

Background: HC-HA is a unique anti-inflammatory matrix different from hyaluronic acid (HA).
Results: Soluble HC-HA induces apoptosis of inflammatory neutrophils and macrophages, and immobilized HC-HA promotes M2 polarization upon LPS/TLR ligation while both enhancing macrophage phagocytosis.
Conclusion: HC-HA exerts its anti-inflammatory action using multiple mechanisms.
Significance: HC-HA is the first known matrix component to polarize M2b.
Keywords: Apoptosis; Cell Adhesion; Cell Proliferation; Inflammation; Macrophages; Phagocytosis; Amniotic Membrane, HC-HA, M1/M2 Phenotype
Abstract
Despite the known anti-inflammatory effect of amniotic membrane, its action mechanism remains largely unknown. HC-HA complex (HC-HA) purified from human amniotic membrane consists of high molecular weight hyaluronic acid (HA) covalently linked to the heavy chain (HC) 1 of inter-α-trypsin inhibitor. In this study, we show that soluble HC-HA also contained pentraxin 3 and induced the apoptosis of both formyl-Met-Leu-Phe or LPS-activated neutrophils and LPS-activated macrophages while not affecting the resting cells. This enhanced apoptosis was caused by the inhibition of cell adhesion, spreading, and proliferation caused by HC-HA binding of LPS-activated macrophages and preventing adhesion to the plastic surface. Preferentially, soluble HC-HA promoted phagocytosis of apoptotic neutrophils in resting macrophages, whereas immobilized HC-HA promoted phagocytosis in LPS-activated macrophages. Upon concomitant LPS stimulation, immobilized HC-HA but not HA polarized macrophages toward the M2 phenotype by down-regulating IRF5 protein and preventing its nuclear localization and by down-regulating IL-12, TNF-α, and NO synthase 2. Additionally, IL-10, TGF-β1, peroxisome proliferator-activated receptor γ, LIGHT (TNF superfamily 14), and sphingosine kinase-1 were up-regulated, and such M2 polarization was dependent on TLR ligation. Collectively, these data suggest that HC-HA is a unique matrix component different from HA and uses multiple mechanisms to suppress M1 while promoting M2 phenotype. This anti-inflammatory action of HC-HA is highly desirable to promote wound healing in diseases heightened by unsuccessful transition from M1 to M2 phenotypes.
Introduction
Inflammation serves as the first host response to real or perceived threats to tissue homeostasis. It is heralded by the recruitment of neutrophils, which eliminate pathogens and damaged tissues before eventually undergoing apoptosis (1–3). These apoptotic neutrophils are then removed by macrophages via phagocytosis, resulting in the restoration and maintenance of anti-inflammatory and tolerogenic milieu (4). On the contrary, delayed neutrophil apoptosis and/or impaired phagocytic clearance of apoptotic neutrophils by macrophages leads to prolonged inflammation that is the hallmark of a number of diseases (5–8). Hence, facilitation of neutrophil apoptosis and phagocytic clearance of apoptotic neutrophils by macrophages is an important strategy to limit inflammation-mediated tissue damage to restore tissue homeostasis (9–11).
Macrophages undergo phenotypic changes to adapt to the transition from proinflammatory to anti-inflammatory states of wound healing (12–15). During the acute inflammatory stage, macrophages adopt the classically activated M1 phenotype that can be reproduced in vitro by TNF and IFN-γ signals or by the Toll-like receptor 4 (TLR4)2 agonist LPS (13), which produce TNF and IFN-β via MyD88- and TIR domain-containing adaptor-inducing IFN-β-dependent pathways, respectively (16). M1 macrophages secrete proinflammatory cytokines such as IL-12, TNF, and IL-6; up-regulate the expression of CD86 and MHC class II (17); and have a decreased phagocytic capacity (18, 19). In contrast, phagocytosis of apoptotic neutrophils is more efficiently executed by M2 macrophages (11, 20, 21), which can further be subdivided into wound-healing (originally “alternative,” M2a), regulatory (M2b), and anti-inflammatory (sometimes “deactivated,” M2c) that generally produce anti-inflammatory cytokines such as IL-10 (12, 22–25) and TGF-β (4, 26) and up-regulate the expression of PPAR-γ (27), LIGHT (TNF superfamily 14), and sphingosine kinase-1 (SPHK1) (12). Failure to transition from M1 to M2 phenotypes results in unresolved inflammation, which is the common denominator of diverse inflammatory diseases (15, 28, 29).
Amniotic membrane (AM) is the innermost layer of the placental membrane, consisting of a simple epithelium, a thick basement membrane, and a subjacent avascular stroma. Over the past decade, cryopreserved human AM has been applied to surgical or injury sites to reduce inflammation and scarring on the eye surface (for reviews see Refs. 30–32). Application of cryopreserved human AM reduces infiltration of neutrophils in the excimer-ablated rabbit corneal stroma while promoting apoptosis of neutrophils (33, 34); traps and causes apoptosis of monocytes and macrophages during the treatment of human corneal epithelial defects (35); induces rapid regression of corneal stromal edema and inflammation by reducing infiltration of lymphocytes (36), neutrophils (37), and macrophages (37, 38); and promotes M2a phenotype in the corneal stroma of murine HSV-1 necrotizing keratitis (39). These in vivo data are supported by in vitro findings that human AM induces apoptosis of IFN-γ activated monocyte/macrophage RAW264.7 cells (40).
To better understand the molecular mechanism of these clinical efficacies of cryopreserved AM, we have previously found that soluble AM extract (AME; e.g., AM extracted by PBS) promotes apoptosis of IFN-γ-, LPS-, and IFN-γ/LPS-activated but not resting macrophages and down-regulates the expression of M1 markers such as TNF-α, IL-6, CD86, and class II MHC while up-regulating the M2 cytokine IL-10 (41). Recently, we have purified HC-HA complex (HC-HA) from soluble AM extract. This complex, formed by a covalent linkage between hyaluronan (HA) and heavy chain 1 (HC1) of inter-α-trypsin inhibitor (IαI) via TSG-6 catalytic action (42), has been shown to be effective in promoting apoptosis of LPS-activated macrophages, inhibiting TGF-β1 promoter activity in corneal stromal fibroblasts (43), and suppressing endothelial tube formation (44). Herein, we further demonstrate that HC-HA is indeed responsible for the anti-inflammatory effect of AM by promoting apoptosis of activated but not resting neutrophils and macrophages and by polarizing the M2 phenotype in LPS-activated macrophages for efficient phagocytosis of apoptotic neutrophils.
EXPERIMENTAL PROCEDURES
Materials
Cesium chloride, EDTA, guanidine hydrochloride, sodium hydroxide, protease inhibitor mixture (a mixture of aprotinin, bestatin hydrochloride, E-64, leupeptin, and pepstatin A), and PMSF were from Sigma. Medical grade HA with a molecular mass of 4000 kDa, Healon®, was from Advanced Medical Optics (Santa Ana, CA). Enzyme-free cell dissociation buffer, PBS, Dulbecco's PBS, DMEM, Iscove's modified Dulbecco's medium, fetal bovine serum, and penicillin/streptomycin were from Invitrogen. Phosphatase inhibitors (sodium fluoride and sodium vanadate) and dialysis tubes (molecular mass cutoff, 3500 Da) were from Fisher Scientific (Pittsburgh, PA). BCA protein assay kit, Sulfo-NHS, and 1-ethyl-3-(3-dimethylaminopropyl) carbidodiimide (EDAC) were from Pierce. The HA quantitative test kit was from Corgenix (Westminster, CO). The CovaLink NH 96-well plate was from Nunc (Thermo Scientific, Rochester, NY). 4–15% gradient acrylamide ready gels and nitrocellulose membranes were from Bio-Rad. The antibodies to interferon regulatory factor 5 (IRF5) (ab33478), HC1 (ab70048), HC2 (ab118257), and bikunin (ab43073) were from Abcam (Cambridge, UK). The antibody to pentraxin 3 (PTX3) (MNB1, ALX-804-463-C100) was from Enzo Life Sciences, Inc. (Farmingdale, NY). Proteins of TSG-6 (2104-TS-050) and PTX3 (1826-TS-025), the antibody to TSG-6 (MAB2104), and the human myeloperoxidase ELISA kit (DMYE00) were from R&D Systems (Minneapolis, MN). Secondary antibodies conjugated with the peroxidase were from DAKO (Carpinteria, CA). The RNeasy mini kit was from Qiagen. cDNA synthesis kit, cell viability kit (MTT), cell proliferation kit (MTT), and cell death detection ELISA were from Roche Applied Science. All primers (Arg-1, FIZZ1, GAPDH, IL-10, IL-12p35, IL-12p40, LIGHT, nitric-oxide synthase 2 (NOS2), PPAR-γ, SPHK1, TNF-α, TGF-β1, and Ym1 for qPCR) were from Applied Biosystems (Carlsbad, CA). ELISAs for murine IL-10, IL-12p70, TGF-β1, and TNF-α were from Biolegend (San Diego, CA). Lymphocyte Poly(R) was from Cedarlane Labs (Burlington, NC). Western Lighting® Chemiluminesence Reagent was from PerkinElmer (Wellesley, MA).
Preparation of AM Extract and Purification of HC-HA Complex
As reported (41, 43), AM extract and HC-HA were prepared aseptically from frozen human AM obtained from Bio-Tissue, Inc. (Miami, FL). After washing off the original storage medium, intact AM was frozen in liquid nitrogen and ground to fine particles (BioPulverizer, Biospec Products, Inc., Bartlesville, OK) followed by homogenization on ice. The supernatant was collected by centrifugation at 48,000 g at 4 °C for 30 min, followed by two runs of ultracentrifugation in CsCl/4 M guanidine HCl at the initial density of 1.35 g/ml (1st run) and 1.40 g/ml (2nd run). Fractions containing HA measured by the HA Quantitative Test Kit but no detectable amount of proteins measured by the BCA assay were designated as HC-HA. These fractions were pooled and dialyzed extensively with sterile distilled water. Thus, we expressed HC-HA concentration based on the HA concentration.
Immobilization of HA and HC-HA
The covalent coupling of HA or HC-HA was performed on the surface of CovaLink NH 96-well as previously reported (43). In brief, CovaLink NH 96-well plates were sterilized in 70% alcohol for 1 h then washed three times with distilled water. In each well, 100 μl of 0.184 mg/ml Sulfo-NHS, 0.123 mg/ml of EDAC, and 20 μg/ml HA or HC-HA in distilled water was added. The plate was incubated at 4 °C overnight or at 25 °C for 2 h before the coupling solution was removed, washed three times with PBS containing 2 m NaCl and 50 mm MgSO4, and washed three times with PBS.
Isolation, Activation, and Apoptosis of Human Neutrophils
Neutrophils were isolated from the peripheral blood of healthy human donors using dextran density [Lymphocyte Poly(R)] centrifugation according to the manufacturer's instruction. Neutrophils were cultivated at 6.3 × 105/cm2 at 37 °C with 5% CO2 in Iscove's modified Dulbecco's medium supplemented with 10% FBS and 100 units/ml penicillin and 100 μg/ml streptomycin. Neutrophils were activated with 1 μm fMLP or 1 μg/ml LPS for 20 h to reduce apoptosis or treated with 20 μm R-roscovitine (a selective inhibitor of cyclin-dependent kinases or CDKs) for 8 h to enhance apoptosis (45).
Cell Viability, Apoptosis, and Proliferation
Cell viability and apoptosis were measured by MTT and Cell Death Detection ELISA, respectively, as reported (41). To measure cell proliferation, resting or 1 μg/ml LPS-stimulated RAW264.7 cells (3.1 × 104/cm2) were treated with 25 μg/ml of soluble HA or HC-HA immediately before seeding on plastic 96-well plates. In parallel, the same density of resting and LPS-stimulated cells was seeded on immobilized HA or HC-HA wells. After incubation for 22 h, cells were labeled with 10 μm BrdU for 2 h prior to Cell Proliferation ELISA to determine the amount of BrdU incorporated into the cellular DNA.
Phagocytosis of Apoptotic Neutrophils
Resting RAW264.7 cells were seeded at 3.1 × 105/cm2 (or 1 × 105 per each well) with or without immobilized HA or HC-HA and simultaneously treated with or without 1 μg/ml LPS for 6 days. After removal of the cell culture medium, the cells were washed twice with Iscove's modified Dulbecco's medium. Afterward, 0.1 ml of apoptotic neutrophils (pretreated with 20 μm roscovitine for 8 h to induce >90% apoptosis, 2 × 105 per well) were added to each well containing differently treated macrophages for 30 min at 37 °C. Apoptotic neutrophils that were not phagocytosed by macrophages were removed. The remaining cells were washed three times with ice-cold PBS and lysed with RIPA buffer (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA,1% Nonidet P-40, 1% sodium deoxycholate, 2.5 mm sodium pyrophosphate,1 mm b-glycerophosphate,1 mm Na3VO4, 1 μg/ml leupeptin). Cell lysates were subjected to human myeloperoxidase ELISA assay. The phagocytic index was expressed as the relative units of the myeloperoxidase activity in each sample normalized with its respective total protein content. This index was arbitrarily defined as 100% in resting RAW264.7 cells on plastic controls with or without immobilization.
Cell Adhesion and Spreading
Resting RAW264.7 cells were harvested using enzyme-free cell dissociation buffer and treated with or without 1 μg/ml LPS, mixed with 25 μg/ml of soluble HA or HC-HA, and immediately seeded on plastic 96-well (3.1 × 105/cm2). Alternatively, these cells were seeded on immobilized control, HA, or HC-HA (3.1 × 105/cm2). For cell attachment assay, cells were incubated for 90 min before unattached cells were removed by three times wash with Dulbecco's PBS. The remaining cells were quantified by CyQuant fluorescent dye using a fluorescence microplate reader with excitation at 485 nm and emission detection at 530 nm. This assay exhibits a linear correlation in 100–10,000 cells. For cell spreading assay, the incubation was continued for 24 h. After 1 h, 2 h, 3 h, and 24 h incubation, the cell morphology was recorded with a converted microscope. Cell dimensions were measured using ImageJ 1.46 software (National Institutes of Health, Bethesda, MD). At least 200 randomly selected cells of each sample were quantified (46).
Extraction of Total RNA and Real Time Quantitative PCR
Total RNAs were extracted using RNeasy Mini Kit (Qiagen, Valencia, CA) and reverse-transcribed using High Capacity Reverse Transcription Kit (Applied Biosystems, Foster City, CA). cDNA of each sample was amplified by qPCR using specific primer-probe mixtures and DNA polymerase in the 7300 Real-time PCR System (Applied Biosystems). The program for real time PCR profile consisted of 10 min of initial activation at 95 °C followed by 40 cycles of 15 s of denaturation at 95 °C and 1 min of annealing and extension at 60 °C. The relative gene expression data were analyzed by the comparative CT method (ΔΔCT). All assays were performed in triplicate; the results were normalized by GAPDH as an internal control.
TGF-β1 Promoter Assay
The expression of the TGF-β1 gene was measured by a promoter assay as described before (43). Briefly, RAW264.7 cells (3.1 × 105/cm2) were seeded on immobilized control, HA, or HC-HA for 16 h. After the medium was removed, cells were infected with adeno-TGF-β1 promoter-luciferase (multiplicity of infection = 100) and adeno-CMV-β-galactosidase (multiplicity of infection = 30) for 48 h. The cells were then lysed in a buffer containing 25 mm Tris phosphate, pH 7.8, 2 mm DTT, 2 mm 1,2-diaminocyclohexane-N,N,N,N-tetraacetic acid, 10% (v/v) glycerol, 1% (v/v) Triton X-100. The supernatant was assayed for both luciferase and β-galactosidase activities. The relative luciferase activity was calculated by dividing the luciferase activity by the corresponding β-galactosidase activity.
Immunofluorescence Staining
RAW264.7 cells (3.1 × 104/cm2) were treated with or without 1 μg/ml LPS and seeded on wells with immobilized control, HA, or HC-HA. After incubation for 4 h, the cells were fixed with 4% paraformaldehyde in PBS for 20 min followed by permeabilization with 0.2% Triton X-100 in PBS for 15 min. After nonspecific binding was blocked with 2% BSA in PBS, the cells were exposed to IRF5 antibody (1 μg/ml in 2% BSA in PBS) overnight at 4 °C. After washing, FITC-conjugated secondary antibody was added for 30 min. The nuclei were counterstained with Hoescht 33342 dye. The images were taken with a LSM 700 confocal microscope (Zeiss, Oberkochen, Germany).
Cytokine ELISAs
Cell supernatants were collected from resting and LPS-stimulated (1 μg/ml) RAW264.7 cells subjected to different conditions for 4 h for TNF-α or 24 h for other cytokines before being quantified by their respective ELISA according to the manufacturer's instructions.
Western Blot
Samples containing equivalent amount of total proteins were treated with or without 0.05 n NaOH at 25 °C for 1 h or digested with or without 20 units/ml hyaluronidase (HAase) at 37 °C for 2 h before electrophoresed on 4–15% (w/v) gradient acrylamide ready gels under denaturing and reducing conditions. Proteins were then transferred to nitrocellulose membrane. The membrane was blocked with 5% (w/v) fat-free milk in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.05% (v/v) Twen-20 followed by sequential incubation with specific primary antibodies and their respective secondary antibodies. Immunoreactive proteins were detected with Western Lighting® chemiluminesence reagent.
Statistical Analysis
Data are represented as the means ± S.D. of three or more independent experiments. Student's t test was performed to test statistical significance from controls. A p value equal to or less than 0.05 was considered statistically significant.
RESULTS
HC1 and PTX3, but Not HC2, Bikunin, and TSG-6, Are Present in HC-HA
We previously reported that HC-HA purified from human AM contains HC1 but not HC2 of IαI (42). To further confirm this, HC-HA purified through two sequential runs of ultracentrifugation in CsCl, 4 m guanidine HCl was subjected to a mild NaOH (0.05 n) treatment to cleave the ester bond between HC and HA (47) or digestion with HAase to release HC. Because this ester bond is also formed between two HCs and the chondroitin sulfate chain of bikunin in the intact IαI molecule (48), we also treated IαI with NaOH as the positive control for HC1 and HC2. As shown in Fig. 1, HC-HA without either NaOH or HAase treatment gave a weak ∼75-kDa band, which was likely released from HC-HA because of degradation of HA and a high molecular mass band around the loading well when probed with an anti-HC1 antibody (Fig. 1A, lane 4). Treatment of HC-HA with either NaOH or HAase eliminated the high molecular mass species but increased the intensity of the ∼75-kDa band (lanes 5 and 6), suggesting that HC1 was covalently linked to HA. In contrast, HC2 was clearly released from IαI with NaOH treatment (∼80 kDa; Fig. 1B, lane 3), but there was no HC2 detected in HC-HA either with NaOH or HAase treatment or no treatment at all (lanes 4-6). This result confirmed that HC-HA purified from human AM lacked HC2. Interestingly, PTX3, a TNF-α-stimulated gene product (49) that is tightly associated with the HC-HA matrix found in the cumulus-oocyte complex (50), was also detected in HC-HA (Fig. 1C). Human PTX3 is a multimeric glycoprotein with a molecular mass of 45 kDa and forms an octameric structure (51). The PTX3 control exhibited monomer, dimer, trimer, and multimer (Fig. 1C, lane 2), but only dimer and multimer were displayed in HC-HA (Fig. 1C, lane 3), of which the latter increased its intensity after NaOH treatment (Fig. 1C, lane 4). This result suggests that PTX3 was also tightly associated with the HC-HA. As reported (42), we did not detect TSG-6 and bikunin using their specific antibodies (data not shown).
FIGURE 1.

HC1 and PTX3 but not HC2 are present in purified HC-HA complex. HC-HA complex was purified through two sequential runs of ultracentrifugation in CsCl, 4 m guanidine HCl from soluble AM extract followed by treatment with or without 0.05 n NaOH at 25 °C for 1 h or digestion with or without 20 units/ml HAase at 37 °C for 2 h. Western blot was run with IαI or PTX3 as a positive control with or without NaOH treatment using antibodies specific to HC1 (A), HC2 (B), or PTX3 (C). The protein marker ladder is shown at the left (lane M, kDa). HC1 and PTX3 but not HC2 were detected in HC-HA complex.
Soluble HC-HA but Not HA Induces Apoptosis in Activated but Not Resting Neutrophils and Macrophages
Previously, we and others have reported that human AM as a temporary graft reduces infiltration of neutrophils into the ablated corneal stroma and promotes apoptosis of neutrophils adherent to AM (33, 34, 37). Because facilitation of neutrophil apoptosis is an important strategy for resolving inflammation (9, 10), we wondered whether HC-HA, purified from AM (43), promoted neutrophil apoptosis. To test this, we isolated neutrophils (with purity > 94% and cell viability > 96%) from the peripheral blood of healthy human volunteers. Consistent with previous reports, neutrophil apoptosis was decreased by 1 μm fMLP (52) or 1 μg/ml LPS (53) but enhanced by 20 μm R-roscovitine (regardless of fMLP or LPS), a cell cycle CDK inhibitor (45, 54), as determined by cell death detection ELISA (Fig. 2A). Compared with the PBS control, addition of 25 μg/ml of soluble HA or HC-HA did not affect apoptosis on resting neutrophils. In contrast, soluble HC-HA but not HA-enhanced apoptosis of fMLP- and LPS-activated neutrophils.
FIGURE 2.

Soluble HC-HA induces apoptosis of activated but not resting neutrophils and macrophages. A, cell apoptosis was measured by cell death detection ELISA (n = 3) in neutrophils obtained from the peripheral blood of healthy human donors after being stimulated without (resting) or with 1 μm fMLP, 1 μg/ml LPS, or 20 μm R-roscovitine (R-ros.) at the same time without or with 25 μg/ml of soluble HA or HC-HA for 20 h. Compared with the control (Ctrl, PBS), apoptosis of resting neutrophils was expectedly reduced by fMLP and LPS (both p < 0.05) but promoted by R-roscovitine (p < 0.05). Soluble HC-HA but not HA promoted apoptosis of fMLP or LPS-activated (both p < 0.05) but not resting neutrophils (p > 0.05). B, RAW264.7 cells (3.1 × 104/cm2) were activated with LPS with or without AME, soluble HA, or HC-HA at a series of HA concentrations. After 24 h, the cell morphology was photographed (scale bar, 25 μm). C, MTT assay showed that cell viability was significantly inhibited by 100 μg/ml HA, 5–100 μg/ml AME, and 5–100 μg/ml HC-HA compared with the control (all p < 0.05, n = 4). D, soluble HC-HA but not HA (both at 25 μg/ml of HA) induced significant cell apoptosis in LPS-activated but not resting macrophages (*, p < 0.01, n = 4).
We have reported that both AME (100 μg/ml total protein) and soluble HC-HA (25 μg/ml) inhibit the viability of LPS-activated RAW264.7 macrophages (41, 43), which elicit a proinflammatory response through TLR4 (55). Thus, we examined whether soluble HC-HA inhibits cell viability more so than AME and soluble HA. Morphologically, soluble HA at 5–50 μg/ml did not cause any noticeable difference compared with the PBS control, but HA at 100 μg/ml reduced the number of large cells (Fig. 2B). In contrast, 5–100 μg/ml of AME or soluble HC-HA dose-dependently decreased the number of large and well spread cells. Consistent with the aforementioned morphological changes, cell viability measured by MTT assay was inhibited by soluble HA only at 100 μg/ml but not at 5–50 μg/ml, compared with the control (Fig. 2C). In contrast, inhibition of cell viability was more significant for soluble HC-HA than AME at 5, 10, 25, and 50 μg/ml HA (Fig. 2C). The decrease of cell viability caused by 25 μg/ml of soluble HC-HA but not HA was correlated with the promotion of apoptosis measured by cell death ELISA (Fig. 2D). Apoptosis in resting macrophages was not significantly induced by either 25 μg/ml of soluble HA or 25 μg/ml of soluble HC-HA (Fig. 2D). These data suggest that soluble HC-HA can exert its anti-inflammatory action by promoting apoptosis of activated but not resting neutrophils and macrophages.
Soluble HC-HA, but Not Immobilized HC-HA, Inhibits Cell Adhesion and Proliferation of LPS-activated Macrophages
The morphological changes of LPS-activated macrophages caused by AME and soluble HC-HA (Fig. 2B) suggest that soluble HC-HA might affect cell adhesion. Indeed, we found that compared with the control, cell attachment of LPS-activated macrophages was inhibited by 25 μg/ml of soluble HC-HA but not HA after 90 min of culturing (Fig. 3A). This effect was correlated with greater inhibition of cell spreading by soluble HC-HA than HA at 1, 2, 3, and 24 h after seeding (Fig. 3B). Furthermore, the aforementioned changes were correlated with greater inhibition of cell proliferation measured by BrdU labeling by 25 μg/ml soluble HC-HA than the control or HA (Fig. 3C). Taken together, these data suggest that inhibition of macrophage viability by soluble HC-HA is mediated by the promotion of apoptosis resulting from the inhibition of cell adhesion, spreading, and proliferation.
FIGURE 3.

Soluble but not immobilized HC-HA inhibits cell adhesion and proliferation of LPS-activated macrophages. A, RAW264.7 cells (3.1 × 104/cm2) were treated with LPS without or with 25 μg/ml of soluble HA or HC-HA for 90 min after seeding. Attached cells were quantitated by CyQuant (*, p < 0.01 for HC-HA versus control (Ctrl) and HC-HA versus HA, n = 3; scale bar, 100 μm). B, spreading (i.e., not round) cells, counted for more than 200 cells at six random fields, were fewer after being treated with soluble 25 μg/ml HC-HA at all time points (*, p < 0.01, n = 3; scale bar, 50 μm). C, cell proliferation measured by 10 μm BrdU for 2 h before the termination of the above culture at 24 h was reduced by soluble HC-HA but not HA (*, p < 0.01for HC-HA versus the control and HC-HA versus HA, n = 4). D, both HA and HC-HA were covalently coupled to the CovaLink NH 96-well. Coupled HA in HA and HC-HA was measured by ELISA using HA-binding protein (top panel, n = 3). The coupled HC1 in HA (there is no HC1 in HA sample, just as the ELISA indicated) and HC-HA was also measured by ELISA using the anti-IαI antibody (bottom panel, n = 3). E, RAW264.7 cells at the same density were seeded on immobilized HA or HC-HA at the maximal binding capacity of 2 μg/well and treated with LPS for 90 min. Cell attachment counted by CyQuant was significantly higher on immobilized HC-HA than the immobilized control and HA (p = 0.000002 and 0.0001, respectively, n = 3), whereas cell attachment on immobilized HA was significantly lower than the control (p = 0.003, n = 3; scale bar, 100 μm). F, RAW264.7 cells (3.1 × 104/cm2) were treated with LPS and seeded on immobilized control, HA, and HC-HA for 24 h. Cell proliferation was measured by BrdU labeling and ELISA. The proliferation was slightly lower on immobilized HC-HA but not significantly compared with immobilized control or HA (p = 0.25 and 0.26, respectively, n = 4).
To confirm that cells directly interact with soluble HC-HA rather than get trapped in solution because of the large size of HC-HA (> 3000 kDa) (41), we tested cell binding to immobilized HC-HA. We immobilized HA or HC-HA on plastic using Sulfo-NHS/EDAC coupling reagents as reported (43, 56). Respective ELISA assays confirmed that 2 μg of HA was the maximal amount of HA or HC-HA that could be immobilized on a CovaLink NH 96-well (Fig. 3D). Similar to what was shown in Fig. 3A, LPS-stimulated cells barely bound to immobilized HA (Fig. 3E), resulting in cell adhesion that was worse than the control. In contrast, cells strongly adhered to immobilized HC-HA, better than the PBS control or HA. Cell viability of LPS-activated macrophages on immobilized HC-HA was not affected compared with those on the immobilized control or HA (data not shown). Furthermore, cell apoptosis of LPS-activated macrophages was not affected on immobilized HC-HA (data not shown), although cell proliferation was slightly, but not significantly, decreased on immobilized HC-HA compared with both immobilized control and HA (Fig. 3F). Collectively, these data demonstrate that LPS-activated macrophages strongly bind to immobilized HC-HA and likely to soluble HC-HA. However, unlike soluble HC-HA, immobilized HC-HA does not affect cell viability, apoptosis, and proliferation.
Preferentially, Phagocytosis of Apoptotic Neutrophils Is Promoted in Resting Macrophages by Soluble HC-HA but Is Promoted in LPS-activated Macrophages by Immobilized HC-HA
Because clearance of apoptotic neutrophils by macrophages is essential for resolving inflammation (57–59), we examined whether soluble and immobilized HC-HA might affect this process. We seeded both resting and LPS-activated macrophages on plastic wells with or without 25 μg/ml of soluble HA, soluble HC-HA, or immobilized control, HA, or HC-HA for 6 days before adding apoptotic neutrophils induced by 20 μm roscovitine. The phagocytic index, measured by human myeloperoxidase ELISA, normalized with total protein content, was significantly decreased by soluble HA but notably increased by soluble HC-HA by 6–7-fold compared with the PBS control (Fig. 4A). This suggests that soluble HC-HA is particularly effective in inducing phagocytosis of apoptotic neutrophils by resting macrophages. We also noted that the phagocytic index was significantly lowered by LPS-activated macrophages (Fig. 4A, ctrl), a finding consistent with the notion that LPS promotes M1 phenotype to down-regulate phagocytosis (19). Under this LPS-activated condition, soluble HA did not but soluble HC-HA still did, although to a lesser extent than that in resting macrophages, increase the phagocytic index by 2-fold (Fig. 4A). In contrast, the phagocytic index by LPS-activated macrophages was decreased by immobilized HA but dramatically increased by 5-fold by immobilized HC-HA (Fig. 4B). These data suggest that phagocytosis of apoptotic neutrophils by macrophages is promoted by soluble and immobilized HC-HA; the effect of the former is more notable in resting macrophages, whereas that in the latter is more dramatic in LPS-activated macrophages.
FIGURE 4.
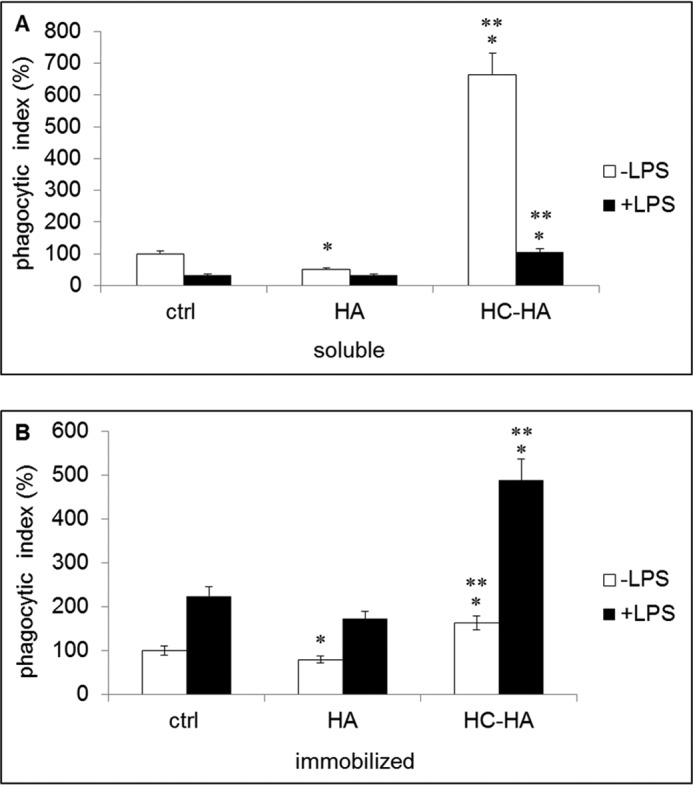
Phagocytosis of apoptotic neutrophils is preferentially promoted in resting macrophages by soluble HC-HA but is preferentially promoted in LPS-activated macrophages by immobilized HC-HA. Resting or LPS-stimulated RAW264.7 cells (3.1 × 104/cm2/96-well plate) were cultivated for 6 days with or without soluble HA or HC-HA or on immobilized HA or HC-HA before the addition of 2 × 105 apoptotic neutrophils induced by 20 μm roscovitine for 30 min. After the removal of remaining neutrophils, the relative phagocytic index was measured by ELISA of human myeloperoxidase in macrophage lysates, normalized by the total protein content. In resting macrophages, the phagocytic index was significantly increased by soluble HC-HA but not HA (A). In contrast, in LPS-activated macrophages, the phagocytic index was significantly increased by immobilized HC-HA but not HA (B). (*, p < 0.05 for HA or HC-HA versus the control (ctrl); **, p < 0.05 for HC-HA versus HA, n = 3).
Immobilized HC-HA Polarizes LPS-activated Macrophages toward the M2 Phenotype
We previously found that AME down-regulates the production of M1 cytokines such as TNF-α and IL-6 but up-regulates the M2 cytokine IL-10 (41). Because M2 macrophages are known to be more effective than M1 macrophages in the phagocytosis of apoptotic neutrophils (19), we speculated the aforementioned enhanced phagocytosis by LPS-activated macrophages on immobilized HC-HA was due to the polarization to M2 phenotype. To test this, we compared the mRNA and protein expression of M1 and M2 markers in LPS-activated macrophages on immobilized control, HA, and HC-HA. Indeed, LPS-activated macrophages on immobilized HC-HA significantly up-regulated the mRNA expression of M2 markers such as IL-10, TGF-β1, PPAR-γ, LIGHT, and SPHK1, whereas they down-regulated M1 markers such as IL-12p40, TNF-α, and inducible NOS2 compared with immobilized control and HA (Fig. 5). The up-regulation of TGF-β1 transcription was confirmed by the TGF-β1 promoter assay (Fig. 6A) and TGF-β1 ELISA (Fig. 6B). ELISA also confirmed the up-regulation of IL-10 protein level by immobilized HC-HA but not immobilized HA (Fig. 6C). In contrast, the protein level of IL-12p70 was abolished, and that of TNF-α was significantly reduced by immobilized HC-HA but not immobilized HA (Fig. 6, D and E, respectively). IRF5 is a transcriptional factor that is highly expressed in M1 macrophages in both mice and humans (60). Phosphorylated IRF5 translocates into the nucleus (61, 62) and activates the transcription of genes encoding IL-12p40 and IL-12p35 while repressing the gene encoding IL-10. In LPS-activated macrophages on immobilized HC-HA, we found that IRF5 protein was down-regulated compared with that on immobilized control and HA (Fig. 6F). Immunolocalization data showed that IRF5 was localized in both the cytoplasm and the nucleus in cells on immobilized control and HA but was excluded from the nucleus on immobilized HC-HA (Fig. 6G). Therefore, the expression and activation of IRF5 in LPS-activated macrophages is down-regulated by immobilized HC-HA.
FIGURE 5.
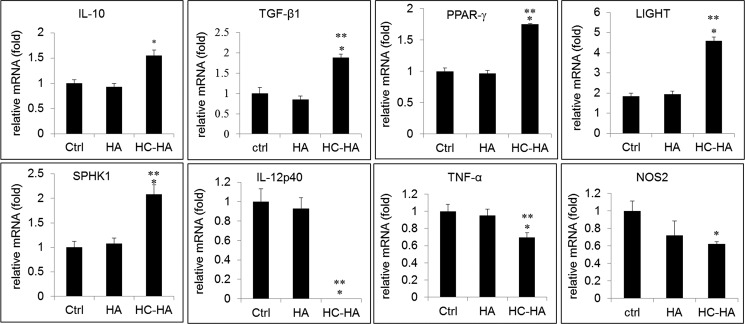
Immobilized HC-HA but not HA up-regulates M2 markers and down-regulates M1 markers. qPCR of total mRNA extracted from RAW264.7 cells (3.1 × 104/cm2) seeded on immobilized HA or HC-HA after being stimulated with LPS (1 μg/ml) for 4 h is shown for M2 markers (IL-10, TGF-β1, PPAR-γ, LIGHT, and SPHK1) and M1 markers (IL-12p40, TNF-α, and NOS2) with GAPDH as the endogenous control (Ctrl) (*, p < 0.05 for HC-HA versus control; **, p < 0.05 for HC-HA versus HA, n = 3).
FIGURE 6.

Immobilized HC-HA up-regulates TGF-β1 and IL-10 protein level but down-regulates IRF5, IL-12p70, and TNF-α protein levels. RAW264.7 cells (3.1 × 104/cm2) were cultivated on immobilized control (Ctrl), HA, or HC-HA after being stimulated with LPS (1 μg/ml) for 4 h (qPCR and immunostaining) or 24 h (Western blot and ELISA). A, the TGF-β1 promoter activity was increased by immobilized HC-HA more so than immobilized HA (p < 0.05, n = 3). B, free active TGF-β1 in the conditioned medium from LPS-activated macrophages on immobilized HC-HA was significantly higher than that on either immobilized control or HA (*, p < 0.05 for HC-HA versus control; **, p < 0.05 for HC-HA versus HA, n = 3). C–E, ELISA of the conditioned medium showed that IL-10 (C) was up-regulated, whereas IL-12p70 (D) and TNF-α (E) were down-regulated by immobilized HC-HA but not by HA (*, p < 0.05 for HC-HA versus control; **, p < 0.05 for HC-HA versus HA, n = 3). F, IRF5 was detected by Western blot and normalized by β-actin. G, IRF5 was excluded from the nucleus shown by immunostaining. The quantitation was performed by divided IRF5-positive cells with total cells in six randomly selected fields.
Because IFN-γ is a primary cytokine inducing proinflammatory response and its priming enhances polarization of M1 macrophages by LPS (63), we also tested whether immobilized HC-HA could promote M2 phenotype in IFN-γ- or IFN-γ primed LPS-activated (IFN-γ/LPS) macrophages. RAW264.7 cells on immobilized control activated by IFN-γ alone increased mRNA expression of IL-12p40, TNF-α, NOS2, but had little effect on mRNA expression of IL-10, TGF-β1, and PPAR-γ. Under the same treatment, cells on immobilized HC-HA did not change the mRNA expression of all markers except increasing PPAR-γ mRNA when compared with those on immobilized control (data not shown). These data suggest that HC-HA is unlikely to change the macrophage phenotype activated by IFN-γ. Activation of RAW264.7 cells on immobilized control by IFN-γ/LPS increased mRNA expression of IL-10, IL-12p40, TNF-α, NOS2, and PPAR-γ but decreased TGF-β1 mRNA. Immobilized HC-HA did not affect mRNA expression of these markers either (data not shown). We then repeated the same experiment without LPS to determine TLR ligation dependence. qPCR analyses showed that mRNA expression of IL-12p40 was undetectable, whereas that of TNF-α, NOS2, IL-10, PPAR-γ, and TGF-β1 on immobilized HC-HA was not significantly changed from the immobilized control and HA (data not shown). ELISA showed that the TGF-β1 protein level on immobilized HC-HA was significantly less than that of control or HA, whereas IL-10 protein level was undetectable (data not shown). Collectively, these data suggest that immobilized HC-HA only polarizes LPS-activated macrophages toward the M2 phenotype in an LPS-TLR4 ligation-dependent manner.
DISCUSSION
It is still not clear whether HC-HA is proinflammatory or anti-inflammatory. One form of HC-HA is SHAP-HA isolated and characterized from synovial fluids of patients with rheumatoid arthritis (64, 65). Interestingly, SHAP-HA-deficient (bikunin knock-out) mice show higher airway hyper-responsiveness to inhaled methacholine and higher late phase response to exposed aerosol ovalbumin, exhibiting an increased number of macrophages and neutrophils but decreased soluble tumor necrosis factor receptor-1 in their bronchoalveolar lavage fluid. This finding suggests that SHAP-HA has an inhibitory role in the development of airway hyper-responsiveness and allergic airway inflammation (66). Although both SHAP-HA and HC-HA purified from human AM are similar complexes, characterized by high molecular mass HA covalently linked to HC (42, 43, 64), there are important differences. In adult cells, SHAP-HA contains both HC1 and HC2 from serum IαI (derived from the liver) (64). However, HC-HA purified from human AM contains only HC1 derived from endogenously synthesized IαI and depends on constitutively expressed TSG-6 in human AM epithelial and stromal cells (Fig. 1A) (42). Although the protein level of the HC-HA complex is undetectable by the BCA assay, the amount of HA and HC1 in HC-HA measured by the respective ELISAs was 1 μg of HA and 36 ng of HC1, respectively (Fig. 3D), yielding a molar ratio of HC1 to HA at 1.9:1 based on the average molecular mass of HA in HC-HA being ∼4,000 kDa (43) and that of HC1 being ∼75 kDa. Furthermore, PTX3 is present in HC-HA (Fig. 1C). Similar to the HC-HA matrix in the cumulus-oocyte complex (50), we noted that PTX3 was tightly associated with the HC-HA purified from AM. Because PTX3 in HC-HA could not be detected by PTX3 ELISA, possibly because of the masking effect by HA and/or HC1, the amount of PTX3 in the HC-HA was estimated by Western blot after NaOH treatment to be 20 ng (Fig. 1C, lane 4) in 2 μg of HC-HA in reference to 100 ng of the control PTX3 (Fig. 1C, lane 2). This yielded a molar ratio of PTX3 to HA in the HC-HA complex of ∼0.9 based on the molecular mass of monomer PTX3 being 45 kDa. Because PTX3 is reported to interact with HC (67), it may modify or mask HC1 activity in HC-HA. In all, our present study provides strong experimental evidence supporting that HC-HA purified from human AM exerts anti-inflammatory action.
The first evidence is that soluble HC-HA but not soluble HA effectively induced apoptosis of activated but not resting neutrophils. Neutrophils isolated from the peripheral blood are short-lived and undergo spontaneous apoptosis upon culturing in vitro (11). Because neutrophils contain a myriad of biological products capable of promoting inflammation by degrading matrix proteins, prolonging neutrophil lifespan by inhibiting or delaying apoptosis with proinflammatory agents such as fMLP (52) and LPS (53) poses a threat to the host tissue integrity. To limit this potential detrimental response, one therapeutic strategy is to facilitate the apoptosis of activated neutrophils. In line with this thinking, agents such as R-roscovitine, a pharmacological inhibitor of cyclin-dependent kinase, have been used to promote neutrophil apoptosis in an attempt to resolve inflammatory conditions (45). Our data showed that soluble high molecular mass HA (>3,000 kDa) did not promote apoptosis of activated neutrophil (Fig. 2). This result is similar to the finding that high molecular mass HA-carboxymethylcellulose membrane does not affect apoptosis of LPS- or TNF-α-activated human peripheral blood neutrophils (68). However, HC-HA promoted apoptosis of both fMLP and LPS-activated neutrophils but not resting neutrophils (Fig. 2). Because promotion of apoptosis of activated macrophages is also an important strategy to control tissue inflammation (69), it is not surprising that soluble HC-HA, but not HA, induced apoptosis of LPS-activated macrophages (Fig. 2). Collectively, these data corroborate the previous studies based on AM (33, 34, 40) and AME (41) while beginning to explain the mechanism of their anti-inflammatory effects via promotion of apoptosis of activated neutrophils (33, 34, 37), macrophages (35, 37, 39), and probably lymphocytes (36).
The mechanism leading to apoptosis induced by soluble HC-HA may be linked to anoikis, a form of apoptosis induced by the detachment of anchorage-dependent cells from the surrounding extracellular matrix (70). This interpretation is supported by the significant inhibition of cell adhesion, spreading, and subsequent proliferation of LPS-activated macrophages by soluble HC-HA (Fig. 3). We also found that macrophages actually bound more strongly to immobilized HC-HA than immobilized HA (Fig. 3). similar to what has been observed for immobilized SHAP-HA (65). Hence, we speculate that such strong binding with soluble HC-HA may have prevented cells from adhering to the plastic, resulting in apoptosis of unattached cells. Similar to the finding in SHAP-HA (65), our preliminary data suggested that LPS-activated macrophages adhere to immobilized HC-HA through CD44, a cell receptor of HA (data not shown). Several studies have shown that adhesion of a number of CD44+ cells to immobilized HA is poor but can be markedly up-regulated by LPS, PMA (71–73), or protein modification of HA (such as HC or TSG-6) (65, 74). Hence, future studies are needed to delineate how immobilized HC-HA might promote cell adhesion via CD44 and whether such action depends on LPS.
The second evidence supporting the anti-inflammatory action of HC-HA is the promotion of phagocytic clearance of apoptotic neutrophils by macrophages, another strategy for resolving inflammation (57–59). Herein, we noted that such phagocytosis by resting macrophages was significantly inhibited by soluble HA, consistent with previous reports showing that soluble HA (>1,000 kDa) inhibits phagocytosis by murine (75) and guinea pig (76, 77) peritoneal macrophages. In contrast, phagocytosis of apoptotic neutrophils by resting macrophages was notably enhanced by soluble HC-HA by 6–7-fold (Fig. 4). Because LPS activates M1 macrophages (13), phagocytosis in these cells is expected to be down-regulated (18, 19). Under this condition, soluble HC-HA still promoted phagocytosis by 2-fold, whereas soluble HA did not (Fig. 4A). Because RAW264.7 cells are known to be heterogeneous (78, 79) and because soluble HC-HA reduced cell viability by promoting apoptosis in LPS-activated macrophages (Fig. 2), future studies are needed to determine whether soluble HC-HA selectively eliminates a subpopulation of RAW264.7 cells while promoting the phagocytic activity of the remaining cells.
The enhanced phagocytosis of apoptotic neutrophils by LPS-activated macrophages on immobilized HC-HA was because of effective polarization to the M2 phenotype (Figs. 5 and 6). The M2 phenotype can be further subcategorized into M2a, M2b, and M2c subtypes. Considering that LPS/immune complex stimulus is known to polarize RAW264.7 cells toward M2b in a TLR4 ligation-dependent manner (12), we propose that immobilized HC-HA under our conditions promoted M2b. This conclusion is supported by our data showing the up-regulation of M2b markers: IL-10, LIGHT, SPHK1 (12), and a low level of IL-12 (13) (Figs. 5 and 6). Previously, a number of factors such as apoptotic cells, immune complex, prostaglandin, adenosine, some ligands for G-protein-coupled receptors, dopamine, histamine, sphingosine-1-phosphate, melanocortin, VIP, adiponectin, and Siglect9 have been found to achieve M2b polarization in conjunction with TLR2 or TLR4 ligation (13, 24, 80–83). Herein, we add HC-HA to this list as the first matrix component that may polarize M2b with concomitant LPS stimulation. Our data also suggest that HC-HA might also polarize RAW264.7 cells into M2c because of up-regulation of TGF-β1 (Figs. 5 and 6). Up-regulation of TGFβ1 is not found in any of the listed soluble factors that promote M2b polarization (23, 84) with exception of phagocytosis of apoptotic cells (85). It has been shown that TGF-β1 alone is inefficient at down-regulating proinflammatory cytokines such as TNF-α, and the effective inhibition requires a synergistic effect with apoptotic cells (86). It is worth noting that M2 macrophage classification is more or less flexible, and variations can be found across the literature with regards to M2b and M2c. For instance, some researchers prefer to categorize M2b and M2c together under the functional label of “regulatory macrophage” (13, 82). The defining property of this category is high IL-10 production relative to IL-12. Interestingly, we noted that immobilized HC-HA also up-regulated PPAR-γ (Fig. 5), a M2a marker typically generated by IL-4/IL-13 (87). The latter finding is consistent with a previous report showing PPAR-γ is up-regulated in corneal macrophages after AM transplanted to corneas infected with herpes simplex virus (39). We suspect that such broad spectrum of M2 polarization by immobilized HC-HA may be linked to the suppression of the nuclear translocation of IRF5 because IRF5 is known to transcriptionally activate IL-12p40 (60), TNF-α (88, 89), and NOS2 (90) but down-regulate IL-10 (60). Because resting macrophages phagocytosed apoptotic neutrophils without TLR ligation (Fig. 4A), we wonder whether phagocytosis of apoptotic cells is not always coupled with M2 polarization. Understanding the molecular mechanism behind the anti-inflammatory action of HC-HA purified from human AM will allow us to deploy it as a new therapeutic agent to facilitate the restoration of tissue homeostasis during wound healing.
Acknowledgment
We thank Angela Tseng for critical reading and editing of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 EY06819, R43 EY21045, and R44 EY017497 (to S. C. G. T.). This work was also supported by a research grant from TissueTech, Inc.
- TLR4
- toll-like receptor 4
- AM
- amniotic membrane
- fMLP
- formyl-Met-Leu-Phe
- HA
- hyaluronan or hyaluronic acid
- HC-HA
- a complex of HC covalently linked with HA
- IαI
- inter-α-inhibitor
- HC
- heavy chain of inter-α-inhibitor
- IRF5
- interferon-regulatory factor 5
- PTX3
- pentraxin 3
- PPAR-γ
- peroxisome proliferator-activated receptor γ
- SHAP-HA
- a complex of serum-derived hyaluronan-associated proteins (SHAP) covalently linked with HA
- SPHK1
- sphingosine kinase-1
- NOS2
- nitric oxide synthase 2 (inducible)
- TSG-6
- TNF-stimulated gene-6
- EDAC
- 1-ethyl-3-(3-dimethylaminopropyl) carbidodiimide
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- qPCR
- quantitative real time PCR
- HAase
- hyaluronidase
- AME
- AM extract.
REFERENCES
- 1. Frasch S. C., Nick J. A., Fadok V. A., Bratton D. L., Worthen G. S., Henson P. M. (1998) p38 mitogen-activated protein kinase-dependent and -independent intracellular signal transduction pathways leading to apoptosis in human neutrophils. J. Biol. Chem. 273, 8389–8397 [DOI] [PubMed] [Google Scholar]
- 2. Pongracz J., Webb P., Wang K., Deacon E., Lunn O. J., Lord J. M. (1999) Spontaneous neutrophil apoptosis involves caspase 3-mediated activation of protein kinase C-delta. J. Biol. Chem. 274, 37329–37334 [DOI] [PubMed] [Google Scholar]
- 3. Khwaja A., Tatton L. (1999) Caspase-mediated proteolysis and activation of protein kinase Cδ plays a central role in neutrophil apoptosis. Blood 94, 291–301 [PubMed] [Google Scholar]
- 4. Fadok V. A., Bratton D. L., Konowal A., Freed P. W., Westcott J. Y., Henson P. M. (1998) Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J. Clin. Invest. 101, 890–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haringman J. J., Gerlag D. M., Zwinderman A. H., Smeets T. J., Kraan M. C., Baeten D., McInnes I. B., Bresnihan B., Tak P. P. (2005) Synovial tissue macrophages. A sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann. Rheum. Dis. 64, 834–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lumeng C. N., Bodzin J. L., Saltiel A. R. (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117, 175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lutgens E., Lievens D., Beckers L., Wijnands E., Soehnlein O., Zernecke A., Seijkens T., Engel D., Cleutjens J., Keller A. M., Naik S. H., Boon L., Oufella H. A., Mallat Z., Ahonen C. L., Noelle R. J., de Winther M. P., Daemen M. J., Biessen E. A., Weber C. (2010) Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J. Exp. Med. 207, 391–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Woollard K. J., Geissmann F. (2010) Monocytes in atherosclerosis. Subsets and functions. Nat. Rev. Cardiol. 7, 77–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gilroy D. W., Lawrence T., Perretti M., Rossi A. G. (2004) Inflammatory resolution. New opportunities for drug discovery. Nat. Rev. Drug Discov. 3, 401–416 [DOI] [PubMed] [Google Scholar]
- 10. Serhan C. N., Brain S. D., Buckley C. D., Gilroy D. W., Haslett C., O'Neill L. A., Perretti M., Rossi A. G., Wallace J. L. (2007) Resolution of inflammation. State of the art, definitions and terms. FASEB J. 21, 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duffin R., Leitch A. E., Fox S., Haslett C., Rossi A. G. (2010) Targeting granulocyte apoptosis. Mechanisms, models, and therapies. Immunol. Rev. 236, 28–40 [DOI] [PubMed] [Google Scholar]
- 12. Edwards J. P., Zhang X., Frauwirth K. A., Mosser D. M. (2006) Biochemical and functional characterization of three activated macrophage populations. J. Leukocyte Biol. 80, 1298–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mosser D. M., Edwards J. P. (2008) Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gordon S., Martinez F. O. (2010) Alternative activation of macrophages. Mechanism and functions. Immunity 32, 593–604 [DOI] [PubMed] [Google Scholar]
- 15. Sica A., Mantovani A. (2012) Macrophage plasticity and polarization. In vivo veritas. J. Clin. Invest. 122, 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yamamoto M., Sato S., Hemmi H., Hoshino K., Kaisho T., Sanjo H., Takeuchi O., Sugiyama M., Okabe M., Takeda K., Akira S. (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643 [DOI] [PubMed] [Google Scholar]
- 17. Mosser D. M. (2003) The many faces of macrophage activation. J. Leukocyte Biol. 73, 209–212 [DOI] [PubMed] [Google Scholar]
- 18. Mosser D. M., Handman E. (1992) Treatment of murine macrophages with interferon-gamma inhibits their ability to bind leishmania promastigotes. J. Leukocyte Biol. 52, 369–376 [DOI] [PubMed] [Google Scholar]
- 19. Michlewska S., Dransfield I., Megson I. L., Rossi A. G. (2009) Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents. Key role for TNF-α. FASEB J. 23, 844–854 [DOI] [PubMed] [Google Scholar]
- 20. Leidi M., Gotti E., Bologna L., Miranda E., Rimoldi M., Sica A., Roncalli M., Palumbo G. A., Introna M., Golay J. (2009) M2 macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than M1 cells in vitro. J. Immunol. 182, 4415–4422 [DOI] [PubMed] [Google Scholar]
- 21. Chihara T., Hashimoto M., Osman A., Hiyoshi-Yoshidomi Y., Suzu I., Chutiwitoonchai N., Hiyoshi M., Okada S., Suzu S. (2012) HIV-1 proteins preferentially activate anti-inflammatory M2-type macrophages. J. Immunol. 188, 3620–3627 [DOI] [PubMed] [Google Scholar]
- 22. Delgado M., Munoz-Elias E. J., Gomariz R. P., Ganea D. (1999) Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide prevent inducible nitric oxide synthase transcription in macrophages by inhibiting NF-κB and IFN regulatory factor 1 activation. J. Immunol. 162, 4685–4696 [PubMed] [Google Scholar]
- 23. Gerber J. S., Mosser D. M. (2001) Reversing lipopolysaccharide toxicity by ligating the macrophage Fcγ receptors. J. Immunol. 166, 6861–6868 [DOI] [PubMed] [Google Scholar]
- 24. Anderson C. F., Mosser D. M. (2002) A novel phenotype for an activated macrophage. The type 2 activated macrophage. J. Leukocyte Biol. 72, 101–106 [PubMed] [Google Scholar]
- 25. Ando M., Tu W., Nishijima K., Iijima S. (2008) Siglec-9 enhances IL-10 production in macrophages via tyrosine-based motifs. Biochem. Biophys. Res. Commun. 369, 878–883 [DOI] [PubMed] [Google Scholar]
- 26. Bastos K. R., Alvarez J. M., Marinho C. R., Rizzo L. V., Lima M. R. (2002) Macrophages from IL-12p40-deficient mice have a bias toward the M2 activation profile. J. Leukocyte Biol. 71, 271–278 [PubMed] [Google Scholar]
- 27. Odegaard J. I., Ricardo-Gonzalez R. R., Goforth M. H., Morel C. R., Subramanian V., Mukundan L., Red Eagle A., Vats D., Brombacher F., Ferrante A. W., Chawla A. (2007) Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duffield J. S. (2003) The inflammatory macrophage. A story of Jekyll and Hyde. Clin. Sci. 104, 27–38 [DOI] [PubMed] [Google Scholar]
- 29. Nathan C., Ding A. (2010) Nonresolving inflammation. Cell 140, 871–882 [DOI] [PubMed] [Google Scholar]
- 30. Tseng S. C., Espana E. M., Kawakita T., Di Pascuale M. A., Li W., He H., Liu T. S., Cho T. H., Gao Y. Y., Yeh L. K., Liu C.-Y. (2004) How does amniotic membrane work? Ocul. Surf. 2, 177–187 [DOI] [PubMed] [Google Scholar]
- 31. Dua H. S., Gomes J. A., King A. J., Maharajan V. S. (2004) The amniotic membrane in ophthalmology. Surv. Ophthalmol. 49, 51–77 [DOI] [PubMed] [Google Scholar]
- 32. Bouchard C. S., John T. (2004) Amniotic membrane transplantation in the management of severe ocular surface disease. Indications and outcomes. Ocul. Surf. 2, 201–211 [DOI] [PubMed] [Google Scholar]
- 33. Park W. C., Tseng S. C. (2000) Modulation of acute inflammation and keratocyte death by suturing, blood and amniotic membrane in PRK. Invest. Ophthalmol. Vis. Sci. 41, 2906–2914 [PubMed] [Google Scholar]
- 34. Wang M. X., Gray T. B., Park W. C., Prabhasawat P., Culbertson W., Forster R., Hanna K., Tseng S. C. (2001) Corneal haze and apoptosis is reduced by amniotic membrane matrix in excimer laser photoablation in rabbits. J. Cataract Refract. Surg. 27, 310–319 [DOI] [PubMed] [Google Scholar]
- 35. Shimmura S., Shimazaki J., Ohashi Y., Tsubota K. (2001) Antiinflammatory effects of amniotic membrane transplantation in ocular surface disorders. Cornea 20, 408–413 [DOI] [PubMed] [Google Scholar]
- 36. Bauer D., Wasmuth S., Hennig M., Baehler H., Steuhl K. P., Heiligenhaus A. (2009) Amniotic membrane transplantation induces apoptosis in T lymphocytes in murine corneas with experimental herpetic stromal keratitis. Invest. Ophthalmol. Vis. Sci. 50, 3188–3198 [DOI] [PubMed] [Google Scholar]
- 37. Bauer D., Wasmuth S., Hermans P., Hennig M., Meller K., Meller D., van Rooijen N., Tseng S. C., Steuhl K. P., Heiligenhaus A. (2007) On the influence of neutrophils in corneas with necrotizing HSV-1 keratitis following amniotic membrane transplantation. Exp. Eye Res. 85, 335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heiligenhaus A., Bauer D., Meller D., Steuhl K. P., Tseng S. C. (2001) Improvement of HSV-1 necrotizing keratitis with amniotic membrane transplantation. Invest. Ophthalmol. Vis. Sci. 42, 1969–1974 [PubMed] [Google Scholar]
- 39. Bauer D., Hennig M., Wasmuth S., Baehler H., Busch M., Steuhl K. P., Thanos S., Heiligenhaus A. (2012) Amniotic membrane induces peroxisome proliferator-activated receptor-gamma positive alternatively activated macrophages. Invest. Ophthalmol. Vis. Sci. 53, 799–810 [DOI] [PubMed] [Google Scholar]
- 40. Li W., He H., Kawakita T., Espana E. M., Tseng S. C. (2006) Amniotic membrane induces apoptosis of interferon-gamma activited macrophages in vitro. Exp. Eye Res. 82, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He H., Li W., Chen S. Y., Zhang S., Chen Y. T., Hayashida Y., Zhu Y. T., Tseng S. C. (2008) Suppression of activation and induction of apoptosis in RAW264.7 cells by amniotic membrane extract. Invest. Ophthalmol. Vis. Sci. 49, 4468–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang S., He H., Day A. J., Tseng S. C. (2012) Constitutive expression of inter-α-inhibitor (IαI) family proteins and tumor necrosis factor-stimulated gene-6 (TSG-6) by human amniotic membrane epithelial and stromal cells supporting formation of the heavy chain-hyaluronan (HC-HA) complex. J. Biol. Chem. 287, 12433–12444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. He H., Li W., Tseng D. Y., Zhang S., Chen S. Y., Day A. J., Tseng S. C. (2009) Biochemical characterization and function of complexes formed by hyaluronan and the heavy chains of inter-α-inhibitor (HC·HA) purified from extracts of human amniotic membrane. J. Biol. Chem. 284, 20136–20146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shay E., He H., Sakurai S., Tseng S. C. (2011) Inhibition of angiogenesis by HC·HA, a complex of hyaluronan and the heavy chain of inter-α-inhibitor, purified from human amniotic membrane. Invest. Ophthalmol. Vis. Sci. 52, 2669–2678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rossi A. G., Sawatzky D. A., Walker A., Ward C., Sheldrake T. A., Riley N. A., Caldicott A., Martinez-Losa M., Walker T. R., Duffin R., Gray M., Crescenzi E., Martin M. C., Brady H. J., Savill J. S., Dransfield I., Haslett C. (2006) Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 12, 1056–1064 [DOI] [PubMed] [Google Scholar]
- 46. Lasunskaia E. B., Campos M. N., de Andrade M. R., Damatta R. A., Kipnis T. L., Einicker-Lamas M., Da Silva W. D. (2006) Mycobacteria directly induce cytoskeletal rearrangements for macrophage spreading and polarization through TLR2-dependent PI3K signaling. J. Leukocyte Biol. 80, 1480–1490 [DOI] [PubMed] [Google Scholar]
- 47. Zhao M., Yoneda M., Ohashi Y., Kurono S., Iwata H., Ohnuki Y., Kimata K. (1995) Evidence for the covalent binding of SHAP, heavy chains of inter-α-trypsin inhibitor, to hyaluronan. J. Biol. Chem. 270, 26657–26663 [DOI] [PubMed] [Google Scholar]
- 48. Enghild J. J., Salvesen G., Hefta S. A., Thøgersen I. B., Rutherfurd S., Pizzo S. V. (1991) Chondroitin 4-sulfate covalently cross-links the chains of the human blood protein pre-α-inhibitor. J. Biol. Chem. 266, 747–751 [PubMed] [Google Scholar]
- 49. Lee G. W., Lee T. H., Vilcek J. (1993) TSG-14, a tumor necrosis factor- and IL-1-inducible protein, is a novel member of the pentaxin family of acute phase proteins. J. Immunol. 150, 1804–1812 [PubMed] [Google Scholar]
- 50. Salustri A., Garlanda C., Hirsch E., De Acetis M., Maccagno A., Bottazzi B., Doni A., Bastone A., Mantovani G., Beck Peccoz P., Salvatori G., Mahoney D. J., Day A. J., Siracusa G., Romani L., Mantovani A. (2004) PTX3 plays a key role in the organization of the cumulus oophorus extracellular matrix and in in vivo fertilization. Development 131, 1577–1586 [DOI] [PubMed] [Google Scholar]
- 51. Inforzato A., Baldock C., Jowitt T. A., Holmes D. F., Lindstedt R., Marcellini M., Rivieccio V., Briggs D. C., Kadler K. E., Verdoliva A., Bottazzi B., Mantovani A., Salvatori G., Day A. J. (2010) The angiogenic inhibitor long pentraxin PTX3 forms an asymmetric octamer with two binding sites for FGF2. J. Biol. Chem. 285, 17681–17692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Benard V., Bohl B. P., Bokoch G. M. (1999) Characterization of Rac and Cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J. Biol. Chem. 274, 13198–13204 [DOI] [PubMed] [Google Scholar]
- 53. Lee A., Whyte M. K., Haslett C. (1993) Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J. Leukocyte Biol. 54, 283–288 [PubMed] [Google Scholar]
- 54. Leitch A. E., Riley N. A., Sheldrake T. A., Festa M., Fox S., Duffin R., Haslett C., Rossi A. G. (2010) The cyclin-dependent kinase inhibitor R-roscovitine down-regulates Mcl-1 to override pro-inflammatory signalling and drive neutrophil apoptosis. Eur. J. Immunol. 40, 1127–1138 [DOI] [PubMed] [Google Scholar]
- 55. Rhee S. H., Hwang D. (2000) Murine TOLL-like receptor 4 confers lipopolysaccharide responsiveness as determined by activation of NFκB and expression of the inducible cyclooxygenase. J. Biol. Chem. 275, 34035–34040 [DOI] [PubMed] [Google Scholar]
- 56. Frost G. I., Stern R. (1997) A microtiter-based assay for hyaluronidase activity not requiring specialized reagents. Anal. Biochem. 251, 263–269 [DOI] [PubMed] [Google Scholar]
- 57. Aderem A., Underhill D. M. (1999) Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17, 593–623 [DOI] [PubMed] [Google Scholar]
- 58. Underhill D. M., Goodridge H. S. (2012) Information processing during phagocytosis. Nat. Rev. Immunol. 12, 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Murray P. J., Wynn T. A. (2011) Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Krausgruber T., Blazek K., Smallie T., Alzabin S., Lockstone H., Sahgal N., Hussell T., Feldmann M., Udalova I. A. (2011) IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238 [DOI] [PubMed] [Google Scholar]
- 61. Barnes B. J., Kellum M. J., Field A. E., Pitha P. M. (2002) Multiple regulatory domains of IRF-5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Mol. Cell. Biol. 22, 5721–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lin R., Yang L., Arguello M., Penafuerte C., Hiscott J. (2005) A CRM1-dependent nuclear export pathway is involved in the regulation of IRF-5 subcellular localization. J. Biol. Chem. 280, 3088–3095 [DOI] [PubMed] [Google Scholar]
- 63. Schroder K., Sweet M. J., Hume D. A. (2006) Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology 211, 511–524 [DOI] [PubMed] [Google Scholar]
- 64. Yingsung W., Zhuo L., Morgelin M., Yoneda M., Kida D., Watanabe H., Ishiguro N., Iwata H., Kimata K. (2003) Molecular heterogeneity of the SHAP-hyaluronan complex. Isolation and characterization of the complex in synovial fluid from patients with rheumatoid arthritis. J. Biol. Chem. 278, 32710–32718 [DOI] [PubMed] [Google Scholar]
- 65. Zhuo L., Kanamori A., Kannagi R., Itano N., Wu J., Hamaguchi M., Ishiguro N., Kimata K. (2006) SHAP potentiates the CD44-mediated leukocyte adhesion to the hyaluronan substratum. J. Biol. Chem. 281, 20303–20314 [DOI] [PubMed] [Google Scholar]
- 66. Zhu L., Zhuo L., Kimata K., Yamaguchi E., Watanabe H., Aronica M. A., Hascall V. C., Baba K. (2010) Deficiency in the serum-derived hyaluronan-associated protein-hyaluronan complex enhances airway hyperresponsiveness in a murine model of asthma. Int. Arch. Allergy Immunol. 153, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Scarchilli L., Camaioni A., Bottazzi B., Negri V., Doni A., Deban L., Bastone A., Salvatori G., Mantovani A., Siracusa G., Salustri A. (2007) PTX3 interacts with inter-α-trypsin inhibitor. Implications for hyaluronan organization and cumulus oophorus expansion. J. Biol. Chem. 282, 30161–30170 [DOI] [PubMed] [Google Scholar]
- 68. Otake K., Uchida K., Yoshiyama S., Inoue M., Okita Y., Watanabe H., Inoue Y., Mohri Y., Miki C., Kusunoki M. (2008) Effects of a hyaluronate-carboxymethylcellulose membrane (Seprafilm) on human polymorphonuclear neutrophil functions. J. Surg. Res. 149, 243–249 [DOI] [PubMed] [Google Scholar]
- 69. Koh T. J., DiPietro L. A. (2011) Inflammation and wound healing. The role of the macrophage. Expert. Rev. Mol. Med. 13, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Frisch S. M., Screaton R. A. (2001) Anoikis mechanisms. Curr. Opin. Cell Biol. 13, 555–562 [DOI] [PubMed] [Google Scholar]
- 71. Lesley J., Schulte R., Hyman R. (1990) Binding of hyaluronic acid to lymphoid cell lines is inhibited by monoclonal antibodies against Pgp-1. Exp. Cell Res. 187, 224–233 [DOI] [PubMed] [Google Scholar]
- 72. McDonald B., McAvoy E. F., Lam F., Gill V., de la Motte C., Savani R. C., Kubes P. (2008) Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J. Exp. Med. 205, 915–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Termeer C., Johannsen H., Braun T., Renkl A., Ahrens T., Denfeld R. W., Lappin M. B., Weiss J. M., Simon J. C. (2001) The role of CD44 during CD40 ligand-induced dendritic cell clustering and maturation. J. Leukocyte Biol. 70, 715–722 [PubMed] [Google Scholar]
- 74. Lesley J., Gál I., Mahoney D. J., Cordell M. R., Rugg M. S., Hyman R., Day A. J., Mikecz K. (2004) TSG-6 modulates the interaction between hyaluronan and cell surface CD44. J. Biol. Chem. 279, 25745–25754 [DOI] [PubMed] [Google Scholar]
- 75. Forrester J. V., Balazs E. A. (1980) Inhibition of phagocytosis by high molecular weight hyaluronate. Immunology 40, 435–446 [PMC free article] [PubMed] [Google Scholar]
- 76. Suzuki Y., Yamaguchi T. (1993) Effects of hyaluronic acid on macrophage phagocytosis and active oxygen release. Agents Actions 38, 32–37 [DOI] [PubMed] [Google Scholar]
- 77. Tamoto K., Nochi H., Tada M., Shimada S., Mori Y., Kataoka S., Suzuki Y., Nakamura T. (1994) High-molecular-weight hyaluronic acids inhibit chemotaxis and phagocytosis but not lysosomal enzyme release induced by receptor-mediated stimulations in guinea pig phagocytes. Microbiol. Immunol. 38, 73–80 [DOI] [PubMed] [Google Scholar]
- 78. Ravasi T., Wells C., Forest A., Underhill D. M., Wainwright B. J., Aderem A., Grimmond S., Hume D. A. (2002) Generation of diversity in the innate immune system. Macrophage heterogeneity arises from gene-autonomous transcriptional probability of individual inducible genes. J. Immunol. 168, 44–50 [DOI] [PubMed] [Google Scholar]
- 79. Cuetara B. L., Crotti T. N., O'Donoghue A. J., McHugh K. P. (2006) Cloning and characterization of osteoclast precursors from the RAW264.7 cell line. In Vitro Cell Dev. Biol. Anim 42, 182–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Strassmann G., Patil-Koota V., Finkelman F., Fong M., Kambayashi T. (1994) Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. J. Exp. Med. 180, 2365–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Erwig L. P., Henson P. M. (2007) Immunological consequences of apoptotic cell phagocytosis. Am. J. Pathol. 171, 2–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fleming B. D., Mosser D. M. (2011) Regulatory macrophages. Setting the threshold for therapy. Eur. J. Immunol. 41, 2498–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Perdiguero E., Kharraz Y., Serrano A. L., Muñoz-Cánoves P. (2012) MKP-1 coordinates ordered macrophage-phenotype transitions essential for stem cell-dependent tissue repair. Cell Cycle 11, 877–886 [DOI] [PubMed] [Google Scholar]
- 84. Haskó G., Pacher P., Deitch E. A., Vizi E. S. (2007) Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol. Ther. 113, 264–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gordon S., Taylor P. R. (2005) Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964 [DOI] [PubMed] [Google Scholar]
- 86. Lucas M., Stuart L. M., Zhang A., Hodivala-Dilke K., Febbraio M., Silverstein R., Savill J., Lacy-Hulbert A. (2006) Requirements for apoptotic cell contact in regulation of macrophage responses. J. Immunol. 177, 4047–4054 [DOI] [PubMed] [Google Scholar]
- 87. Stein M., Keshav S., Harris N., Gordon S. (1992) Interleukin 4 potently enhances murine macrophage mannose receptor activity. A marker of alternative immunologic macrophage activation. J. Exp. Med. 176, 287–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Paun A., Reinert J. T., Jiang Z., Medin C., Balkhi M. Y., Fitzgerald K. A., Pitha P. M. (2008) Functional characterization of murine interferon regulatory factor 5 (IRF-5) and its role in the innate antiviral response. J. Biol. Chem. 283, 14295–14308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Krausgruber T., Saliba D., Ryzhakov G., Lanfrancotti A., Blazek K., Udalova I. A. (2010) IRF5 is required for late-phase TNF secretion by human dendritic cells. Blood 115, 4421–4430 [DOI] [PubMed] [Google Scholar]
- 90. Paun A., Bankoti R., Joshi T., Pitha P. M., Stäger S. (2011) Critical role of IRF-5 in the development of T helper 1 responses to Leishmania donovani infection. PLoS Pathog. 7, e1001246. [DOI] [PMC free article] [PubMed] [Google Scholar]