly higher (p < 0.001) than the rate catalyzed by GSTO1-1A140 (Fig. 5A). To further validate the finding, we measured the level of glutathionylation in lymphoblastoid cells homozygous for the Ala-140 wild type and the Asp-140 variant. Cells expressing GSTO1-1 D140 had significantly higher basal glutathionylation levels (p < 0.05) and consistently exhibited higher glutathionylation levels after GSNO treatment (Fig. 5B).
FIGURE 5.
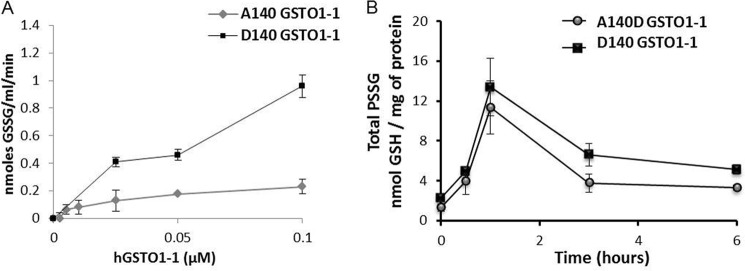
The Asp-140 variant of GSTO1 catalyzes increased GSSG formation and increased glutathionylation of cellular protein. A, GSTO1-1A140 catalyzes the formation of GSSG from glutathione thiyl radicals (GS·) at a significantly higher rate than GSTO1-1A140 (mean of three determinations ± S.E.). B, total protein glutathionylation after GSNO treatment is higher in cells expressing the GSTO1-1D140 variant.
DISCUSSION
A comparison of the structures of GSTO1, GSTO2, CLIC2, and glutaredoxin shown in Fig. 6 reveals several conserved features including their thioredoxin-like domains and a glutathione binding site where glutathione can form a disulfide bond with a conserved active site cysteine residue positioned near the amino-terminal end of helix 1. In GSTO1-1, GSTO2-2, and glutaredoxin these structural similarities translate into functional similarity indicated by their shared glutaredoxin (thioltransferase) activity. The close structural and functional similarity suggested that GSTO1-1 and GSTO2-2 may also catalyze glutathionylation reactions. In our initial studies we found that GSTO1-1, but not GSTO2-2, was capable of deglutathionylating a model peptide and that the activity was dependent on Cys-32, a residue that is also required for the glutaredoxin and S-phenacylglutathione reductase activities of GSTO1-1 (16, 20). We determined that this reaction is physiologically relevant by comparing the level of glutathionylation in the GSTO1-1-deficient T47-D breast cancer cell line with a derivative cell line that expresses recombinant GSTO1-1. Our finding that the expression of GSTO1-1 in T47-D cells causes a significant reduction in the level of intracellular glutathionylation confirmed the results obtained with the glutathionylated peptide. We suggest that the reaction proceeds by the following path where the model peptide represents a protein substrate and the enzyme is recycled by the action of GSH,
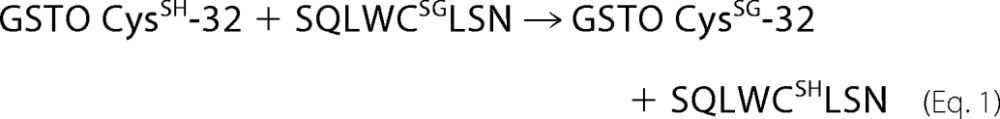 |
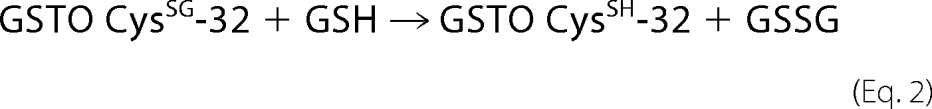 |
FIGURE 6.

The structure of the active sites of GSTO1-1 and Grx1 are well conserved. A, the structures of GSTO1, GSTO2, CLIC2, and Grx1 shown in a schematic are drawn from the coordinates in PDB files 1EEM, 3Q18, 2R4V, and 1GRX, respectively. In the conserved thioredoxin fold, domain β-sheet is shown in magenta, and helicies are shown in cyan. The carboxyl-terminal helical domain of GSTO1, GSTO2, and CLIC2 is shown in green. Glutathione is shown in stick format occupying a similar position in GSTO1 and Grx1 and forming a disulfide with the active site cysteine residue positioned near the amino-terminal end of helix 1. In CLIC2 and Grx1 the active site Cys residues form disulfide bonds with neighboring Cys residues. B shows the conservation of active site residues. Residues shaded pink are fully conserved, residues shaded in pink are very similar, and those residues shaded in blue are weakly similar.
Although GSTO2 has 68% sequence identity with GSTO1 and a conserved cysteine residue in its active site (57), it did not exhibit any deglutathionylation activity. We previously reported significant differences in the substrate/activity profile of GSTO2-2 compared with GSTO1-1. For example, whereas both enzymes exhibit glutaredoxin activity, GSTO2-2 has comparatively very high dehydroascorbate reductase activity and no S-phenacylglutathione reductase activity (16, 18, 58). Thus the observation that GSTO2-2 does not catalyze deglutathionylation is not without precedent. It is also notable that the GSTO1-C32A mutant showed increased dehydroascorbate reductase activity over the wild type enzyme, suggesting that Cys-32 is not required for this activity. Previous studies have shown that this mutant also has increased activity with 1-chloro-2,4-dinitrobenzene as a substrate (16). Clearly other components of the active site must contribute to the stabilization of GSH as a reactive thiolate ion. This has been shown to be the case in GSTA1-1 where Arg-15 contributes significantly to catalysis (59, 60) in the presence or absence of Tyr-8, the primary active site residue.
Immunoblotting shown in Fig. 2C suggested that there may be some substrate specificity and that some proteins may be more susceptible to deglutathionylation by GSTO1-1 than others. Mass spectrometry identified four proteins including β-actin that appear to be deglutathionylated by GSTO1-1 (Table 2). Immunoprecipitation of β-actin from T47-D cells confirmed the mass spectrometry and clearly demonstrated that it is specifically deglutathionylated by GSTO1-1. The deglutathionylation of β-actin catalyzed by GSTO1-1 was also shown to have a direct effect on the G/F actin ratio confirming the physiological significance of this deglutathionylation reaction. The substrate specificity suggested by the immunoblot in Fig. 2C is not surprising as glutathionylation is known to regulate the activity of specific proteins and metabolic pathways (3, 8, 12), and substrate specificity may be an important differentiating factor between the actions of glutaredoxin, thioredoxin, sulfiredoxin, and GSTP1–1 that have all been reported to catalyze glutathionylation cycle reactions. As a regulatory mechanism, glutathionylation has been likened to phosphorylation that is dependent on the actions of a plethora of specific kinases and phosphatases. The addition of GSTO1-1 to the catalogue of enzymes participating in specific glutathionylation cycle reactions clearly supports the view that glutathionylation is an important and specific regulatory process. The determination of the different protein substrate specificities for each of the enzymes involved in glutathionylation cycle reactions requires considerable further investigation.
We also considered the possibility that GSTO1-1 or GSTO2-2 may also contribute to the forward glutathionylation reaction; however, the data derived from the peptide assay suggest that this reaction is minor under those in vitro conditions. Interestingly it is clear from the data in Fig. 4b that GSTO1-1 plays a role in the rapid protein glutathionylation that occurs when cells are exposed to GSNO. It has been proposed that GSNO may enter cells indirectly via the transnitrosation of cysteine to S-nitrosocysteine that is taken up into cells by the L-system amino acid transporter (Fig. 7). A further transnitrosation step with GSH can regenerate GSNO (61). Previous studies have shown that GSNO is not a good glutathionylating agent in vitro (54, 55), and our data (not shown) indicated that it is not a glutathionylating substrate for GSTO1-1 in the peptide fluorescence assay. Thus the effect of GSTO1-1 on the protein glutathionylation that occurs when cells are treated with GSNO appears to be indirect. We found that GSNO treatment significantly elevated GSSG concentrations in T47-D cells expressing GSTO1-1 compared with untransfected T47-D cells. This change in the redox balance could lead to increased protein S-glutathionylation. The degradation of GSNO is dependent on the action of alcohol dehydrogenase class III and can generate a number of different intermediates (Fig. 7). In the presence of high glutathione concentrations the formation of GSSG is favored (62). In contrast, at low GSH concentrations the formation of glutathione disulfide-S-oxide occurs. Glutathione disulfide-S-oxide has been identified as a strong glutathionylating agent. Our data suggest that GSTO1-1 may play a role in the degradation of GSNO to glutathionylating intermediates. However, apart from the role played by alcohol dehydrogenase class III, none of the other reactions has thus far been attributed to the activity of a particular enzyme (Fig. 7).
FIGURE 7.
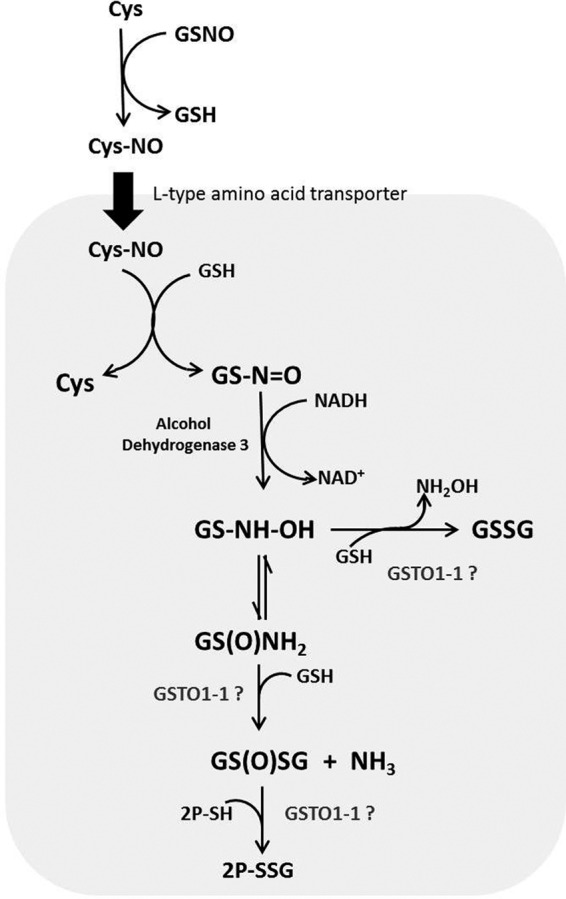
Pathways for the GSH-dependent metabolism of GSNO. GSNO can be taken into cells by transnitrosation with cysteine and the L-system amino acid transporter (61). Hypothetically GSTO1-1 could catalyze the formation of glutathione disulfide from S-(N-hydroxyamino)glutathione (GS-NH-OH) or the formation of glutathione disulfide-S-oxide (GS(O)SG; from glutathione sulfinamide (GS(O)NH2). Alternatively, GSTO1-1 could catalyze the final glutathionylation of proteins (P-SSG) by glutathione disulfide-S-oxide.
Glutaredoxin has been shown to use glutathione thiyl radicals to catalyze the formation of GSSG and to glutathionylate proteins. Our observation that GSTO1-1 can also generate GSSG from glutathione thiyl radicals suggests that, as occurs with the glutaredoxin-catalyzed reaction, protein thiols may also be a target for glutathionylation via this mechanism. Thus GSTO1-1 may contribute to the forward glutathionylation of proteins in the presence of suitable substrates.
Kinetic analysis of the deglutathionylation reaction with the glutathionylated peptide revealed that the Ala-140 allelic variant of GSTO1 has a significantly higher specific activity than the Asp-140 variant. In contrast the Asp-140 variant has a higher activity in the forward glutathionylation reaction with glutathione thiyl radicals as a substrate. Because glutathionylation can influence protein structure and function, the difference in activity and the potential difference in protein specificity between the allelic variants of GSTO1 could provide a plausible mechanism to explain the associations between this genetic polymorphism and a range of disorders (22, 24, 32, 33). For example, the A140D polymorphism is one of the major drivers of the association between the age at onset of both Alzheimer and Parkinson diseases with GSTO1 (22, 23), and it is conceivable that differences in the glutathionylation state of specific proteins expressed in the brain could contribute to a similar effect on these two distinct neurological disorders.
The experiments reported here have revealed the previously unrecognized contribution of GSTO1-1 to the deglutaththionylation of proteins. Because GSTO1-1 is widely expressed in many tissues, it appears to be capable of playing a major role in the glutathionylation cycle. In addition, the differences in the deglutathionylation/glutathionylation reaction kinetics that we observed between different allelic variants of GSTO1 could translate into distinct genetic effects on those enzymes and pathways that are regulated by GSTO1-1.
Footnotes
- GSTO
- GST Omega class
- GSNO
- nitrosoglutathione
- CLIC-2
- chloride intracellular channel 2 protein
- GSTK
- GST Kappa class
- GS
- glutathione
- Grx1
- glutaredoxin 1
- PSSG
- protein-glutathione mixed disulfide.
REFERENCES
- 1. Cooper A. J., Pinto J. T., Callery P. S. (2011) Reversible and irreversible protein glutathionylation. Biological and clinical aspects. Expert. Opin. Drug Metab. Toxicol. 7, 891–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dalle-Donne I., Rossi R., Colombo G., Giustarini D., Milzani A. (2009) Protein S-glutathionylation. A regulatory device from bacteria to humans. Trends Biochem. Sci. 34, 85–96 [DOI] [PubMed] [Google Scholar]
- 3. Pastore A., Piemonte F. (2012) S-Glutathionylation signaling in cell biology. Progress and prospects. Eur. J. Pharm. Sci. 46, 279–292 [DOI] [PubMed] [Google Scholar]
- 4. Jarry A., Charrier L., Bou-Hanna C., Devilder M. C., Crussaire V., Denis M. G., Vallette G., Laboisse C. L. (2004) Position in cell cycle controls the sensitivity of colon cancer cells to nitric oxide-dependent programmed cell death. Cancer Res. 64, 4227–4234 [DOI] [PubMed] [Google Scholar]
- 5. Bedhomme M., Adamo M., Marchand C. H., Couturier J., Rouhier N., Lemaire S. D., Zaffagnini M., Trost P. (2012) Glutathionylation of cytosolic glyceraldehyde-3-phosphate dehydrogenase from the model plant Arabidopsis thaliana is reversed by both glutaredoxins and thioredoxins in vitro. Biochem. J. 445, 337–347 [DOI] [PubMed] [Google Scholar]
- 6. Anathy V., Roberson E., Cunniff B., Nolin J. D., Hoffman S., Spiess P., Guala A. S., Lahue K. G., Goldman D., Flemer S., van der Vliet A., Heintz N. H., Budd R. C., Tew K. D., Janssen-Heininger Y. M. (2012) Oxidative processing of latent Fas in the endoplasmic reticulum controls the strength of apoptosis. Mol. Cell Biol. 32, 3464–3478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Allen E. M., Mieyal J. J. (2012) Protein-thiol oxidation and cell death. Regulatory role of glutaredoxins. Antioxid. Redox Signal. 17, 1748–1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lei K., Townsend D. M., Tew K. D. (2008) Protein cysteine sulfinic acid reductase (sulfiredoxin) as a regulator of cell proliferation and drug response. Oncogene 27, 4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carletti B., Passarelli C., Sparaco M., Tozzi G., Pastore A., Bertini E., Piemonte F. (2011) Effect of protein glutathionylation on neuronal cytoskeleton. A potential link to neurodegeneration. Neuroscience 192, 285–294 [DOI] [PubMed] [Google Scholar]
- 10. Lindahl M., Mata-Cabana A., Kieselbach T. (2011) The disulfide proteome and other reactive cysteine proteomes. Analysis and functional significance. Antioxid. Redox Signal. 14, 2581–2642 [DOI] [PubMed] [Google Scholar]
- 11. Mieyal J. J., Gallogly M. M., Qanungo S., Sabens E. A., Shelton M. D. (2008) Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 10, 1941–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shelton M. D., Chock P. B., Mieyal J. J. (2005) Glutaredoxin. Role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 7, 348–366 [DOI] [PubMed] [Google Scholar]
- 13. Townsend D. M., Tew K. D., He L., King J. B., Hanigan M. H. (2009) Role of glutathione S-transferase Pi in cisplatin-induced nephrotoxicity. Biomed. Pharmacother. 63, 79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Board P. G., Coggan M., Chelvanayagam G., Easteal S., Jermiin L. S., Schulte G. K., Danley D. E., Hoth L. R., Griffor M. C., Kamath A. V., Rosner M. H., Chrunyk B. A., Perregaux D. E., Gabel C. A., Geoghegan K. F., Pandit J. (2000) Identification, characterization, and crystal structure of the omega class glutathione transferases. J. Biol. Chem. 275, 24798–24806 [DOI] [PubMed] [Google Scholar]
- 15. Whitbread A. K., Tetlow N., Eyre H. J., Sutherland G. R., Board P. G. (2003) Characterization of the human omega class glutathione transferase genes and associated polymorphisms. Pharmacogenetics 13, 131–144 [DOI] [PubMed] [Google Scholar]
- 16. Whitbread A. K., Masoumi A., Tetlow N., Schmuck E., Coggan M., Board P. G. (2005) Characterization of the omega class of glutathione transferases. Methods Enzymol. 401, 78–99 [DOI] [PubMed] [Google Scholar]
- 17. Yin Z. L., Dahlstrom J. E., Le Couteur D. G., Board P. G. (2001) Immunohistochemistry of omega class glutathione S-transferase in human tissues. J. Histochem. Cytochem. 49, 983–987 [DOI] [PubMed] [Google Scholar]
- 18. Schmuck E. M., Board P. G., Whitbread A. K., Tetlow N., Cavanaugh J. A., Blackburn A. C., Masoumi A. (2005) Characterization of the monomethylarsonate reductase and dehydroascorbate reductase activities of omega class glutathione transferase variants. Implications for arsenic metabolism and the age-at-onset of Alzheimer's and Parkinson's diseases. Pharmacogenet. Genomics 15, 493–501 [DOI] [PubMed] [Google Scholar]
- 19. Board P. G., Chelvanayagam G., Jermiin L. S., Tetlow N., Tzeng H. F., Anders M. W., Blackburn A. C. (2001) Identification of novel glutathione transferases and polymorphic variants by expressed sequence tag database analysis. Drug Metab. Dispos. 29, 544–547 [PubMed] [Google Scholar]
- 20. Board P. G., Anders M. W. (2007) Glutathione transferase omega 1 catalyzes the reduction of S-(phenacyl)glutathiones to acetophenones. Chem. Res. Toxicol. 20, 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kodym R., Calkins P., Story M. (1999) The cloning and characterization of a new stress response protein. A mammalian member of a family of theta class glutathione S-transferase-like proteins. J. Biol. Chem. 274, 5131–5137 [DOI] [PubMed] [Google Scholar]
- 22. Li Y. J., Oliveira S. A., Xu P., Martin E. R., Stenger J. E., Scherzer C. R., Hauser M. A., Scott W. K., Small G. W., Nance M. A., Watts R. L., Hubble J. P., Koller W. C., Pahwa R., Stern M. B., Hiner B. C., Jankovic J., Goetz C. G., Mastaglia F., Middleton L. T., Roses A. D., Saunders A. M., Schmechel D. E., Gullans S. R., Haines J. L., Gilbert J. R., Vance J. M., Pericak-Vance M. A. (2003) Glutathione S-transferase omega-1 modifiesage-at-onset of Alzheimer disease and Parkinson disease. Hum. Mol. Genet. 12, 3259–3267 [DOI] [PubMed] [Google Scholar]
- 23. Li Y. J., Scott W. K., Zhang L., Lin P. I., Oliveira S. A., Skelly T., Doraiswamy M. P., Welsh-Bohmer K. A., Martin E. R., Haines J. L., Pericak-Vance M. A., Vance J. M. (2006) Revealing the role of glutathione S-transferase omega in age-at-onset of Alzheimer and Parkinson diseases. Neurobiol. Aging 27, 1087–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Capurso C., Panza F., Seripa D., Frisardi V., Imbimbo B. P., Verdile G., Vendemiale G., Pilotto A., Solfrizzi V. (2010) Polymorphisms in glutathione S-transferase omega-1 gene and increased risk of sporadic Alzheimer disease. Rejuvenation Res. 13, 645–652 [DOI] [PubMed] [Google Scholar]
- 25. Li Y. J., Scott W. K., Hedges D. J., Zhang F., Gaskell P. C., Nance M. A., Watts R. L., Hubble J. P., Koller W. C., Pahwa R., Stern M. B., Hiner B. C., Jankovic J., Allen F. A., Jr., Goetz C. G., Mastaglia F., Stajich J. M., Gibson R. A., Middleton L. T., Saunders A. M., Scott B. L., Small G. W., Nicodemus K. K., Reed A. D., Schmechel D. E., Welsh-Bohmer K. A., Conneally P. M., Roses A. D., Gilbert J. R., Vance J. M., Haines J. L., Pericak-Vance M. A. (2002) Age at onset in two common neurodegenerative diseases is genetically controlled. Am. J. Hum. Genet. 70, 985–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kölsch H., Linnebank M., Lütjohann D., Jessen F., Wüllner U., Harbrecht U., Thelen K. M., Kreis M., Hentschel F., Schulz A., von Bergmann K., Maier W., Heun R. (2004) Polymorphisms in glutathione S-transferase omega-1 and AD, vascular dementia, and stroke. Neurology 63, 2255–2260 [DOI] [PubMed] [Google Scholar]
- 27. Kölsch H., Larionov S., Dedeck O., Orantes M., Birkenmeier G., Griffin W. S., Thal D. R. (2007) Association of the glutathione S-transferase Omega-1 Ala140Asp polymorphism with cerebrovascular atherosclerosis and plaque-associated interleukin-1α Expression. Stroke 38, 2847–2850 [DOI] [PubMed] [Google Scholar]
- 28. van de Giessen E., Fogh I., Gopinath S., Smith B., Hu X., Powell J., Andersen P., Nicholson G., Al Chalabi A., Shaw C. E. (2008) Association study on glutathione S-transferase omega 1 and 2 and familial ALS. Amyotroph. Lateral Scler. 9, 81–84 [DOI] [PubMed] [Google Scholar]
- 29. Laliberte R. E., Perregaux D. G., Hoth L. R., Rosner P. J., Jordan C. K., Peese K. M., Eggler J. F., Dombroski M. A., Geoghegan K. F., Gabel C. A. (2003) Glutathione S-transferase omega 1-1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1β posttranslational processing. J. Biol. Chem. 278, 16567–16578 [DOI] [PubMed] [Google Scholar]
- 30. Zakharyan R. A., Sampayo-Reyes A., Healy S. M., Tsaprailis G., Board P. G., Liebler D. C., Aposhian H. V. (2001) Human monomethylarsonic acid (MMA(V)) reductase is a member of the glutathione S-transferase superfamily. Chem. Res. Toxicol. 14, 1051–1057 [DOI] [PubMed] [Google Scholar]
- 31. Harju T. H., Peltoniemi M. J., Rytilä P. H., Soini Y., Salmenkivi K. M., Board P. G., Ruddock L. W., Kinnula V. L. (2007) Glutathione S-transferase omega in the lung and sputum supernatants of COPD patients. Respir. Res. 8, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yanbaeva D. G., Wouters E. F., Dentener M. A., Spruit M. A., Reynaert N. L. (2009) Association of glutathione S-transferase omega haplotypes with susceptibility to chronic obstructive pulmonary disease. Free Radic. Res. 43, 738–743 [DOI] [PubMed] [Google Scholar]
- 33. Wilk J. B., Walter R. E., Laramie J. M., Gottlieb D. J., O'Connor G. T. (2007) Framingham heart study genome-wide association. Results for pulmonary function measures. BMC Med. Genet. 8, S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pongstaporn W., Pakakasama S., Sanguansin S., Hongeng S., Petmitr S. (2009) Polymorphism of glutathione S-transferase Omega gene. Association with risk of childhood acute lymphoblastic leukemia. J. Cancer Res. Clin. Oncol. 135, 673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pongstaporn W., Rochanawutanon M., Wilailak S., Linasamita V., Weerakiat S., Petmitr S. (2006) Genetic alterations in chromosome 10q24.3 and glutathione S-transferase omega 2 gene polymorphism in ovarian cancer. J. Exp. Clin. Cancer Res. 25, 107–114 [PubMed] [Google Scholar]
- 36. Wang Y. H., Yeh S. D., Shen K. H., Shen C. H., Juang G. D., Hsu L. I., Chiou H. Y., Chen C. J. (2009) A significantly joint effect between arsenic and occupational exposures and risk genotypes/diplotypes of CYP2E1, GSTO1, and GSTO2 on risk of urothelial carcinoma. Toxicol. Appl. Pharmacol. 241, 111–118 [DOI] [PubMed] [Google Scholar]
- 37. Andonova I. E., Justenhoven C., Winter S., Hamann U., Baisch C., Rabstein S., Spickenheuer A., Harth V., Pesch B., Brüning T., Ko Y. D., Ganev V., Brauch H. (2010) No evidence for glutathione S-transferases GSTA2, GSTM2, GSTO1, GSTO2, and GSTZ1 in breast cancer risk. Breast Cancer Res. Treat. 121, 497–502 [DOI] [PubMed] [Google Scholar]
- 38. Chariyalertsak S., Purisa W., Sangrajrang S. (2009) Role of glutathione S-transferase omega gene polymorphisms in breast-cancer risk. Tumori 95, 739–743 [DOI] [PubMed] [Google Scholar]
- 39. Masoudi M., Saadat I., Omidvari S., Saadat M. (2009) Genetic polymorphisms of GSTO2, GSTM1, and GSTT1 and risk of gastric cancer. Mol. Biol. Rep. 36, 781–784 [DOI] [PubMed] [Google Scholar]
- 40. Granja F., Morari E. C., Assumpção L. V., Ward L. S. (2005) GSTO polymorphism analysis in thyroid nodules suggest that GSTO1 variants do not influence the risk for malignancy. Eur. J. Cancer Prev. 14, 277–280 [DOI] [PubMed] [Google Scholar]
- 41. Baker R. T., Catanzariti A. M., Karunasekara Y., Soboleva T. A., Sharwood R., Whitney S., Board P. G. (2005) Using deubiquitylating enzymes as research tools. Methods Enzymol. 398, 540–554 [DOI] [PubMed] [Google Scholar]
- 42. Peltoniemi M. J., Karala A. R., Jurvansuu J. K., Kinnula V. L., Ruddock L. W. (2006) Insights into deglutathionylation reactions. Different intermediates in the glutaredoxin and protein disulfide isomerase catalyzed reactions are defined by the γ-linkage present in glutathione. J. Biol. Chem. 281, 33107–33114 [DOI] [PubMed] [Google Scholar]
- 43. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 44. Menon D., Board P. G. (2013) A fluorometric method to quantify protein glutathionylation using glutathione derivatization with 2,3-naphthalenedicarboxaldehyde. Anal. Biochem. 433, 132–136 [DOI] [PubMed] [Google Scholar]
- 45. Robinson A., Huttley G. A., Booth H. S., Board P. G. (2004) Modelling and bioinformatics studies of the human Kappa-class glutathione transferase predict a novel third glutathione transferase family with similarity to prokaryotic 2-hydroxychromene-2-carboxylate isomerases. Biochem. J. 379, 541–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Starke D. W., Chock P. B., Mieyal J. J. (2003) Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J. Biol. Chem. 278, 14607–14613 [DOI] [PubMed] [Google Scholar]
- 47. Rahman I., Kode A., Biswas S. K. (2006) Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 1, 3159–3165 [DOI] [PubMed] [Google Scholar]
- 48. Findlay V. J., Townsend D. M., Morris T. E., Fraser J. P., He L., Tew K. D. (2006) A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 66, 6800–6806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Board P. G., Coggan M., Watson S., Gage P. W., Dulhunty A. F. (2004) CLIC-2 modulates cardiac ryanodine receptor Ca2+ release channels. Int. J. Biochem. Cell Biol. 36, 1599–1612 [DOI] [PubMed] [Google Scholar]
- 50. Cromer B. A., Gorman M. A., Hansen G., Adams J. J., Coggan M., Littler D. R., Brown L. J., Mazzanti M., Breit S. N., Curmi P. M., Dulhunty A. F., Board P. G., Parker M. W. (2007) Structure of the Janus protein human CLIC2. J. Mol. Biol. 374, 719–731 [DOI] [PubMed] [Google Scholar]
- 51. Schmuck E., Cappello J., Coggan M., Brew J., Cavanaugh J. A., Blackburn A. C., Baker R. T., Eyre H. J., Sutherland G. R., Board P. G. (2008) Deletion of Glu-155 causes a deficiency of glutathione transferase Omega 1-1 but does not alter sensitivity to arsenic trioxide and other cytotoxic drugs. Int. J. Biochem. Cell Biol. 40, 2553–2559 [DOI] [PubMed] [Google Scholar]
- 52. Zhou H., Brock J., Casarotto M. G., Oakley A. J., Board P. G. (2011) Novel folding and stability defects cause a deficiency of human glutathione transferase omega 1. J. Biol. Chem. 286, 4271–4279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Townsend D. M., Manevich Y., He L., Hutchens S., Pazoles C. J., Tew K. D. (2009) Novel role for glutathione S-transferase pi. Regulator of protein S-glutathionylation following oxidative and nitrosative stress. J. Biol. Chem. 284, 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barglow K. T., Knutson C. G., Wishnok J. S., Tannenbaum S. R., Marletta M. A. (2011) Site-specific and redox-controlled S-nitrosation of thioredoxin. Proc. Natl. Acad. Sci. U.S.A. 108, E600–E606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tao L., English A. M. (2004) Protein S-glutathiolation triggered by decomposed S-nitrosoglutathione. Biochemistry 43, 4028–4038 [DOI] [PubMed] [Google Scholar]
- 56. de Luca A., Moroni N., Serafino A., Primavera A., Pastore A., Pedersen J. Z., Petruzzelli R., Farrace M. G., Pierimarchi P., Moroni G., Federici G., Sinibaldi Vallebona P., Lo Bello M. (2011) Treatment of doxorubicin-resistant MCF7/Dx cells with nitric oxide causes histone glutathionylation and reversal of drug resistance. Biochem. J. 440, 175–183 [DOI] [PubMed] [Google Scholar]
- 57. Zhou H., Brock J., Liu D., Board P. G., Oakley A. J. (2012) Structural insights into the dehydroascorbate reductase activity of human omega-class glutathione transferases. J. Mol. Biol. 420, 190–203 [DOI] [PubMed] [Google Scholar]
- 58. Board P. G., Coggan M., Cappello J., Zhou H., Oakley A. J., Anders M. W. (2008) S-(4-Nitrophenacyl)glutathione is a specific substrate for glutathione transferase omega 1-1. Anal. Biochem. 374, 25–30 [DOI] [PubMed] [Google Scholar]
- 59. Stenberg G., Board P. G., Mannervik B. (1991) Mutation of an evolutionarily conserved tyrosine residue in the active site of a human class Alpha glutathione transferase. FEBS Lett. 293, 153–155 [DOI] [PubMed] [Google Scholar]
- 60. Björnestedt R., Stenberg G., Widersten M., Board P. G., Sinning I., Jones T. A., Mannervik B. (1995) Functional significance of arginine 15 in the active site of human class alpha glutathione transferase A1-1. J. Mol. Biol. 247, 765–773 [DOI] [PubMed] [Google Scholar]
- 61. Broniowska K. A., Diers A. R., Hogg N. (2013) S-nitrosoglutathione. Biochim. Biophys. Acta 1830, 3173–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Staab C. A., Alander J., Brandt M., Lengqvist J., Morgenstern R., Grafström R. C., Höög J. O. (2008) Reduction of S-nitrosoglutathione by alcohol dehydrogenase 3 is facilitated by substrate alcohols via direct cofactor recycling and leads to GSH-controlled formation of glutathione transferase inhibitors. Biochem. J. 413, 493–504 [DOI] [PubMed] [Google Scholar]