

Background: FOF1 ATP synthases are rotary nanomachines transporting protons across the membrane during catalysis.
Results: Escherichia coli FOF1 is assembled from subcomplexes with subunit δ functioning as clamp to generate stable FO.
Conclusion: δ guarantees that the open proton channel is concomitantly assembled within coupled FOF1.
Significance: We investigate how a proton-translocating unit is assembled while simultaneously maintaining the low membrane proton permeability essential for viability.
Keywords: ATP Synthase, Escherichia coli, F1Fo ATPase, H+-ATPase, Membrane Proteins, Protein Assembly, Protein Cross-linking
Abstract
The ATP synthase (FOF1) of Escherichia coli couples the translocation of protons across the cytoplasmic membrane to the synthesis or hydrolysis of ATP. This nanomotor is composed of the rotor c10γϵ and the stator ab2α3β3δ. To study the assembly of this multimeric enzyme complex consisting of membrane-integral as well as peripheral hydrophilic subunits, we combined nearest neighbor analyses by intermolecular disulfide bond formation or purification of partially assembled FOF1 complexes by affinity chromatography with the use of mutants synthesizing different sets of FOF1 subunits. Together with a time-delayed in vivo assembly system, the results demonstrate that FOF1 is assembled in a modular way via subcomplexes, thereby preventing the formation of a functional H+-translocating unit as intermediate product. Surprisingly, during the biogenesis of FOF1, F1 subunit δ is the key player in generating stable FO. Subunit δ serves as clamp between ab2 and c10α3β3γϵ and guarantees that the open H+ channel is concomitantly assembled within coupled FOF1 to maintain the low membrane proton permeability essential for viability, a general prerequisite for the assembly of multimeric H+-translocating enzymes.
Introduction
FOF1 ATP synthases comprise different structural and functional entities that couple the translocation of ions across the membrane to the synthesis or hydrolysis of ATP via a rotary mechanism. In Escherichia coli, the membrane-integrated, H+-translocating FO complex consists of subunits ab2c10, whereas the peripherally associated F1 part with its catalytic centers comprises subunits α3β3γδϵ. As the rotor c10γϵ is driven by the transport of protons through two half-channels formed by subunit a as well as the c10 ring in FO, the rotation of γϵ causes conformational changes in the catalytic nucleotide-binding sites within the α3β3 hexamer of F1, thereby provoking ATP synthesis and its release. To counter the tendency of the α3β3 to follow the rotation of the rotor, a peripheral stalk consisting of b2δ stabilized by subunit a acts as a stator and holds α3β3 in position (1, 2).
In E. coli, insertion of subunit c into the membrane depends on YidC insertase (3), and formation of a c10 subcomplex is independent of the presence of other FOF1 subunits (4). Recent studies on AtpI revealed a participation probably in a chaperone-like manner in the assembly of a stable c ring (5–7). Insertion of subunits a and b into the membrane involves the Sec translocon, the signal recognition particle pathway, and for subunit a also YidC (8–10). Furthermore, a stable insertion of subunit a into the membrane is strictly dependent upon the co-insertion of the other FO subunits, whereas b and c are inserted independently (11). Overexpression of subunits α, β, and γ allowed complex formation with high ATPase activity in the cytoplasm (12), and for interaction between subunits δ and α, the assembly of α with other F1 subunits is a prerequisite (13).
The different subunits of E. coli ATP synthase are translated from a polycistronic atp mRNA, and a balanced stoichiometry is obtained by translational coupling between the cistrons as well as regulation by mRNA secondary structure (14, 15). In most bacteria, the cistrons are arranged in clusters separating those for FO from those for F1, an arrangement fitting well with the proposal that both have been evolved from functionally unrelated ancestor protein complexes (16–18). Furthermore, this suggests that ATP synthases are probably assembled from subcomplexes, and studies on the assembly of the yeast mitochondrial ATP synthase support this assumption (19).
The goal of this study was to gain insight into the assembly pathway of the FO complex of the E. coli ATP synthase with special emphasis on the H+-translocating unit. Accordingly, the analyses of single-subunit knock-out mutants Δa, Δb, and Δδ, in which the synthesis of subunits a, b, and δ, respectively, is prevented due to insertion of an early stop codon into the corresponding gene, were combined with intermolecular disulfide bond formation of cysteine-substituted subunits or affinity purification of partially assembled FOF1 complexes. Δa allowed the formation of FOF1−a in amounts comparable with wild type. Δδ revealed the presence of an ab2 as well as a c10α3β3γϵ subcomplex; both could be assembled into a functional ATP synthase by a time-delayed synthesis of subunit δ. Δb only contained the FOF1 core complex c10α3β3γϵ. In each case, subunit δ was essential for the integration of the b dimer into the core complex independent of the interaction of b2 with subunit a. This demonstrates that δ functions as a clamp between ab2 and c10α3β3γϵ to generate the H+-translocating machinery within FO.
EXPERIMENTAL PROCEDURES
Mutagenesis
All plasmids used are listed in Table 1. In most cases, a two-step PCR method was used to generate the early stop codons and the cysteine substitutions in the individual subunits. For cysteine substitutions, the plasmids used as template DNA contain alanine codons instead of the endogenous cysteine codons in the atp genes mutated. Mutant PCR fragments were transferred into pBWU13 or its derivatives using single or double cutters for restriction. To study subunit interactions in the absence of other FOF1 subunits, atpF or atpEF were cloned into plasmid pET-22b via NdeI/EcoRI with the EcoRI site directly located downstream of the corresponding stop codon. For expression of the atp genes under control of the inducible/repressible promoter ParaBAD, a KpnI site was introduced in pFV2 downstream of the weak, constitutive atp promoter P3 (29). Subsequently, the atp operon was cloned into pBAD33 via KpnI (49 bp upstream of the stop codon of atpI) and XbaI (320 bp downstream of the 3′-end of atpC). The atpH gene was cloned into plasmid pET-22b via NdeI/EcoRI, and the start codon was subsequently exchanged to TTG. The presence of each mutation was confirmed by DNA sequencing.
TABLE 1.
E. coli strains and plasmids
| E. coli strains and plasmids | Genotype/Description | Source/Reference |
|---|---|---|
| Strains | ||
| DK8 | HfrPO1, bglR, thi1, relA1, ilv::Tn10 (TetR), ΔatpBEFHAGDC | Ref. 20 |
| HB1(DE3) | F−, dcm, ompT, hsdS (rb− mb−), gal, λ(DE3), ilv::Tn10 (TetR), ΔatpBEFHAGDC | Ref. 4 |
| Plasmids | ||
| pBWU13 derivatives | ||
| pBWU13 | ApR, pMB1 origin, atpI′BEFHAGDC | Ref. 21 |
| pBH1 | His12 tag N-terminally to a | Ref. 22 |
| pBH7 | His6 (MRGSHHHHHHG) tag N-terminally to β | This study |
| pBH16 | bQ10C, bC21A | This study |
| pBH17 | bS139C, bC21A | This study |
| pBH20.1 | bE155C, bC21A | This study |
| pBH26.1 | αA334C, γL262C, αC47A, αC90A, αC193A, αC243A, γC87A, γC112A | This study |
| pBH29 | βD380C, βC137A, γC112A | This study |
| pBH55.bα | aW231end, bA92C, αR477C, bC21A, αC47A, αC90A, αC193A, αC243A | This study |
| pBH55.bβ | aW231end, bA92C, βQ351C, bC21A, βC137A | This study |
| pBH55.β-His | aW231end, His6 tag N-terminally to β | This study |
| pBH56.a-His12 | δY11end, His12 tag N-terminally to a | This study |
| pBH56.bβ | δY11end, bA92C, βQ351C, bC21A, βC137A | This study |
| pBH56.β-His | δY11end, His6 tag N-terminally to β | This study |
| pBH152 | aN238C, bA13C, bE155C, bC21A | This study |
| pBH152.Δδ | δY11end, aN238C, bA13C, bE155C, bC21A | This study |
| pBWU13.Δb | bI7end | This study |
| pBWU13.Δδ | δY11end | This study |
| pBWU13.NOC | cA21C, cM65C, bC21A | Ref. 23 |
| pHB3 | bN2C, cV74C, bC21A | This study |
| pHB5 | aW231end | This study |
| pHB15 | aW231end, bN2C, cV74C, bC21A | This study |
| pHB17 | aW231end, bL65C, bC21A | This study |
| pJGA3 | bI7end, αP280C, γA285C, αC47A, αC90A, αC193A, αC243A, γC87A, γC112A | This study |
| pJGA4 | bI7end, βD380C, βC137A, γC112A | This study |
| pJGA5.1 | bI7end, cA21C, cM65C | This study |
| pJPKB.βQ351C | bA92C, βQ351C, bC21A, βC137A | Ref. 48 |
| pKB1.αR477C | bA92C, αR477C, bC21A, αC47A, αC90A, αC193A, αC243A | Ref. 48 |
| pKB7.aN238C | aN238C, bA13C, bC21A | This study |
| pRE2 | δY11end, bN2C, cV74C, bC21A | This study |
| pRE3 | δY11end, cA21C, cM65C, bC21A | This study |
| pRE4 | δY11end, bL65C, bC21A | This study |
| pRE8 | δY11end, αP280C, γA285C, αC47A, αC90A, αC193A, αC243A, γC87A, γC112A | This study |
| pRE10 | δY11end, βD380C, βC137A, γC112A | This study |
| pRE14 | δY11end, bA92C, αR477C, bC21A, αC47A, αC90A, αC193A, αC243A | This study |
| pSP2.a238/b13 | δY11end, aN238C, bA13C, bC21A | This study |
| pSTK3 | bC21A | This study |
| pSTK8 | bL65C, bC21A | This study |
| pET-22b derivatives | ||
| pET-22b | ApR, lacI, pMB1 origin | Novagen |
| pET-atpF1 | atpF, bQ10C, bC21A | This study |
| pET-atpF2 | atpF, bS139C, bC21A | This study |
| pET-atpF3 | atpF, bL65C, bC21A | This study |
| pET-atpEF1 | atpEF, bN2C, cV74C, bC21A | This study |
| pET22-atpH-TTG | atpH with TTG as start codon | This study |
| pBAD33 derivatives | ||
| pBAD33 | CmR, araC, pACYC184 origin | Ref. 24 |
| pBAD33.atp | atpI′BEFHAGDC | This study |
| pBAD33.Δδ | atpI′BEFH*AGDC, δY11end | This study |
| Miscellaneous | ||
| pBR322 | ApR, TcR, pMB1 origin | New England Biolabs |
| pDF163.b2-c74 | ApR, pMB1 origin, atpI′BEFH, bN2C, cV74C, bC21A | Ref. 25 |
| pFV2 | ApR, pACYC177 origin, atpI′BEFHAGDC, f-MRGSHHHHHHG N-terminally to β | Ref. 26 |
| pMM10 | ApR, pACYC177 origin, atpI′BEFHAGDC, αP280C, γA285C, γK108C, Cys-less, f-MRGSHHHHHHG N-terminally to β | Ref. 27 |
| pSW3 | ApR, pACYC177 origin, atpI′BEFHAGDC, βD380C, γC87, γK108C, Cys-less, f-MRGSHHHHHHG N-terminally to β | Ref. 27 |
| pT7Pol26 | KanR, lacI, T7gene1 | Ref. 28 |
Bacterial Strains and Growth Conditions
E. coli strains used are listed in Table 1. The atp deletion strain HB1(DE3) was obtained by P1 co-transduction of ΔatpBEFHAGDC and ilv::Tn10 (TetR) using DK8 (20) as donor and BL21(DE3) (Novagen) as recipient strain (4). E. coli strain DK8 transformed with plasmid pBWU13 or its derivatives or strain HB1(DE3) transformed with pET-22b derivatives were grown in minimal medium with 0.5% (v/v) glycerol or in LB medium with 100 μg/ml ampicillin as described (4). All DK8 cells transformed with pBWU13 derivatives grew on succinate as a nonfermentable carbon source indicating a functional oxidative phosphorylation system, whereas no growth was observed in knock-out mutants (data not shown).
Time-delayed in Vivo Assembly of Subunit δ into Preformed FOF1−δ
The time-delayed in vivo assembly system used to study the time-delayed integration of subunit δ into preformed FOF1 subcomplexes missing subunit δ has been characterized in detail by Brockmann et al. (30). DK8 transformed with three different plasmids was grown in TYGPN medium (31) at 37 °C with 100 μg/ml ampicillin, 30 μg/ml chloramphenicol, and 50 μg/ml kanamycin. The medium was preincubated overnight with 10 units/ml β-galactosidase from Kluyveromyces lactis (Sigma) for removal of lactose known to be present in varying amounts in yeast extract used for preparation of TYGPN medium (TaKaRa Single Protein Production System, TaKaRa Bio Inc.). After inoculation, the medium was supplemented with 0.03% (w/v) arabinose for induction of ParaBAD-controlled atp genes. At OD578 nm = 0.3, the araBAD promoter was repressed by adding 0.5% (w/v) glucose and 0.045% (w/v) d-fucose (33, 24). After degradation of atp mRNA within 20 min, the expression of atpH and T7 gene1 (28) was induced by the addition of 0.1 mm IPTG6 for 1 h.
RNA Extraction, cDNA Synthesis, and rt-RT-PCR
2.5 × 108 cells (as calculated according to the finding of Neidhardt et al. (34) that 1 ml of cells contains 109 viable cells at OD = 1.0) were mixed with 2 volumes of RNAprotect bacteria reagent (Qiagen), incubated for 5 min at room temperature, harvested at 5,000 × g, and stored at −20 °C. RNA extraction, synthesis of cDNA, and rt-RT-PCR were performed as described (30, 35) using the following primer pairs: atpE′F (5′-CAGGCGCAGGCGGAAATTG-3′ (bp 1626–1645) and 5′-CCGTAATAAATTCAGACATCAGCCCC-3′ (bp 1821–1796)) and atpA (5′-GCGAACTGATCAAGCAGCGC-3′ (bp 2374–2393) and 5′-ACCCATAACAACCGCACCTAC-3′ (bp 2579–2559)). The primer annealing sites are indicated in parentheses according to the numbering of plasmid pBWU13 (21), starting with 1 at the second HindIII restriction site in atpI. The calculated threshold cycle values were normalized against those of housekeeping gene rpsL (5′-GGTACGCAAACCACGTGCTCG-3′ and 5′-CAGGTTGTGACCTTCACCACC-3′) (30).
Copper Cross-linking
Inverted membrane vesicles were prepared in the presence of protease inhibitor mix without EDTA (Sigma) according to Krebstakies et al. (23) using TMG buffer (50 mm Tris-HCl, pH 7.5, 10 mm MgSO4, 10% (v/v) glycerol). Cross-linking of membrane vesicles was carried out as described by Ballhausen et al. (4), incubating 5 mg/ml membrane protein in the presence of 1.5 mm copper-1,10-phenanthroline (CuP) in TMG, pH 7.5 or pH 8.2, for 1 h at 23 °C. The reaction was stopped by the addition of 50 mm EDTA and 25 mm N-ethylmaleimide, and after 20 min, a quarter volume of 4× SDS sample buffer without 2-mercaptoethanol (36) was added.
Purification of Polyhistidine-tagged Proteins
For isolation of FOF1 via a His6 tag fused N-terminally to subunit β, membranes were prepared from a 3-liter cell culture according to Wise et al. (37), and membrane proteins were extracted as described by Pänke et al. (38). After centrifugation, the solubilized proteins were incubated for 1.5 h on ice with 1 ml of Ni2+-nitrilotriacetic acid-Sepharose FF (GE Healthcare). Subsequently, the matrix was washed with 50 mm Tris-HCl, pH 7.5, 5 mm MgCl2, 1% (w/v) n-octyl-β-d-glucopyranoside, 20 mm imidazole, 10% (v/v) glycerol and eluted with the same buffer containing 150 mm imidazole.
The solubilization of FOF1 with a His12 tag fused N-terminally to subunit a was performed according to Krebstakies et al. (22). 5 mg of membranes prepared in TMG buffer were pelleted, resuspended in 50 mm Tris-HCl, pH 7.5, 10% (v/v) glycerol, 0.1 mm PMSF, 1.4% (w/v) n-dodecyl-β-d-maltoside, and stirred for 30 min at 16 °C. After centrifugation, the supernatant was adjusted to 150 mm NaCl, 0.1 mm PMSF, and 20 mm imidazole (pH 7.5) and incubated end-over-end for 15 min with the matrix of a His SpinTrapTM column (GE Healthcare). Handling was performed as recommended by the supplier using 50 mm Tris-HCl, pH 7.5, 10% (v/v) glycerol, 0.1 mm PMSF, 150 mm NaCl, 20 mm imidazole, 1% (w/v) sodium cholate as binding buffer and the same buffer containing 500 mm imidazole as elution buffer. All buffers used included protease inhibitor mix without EDTA.
Assays
Protein concentrations were determined with the BCA assay (Pierce). Proteins were separated by SDS-PAGE using 10% separating gels (39) with PageRulerTM prestained protein ladder (Fermentas) as standard. Immunoblotting was performed according to Birkenhäger et al. (40). For immunolabeling, polyclonal antibodies raised in mice (anti-α, anti-β, anti-δ, anti-ϵ) or rabbits (anti-α, anti-β, anti-γ, anti-b, anti-c) and monoclonal antibodies raised in rat (anti-γ) or mice (anti-a, anti-b, anti-c) against the individual subunits of FOF1 were used as primary antibodies as indicated in the figures. IRDyeTM800DX-labeled goat-anti-mouse IgG, IRDyeTM800DX-labeled goat-anti-rat IgG, or IRDyeTM700DX-labeled goat-anti-rabbit IgG (Rockland) was applied as secondary antibody and detected with the two-channel system Odyssey (LI-COR). The secondary antibodies are affinity-purified for low cross-reactivities, and they allowed the simultaneous detection of fluorescence (shown in red or green) after immunolabeling of two proteins on one blot membrane. In the case of cross-linking of two different proteins, an overlay of both labels appears as a yellow band. Due to the varying avidities of the antibodies used, the color of the yellow bands may change to orange or lime green.
ATPase activities of membrane vesicles were determined as described (41). ATP- and NADH-driven proton pumping via ACMA fluorescence quenching was performed according to Birkenhäger et al. (40).
RESULTS
Formation of b Dimer Independent of Other FOF1 Subunits
To investigate the formation of the b dimer in the absence of other FOF1 subunits, we performed nearest neighbor analyses by use of disulfide bond formation after substituting a single amino acid residue within the polypeptide chain to cysteine. We have chosen three cysteine substitutions that are spread over the polypeptide chain, namely bQ10C, bL65C, and bS139C, and compared the b-b formation in the presence and absence of other FOF1 subunits. CuP was applied for generation of disulfide bonds using membranes with a bC21A substitution as a Cys-less control. Cross-linking was analyzed by immunoblotting after non-reducing SDS-PAGE (Fig. 1A). The cross-linking yields obtained for b-b assembled in FOF1 (Fig. 1A, left) were comparable with those described previously (42–44). For bS139C, a nearly complete removal of monomeric b was observed, indicating a high cross-linking yield; however, the signal of b-b was very low, and the apparent molecular mass was lower than that observed for the other b-b cross-linking products, as has also been observed by McLachlin and Dunn (44). Both results imply a different protein folding within b-b for bS139C compared with dimerization by residues bQ10C or bL65C.
FIGURE 1.
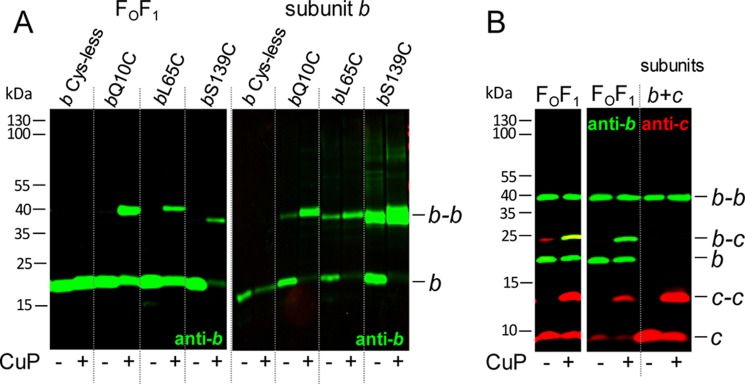
Comparison of cross-linked subunit b or subunits b and c in membranes of cells expressing the atp operon (FOF1) and cells exclusively expressing atpF (subunit b) or atpEF (subunits b and c). A, cross-linking of subunit b. FOF1, DK8 transformed with pBWU13 derivatives (b Cys-less, pSTK3; bQ10C, pBH16; bL65C, pSTK8; bS139C, pBH17) were grown in minimal medium with 0.5% (v/v) glycerol and harvested at OD = 0.8–1.0. subunit b, cells of E. coli HB1(DE3) transformed with pET-22b derivatives (bQ10C, pET22-atpF1; bL65C, pET22-atpF3; bS139C, pET22-atpF2) were grown in LB medium with ampicillin to OD = 0.6 prior to induction of atpF gene expression with 1 mm IPTG for 1 h. Membranes (20 μg of protein/lane) containing cysteine-substituted subunit b were separated after cross-linking with CuP at pH 8.2 by non-reducing SDS-PAGE and analyzed by immunoblotting. B, cross-linking of subunits b and c. FOF1, DK8 transformed with pHB3 (bN2C/cV74C); subunits b + c, cells of HB1(DE3) transformed with pET22-atpEF1 (bN2C/cV74C) were grown and analyzed as described in A. Using the Odyssey system, the intensities of the fluorescence detected in the individually immunolabeled bands are adjusted between a minimum and a maximum for each blot membrane scanned and separately for both detection channels. Due to the extremely high intensity of the red-labeled c and c-c bands of the membranes containing only subunits b and c (right), it was not possible to verify the presence of subunits b and c in the b-c cross-link band of FOF1-containing membranes. Therefore, the left panel shows FOF1 of the same immunoblot after cropping the right part of the right panel to readjust the intensities of the fluorescence.
In the absence of other FOF1 subunits, the immunoblot analysis also showed b-b formation with high cross-linking yields (Fig. 1A, right). In the presence of CuP, monomeric b was completely missing, and the cross-linking products were detected in comparable rates using the same antibody as described above. However, for bS139C, the cross-linking product was split into two bands, indicating the presence of two different conformations for b-b, both comparable with the different apparent molecular masses found for cross-linking of subunit b in FOF1. Furthermore, for bL65C and bS139C, dimer formation could already be observed even without CuP as oxidant, suggesting that the interacting SH-groups are even susceptible to oxidation by oxygen. In conclusion, membrane-bound b subunits showed dimer formation independent of the presence of other FOF1 subunits.
No Interaction between b Dimer and the c10 Ring in the Absence of Other FOF1 Subunits
In the absence of other FOF1 subunits, individually synthesized, membrane-bound subunits b and c have been shown to be organized in homooligomeric subcomplexes; namely, the c subunits are arranged in a ring of 10 monomers (4), whereas the b subunits are present in dimeric form (Fig. 1A). To answer the question of whether both subcomplexes interact with each other, as observed in FOF1 using the cV74C/bN2C variant (25), the same experimental design as described above was used. For expression of individual subunits bN2C and cV74C, the modified genes atpEF were cloned in the same gene order, retaining the intergenic region as in the atp operon, and protein interaction was examined in membranes by oxidation with CuP. Despite massive overproduction of subunits b and c in the membranes due to the expression of these subunits with the pET system, no b-c cross-linking product could be detected in the corresponding immunoblot analysis (Fig. 1B), whereas b-c cross-linking was present in FOF1-containing membranes, as expected. In addition, c-c as well as b-b were monitored as a cross-linking side reaction. Although the proportion of monomeric and dimeric c subunits was comparable in both samples, although the expression level of subunit c is completely different, the dimerization of subunit b even in the absence of CuP as oxidant was unusually high (Fig. 1B), indicating that the flexible N-terminal region forced b-b formation when subunit c was not in close vicinity. In summary, the data demonstrate that the individually assembled membrane-bound subunit b dimer has no contact with the oligomeric c10 ring when both subunits are expressed simultaneously in the absence of other FOF1 subunits.
Presence of FOF1 Subunits in Membranes of Single-subunit Knock-out Mutants Δa, Δb, and Δδ
Subunit a and subunit δ are both known to be tightly bound to the subunit b dimer to stabilize the stator part of FOF1 versus the strong forces present during rotation of c10γϵ in catalysis. Quantitative binding experiments revealed Kd values in the range of 2–3 nm for the bδ interaction in FOF1 (22), and an ab2 subcomplex can be purified from wild type membranes via an N-terminal His12 tag fused to subunit a (41). To elucidate the need of subunit a and/or δ for the assembly of the b dimer into the FOF1 complex, we have constructed single-subunit knock-out mutants, in which one of the FOF1 subunits, namely a, b, or δ, is no longer synthesized. For that purpose, the ΔatpB-C strain DK8 was transformed with pBWU13 derivatives (Table 1) in which the synthesis of one of the eight FOF1 subunits was prevented due to insertion of an early stop codon into the corresponding gene of the otherwise unchanged atp operon. In mutant Δδ, the stop codon was inserted at codon 11 of the atpH gene (δY11end) and in Δb at codon 7 of atpF (bI7end), whereas in Δa the stop codon was introduced at codon position aW231, a mutation that has been well characterized (11, 45). Characterization of membranes of mutants Δa and Δδ by immunoblotting revealed the presence of all FOF1 subunits except the one knocked out (Fig. 2A), indicating that the insertion of the additional stop codon induced no polar effect on the expression of the cistrons located downstream to the mutation in the polycistronic atp mRNA. However, it is important to note that no truncated subunit aW231end could be observed in all experiments performed, as has been discussed in detail by Hermolin and Fillingame (11).
FIGURE 2.

Presence of FOF1 subunits in membrane vesicles of single-subunit knock-out mutants Δa, Δb, and Δδ compared with wild type. A, detection of the different FOF1 subunits to verify their presence in membrane vesicles. DK8 transformed with pBWU13 (WT), pHB5 (Δa), pBWU13.Δb (Δb), and pBWU13.Δδ (Δδ), respectively, was grown in LB medium with ampicillin and harvested at OD = 0.8–1.0. Membranes were prepared in TMG buffer in the presence of protease inhibitor mix without EDTA and analyzed by immunoblotting. B, Cu2+-catalyzed cross-linking within the FOF1 core complex c10α3β3γϵ of single-subunit knock-out mutants Δb and Δδ compared with wild type. DK8 transformed with pBWU13 derivatives was grown, membranes were prepared, and after cross-linking with CuP at pH 7.5, proteins were separated by non-reducing SDS-PAGE and analyzed by immunoblotting. Top, formation of a cross-linked c10 ring using cysteine pair cA21C/cM65C (WT, pBWU13.NOC; Δb, pJGA5.1; Δδ, pRE3) immunolabeled with monoclonal anti-c antibody GDH 9-2A2. Due to an incomplete cross-linking, all intermediate products are visible. Bottom, formation of α-γ and β-γ via cysteine substitutions αP280C/γL262C and βD380C/γC87, respectively (α-γ: WT, pBH26.1; Δb, pJGA3; Δδ, pRE8; β-γ: WT, pBH29; Δb, pJGA4; Δδ, pRE10). Immunolabeling was performed with polyclonal mouse (anti-α, anti-β; green) and polyclonal rabbit (anti-γ; red) antibodies.
In contrast, in membranes of Δb, also subunits a and δ, which are tightly bound to the b dimer in FOF1, are completely missing in addition to subunit b. Such an interdependence between FOF1 subunits during assembly has been previously described for subunit a to be interdependent on subunits b and c (11, 46).
The immunoblot analysis (Fig. 2A) showed that in membranes of mutant Δa, the amount of FOF1 subunits present was comparable with wild type, whereas it was largely reduced in membranes of Δb and Δδ. To obtain roughly comparable signals in immunoblotting, we therefore increased the amount of protein for Δδ by a factor of 7. This fits very well for all FOF1 subunits except subunit c, which now revealed largely increased amounts of protein compared with wild type. For subunit c, comparable intensities for wild type and Δδ were obtained by separation of identical amounts of membrane protein, as can be observed for the Δb mutant as well. Furthermore, intermolecular cross-linking via oxidation of bicysteine-substituted subunit c (cA21C/cM65C) (4, 47) produced an equal pattern of cross-link formation with all intermediate oligomers possible, stopping with the formation of decamers in either case (Fig. 2B). These observations demonstrate that the ringlike structure of the subunit c oligomer is not only independently assembled in the membrane (compare Refs. 4 and 47) but maintains high stability against degradation by proteases and is, therefore, present in Δδ and Δb in amounts comparable with wild type in contrast to the other subunits of FOF1, indicating the presence of free c10 subcomplexes.
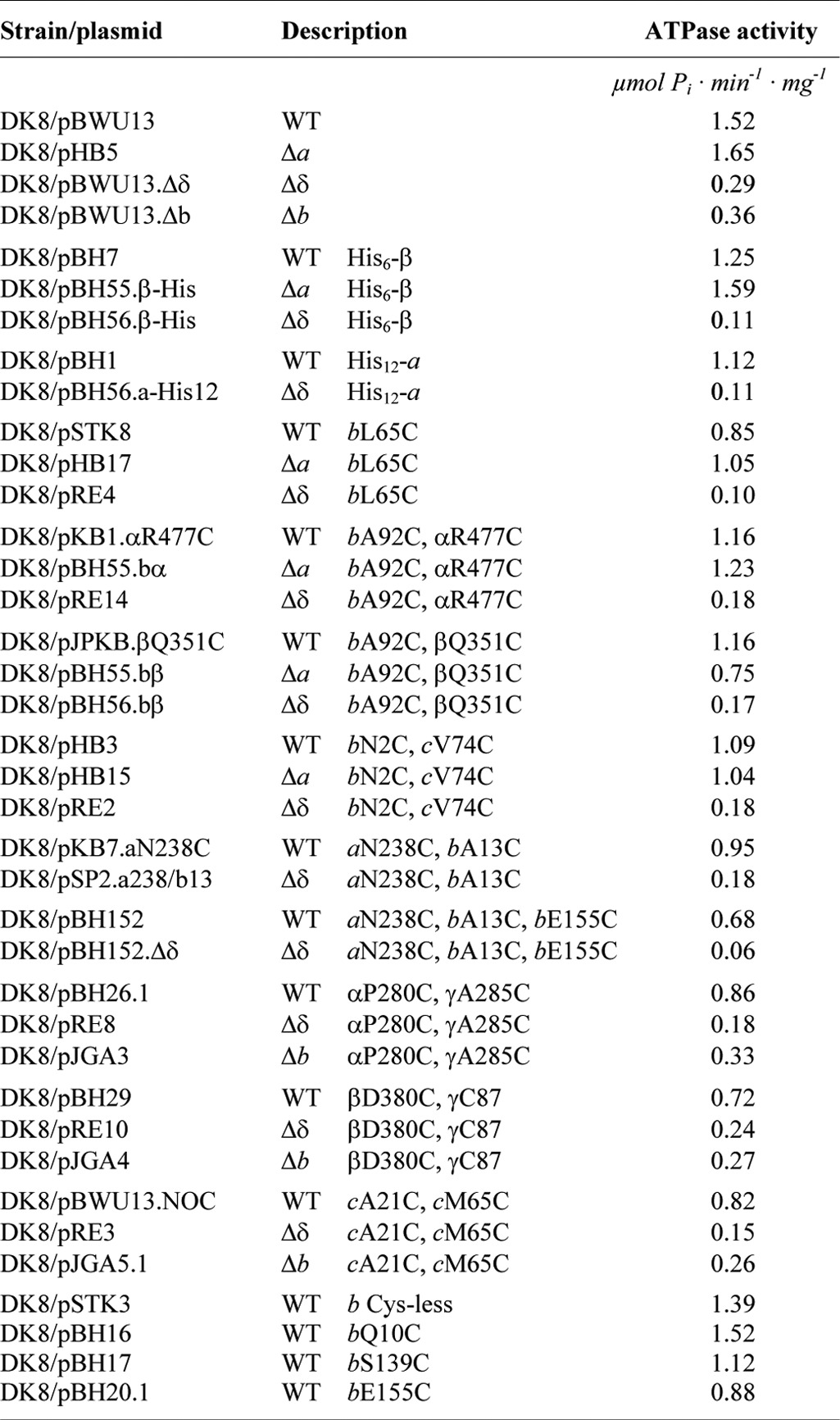
The ATPase activities of membrane vesicles of the knock-out mutants compared with wild type exhibited a similar behavior (Table 2). Although subunit a is missing and the proton pathway cannot be established (30), the ATPase activity of Δa is in a range comparable with wild type activities, providing evidence for a stable assembly of the remaining FOF1 subunits. In membranes of Δδ and Δb, the ATPase activities were reduced by a factor of 5–7, corresponding well to the amount of FOF1 proteins present in the membrane. In addition, the interaction areas of the α3β3 hexamer with subunit γ were verified by disulfide bond formation. Membranes containing subunit pairs with single cysteine substitutions were applied, confirming contact sites between subunits α-γ (αA334C/γL262C) and β-γ (βD380C/γC87) (Fig. 2B) comparable with wild type (27). Therefore, it can be concluded that at least an FOF1 core complex composed of subunits c10α3β3γ exhibiting ATPase activity is assembled in membranes of Δδ and Δb.
TABLE 2.
ATPase activities of E. coli strain DK8/pBWU13 and its Δa, Δb, and Δδ derivatives
Cross-linking of Subunit b with Its Interacting Subunits α, β, and c in Mutants Δa and Δδ
In wild type FOF1, subunit b has contact with subunits a and c of FO and subunits α, β, and δ of F1. To characterize in more detail the status of b present in membranes of Δa and Δδ, nearest neighbor analyses were performed with wild type and mutant membranes containing the amino acid substitution pairs bL65C, bA92C/αR477C, bA92C/βQ351C, or bN2C/cV74C (Fig. 3); the localization of these residues within FOF1 is summarized in Fig. 3A. To facilitate reading, the terms wild type (WT), Δa, and Δδ exclusively refer to the mutations leading to partially assembled FOF1 complexes independent of the presence or absence of cysteine substitutions. All cysteine pairs used are well characterized to generate disulfide bond formation in wild type with high yield (Fig. 3, B–E; compare Refs. 42, 44, and 48) and were applied as indicators for stable interactions between subunits in partially assembled FOF1 complexes. The formation of b-b was comparable in Δa and Δδ with that observed for solely expressed subunit b (Fig. 3B). In Δa, intense b-α and b-β heterodimer formation was observed after oxidation with CuP, indicating a proximal localization between b2 and α3β3 in this partially assembled FOF1 complex (Fig. 3, D and E, left). However, no cross-linking could be obtained between subunits b and c in Δa, suggesting that the right positioning of the amino acid residues of both subunits involved needs the presence of subunit a (Fig. 3C, left). Both cysteine residues are located within terminal regions of the polypeptide chains, which are in general more flexible and, therefore, in most cases a benefit for cross-link formation. However, in this case, a stabilization by subunit a seems to be necessary for disulfide bond formation between b and c, and instead b-b formation is favored.
FIGURE 3.

Cu2+-catalyzed cross-linking of subunit b to subunit α, β, or c in membranes of knock-out mutants Δa and Δδ compared with wild type. DK8 transformed with pBWU13 derivatives was grown, and membranes were prepared as described in the legend of Fig. 2. Membranes of wild type, Δa, and Δδ, containing FOF1 subunits individually substituted with cysteines, were separated after cross-linking with CuP at pH 7.5 by non-reducing SDS-PAGE and analyzed by immunoblotting. A, structural homology model of c10α3β3γϵ of E. coli ATP synthase. The model is composed of several partial structures combined with biochemical data as described in detail by Junge et al. (1) and drawn by RasMol version 2.7.2.1.1 to mark the positions of the cross-linking pairs used, with b2 illustrated as yellow rectangles. B, formation of b dimer via cysteine substitution bL65C (WT, pSTK8; Δa, pHB17; Δδ, pRE4). C, cross-linking between subunits b and c using cysteine pair bN2C/cV74C (WT, pHB3; Δa, pHB15; Δδ, pRE2). D, cross-linking between subunits b and α using cysteine pair bA92C/αR477C (WT, pKB1.αR477C; Δa, pBH55.bα; Δδ, pRE14). E, cross-linking between subunits b and β via cysteine pair bA92C/βQ351C (WT, pJPKB.βQ351C; Δa, pBH55.bβ; Δδ, pBH56.bβ). B–E, left, WT, 20 μg/lane; Δa, 20 μg/lane. B–E, right, WT, 5 μg/lane; Δδ, 35 μg/lane. unsp., unspecific.
In contrast, in Δδ, no cross-linking of subunit b to any of its nearest neighbors could be observed, indicating that in the absence of δ, b2 had no contact with a partially assembled FOF1 core complex, at least within the area tested. In general, the ATPase activities observed were within the same range as those measured in the absence of the cysteine substitutions necessary for cross-linking (Table 2).
In the case of b-α cross-linking, as a side reaction, an α-α cross-linking was observed, which was analyzed by Brandt et al. (48) to be an interenzyme cross-link reaction. Substitution of a cysteine residue in α resulted in incorporation of three thiol groups into FOF1. While one thiol group is proximal to the b dimer (b-α cross-linking in wild type or Δa FOF1), the other two, located at the surface of the hexamer, will be freely available for other reactions. Because wild type membranes are rich in FOF1, such an intermolecular cross-linking is not unexpected. However, in membranes of mutant Δδ, the amount of FOF1 in the membrane was reduced approximately by a factor of 7. Therefore, the probability of interenzyme cross-linking was also reduced, and the amount of α-α varied from experiment to experiment (data not shown).
Taken together, the results revealed that the b dimer can be assembled with the core FOF1 complex in the absence of subunit a, forming a stable complex with interactions in the F1 part comparable with functional FOF1, whereas an integration of b2 into FOF1 is not possible in the absence of subunit δ.
Purification of Partially Assembled FOF1 Complexes via N-terminally His6-tagged Subunit β from Mutants Δa and Δδ
To further characterize the partially assembled core FOF1 complex formed in Δa and Δδ, a His6 tag was fused to the N terminus of subunit β, usually applied to purify ATP synthase complexes from detergent-solubilized wild type membranes (26, 38). For Δa and Δδ, this approach was used to determine which FOF1 subunits are stably associated with subunit β. After purification via Ni2+-nitrilotriacetic acid-agarose chromatography, the purified complexes were analyzed by immunoblotting in comparison with their corresponding membranes using antibodies against subunits b and γ. Both subunits were in contact with subunit β in wild type FOF1 but did not interact with each other (Fig. 4A). In membranes, both subunits were present in comparable amounts after adjustment of the amount of membrane protein of Δδ by a factor of 7, as described above (compare Fig. 2A and Table 2). After purification of FOF1, subunit γ was again present in comparable amounts in both mutants, as expected, without any adjustment. In contrast, for subunit b, a different picture emerged; whereas in Δa the amount of b was comparable with wild type FOF1, subunit b was not co-purified and, therefore, was completely missing in the subcomplex purified via His6-β by affinity chromatography from Δδ. To further characterize the subcomplexes isolated using His6-β as the bait, immunoblot analyses were carried out for all FOF1 subunits (Fig. 4B). In Δa, all subunits except a, which was knocked out, were present in amounts comparable with wild type, verifying that an FOF1−a complex can be stably assembled.
FIGURE 4.
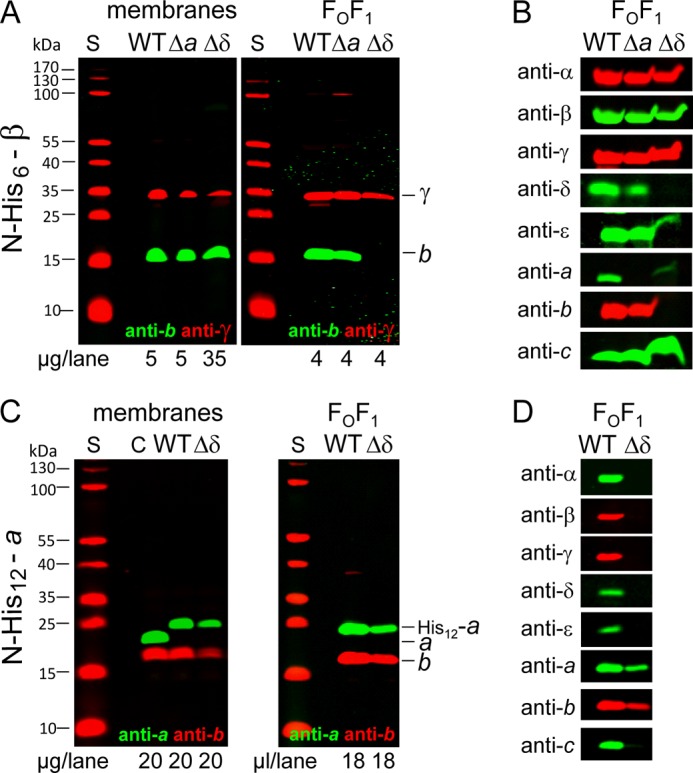
Purification of partially assembled FOF1 of mutants Δa and Δδ by affinity chromatography in comparison with wild type. A and C, comparison of membranes and purified FOF1 complexes. B and D, detection of the different FOF1 subunits to verify their presence in the complexes purified. DK8 transformed with pBWU13 derivatives was grown as described in the legend of Fig. 2. Membranes were prepared and solubilized, and FOF1 complexes were purified by affinity chromatography via a His6 tag (A and B) present N-terminal to subunit β (WT, pBH7; Δa, pBH55.β-His; Δδ, pBH56.β-His) or a His12 tag (C and D) fused to the N terminus of subunit a (WT, pBH1; Δδ, pBH56.a-His12). Membranes and purified FOF1 were analyzed by immunoblotting. S, molecular mass standard; C, subunits a and b detected in membranes of DK8/pBWU13.
In contrast, in Δδ, subunits δ and b could not be detected, and in addition, subunit a was only present in extremely low amounts and, therefore, barely visible. The results demonstrate that although subunits a and b are present in the cytoplasmic membrane of Δδ, they cannot be assembled into FOF1 in the absence of subunit δ, as has also been observed for subunit b by cross-linking (compare Fig. 3). Nevertheless, a protein complex, which comprises subunits α, β, γ, ϵ, and c, could be isolated from Δδ membranes by use of His6-β as ligand in affinity chromatography, implying the presence of an c10α3β3γϵ subcomplex.
Purification of Partially Assembled FOF1 Subcomplexes via N-terminally His12-tagged Subunit a from Mutant Δδ
Accordingly, in the next step, we studied the status of subunit a present in the membrane of Δδ by affinity chromatography using a His12 tag N-terminally fused to subunit a (23, 41). A modified purification protocol was applied to membranes of wild type and Δδ. The immunoblot analysis using anti-a and anti-b antibodies simultaneously revealed the presence of both subunits in the eluate for wild type and Δδ (Fig. 4C). In addition, the eluates obtained were screened for the presence of other FOF1 subunits by immunoblotting (Fig. 4D). Although from wild type membranes, the entire ATP synthase complex was purified, only subunit b could be co-purified from membranes of Δδ using His12-a as bait, indicating that a separate subcomplex is formed. Taken together, in the absence of subunit δ, two FOF1 subcomplexes are independently present in the cytoplasmic membrane, one comprising subunits α, β, γ, ϵ, and c and the other containing the residual subunits a and b.
Formation of an ab2 Subcomplex in Membranes of Mutant Δδ
To verify the presence of an independent ab2 subcomplex in Δδ, we used an experimental set-up in which two cysteine pairs were combined for cross-linking of two different subunit contact areas. As shown in Fig. 5A for wild type membranes, in the presence of CuP as oxidant, bE155C located in the C-terminal region of subunit b was able to generate a b-b product, whereas the cysteine pair bA13C/aN238C located in the transmembrane region of both subunits formed a disulfide bond between subunit a and one of the b subunits (b-a). After combining both cross-linking pairs, an a-b-b cross-linked complex was obtained in high yield in the presence of CuP (b-b/b-a), exhibiting a molecular mass of ∼65 kDa, as expected. Due to the presence of the cysteine substitutions, the ATPase activity was reduced by a factor of approximately 2 (Table 2); however, the DCCD sensitivity remained unchanged (78.4% inhibition), indicating a tight coupling between F1 and FO in those membranes.
FIGURE 5.
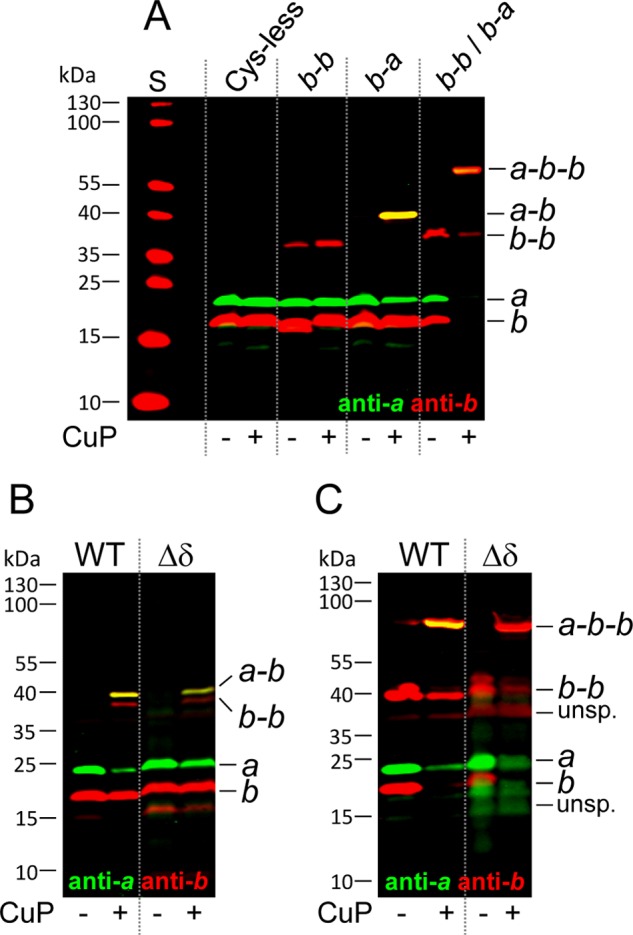
Cross-linking between subunits a and b in membranes of Δδ compared with wild type. Cell growth, membrane preparation, and cross-linking at pH 8.2 were performed as described in the legend of Fig. 3. Membranes were separated by non-reducing SDS-PAGE and analyzed by immunoblotting. A, generation of an a-b-b subcomplex by combining cross-linking pairs bA13C/aN238C and bE155C (Cys-less, pSTK3; b-b, pBH20.1; b-a, pKB7.aN238C; b-b/b-a, pBH152) using 20 μg protein/lane. B, cross-linking between subunits b and a using the cysteine pair bA13C/aN238C in mutant Δδ (WT, pKB7.aN238C; Δδ, pSP2.a238/b13). C, formation of a cross-linked a-b-b subcomplex in mutant Δδ (WT, pBH152; Δδ, pBH152.Δδ) using different amounts of protein (WT, 5 μg/lane; Δδ, 35 μg/lane). S, molecular mass standard; unsp., unspecific.
After introducing the corresponding cysteine residues into subunits a and b of mutant Δδ, the addition of CuP allowed the formation of an a-b cross-linking product in the presence of cysteine residues bA13C/aN238C (Fig. 5B) and, furthermore, the formation of a tertiary a-b-b product when, in addition, cysteine residue bA155C was provided (Fig. 5C), suggesting that an ab2 subcomplex was purified using His12-a for affinity chromatography. In each case, b-b was observed as byproduct. In summary, membranes of Δδ contained two FOF1 subcomplexes, an ab2 subcomplex and a c10α3β3γϵ subcomplex, both verified by a combination of affinity purification and zero length cross-linking and the latter exhibiting ATPase activity, whereas in membranes of Δa, an FOF1 complex lacking only subunit a (FOF1−a) was assembled, showing ATPase activities comparable with wild type FOF1.
Time-delayed in Vivo Assembly of Subunit δ into Preformed FOF1 Subcomplexes Generating a Functional ATP Synthase
The presence of the two subcomplexes, ab2 and α3β3γϵc10, in membranes of Δδ, just missing knocked out subunit δ, implies that δ is probably the last subunit being assembled into FOF1, as has been suggested by Rak et al. (19) for yeast mitochondrial FOF1. To answer the question, we applied our time-delayed in vivo assembly system (30) for a time-delayed integration of subunit δ into preformed FOF1 subcomplexes missing subunit δ.
The experimental design of the time-delayed in vivo assembly system is depicted in Fig. 6. After inoculation, ParaBAD was induced by arabinose to allow synthesis of FOF1 subunits except δ (FOF1−δ). During this growth phase, the lac operator-controlled promoters were repressed (compare Fig. 8B) due to preincubation of the medium with β-galactosidase for removal of residual lactose and the use of TTG as a start codon for atpH. At OD = 0.3, ParaBAD was repressed by the simultaneous addition of the catabolite repressor glucose and the anti-inducer d-fucose, a non-metabolizable analog of l-arabinose, and, after further growth for 20 min, nearly all atp mRNA present in the cell was degraded (see below). Due to this time delay, the de novo biosynthesis of FOF1 subunits was completely prevented. Subsequently, IPTG induction of lac operator-controlled promoters enabled the individual synthesis of subunit δ, which might be assembled together with the preformed subcomplexes ab2 and c10α3β3γϵ (FOF1−δ+δ) to generate functional FOF1.
FIGURE 6.

Design of the time-delayed in vivo assembly system and its different states during cell growth. ΔatpB-C strain DK8 was transformed with three plasmids bearing different resistance genes and different origins to gain compatibility: (i) a pBAD33 derivative (p15A ori; CmR) with structural genes of the atp operon carrying an early stop codon (δY11end) in atpH (atpBEFH*AGDC) under control of ParaBAD; (ii) a pET-22b derivative (pMB1 ori, ApR) carrying the atpH gene under control of the IPTG-inducible T7lac promoter using the weak start codon TTG for atpH; (iii) a pSC101 derivative (KanR) containing T7 gene1 under control of the IPTG-inducible T5N25lac promoter, encoding the RNA polymerase specific for promoters of phage T7. Left, cells were inoculated with OD = 0.05; ParaBAD was induced by arabinose to allow expression of the atpBEFH*AGDC genes, and the FOF1 subunits except for subunit δ (FOF1−δ) were synthesized. During this growth phase, the lac operator-controlled promoters are completely repressed. Middle, at OD = 0.3, ParaBAD is repressed by the simultaneous addition of glucose and d-fucose, and after further growth for 20 min, the atp mRNA present within the cell is completely degraded. Due to this time delay, the de novo biosynthesis of FOF1 subunits is prevented. Right, time-delayed IPTG induction of lac operator-controlled T7 and T5N25 promoters for 1 h enables the synthesis of subunit δ assembling together with the preformed FOF1 subcomplexes ab2 and c10α3β3γϵ (FOF1−δ+δ), a functional FOF1 complex. Red cross, repressed or uninduced state of promoters.
FIGURE 8.

Time-delayed in vivo assembly system of subunit δ into preformed FOF1 subcomplexes yielding functional FOF1. A–D, DK8 carrying plasmids pBAD33.atp, pET-22b, and pT7POL26 (FOF1) or pBAD33.Δδ, pET22-atpH-TTG, and pT7POL26 (FOF1−δ; FOF1−δ+δ) was grown as described under “Experimental Procedures.” Seven independent cell batches were prepared in parallel containing the additives indicated. The data represent average values of three independent measurements. A, level of atp mRNA. The amount of atp mRNA was determined via rt-RT-PCR using primer pairs atpE′F (dark gray) and atpA (light gray). The amount of atp mRNA present in the FOF1 sample grown with Ara was set to 100%. B, immunoblot analysis of membrane vesicles (20 μg of protein/lane) as indicated. C, ATPase activity of membrane vesicles. Gray and white portions of the bars represent DCCD-sensitive and DCCD-insensitive fractions of ATP hydrolysis, respectively. D, ATP-driven proton translocation of membrane vesicles. The relative magnitude of ACMA fluorescence quenching induced by ATP is shown. Error bars, S.E.
The stability of both subcomplexes formed was controlled by immunoblotting applying antibodies against subunits a, b, α, and γ to analyze both subcomplexes individually (Fig. 7A). Cells of FOF1−δ were grown to OD = 0.3 in the presence of arabinose (Ara) before the addition of glucose and d-fucose (Glu/Fuc), and in defined time intervals, samples were taken for analysis of FOF1 subunits in cell lysates. Whereas the amount of α and γ was stable over a period of at least 60 min, the immunodetection of a and b revealed a continuous degradation of ab2, starting at the latest after 30 min, and implies that the ab2 subcomplex is more susceptible to degradation by proteases than the core complex c10α3β3γϵ. In parallel, the decomposition of the atp mRNA was controlled by rt-RT-PCR, revealing a complete degradation 15 min after the addition of Glu/Fuc (Fig. 7B). Therefore, the delay time for the degradation of atp mRNA, being simultaneously the starting point for the synthesis of δ, was set to 20 min.
FIGURE 7.
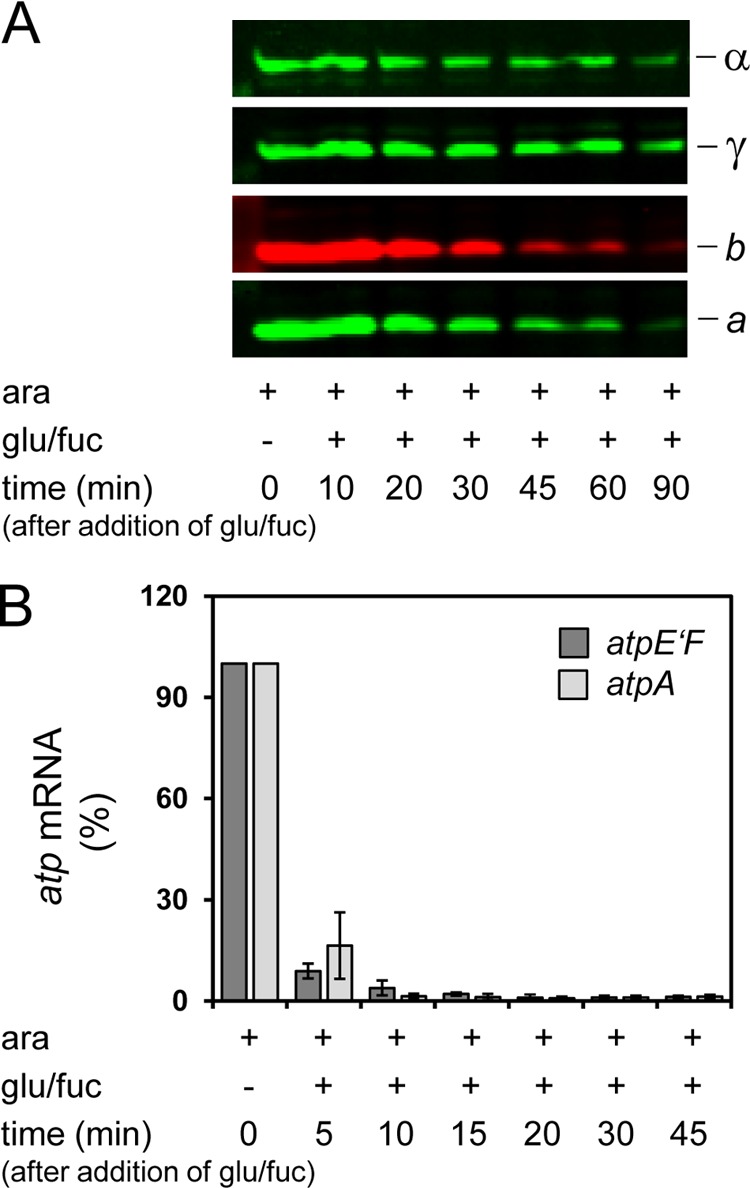
Stability of FOF1−δ (A) and degradation of atp mRNA (B) after repression of ParaBAD controlling expression of atpBEFH*AGDC. DK8 carrying plasmids pBAD33.Δδ, pET22.atpH-TTG and pT7POL26 (FOF1−δ) was grown as described under “Experimental Procedures.” At each time point indicated, cells were harvested for immunoblot analysis (A) and isolation of RNA (B). A, stability of FOF1−δ. After resuspension of cell lysates in sample loading buffer, cells were incubated for 5 min at 99 °C. The amount of cell extract (20 μg/lane) was calculated according to the determination of Neidhardt et al. (34) that 160 μg of protein is present per ml of growth medium at OD = 1. Proteins were separated by SDS-PAGE and detected by immunolabeling as indicated. B, degradation of atp mRNA. rt-RT-PCR was performed using primer pairs atpE′F (dark gray) and atpA (light gray). The amount of atp mRNA present in the FOF1 sample grown with Ara was set to 100%. Error bars, S.E.
Different cell batches of FOF1−δ were grown, each with the additives added at the time points addressed in Fig. 6. As a control, samples expressing wild type FOF1 were handled correspondingly. Under Ara-induced conditions, atp mRNA was present within the cells, as shown by rt-RT-PCR (Fig. 8A), although the amount was reduced in FOF1−δ by a factor of approximately 3 compared with wild type. In samples containing Glu/Fuc independent of the presence of Ara, atp mRNA was almost completely absent (0.3–2%), revealing that the synthesis of FOF1 subunits is prohibited.
Immunoblotting using antibodies against subunits b and γ as representatives of the two subcomplexes formed also revealed a reduction for FOF1−δ compared with FOF1 under Ara-induced conditions (Fig. 8B). In the presence of Glu/Fuc but in the absence of Ara, no FOF1 subunits were detectable in FOF1−δ, whereas extremely low amounts could be observed in wild type FOF1, which were quantitated to be below 2% by determining ATPase activity (Fig. 8C) but remained below the detection limit for ATP-driven H+ translocation (Fig. 8D). Samples induced with Ara and subsequently repressed by Glu/Fuc showed reduced but significant signals by immunolabeling, and again the ATPase activities observed correlated very well (Fig. 8, B and C). In addition, the extent of subunit degradation after repression with Glu/Fuc (in total for 80 min) was comparable with that observed in Fig. 7A. Subunit δ was not detectable in membranes of FOF1−δ; however, after expression of atpH, it was overproduced beyond stoichiometric amounts (FOF1−δ+δ; Fig. 8B). Importantly, a stabilizing effect, especially on subunit b, could be observed due to induction of atpH expression, whereas the amount of γ as well as membrane-bound ATPase activity remained unchanged (Fig. 8, B and C). After repression of atp transcription by Glu/Fuc, degradation of subunits was observed for FOF1 as well as FOF1−δ+δ. Nevertheless, in the absence of subunit δ (FOF1−δ), the degradation of b is increased, supporting the notion that δ protects the ab2 subcomplex against proteolytic digestion probably by its integration into the holoenzyme, whereas the stability of the c10α3β3γϵ remained unaffected.
The acidification of the lumen of membrane vesicles by ATP-driven H+ translocation was measured using ACMA as a pH-sensitive dye and applied to determine the assembly of a functional FOF1 complex from subcomplexes (Fig. 8D). Under Ara-induced conditions, the fluorescence quenching rates obtained for FOF1 were within the range expected (40, 49). After the addition of Glu/Fuc in the presence of Ara, the quenching rate was only slightly decreased, although the ATPase activity revealed that the amount of FOF1 in the membrane had been reduced to one-third, a discrepancy only at first glance that is probably due to a less dense protein packing within the membrane and, therefore, a significantly decreased membrane proton permeability (23, 50), leading to a stronger acidification of the vesicle lumen. Under repressed conditions, no H+ transport could be observed for FOF1 as well as FOF1−δ, although H+ pumping via the respiratory chain with NADH as substrate revealed intactness of the vesicles (data not shown), indicating a tight repression of ParaBAD by Glu/Fuc. In contrast, FOF1−δ showed extremely low H+ pumping rates (below 3%) in the presence of Ara. However, these rates were independent of the presence/absence of Glu/Fuc, implying that now and then spontaneously an H+-conducting unit could be formed from the two individual subcomplexes containing subunits a and c, an assumption corroborated by the complete absence of ATP-driven H+ transport in otherwise identical experiments performed with FOF1−a (30); an observation that requires further investigations to clarify whether subunit δ increases the affinity of ab2 for the FOF1 core complex or whether it changes the conformation of subunit a to an active one. However, after synthesis of subunit δ (FOF1−δ+δ) the quenching rate increased to 21.4%, demonstrating that functional FOF1 was assembled (Fig. 8D). The rate obtained fits well with the corresponding ATPase activity, and furthermore, both values are in good correlation with the activities of wild type FOF1 (in the presence of Ara and Glu/Fuc), indicating that both enzyme complexes independent of their assembly pathway exhibit comparable ATPase activities. Furthermore, ATP-driven H+ transport was completely abolished after preincubation of FOF1 as well as FOF1−δ+δ membranes with DCCD (data not shown). DCCD, which modifies specifically the protonated carboxyl groups of Asp-61 of subunit c at pH 8.0, inhibits the rotation of the c10 ring and, thereby, H+ translocation. Taken together, a functional FOF1 complex was assembled from subcomplexes ab2 and c10α3β3γϵ by time-delayed synthesis of subunit δ.
DISCUSSION
Generation of the H+-translocating Unit
The FO complex of E. coli ATP synthase is composed of subunits a, b, and c present as ab2c10. Wild type membranes depleted of F1 revealed an FO complex functional in H+ conduction as well as rebinding of F1 (40, 49). Furthermore, purification of FO subunits and reconstitution into liposomes revealed the same characteristics (51, 52). In contrast, after synthesis of FO subunits in the absence of F1, only low levels of proton permeability were observed in membranes as well as after reconstitution into liposomes, implying a dependence on F1 for the proton translocation by FO (53, 54), although the additional presence of subunit δ enhances the proton conduction through FO (55, 56).
Our results indicate that subunit δ is the key player in generating the functional H+-translocating unit composed of ac10. The position of b2 in FOF1 is restricted by its specific interactions with subunits a and δ at both terminal regions. The characterization of partially assembled FOF1 in Δa and Δδ, however, revealed that both interactions do not influence each other; the binding of a and δ to b2 are two unrelated processes apparently without major conformational changes. Whereas in Δa, b2 interacts with δ, thereby enabling its integration into FOF1, in Δδ, a specific ab2 subcomplex is formed by specific interaction with subunit a. Moreover, time-delayed binding of the second interaction partner (subunit δ (Fig. 8) or subunit a (30)) is possible without any preassigned assembly sequence, leading to the formation of a functional FOF1 complex in each case. As a consequence, it can be concluded that subunit δ is functioning as a clamp to induce a first contact between ab2 and c10 (via α3β3γϵ) to generate the H+-conducting unit within the membrane because the exclusive presence of both subcomplexes in Δδ is not sufficient to constitute an open H+ channel. By this arrangement, two important characteristics of FOF1 are respected. First, an intermediate assembly product exhibiting uncontrolled H+ conduction is avoided, which would lead to a breakdown of the proton motive force and, therefore, be lethal for the cell. Instead, it is guaranteed that an open H+ channel is concomitantly assembled within a coupled FOF1 complex, thereby preventing membrane proton permeability. Second, due to the rotation of the c10 ring versus the stator part ab2, it is essential for function that the affinity between stator and rotor subunits in FO is low. In addition, the recent 9.7 Å cryoelectron microscopy structure of the A-type ATP synthase of Thermus thermophilus (57) revealed a surprisingly small contact area between the rotor ring and the I protein (equivalent to subunit a of F-type ATP synthases). This low affinity is elegantly bypassed by subunit δ holding both complexes of FO, ab2 as well as c10 (via α3β3γϵ), in position, with b2 providing the necessary stiffness.
Nonetheless, as described above, once the FO complex has been generated, a depletion of F1 is possible without major changes in its H+-conducting capabilities, indicating that the first contact between ab2 and c10 enabled by δ triggers a kind of induced fit within subunits a and c that later on facilitates binding between FO subunits in the absence of F1. However, whether subunit δ simply increases the affinity of ab2 for the rest of the complex or whether it changes the conformation of FO to an active one, as suggested previously (55, 56), remains to be solved in further investigations.
Assembly of E. coli ATP Synthase from Subcomplexes
Our results support the notion that E. coli FOF1 is assembled from different subcomplexes (summarized in Fig. 9). The formation of b2 is independent of the presence of other FOF1 subunits, as shown by zero length cross-linking. With the same experimental design, it has recently been demonstrated that the c10 ring is also stably organized in the absence of other FOF1 subunits (4). Furthermore, a simultaneous but exclusive expression of subunits b and c in stoichiometric amounts allowed no b-c cross-link formation using a contact site characterized for wild type FOF1 (25). Although both subunits are overexpressed, no interaction between subunits b and c could be observed in the absence of other FOF1 subunits, which is in contrast to the model for assembly of E. coli FOF1 proposing a b2c10 subcomplex as an intermediate product (59, 60) but in accordance with models proposed for yeast mitochondrial ATP synthase (19, 58, 61).
FIGURE 9.
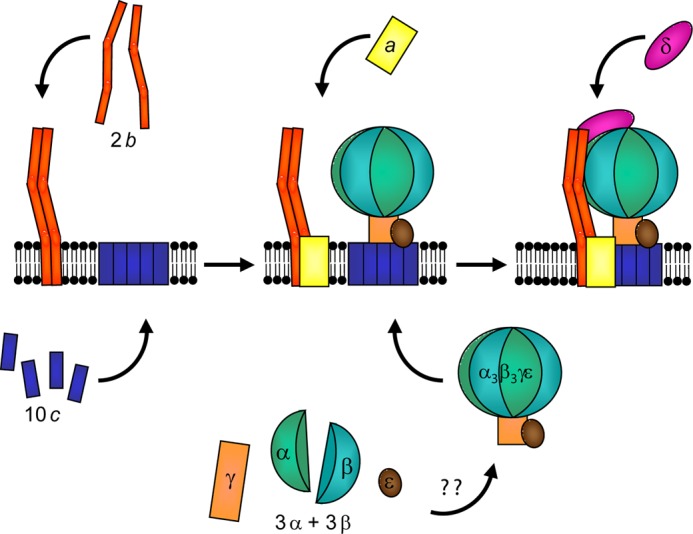
Model for the assembly of E. coli ATP synthase. The assembled c10 ring and b2 are independently present in cytoplasmic membranes. Formation of α3β3γϵ is not known in detail for E. coli. For FOF1 from yeast mitochondria, it has been proposed that two subcomplexes, α3β3 hexamer and γϵ, are assembled, which combine to form α3β3γϵ, before it subsequently binds to the membrane-bound c10 ring (58, 19). The membrane insertion of subunit a is interdependent on the presence of b and c. Integration of b2 into the complex is dependent on subunit δ, whereas binding of a as well as δ to b2 possibly has no preferred sequence.
Characterization of Δδ and Δb revealed the presence of the membrane-bound core complex c10α3β3γϵ possessing ATPase activity with rates comparable with wild type in relation to the amount of protein present. Furthermore, due to the absence of subunits a, b, and δ in Δb, it can be concluded that core complex formation occurs independently of other subcomplexes. Until now, only little experimental data regarding the order of the assembly of the E. coli F1 part have been available. However, in vitro reconstitution experiments (13, 62, 63) support an assembly in the cytoplasm prior to its binding to the c ring comparable with that described for yeast mitochondrial F1 (19, 64, 65). Therefore, an assembly of the FOF1 core complex from two individual subcomplexes c10 and α3β3γϵ is proposed, with α3β3γ forming the minimal unit exhibiting ATPase activities (12, 62, 66) and subunit ϵ being necessary for a stable binding to the membrane via c10 (63, 67). Preliminary experiments on chromosomally expressed ATP synthase containing His6-β enabled the purification of subunits α, β, γ, and ϵ exhibiting ATPase activity.7
In mutant Δδ, a separate ab2 subcomplex is present in the membrane in addition to the ATP-hydrolyzing FOF1 core complex. Furthermore, time-delayed synthesis of subunit δ enabled a subsequent assembly of functional ATP synthase, supporting the notion that both subcomplexes are present as native assembly intermediates. In addition, a reduction in synthesis of subunit c due to a point mutation in the ribosome binding site of atpE revealed the presence of free ab2 subcomplexes that can be reconstituted in liposomes with subunit c after purification to form FO complexes functional in passive H+ translocation as well as F1 binding (23). Due to the plasmid-encoded overexpression of FOF1 subunits, free excess subunits are inevitably present in the cell, and therefore, the assembly of active FOF1 complexes from these isolated subunits during time-delayed synthesis of subunit δ is at least conceivable. However, the observation that the amount of subunit γ present in the membrane as well as the membrane-bound ATPase activity remained unchanged after time-delayed expression of subunit δ revealed that ab2 as well as c10α3β3γϵ are true assembly intermediates instead of dead end products of incomplete assembly.
Last Subunit Being Assembled into FOF1
Whereas in Δa, a single subcomplex just missing subunit a is assembled, in Δδ, two subcomplexes, ab2 and c10α3β3γϵ, are present in the membrane. Furthermore, in both cases, a time-delayed synthesis of the missing subunit enabled the formation of functional ATP synthase, suggesting that each subunit can be assembled as the last subunit into preformed subcomplexes. Comparable results have been described for Bacillus PS3 as well as mitochondrial FOF1. Thermophilic Bacillus PS3 FOF1 lacking subunit a (FOF1−a) can be isolated in stable form from E. coli membranes, and after co-reconstitution with independently purified or cell-free synthesized subunit a, both components form a functional enzyme complex in liposomes (68, 69). In addition, from human ρ0 cells, a stable FOF1 complex lacking subunits a (Atp6p) and A6L (an additional subunit not present in bacterial FOF1) was purified (70). On the other hand, the data of Rak et al. (19) revealed that FOF1 is assembled in a modular way from at least three different modules, F1, the c ring, and a stator subcomplex. No information is given, however, for the assembly of OSCP (homologous to bacterial δ) into FOF1, although it has been deduced that OSCP might may be last subunit being assembled.
This seems to be contradictory at first sight; however, it is probably due to the manipulative conditions applied. In each case, the use of mutants provokes an arrest in the assembly pathway at a certain point, whereas in wild type cells, all subunits are nearly isochronously at hand by their simultaneous translation from polycistronic mRNA (15, 71). Apparently, subunit a as well as subunit δ could bind to preformed b2, probably simultaneously due to their autonomous interaction, therefore implying that in vivo a subcomplex of ab2δ is formed prior to its binding to c10α3β3γϵ. This is further supported by the observation that in Δb in addition to b, also subunits a and δ were absent. In vitro studies with purified subunit δ and the hydrophilic part of subunit b (comprising amino acid residues 34–156) revealed the formation of a b2δ complex (72). On the other hand, subunit δ is highly susceptible to proteolytic digestion and, therefore, rapidly degraded in most single-subunit knock-out mutants producing partially assembled FOF1 complexes.7 A proteolytic fragment of subunit δ comprising amino acid residues 1–134 has been characterized (73), although the corresponding protease has not yet been identified. In addition, in the absence of subunit δ, ab2 as well as α3β3γϵ were also degraded by proteases in appreciable amounts. Certainly, this rapid turnover indicates a tight regulation of δ and underlines its importance in generating functional FOF1. Whether δ makes the first contact with its C-terminal region with b2 (74), forming an ab2δ subcomplex, or whether it is first bound with its N-terminal region to the FOF1 core complex via subunit α (13, 32), forming a c10α3β3γδϵ subcomplex, is the next question to be investigated.
Acknowledgments
We are grateful to Drs. S. D. Dunn (University of Western Ontario), S. B. Vik (Southern Methodist University), R. H. Fillingame (University of Wisconsin Medical School), R. D. Simoni (Stanford University), and S. Engelbrecht (University of Osnabrück) for kindly providing antibodies and plasmids; S. Konrad, J. Garrelfs, and H. Brookman for aid in generating some of the plasmids; and Drs. K. Altendorf and J.-C. Greie for critical reading of the manuscript.
This work was supported by Deutsche Forschungsgemeinschaft Grant DE482/1-1.
G. Deckers-Hebestreit, unpublished observation.
- IPTG
- isopropyl-β-d-thiogalactopyranoside
- ACMA
- 9-amino-6-chloro-2-methoxyacridine
- Ara
- l-arabinose
- CuP
- copper-1,10-phenanthroline
- DCCD
- N,N′-dicyclohexylcarbodiimide
- Fuc
- d-fucose
- OD
- optical density measured at 578 nm
- OSCP
- oligomycin sensitivity-conferring protein
- rt-RT-PCR
- real-time RT-PCR
- TMG
- Tris/magnesium/glycerol.
REFERENCES
- 1. Junge W., Sielaff H., Engelbrecht S. (2009) Torque generation and elastic power transmission in the rotary FOF1-ATPase. Nature 459, 364–370 [DOI] [PubMed] [Google Scholar]
- 2. von Ballmoos C., Wiedenmann A., Dimroth P. (2009) Essentials for ATP synthesis by F1FO ATP synthases. Annu. Rev. Biochem. 78, 649–672 [DOI] [PubMed] [Google Scholar]
- 3. van der Laan M., Bechtluft P., Kol S., Nouwen N., Driessen A. J. (2004) FOF1 ATP synthase subunit c is a substrate of the novel YidC pathway for membrane protein biogenesis. J. Cell Biol. 165, 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ballhausen B., Altendorf K., Deckers-Hebestreit G. (2009) Constant c10 ring stoichiometry in the Escherichia coli ATP synthase analyzed by cross-linking. J. Bacteriol. 191, 2400–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Suzuki T., Ozaki Y., Sone N., Feniouk B. A., Yoshida M. (2007) The product of uncI gene in F1FO-ATP synthase operon plays a chaperone-like role to assist c-ring assembly. Proc. Natl. Acad. Sci. U.S.A. 104, 20776–20781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brandt K., Müller D. B., Hoffmann J., Hübert C., Brutschy B., Deckers-Hebestreit G., Müller V. (2013) Functional production of the Na+ F1FO ATP synthase from Acetobacterium woodii in Escherichia coli requires the native AtpI. J. Bioenerg. Biomembr. 45, 15–23 [DOI] [PubMed] [Google Scholar]
- 7. Liu J., Hicks D. B., Krulwich T. A. (2013) Roles of AtpI and two YidC-type proteins from alkaliphilic Bacillus pseudofirmus OF4 in ATP synthase assembly and nonfermentative growth. J. Bacteriol. 195, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kol S., Majczak W., Heerlien R., van der Berg J. P., Nouwen N., Driessen A. J. (2009) Subunit a of the F1FO ATP synthase requires YidC and SecYEG for membrane insertion. J. Mol. Biol. 390, 893–901 [DOI] [PubMed] [Google Scholar]
- 9. Yi L., Jiang F., Chen M., Cain B., Bolhuis A., Dalbey R. E. (2003) YidC is strictly required for membrane insertion of subunits a and c of the F1FO ATP synthase and SecE of the SecYEG translocase. Biochemistry 42, 10537–10544 [DOI] [PubMed] [Google Scholar]
- 10. Yi L., Celebi N., Chen M., Dalbey R. E. (2004) Sec/SRP requirements and energetics of membrane insertion of subunits a, b, and c of the Escherichia coli F1FO ATP synthase. J. Biol. Chem. 279, 39260–39267 [DOI] [PubMed] [Google Scholar]
- 11. Hermolin J., Fillingame R. H. (1995) Assembly of FO sector of Escherichia coli H+ ATP synthase. Interdependence of subunit insertion into the membrane. J. Biol. Chem. 270, 2815–2817 [DOI] [PubMed] [Google Scholar]
- 12. Koebmann B. J., Westerhoff H. V., Snoep J. L., Nilsson D., Jensen P. R. (2002) The glycolytic flux in Escherichia coli is controlled by the demand for ATP. J. Bacteriol. 184, 3909–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Senior A. E., Muharemagić A., Wilke-Mounts S. (2006) Assembly of the stator in Escherichia coli ATP synthase. Complexation of α subunit with other F1 subunits is prerequisite for δ subunit binding to the N-terminal region of α. Biochemistry 45, 15893–15902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pati S., DiSilvestre D., Brusilow W. S. (1992) Regulation of the Escherichia coli uncH gene by mRNA secondary structure and translational coupling. Mol. Microbiol. 6, 3559–3566 [DOI] [PubMed] [Google Scholar]
- 15. McCarthy J. E. (1988) Expression of the unc genes in Escherichia coli. J. Bioenerg. Biomembr. 20, 19–39 [DOI] [PubMed] [Google Scholar]
- 16. Walker J. E., Cozens A. L. (1986) Evolution of ATP synthase. Chem. Scripta 26B, 263–272 [Google Scholar]
- 17. Gomis-Rüth F. X., Moncalián G., Pérez-Luque R., González A., Cabezón E., de la Cruz F., Coll M. (2001) The bacterial conjugation protein TrwB resembles ring helicases and F1-ATPase. Nature 409, 637–641 [DOI] [PubMed] [Google Scholar]
- 18. Mulkidjanian A. Y., Makarova K. S., Galperin M. Y., Koonin E. V. (2007) Inventing the dynamo machine. The evolution of the F-type and V-type ATPases. Nat. Rev. Microbiol. 5, 892–899 [DOI] [PubMed] [Google Scholar]
- 19. Rak M., Gokova S., Tzagoloff A. (2011) Modular assembly of yeast mitochondrial ATP synthase. EMBO J. 30, 920–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klionsky D. J., Brusilow W. S., Simoni R. D. (1984) In vivo evidence for the role of the ϵ subunit as an inhibitor of the proton-translocating ATPase of Escherichia coli. J. Bacteriol. 160, 1055–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moriyama Y., Iwamoto A., Hanada H., Maeda M., Futai M. (1991) One-step purification of Escherichia coli H+-ATPase (FOF1) and its reconstitution into liposomes with neurotransmitter transporters. J. Biol. Chem. 266, 22141–22146 [PubMed] [Google Scholar]
- 22. Krebstakies T., Zimmermann B., Gräber P., Altendorf K., Börsch M., Greie J. (2005) Both rotor and stator subunits are necessary for efficient binding of F1 to FO in functionally assembled Escherichia coli ATP synthase. J. Biol. Chem. 280, 33338–33345 [DOI] [PubMed] [Google Scholar]
- 23. Krebstakies T., Aldag I., Altendorf K., Greie J.-C., Deckers-Hebestreit G. (2008) The stoichiometry of subunit c of Escherichia coli ATP synthase is independent of its rate of synthesis. Biochemistry 47, 6907–6916 [DOI] [PubMed] [Google Scholar]
- 24. Guzman L. M., Belin D., Carson M. J., Beckwith J. (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones P. C., Hermolin J., Jiang W., Fillingame R. H. (2000) Insights into the rotary catalytic mechanism of FOF1 ATP synthase from the cross-linking of subunits b and c in the Escherichia coli enzyme. J. Biol. Chem. 275, 31340–31346 [DOI] [PubMed] [Google Scholar]
- 26. Ishmukhametov R. R., Galkin M. A., Vik S. B. (2005) Ultrafast purification and reconstitution of His-tagged cysteine-less Escherichia coli F1FO ATP synthase. Biochim. Biophys. Acta 1706, 110–116 [DOI] [PubMed] [Google Scholar]
- 27. Gumbiowski K., Cherepanov D., Muller M., Panke O., Promto P., Winkler S., Junge W., Engelbrecht S. (2001) F-ATPase. Forced full rotation of the rotor despite covalent cross-link with the stator. J. Biol. Chem. 276, 42287–42292 [DOI] [PubMed] [Google Scholar]
- 28. Mertens N., Remaut E., Fiers W. (1995) Tight transcriptional control mechanism ensures stable high-level expression from T7 promoter-based expression plasmids. Biotechnology 13, 175–179 [DOI] [PubMed] [Google Scholar]
- 29. Nielsen J., Hansen F. G., Hoppe J., Friedl P., von Meyenburg K. (1981) The nucleotide sequence of the atp genes coding for the FO subunits a, b, c and the F1 subunit δ of the membrane-bound ATP synthase of Escherichia coli. Mol. Gen. Genet. 184, 33–39 [DOI] [PubMed] [Google Scholar]
- 30. Brockmann B., Koop genannt Hoppmann K. D., Strahl H., Deckers-Hebestreit G. (2013) Time-delayed in vivo assembly of subunit a into preformed Escherichia coli FOF1 ATP synthase. J. Bacteriol. 10.1128/JB.00468-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Ed., p. A2.2, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 32. Dunn S. D., Heppel L. A., Fullmer C. S. (1980) The NH2-terminal portion of the α subunit of Escherichia coli F1 ATPase is required for binding the δ subunit. J. Biol. Chem. 255, 6891–6896 [PubMed] [Google Scholar]
- 33. Cagnon C., Valverde V., Masson J.-M. (1991) A new family of sugar-inducible expression vectors for Escherichia coli. Protein Eng. 4, 843–847 [DOI] [PubMed] [Google Scholar]
- 34. Neidhardt F. C., Ingraham J. L., Schaechter M. (1990) Physiology of the Bacterial Cell. A Molecular Approach, pp. 1–29, Sinauer Associates, Sunderland, MA [Google Scholar]
- 35. Strahl H., Greie J.-C. (2008) The extremely halophilic archaeon Halobacterium salinarum R1 responds to potassium limitation by expression of the K+-transporting KdpFABC P-type ATPase and by a decrease in intracellular K+. Extremophiles 12, 741–752 [DOI] [PubMed] [Google Scholar]
- 36. Douglas M., Finkelstein D., Butow R. A. (1979) Analysis of products of mitochondrial protein synthesis in yeast. Genetic and biochemical aspects. Methods Enzymol. 56, 58–66 [DOI] [PubMed] [Google Scholar]
- 37. Wise J. G. (1990) Site-directed mutagenesis of the conserved β subunit tyrosine 331 of Escherichia coli ATP synthase yields catalytically active enzymes. J. Biol. Chem. 265, 10403–10409 [PubMed] [Google Scholar]
- 38. Pänke O., Gumbiowski K., Junge W., Engelbrecht S. (2000) F-ATPase. Specific observation of the rotating c subunit oligomer of EFOEF1. FEBS Lett. 472, 34–38 [DOI] [PubMed] [Google Scholar]
- 39. Schägger H., von Jagow G. (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 40. Birkenhäger R., Greie J.-C., Altendorf K., Deckers-Hebestreit G. (1999) FO complex of the Escherichia coli ATP synthase. Not all monomers of the subunit c oligomer are involved in F1 interaction. Eur. J. Biochem. 264, 385–396 [DOI] [PubMed] [Google Scholar]
- 41. Stalz W.-D., Greie J.-C., Deckers-Hebestreit G., Altendorf K. (2003) Direct interaction of subunits a and b of the FO complex of Escherichia coli ATP synthase by forming an ab2 subcomplex. J. Biol. Chem. 278, 27068–27071 [DOI] [PubMed] [Google Scholar]
- 42. Dmitriev O., Jones P. C., Jiang W., Fillingame R. H. (1999) Structure of the membrane domain of subunit b of the Escherichia coli FOF1 ATP synthase. J. Biol. Chem. 274, 15598–15604 [DOI] [PubMed] [Google Scholar]
- 43. Greie J.-C., Deckers-Hebestreit G., Altendorf K. (2000) Subunit organization of the stator part of the FO complex from Escherichia coli ATP synthase. J. Bioenerg. Biomembr. 32, 357–364 [DOI] [PubMed] [Google Scholar]
- 44. McLachlin D. T., Dunn S. D. (1997) Dimerization interactions of the b subunit of the Escherichia coli F1FO-ATPase. J. Biol. Chem. 272, 21233–21239 [DOI] [PubMed] [Google Scholar]
- 45. Eya S., Maeda M., Futai M. (1991) Role of the carboxyl terminal region of H+-ATPase (FOF1) a subunit from Escherichia coli. Arch. Biochem. Biophys. 284, 71–77 [DOI] [PubMed] [Google Scholar]
- 46. Pierson H. E., Uhlemann E.-M., Dmitriev O. Y. (2011) Interaction with monomeric subunit c drives insertion of ATP synthase subunit a into the membrane and primes a-c complex formation. J. Biol. Chem. 286, 38583–38591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jiang W., Hermolin J., Fillingame R. H. (2001) The preferred stoichiometry of c subunits in the rotary motor sector of Escherichia coli ATP synthse is 10. Proc. Natl. Acad. Sci. U.S.A. 98, 4966–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brandt K., Maiwald S., Herkenhoff-Hesselmann B., Gnirss K., Greie J.-C., Dunn S. D., Deckers-Hebestreit G. (July 11, 2013) Individual interactions of the b subunits within the stator of the Escherichia coli ATP synthase. J. Biol. Chem. 10.1074/jbc.M113.465633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Deckers-Hebestreit G., Simoni R. D., Altendorf K. (1992) Influence of subunit-specific antibodies on the activity of the FO complex of the ATP synthase of Escherichia coli. I. Effects of subunit b-specific polyclonal antibodies. J. Biol. Chem. 267, 12364–12369 [PubMed] [Google Scholar]
- 50. Solomon K. A., Brusilow W. S. (1988) Effect of an uncE ribosome-binding site mutation on the synthesis and assembly of the Escherichia coli proton-translocating ATPase. J. Biol. Chem. 263, 5402–5407 [PubMed] [Google Scholar]
- 51. Schneider E., Altendorf K. (1984) Subunit b of the membrane moiety (FO) of ATP synthase (F1FO) from Escherichia coli is indispensable for H+ translocation and binding of the water-soluble F1 moiety. Proc. Natl. Acad. Sci. U.S.A. 81, 7279–7283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schneider E., Altendorf K. (1985) All three subunits are required for the reconstitution of an active proton channel (FO) of Escherichia coli ATP synthase (F1FO). EMBO J. 4, 515–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pati S., Brusilow W. S. (1989) The roles of the α and γ subunits in proton conduction through the FO sector of the proton-translocating ATPase of Escherichia coli. J. Biol. Chem. 264, 2640–2644 [PubMed] [Google Scholar]
- 54. Pati S., Brusilow W. S., Deckers-Hebestreit G., Altendorf K. (1991) Assembly of the FO proton channel of the Escherichia coli F1FO ATPase. Low proton conductance of reconstituted FO sectors synthesized and assembled in the absence of F1. Biochemistry 30, 4710–4714 [DOI] [PubMed] [Google Scholar]
- 55. Fillingame R. H., Porter B., Hermolin J., White L. K. (1986) Synthesis of the Escherichia coli H+-ATPase does not require synthesis of the α and β subunits in F1. J. Bacteriol. 165, 244–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Monticello R. A., Brusilow W. S. (1994) Role of the δ subunit in enhancing proton conduction through the FO of the Escherichia coli F1FO ATPase. J. Bacteriol. 176, 1383–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lau W. C., Rubinstein J. L. (2012) Subnanometre-resolution structure of the intact Thermus thermophilus H+-driven ATP synthase. Nature 481, 214–218 [DOI] [PubMed] [Google Scholar]
- 58. Kucharczyk R., Zick M., Bietenhader M., Rak M., Couplan E., Blondel M., Caubet S.-D., di Rago J.-P. (2009) Mitochondrial ATP synthase disorders. Molecular mechanisms and the quest for curative therapeutic approaches. Biochim. Biophys. Acta 1793, 186–199 [DOI] [PubMed] [Google Scholar]
- 59. Price C. E., Driessen A. J. (2010) Biogenesis of membrane bound respiratory complexes in Escherichia coli. Biochim. Biophys. Acta 1803, 748–766 [DOI] [PubMed] [Google Scholar]
- 60. Dalbey R. E., Wang P., Kuhn A. (2011) Assembly of bacterial inner membrane proteins. Annu. Rev. Biochem. 80, 161–187 [DOI] [PubMed] [Google Scholar]
- 61. Rak M., Zeng X., Brière J. J., Tzagoloff A. (2009) Assembly of FO in Saccharomyces cerevisiae. Biochim. Biophys. Acta 1793, 108–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dunn S. D., Futai M. (1980) Reconstitution of a functional coupling factor from the isolated subunits of Escherichia coli F1 ATPase. J. Biol. Chem. 255, 113–118 [PubMed] [Google Scholar]
- 63. Sternweis P. C. (1978) The ϵ subunit of Escherichia coli coupling factor 1 is required for its binding to the cytoplasmic membrane. J. Biol. Chem. 253, 3123–3128 [PubMed] [Google Scholar]
- 64. Schatz G. (1968) Impaired binding of mitochondrial adenosine triphosphatase in the cytoplasmic “petite” mutant of Saccharomyces cerevisiae. J. Biol. Chem. 243, 2192–2199 [PubMed] [Google Scholar]
- 65. Tzagoloff A. (1969) Assembly of the mitochondrial membrane system. II. Synthesis of the mitochondrial adenosine triphosphatase, F1. J. Biol. Chem. 244, 5027–5033 [PubMed] [Google Scholar]
- 66. al-Shawi M. K., Parsonage D., Senior A. E. (1990) Adenosine triphosphatase and nucleotide binding activity of isolated β-subunit preparations from Escherichia coli F1FO-ATP synthase. J. Biol. Chem. 265, 5595–5601 [PubMed] [Google Scholar]
- 67. Feniouk B. A., Suzuki T., Yoshida M. (2006) The role of subunit epsilon in the catalysis and regulation of FOF1-ATP synthase. Biochim. Biophys. Acta 1757, 326–338 [DOI] [PubMed] [Google Scholar]
- 68. Ono S., Sone N., Yoshida M., Suzuki T. (2004) ATP synthase that lacks FOa-subunit. Isolation, properties, and indication of FOb2-subunits as an anchor rail of a rotating c-ring. J. Biol. Chem. 279, 33409–33412 [DOI] [PubMed] [Google Scholar]
- 69. Kuruma Y., Suzuki T., Ono S., Yoshida M., Ueda T. (2012) Functional analysis of membraneous FO-a subunit of F1FO-ATP synthase by in vitro protein synthesis. Biochem. J. 442, 631–638 [DOI] [PubMed] [Google Scholar]
- 70. Wittig I., Meyer B., Heide H., Steger M., Bleier L., Wumaier Z., Karas M., Schägger H. (2010) Assembly and oligomerization of human ATP synthase lacking mitochondrial subunits a and A6L. Biochim. Biophys. Acta 1797, 1004–1011 [DOI] [PubMed] [Google Scholar]
- 71. Brusilow W. S. (1993) Assembly of the Escherichia coli F1FO ATPase, a large multimeric membrane-bound enzyme. Mol. Microbiol. 9, 419–424 [DOI] [PubMed] [Google Scholar]
- 72. Dunn S. D., Chandler J. (1998) Characterization of a b2δ complex from Escherichia coli ATP synthase. J. Biol. Chem. 273, 8646–8651 [DOI] [PubMed] [Google Scholar]
- 73. Wilkens S., Dunn S. D., Chandler J., Dahlquist F. W., Capaldi R. A. (1997) Solution structure of the N-terminal domain of the δ subunit of the E. coli ATP synthase. Nat. Struct. Biol. 4, 198–201 [DOI] [PubMed] [Google Scholar]
- 74. McLachlin D. T., Bestard J. A., Dunn S. D. (1998) The b and δ subunits of the Escherichia coli ATP synthase interact via residues in their C-terminal regions. J. Biol. Chem. 273, 15162–15168 [DOI] [PubMed] [Google Scholar]