

Background: NAMPT inhibitors showed antitumor activity in preclinical cancer models, but no tumor remission occurred in clinical studies.
Results: Cells treated with a NAMPT inhibitor are rescued by low NAD+e or NAD+ precursors, depending on CD38 and CD73 expression.
Conclusion: CD73 enables, whereas CD38 impairs, extracellular NMN utilization by cells for NAD+ biosynthesis.
Significance: Combining CD73 and NAMPT inhibition could represent a new anti-cancer strategy.
Keywords: Cancer Therapy, Cell Death, NAD, NAD Biosynthesis, Nicotinamide, CD73, Nicotinamide Mononucleotide, Nicotinamide Riboside
Abstract
NAD+ is mainly synthesized in human cells via the “salvage” pathways starting from nicotinamide, nicotinic acid, or nicotinamide riboside (NR). The inhibition with FK866 of the enzyme nicotinamide phosphoribosyltransferase (NAMPT), catalyzing the first reaction in the “salvage” pathway from nicotinamide, showed potent antitumor activity in several preclinical models of solid and hematologic cancers. In the clinical studies performed with FK866, however, no tumor remission was observed. Here we demonstrate that low micromolar concentrations of extracellular NAD+ or NAD+ precursors, nicotinamide mononucleotide (NMN) and NR, can reverse the FK866-induced cell death, this representing a plausible explanation for the failure of NAMPT inhibition as an anti-cancer therapy. NMN is a substrate of both ectoenzymes CD38 and CD73, with generation of NAM and NR, respectively. In this study, we investigated the roles of CD38 and CD73 in providing ectocellular NAD+ precursors for NAD+ biosynthesis and in modulating cell susceptibility to FK866. By specifically silencing or overexpressing CD38 and CD73, we demonstrated that endogenous CD73 enables, whereas CD38 impairs, the conversion of extracellular NMN to NR as a precursor for intracellular NAD+ biosynthesis in human cells. Moreover, cell viability in FK866-treated cells supplemented with extracellular NMN was strongly reduced in tumor cells, upon pharmacological inhibition or specific down-regulation of CD73. Thus, our study suggests that genetic or pharmacologic interventions interfering with CD73 activity may prove useful to increase cancer cell sensitivity to NAMPT inhibitors.
Introduction
Nicotinamide adenine dinucleotide (NAD+), besides being the prevalent coenzyme of oxidoreductase reactions, is involved in several signaling processes, as a donor of ADP-ribose (ADPR)2 moieties in mono- and poly-ADP-ribosylation reactions, as a precursor of Ca2+-mobilizing second messengers (1, 2), and as a mediator of sirtuin-catalyzed protein deacetylation reactions (3–5). NAD+ is synthesized in human cells either via the “de novo” pathway, starting from tryptophan, or through any of the “salvage” pathways, starting from nicotinamide (NAM), nicotinic acid, or nicotinamide riboside (NR) (6, 7).
The NAM salvage pathway involves the enzyme NAM phosphoribosyltransferase (NAMPT), which catalyzes the synthesis of nicotinamide mononucleotide (NMN) and inorganic pyrophosphate using NAM and 5-phosphoribosyl-1-pyrophosphate as substrates. In the next step, NMN is converted to NAD+ by the enzyme NMN-adenylyltransferase with ATP consumption (8–10).
The key contribution of NAMPT-mediated NAD+ biosynthesis in many cell types was unveiled by the use of FK866, a NAMPT inhibitor, which severely decreases NAD+ levels (11). A prominent role for NAMPT in different physiological processes and diseases has been described since, and a number of therapeutic options for NAMPT inhibitors have been proposed in cancer, inflammation, and cardiovascular disease (11–13).
More specifically, the NAMPT inhibitors FK866 and CHS828 (or its pro-drug GMX1777), as well as other NAMPT inhibitors that were subsequently identified, showed potent antitumor activity in several preclinical models of solid and hematologic cancers (11, 14–20) because NAMPT inhibition leads to ATP shortage and to consequent cell death (11, 12). Moreover, NAMPT inhibition was found to increase the efficacy of standard chemotherapeutics: histone deacetylase inhibitors, tumor necrosis factor-related apoptosis-inducing ligand, and poly(ADP-ribose) polymerase inhibitors (21–24).
Clinical studies of CHS828 and FK866 have been performed on more than 100 patients with advanced solid tumors (25, 26). The main forms of reported toxicity for these agents were thrombocytopenia, gastrointestinal symptoms (especially for CHS828), and lymphopenia (for FK866). Regrettably, no objective tumor remission was observed, although a few cases of disease stabilization could be recorded with FK866 (25), which led to the conclusion that FK866 and CHS828 show limited antitumor activity as single agents and that their benefit should be sought in combination treatments.
A plausible explanation for the failure of NAMPT inhibition as an anti-cancer therapy could be the fact that extracellular NAD+ or extracellular NAD+ precursors, including the vitamin B3 forms nicotinic acid, NMN, and NR, can prevent FK866-induced cell death by overcoming blockade of NAD+ biosynthesis from NAM (12, 27, 28). Indeed, NAD+ has been detected in mammal plasma and fluids (29–31), whereas NR has been found in cow milk (32). NMN has also been detected in plasma (33, 34), although it remains controversial whether extracellular NMN is synthesized extracellularly by NAMPT (35). How extracellular NMN could support intracellular NAD+ synthesis is also not clear because a transport mechanism for NMN on the plasma membrane of intact cells has not been demonstrated so far, although it has been postulated in certain cell types and tissues (33, 36, 37). The ectoenzyme CD38 degrades NMN and generates NAM (38), which can cross the plasma membrane (39) and fuel NAD+ synthesis via NAMPT activity. Recently, the ectoenzyme CD73, a 5′-nucleotidase mainly active on adenosine monophosphate (AMP), has been demonstrated by experiments with the purified recombinant protein to be able to dephosphorylate NMN to produce NR (40). The latter, in turn, can cross the plasma membrane through dipyridamole-sensitive nucleoside transporters (27) and be phosphorylated intracellularly to NMN by specific nicotinamide riboside kinases (3, 27, 32), thus contributing to NAD+ biosynthesis.
The aim of this study was to better understand the role of NAD+-degrading ectoenzymes, such as CD38 and CD73, in providing ectocellular NAD+ precursors for NAD+ biosynthesis and in modulating cell susceptibility to FK866. We demonstrate that endogenous CD73 enables, whereas CD38 impairs, the utilization of extracellular NMN as a precursor for intracellular NAD+ biosynthesis in human cells by converting NMN to NR. Our results highlight the possibility of implementing antitumor therapies based on combined inhibition of NAMPT and CD73.
EXPERIMENTAL PROCEDURES
Drugs
All chemicals were purchased from Sigma-Aldrich unless otherwise stated. FK866 was generously provided by the NIMH, National Institutes of Health, Chemical Synthesis and Drug Supply Program. In this study, FK866 was used at a final concentration of 30 nm. This is the minimum concentration resulting, after 72-h incubation, in >80% cell death, a necessary condition to evaluate the rescue effects from the FK866-induced cell death in different experimental conditions. The mimetic peptide Gap26 (VCYDKSFPISHVR; 0.25 mg/ml) and scrambled Gap26 (PSFDSRHCIVKYV; 0.25 mg/ml) were synthesized as described (41).
Synthesis of NR
To prepare NR, 25 μmol of NAD+ was treated with 5 units of Crotalus adamanteus venom nucleotide pyrophosphatase (Sigma-Aldrich) and 40 units of calf intestinal alkaline phosphatase (Sigma-Aldrich) for 20 h at 37 °C, in 100 mm Tris/HCl, pH 8.0, containing 100 mm MgCl2 (final volume, 0.3 ml). The reaction was stopped with 150 μl of 1.2 m HClO4, and after 15 min on ice, the sample was centrifuged for 5 min at 12,000 × g. Aliquots (400 μl) of the supernatant were neutralized with 80 μl of 1 m K2CO3, kept on ice for 10 min, and centrifuged as described above. Fifty-μl aliquots of the supernatants were loaded onto a Phenomenex C18 Kinetex column (2.6 μm, 4.6 × 150 mm). Elution conditions were as follows: 2 min at 100% buffer A (5 mm ammonium formate, pH 3.3), 7 min up to 100% buffer B (5 mm ammonium formate, pH 3.3, 25% acetonitrile), holding at 100% buffer B for 1 min, returning to 100% buffer A in 1 min, and holding at 100% buffer A for 6 min. Flow rate was maintained at 0.75 ml/min, and temperature was fixed at 25 °C. Eluates containing NR (retention time, 4 min) were pooled, lyophilized, and stored at −20 °C. Purity was verified by HPLC analysis of 5 nmol of NR, after resuspension of the lyophilized sample in water and spectrophotometric quantitation (A259 = 4305 m−1 cm−1).
Cell Culture
U87 (human glioblastoma), A549 (human lung adenocarcinoma), and PC3 (human prostate cancer) cells, obtained from the American Type Culture Collection (ATCC, Manassas, VA), were grown in RPMI 1640 supplemented with 10% fetal calf serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) at 37 °C in a humidified atmosphere with 5% CO2.
Assays of Ectocellular NAD+- or AMP-degrading Enzyme Activities
Ectocellular enzymatic activities of CD73 and CD38 were assayed by incubating 1 × 106 U87 and A549 intact cells in 0.6 ml of Hanks' buffered saline solution in the presence of the substrates AMP or NAD+ (0.4 mm). At various times (0, 1, 5, 30, and 120 min), 100-μl aliquots of the incubations were withdrawn and briefly centrifuged, and 2.5% trichloroacetic acid (TCA) was added to 90-μl supernatants. Samples were centrifuged, and the excess TCA was removed with diethylether. The amounts of nucleotides produced were determined by the phosphate HPLC analysis described previously (40).
Assay of Enzymatic Activities of Soluble Human Recombinant CD38
Human recombinant CD38 (0.1 μg) was incubated at 37 °C in the presence of 0.25 mm NAD+, NMN, or NR in a total volume of 0.5 ml of 10 mm Tris-HCl, pH 6.5. At various times (0, 0.5, 3, and 15 min), 100-μl aliquots were withdrawn, and enzymatic reactions were stopped by the addition of 5 μl of 50% (v/v) TCA. Controls without the enzyme were always processed in parallel to correct for non-enzymatic NAM generation. The products ADPR and NAM were analyzed and quantified by analytical phosphate HPLC (40).
Degradation of Extracellular NAD+
U87 and A549 cells were seeded in 96-well plates (5 × 105 cells/well). After 24 h, culture medium was removed, and fresh medium, containing NAD+ at different concentrations (10, 3, or 1 μm), was added in different wells. Medium was collected in triplicate at different times (0, 10, 60, and 180 min), and perchloric acid (PCA) was added (0.6 m, final concentration). The NAD+ concentration was evaluated with an enzymatic cycling assay (42).
Expression Vectors for Human CD73 and CD38
Construction of expression vector for human CD73 (40) and for human CD38 (43) was performed as described, followed by cloning in pcDNA3.1.
U87 cells were transfected in parallel with pcDNA3.1/V5-HisTOPO® or pcDNA3.1 (empty, control plasmid), with CD73-pcDNA3.1/V5-His TOPO® (CD73 plasmid), or with CD38-pcDNA3.1 (CD38 plasmid), respectively, using the Nucleofector system (Amaxa, Koeln, Germany), with the Cell Line Nucleofector Kit T, program X-001, following the manufacturer's instructions. Thereafter, 2 × 105 cells were seeded onto 35 × 10-mm tissue culture dishes for mRNA quantification, 1 × 106 cells were seeded onto 60 × 15-mm tissue culture dishes for Western blot analysis, and 2 × 105 cells/well were seeded in 12-well plates for the determination of intracellular NAD+ levels. All experiments were performed 24 h after transfection.
CD73 and CD38 siRNA Gene Silencing
A549 cells were transfected with StealthTM RNAi (Invitrogen) targeting human CD38 (Oligo ID HSS107326) and CD73 (Oligo ID HSS101578). Cells were electroporated with 2 μm duplex siRNA with the Nucleofector system, using the Cell Line Nucleofector Kit T, program X-001, following the manufacturer's instructions. As a control, A549 cells were transfected with StealthTM RNAi negative control duplex (Invitrogen). Cells were seeded as follows: 2 × 105 cells onto 35 × 10-mm tissue culture dishes for mRNA quantification; 1 × 106 cells onto 60 × 15-mm tissue culture dishes for Western blot analysis; 2 × 105 cells/well in 12-well plates for the determination of intracellular NAD+ levels; 5 × 103 cells/well in 96-well plates for viability assays. All experiments were carried out 24 h after transfection.
qPCR Analyses
Twenty-four h after transfection, total RNA was extracted from U87 and A549 cells using the RNeasy Micro Plus kit (Qiagen, Milan, Italy); total RNA extracted from cells transfected with CD38 or CD73 plasmid was further treated with the RNase-free DNase set (Qiagen) according to the manufacturer's instructions in order to rule out any possible contamination by plasmid. Quality and quantity of RNA were analyzed using a NanoDrop spectrophotometer (Nanodrop Technologies, Wilmington, DE). cDNA (0.5 μg) was synthesized by using the iScript cDNA synthesis kit (Bio-Rad). PCR primers were designed by using Beacon Designer version 2.0 software (Bio-Rad), and their sequences were as follows: human CD73, 5′-GCTCGGCTCTTCACCAAG-3′ (forward) and 5′-TCAGTCCTTCCACACCATTAT-3′ (reverse); human CD38, 5′-GGACACGCTGCTAGGCTACC-3′ (forward) and 5′-CATCACATGGACCACATCACAGG-3′ (reverse); human GAPDH, 5′-CCTGTTCGACAGTCAGCCG-3′ (forward) and 5′-CGACCAAATCCGTTGACTCC-3′ (reverse); human HPRT1, 5′-GGTCAGGCAGTATAATCCAAAG-3′ (forward) and 5′-TTCATTATAGTCAAGGGCATATCC-3′ (reverse).
Statistical analyses of the qPCR were obtained using iQ5 Optical System software version 2.0 (Bio-Rad) based on the 2−ΔΔCt method, which calculated relative changes in gene expression of the target normalized to GAPDH and HPRT1. Experiments were repeated three times in triplicate.
Western Blot Analyses
Twenty-four h after transfection, cells were washed with cold PBS, collected, and centrifuged at 700 × g for 10 min. Cell pellets were lysed in cold lysis buffer (50 mm Tris-HCl, 150 mm NaCl, and 1% Nonidet P-40, pH 7.4), containing protease and phosphatase inhibitor mixtures. Total protein concentrations were determined by the Bradford method (Bio-Rad). Identical amounts of lysate proteins (20 μg/sample) were resuspended in SDS sample buffer containing 10% β-mercaptoethanol, loaded onto SDS 10% polyacrylamide gels, and then electrophoretically separated and transferred to Immun-Blot PVDF membranes (Bio-Rad). Membranes were blocked with 5% nonfat dry milk in PBS for 1 h at room temperature and visualized with the following antibodies: anti-CD73 (Sc130006, Santa Cruz Biotechnology, Inc., Dallas TX), anti-V5 epitope (Invitrogen), anti-CD38 (C-1586, Sigma-Aldrich), anti-γ-tubulin (Cell Signaling Technology, Danvers, MA), and anti-β-actin (Santa Cruz Biotechnology). Secondary Abs were horseradish peroxidase-conjugated (GE Healthcare). Western blots were developed with the ECL-PLUS kit (GE Healthcare), according to the manufacturer's instructions. Band detection and densitometry were performed using the Chemi-Doc System and the Quantity One software package (Bio-Rad).
Determination of Intracellular NAD+ Levels
U87, A549, and PC3 cells were plated at a density of 2 × 105 cells/well in 12-well plates and cultured in 500 μl of complete RPMI 1640 in the presence or absence of 30 nm FK866 and, depending on the experimental setting, supplemented twice a day (at 9 a.m. and at 6 p.m.) for 3 days, with or without 10 μm NAD+, NMN, or NR. Then cells were harvested and lysed in 0.1 ml of 0.6 m PCA at 4 °C. Cell extracts were centrifuged for 3 min at 16,000 × g, the supernatants were collected, and an aliquot was diluted 200-fold in 100 mm sodium phosphate buffer, pH 8.0, for determination of NAD+ content, as described (42). NAD+ values were normalized to protein concentrations, determined by Bradford assay.
Cell Viability Assays
To determine the EC50 values of NAD+ in rescuing FK866-induced cell death, U87, A549, and PC3 cells (5 × 105/well in 96-well plates) were incubated with 30 nm FK866 and supplemented (or not) twice a day (at 9 a.m. and at 6 p.m.) for 3 days with different concentrations of NAD+ (ranging from 0 to 20 μm). In other experimental settings, 5 × 105 U87 or A549 cells/well were plated in 96-well plates in the presence or absence of 30 nm FK866, with or without 1 mm octanol, 10 μm Gap26, 10 μm scrambled Gap26, or 1 μm adenosine 5′-(α,β-methylene)diphosphate (APCP) and supplemented twice a day (at 9 a.m. and at 6 p.m.) for 3 days, with or without 10 μm NAD+, NMN, NR, NAM, AMP, ADPR, or adenosine or their combinations. Cell viability was measured as described (44).
Statistical Analyses
All parameters were tested by paired t test or one-way analysis of variance followed by Tukey test; p values of <0.05 were considered significant. In the figure legends, only relevant comparisons are shown.
RESULTS
Low Micromolar Concentrations of Extracellular NAD+ Rescue FK866-incubated Cells from Death
The addition of extracellular NAD+ or of the NAD+ precursor NMN or NR prevents FK866-induced cell death (23, 27, 28). In order to evaluate the lowest extracellular concentration of NAD+ sufficient to protect from FK866-induced cell death, three different human tumor cell lines, U87 (glioblastoma), PC3 (prostate cancer), and A549 (lung adenocarcinoma), were incubated for 72 h in the presence or absence of 30 nm FK866, and extracellular NAD+ (NAD+e) was added twice a day at final concentrations ranging between 0.15 and 20 μm. NAD+ was already effective at opposing FK866 anticancer activity at concentrations in the low micromolar range (Fig. 1A), although different degrees of protection were observed on the three cell lines: the EC50 values for the NAD+-rescuing activity were 0.25, 0.87, and 12.48 μm for U87, PC3, and A549 cells, respectively.
FIGURE 1.

Extracellular micromolar NAD+ improves cell viability and increases intracellular NAD+ content in FK866-treated cells. A, A549, PC3, and U87 cell viability was evaluated by a sulforhodamine B colorimetric assay (SBC) after a 72-h incubation of cells in complete medium in the presence (or not) of 30 nm FK866, with the addition (twice a day) of 0, 0.15, 0.30, 0.6, 1.2, 2.5, 5, 10, or 20 μm NAD+. Results are expressed as a percentage of cell growth relative to untreated cells. In the absence of NAD+, FK866 treatment resulted in 8.6 ± 0.4, 11.9 ± 0.4, and 11.4 ± 1.4% cell viability in PC3, A549, and U87 cells, respectively. EC50 values for the NAD+-rescuing activity were calculated from the non-linear regression curves with GraphPad Prism version 5. R2 values were 0.981 for PC3, 0.993 for A549, and 0.966 for U87 cells. B, A549, PC3, and U87 cells were treated for 72 h with or without 30 nm FK866 in the presence or absence of 10 μm NAD+ (added twice a day). Cells were harvested and lysed in 0.6 m PCA, and NAD+ content was measured in neutralized extracts. NAD+ values, normalized to protein content, are expressed as relative to untreated, control cells. Data are expressed as mean ± S.D. (error bars) (n = 3). The intracellular NAD+ content was 6.2 ± 1.1, 7.1 ± 0.9, and 11.6 ± 1.9 nmol/mg protein in A549, PC3, and U87 cells, respectively. *, p < 0.05; #, p < 0.01; §, p < 0.001 compared with the corresponding FK866-treated cells. C, A549, PC3, and U87 cell viability was evaluated by SBC after a 72-h incubation of the cells in complete medium without (control) or with 30 nm FK866 or with FK866 supplemented with either 1 mm octanol or 10 μm NAD+ (added twice a day). Results are expressed as a percentage of cell growth relative to untreated cells. Data are expressed as mean ± S.D. (n = 3). No statistical difference was observed between cells treated with FK866 and NAD+ in the presence or absence of octanol. D, A549, PC3, and U87 cell viability was evaluated by SBC after a 72-h incubation of the cells in complete medium with or without 30 nm FK866, 10 μm Gap26, or a scrambled peptide (scr) and 10 μm NAD+ (added twice a day). Results are expressed as a percentage of cell growth relative to untreated cells. Data are expressed as mean ± S.D. (n = 3). No statistical difference was observed between cells treated with FK866 and NAD+ in the presence or absence of scrambled peptide or Gap26.
Intracellular NAD+ Levels ([NAD+]i) increased significantly in cells incubated in the presence of NAD+e (Fig. 1B), in agreement with their reduced mortality; in U87 cells incubated with FK866 and 10 μm NAD+e, the [NAD+]i was similar to that measured in control cells (cultured without FK866). In PC3 and A549 cells, treatment with 10 μm NAD+e also significantly increased the [NAD+]i, although below control values, in agreement with the higher EC50 of NAD+e on viability in these cell lines (Fig. 1A).
Extracellular NAD+ can cross the plasma membrane of intact cells through Cx43 hemichannels (45). In order to evaluate whether NAD+-mediated rescue from FK866-induced mortality was attributable to NAD+ influx, cells were incubated with extracellular NAD+ in the absence or presence of octanol (a known gap junction inhibitor and disruptor of permeability (45–46)); of a Cx43-specific mimetic peptide (Gap26), which specifically impairs hemichannels opening (47); or of a scrambled peptide used as negative control. As shown in Fig. 1, C and D, inhibition of Cx43 permeability did not significantly impair the NAD+-related cell growth-rescuing activity, indicating that NAD+e influx into cells is not required for the reversal of FK866-induced cell death.
Degradation of Extracellular NAD+
Extracellular NAD+, added in the same experimental conditions used to evaluate viability and at concentrations able to rescue cells from the FK866-induced death, was rapidly degraded by both A549 and U87 cells (Fig. 2A). In an attempt to identify the main products generated from NAD+ by intact A549 and U87 cells, we incubated the cells in the presence of 0.4 mm NAD+, and the supernatants were analyzed by HPLC. A549 cells converted NAD+ to ADPR at a rate of 2.11 nmol/min/mg protein, whereas AMP production was not detectable. On the contrary, U87 cells mainly converted NAD+ to AMP (0.22 nmol/min/mg protein) and, to a lesser extent, to ADPR (0.07 nmol/min/mg protein) (Table 1). In mammalian cells, NAD+ conversion to ADPR and NAM is catalyzed by the multifunctional ectoenzyme CD38, which has a high NAD+-glycohydrolase activity (48). qPCR analyses showed that mRNA for CD38 was barely detectable in U87 cells, whereas it was present in A549 cells, in line with the higher NAD+-glycohydrolase activity of these cells. NAD+ conversion to AMP is due to ectocellular nucleotide pyrophosphatases, catalyzing the generation of NMN and AMP (49). Recently, human recombinant CD73, a 5′-nucleotidase active on AMP (50), was reported to degrade NAD+ to NMN and AMP and also to dephosphorylate NMN to generate NR (40). In order to evaluate CD73 ectoenzymatic activities, AMP was added to intact cells; production of adenosine was higher in A549 than in U87 cells (Table 1). qPCR analysis demonstrated 60% higher expression of the CD73 mRNA in A549 than in U87 cells. Western blot analysis of cell lysates with an anti-CD73 antibody revealed a 6 times more intense band in A549 than in U87 cells (not shown).
FIGURE 2.

NMN and NR prevent FK866-induced cell death. A, U87 and A549 cells were seeded in 96-well plates (5 × 103 cells/well) and incubated in complete medium with NAD+ at different concentrations (10, 3, or 1 μm). At the times indicated, medium was collected in triplicate, and PCA was added (0.6 m final concentration). The extracellular NAD+ concentration at different times was evaluated with an enzymatic cycling assay. Results are the mean of three different experiments (S.D. is not shown for sake of clarity). U87 cells (B) or A549 cells (C) were seeded in 96-well plates (5 × 103 cells/well), and cell viability was evaluated by SBC after a 72-h incubation with or without 30 nm FK866 and with or without a 10 μm concentration of NAD+, AMP, NMN, AMP and NMN, ADPR, NAM, ADPR and NAM, adenosine, NR, or adenosine and NR. Results are expressed as a percentage of cell growth compared with untreated cells. Data are expressed as mean ± S.D. (error bars) (n = 3). §, p < 0.001; °, p < 0.001 compared with FK866-treated cells, in the absence of other supplementations; †, p < 0.001 compared with FK866 + NAD+-treated cells. D, A549 and U87 cells (2 × 105 cells/well in 12-well plates) were treated for 72 h with or without 30 nm FK866 in the presence or absence of 10 μm NMN or NR (added twice a day). Cells were harvested and lysed in 0.6 m PCA, and NAD+ content was measured in neutralized extracts. NAD+ values were normalized on protein content. Data are expressed as the mean ± S.D. (n = 4 and n = 3 for A549 and U87 cells, respectively). #, p < 0.01; °, p < 0.001 compared with the corresponding control cells; §, p < 0.0001; †, p < 0.001 compared with the corresponding FK866-treated cells. Inset, intracellular NAD+ content in A549 and U87 treated with 30 nm FK866 and 10 μm NMN or NR.
TABLE 1.
Main products of ectoenzyme activities in A549 and U87 cells incubated with NAD+ or AMP
Intact U87 and A549 cells were incubated with 0.4 mm NAD+ or AMP. The generation of the indicated products was evaluated by HPLC analyses. ND, not detectable.
| ADPR (NAD+ substrate) | AMP (NAD+ substrate) | Adenosine (AMP substrate) | |
|---|---|---|---|
| nmol/min/mg protein | nmol/min/mg protein | nmol/min/mg protein | |
| A549 | 2.11 | ND | 5.76 |
| U87 | 0.07 | 0.22 | 1.51 |
The facts that supplementation with NAD+e rescues FK866-treated cells from death and that ectocellular enzymes on these cells rapidly degrade NAD+e suggested a possible role for these degradation products in NAD+e-rescuing ability. To identify the NAD+ metabolites able to reverse FK866-induced toxicity, U87 (Fig. 2B) and A549 (Fig. 2C) cells were incubated in the presence of AMP, NMN, or both; with ADPR, NAM, or both; or with NR, adenosine, or both. All of these nucleotides or their paired combinations were added twice a day at a 10 μm final concentration, as in the experiments with NAD+ supplementation. As shown in Fig. 2, B and C, none of these combinations affected cell viability in the absence of FK866 (white bars). As expected, the addition of NAM at micromolar concentration did not have any preventative effect on FK866-induced cell death (Fig. 2, B and C). Likewise, adenine-containing AMP, ADPR or adenosine failed to prevent the FK866-induced cell death. On the contrary, both NMN and NR rescued U87 cells almost completely, and the concomitant presence of AMP or adenosine did not enhance their effect (Fig. 2, B and C). Rescue of A549 cells from FK866-induced death was also observed in the presence of extracellular NMN or NR, although to a lower extent, with NR being more effective (Fig. 2C). Results obtained on cell viability in the presence of NMN or NR were in good agreement with the different intracellular NAD+ content of U87 and A549 cells treated with FK866 in the presence of either of the two NAD+ precursors; indeed, extracellular NMN or NR supplementation restored the intracellular NAD+ content in U87, whereas the [NAD+]i was just slightly, although still significantly, enhanced by the two NAD+ precursors in A549 (Fig. 2D). In agreement with the higher cell viability, the [NAD+]i was increased to a greater extent in the presence of extracellular NR than of NMN (Fig. 2D).
Role of CD38 and CD73 in the Generation of Ectocellular Precursors for Intracellular NAD+ Synthesis
We reasoned that the different EC50 values for extracellular NAD+ in counteracting FK866-induced cell death in A549 compared with U87 cells could be due to the different expression of ectocellular CD38 and CD73 in these cell lines. NAD+ cleavage by CD38 would result in NAM production, which is ineffective as NAD+ precursor in cells treated with FK866. The fact that NMN is less effective than NR in rescuing A549 cells could also be explained by the fact that NMN is a substrate for CD38, resulting in NAM production (38). We confirmed this property by measuring NMN degradation by human recombinant CD38 (Table 2). On the contrary, NR is a poor substrate of CD38 enzymatic activity (Table 2).
TABLE 2.
Degradation of NAD+, NMN, or NR by human recombinant CD38
Human recombinant CD38 (hrCD38) was incubated with 0.4 mm NAD+, NMN or NR. The generation of NAM was evaluated by HPLC analyses.
| NAM |
|||
|---|---|---|---|
| NAD+ substrate | NMN substrate | NR substrate | |
| nmol/min/mg | |||
| Human recombinant CD38 | 19,890 | 46,120 | 413 |
In order to dissect the role of CD38 in the ectocellular generation of NAD+ metabolites, which can be used by the cells as NAD+ precursors, CD38 expression was specifically silenced by the use of siRNA. qPCR and Western blot analyses confirmed that CD38 expression was almost completely abrogated in A549 cells transfected with CD38 siRNA (Fig. 3A, inset). The [NAD+]i was significantly reduced in CD38-silenced cells, in agreement with previous studies demonstrating a modulation of NAD+ levels by CD38 expression (43). The CD38-silenced cells were treated (or not) with FK866, and the effect of exogenous NMN on the [NAD+]i was compared with that on control cells, transfected in the presence of a negative control siRNA. Replenishment of intracellular NAD+ by exogenous NMN in control and FK866-treated cell types proved to be significantly more efficient in CD38-silenced cells (Fig. 3A). Next, CD38 was overexpressed in U87 cells by transfection with CD38-pcDNA3.1 plasmid. As a control, cells were transfected in the presence of an empty plasmid. As shown in Fig. 3B, NMN-mediated rescue of the FK866-induced decrease of the [NAD+]i was almost completely abolished in CD38-overexpressing U87 cells. These results indicate that extracellular NMN degradation by CD38 prevents its utilization as NAD+ precursor, because NAM cannot be converted to NMN in the FK866-treated cells.
FIGURE 3.

CD38 expression affects intracellular NAD+ synthesis triggered by extracellular NMN. A, A549 cells were transfected with specific siRNA for CD38 or with negative control siRNA (siRNA control) and seeded in 12-well plates (2 × 105 cells/well). Cells were treated for 72 h with or without 30 nm FK866 in the presence or absence of 10 μm NMN (added twice a day). Cells were harvested and lysed in 0.6 m PCA, and NAD+ content was measured in neutralized extracts. NAD+ values were normalized to protein content. Data are expressed as mean ± S.D. (error bars) (n = 3). *, p < 0.05; #, p < 0.01 compared with untreated siRNA control cells; †, p < 0.01 compared with untreated siRNA CD38 cells; °, p < 0.001 compared with FK866-treated cells; §, p < 0.001 compared with FK866 + NMN-treated siRNA control cells. Inset, 24 h after transfection; a, qPCR analysis was performed, and results are normalized on the reference genes GAPDH and HPRT1 and compared with negative control; b, Western blot analysis of CD38 protein level was performed (results from one representative experiment is shown). B, U87 cells were transfected in parallel with pcDNA3.1 (empty plasmid) or with CD38-pcDNA3.1 (CD38 plasmid) using the Nucleofector system and seeded in 12-well plates (2 × 105 cells/well). Cells were then treated for 72 h with or without 30 nm FK866 in the presence or absence of 10 μm NMN (added twice a day). Cells were harvested and lysed in 0.6 m PCA, and NAD+ content was measured in neutralized extracts. NAD+ values were normalized to protein content. Data are expressed as mean ± S.D. (n = 3). *, p < 0.05; #, p < 0.01 compared with untreated, empty plasmid cells; §, p < 0.001 compared with FK866-treated empty plasmid cells and with FK866 + NMN-treated CD38 plasmid cells. Inset, 24 h after transfection, Western blot analysis of CD38 protein level was performed (one representative experiment of three comparable ones is shown).
We also investigated whether ectocellular conversion of NAD+ and NMN to NR by CD73 (40) could facilitate NAD+ biosynthesis. Indeed, extracellular NR can cross the plasma membrane and be phosphorylated by NR kinases in mammalian cells to generate intracellular NMN and, eventually, NAD+ (32). CD73 silencing impaired NMN utilization as an NAD+ precursor in FK866-treated A549 cells (Fig. 4A). Conversely, when CD73 was overexpressed, NMN-induced NAD+ synthesis in FK866-treated cells was significantly higher than in cells transfected with an empty plasmid (Fig. 4B). Altogether, these results indicate that the conversion of extracellular NMN to intracellular NAD+ is impaired by CD38, whereas it is favored by CD73.
FIGURE 4.

CD73 expression affects intracellular NAD+ synthesis triggered by extracellular NMN. A, A549 cells were transfected with specific siRNA for CD73 or with negative control (siRNA control) and seeded in 12-well plates (2 × 105 cells/well). Cells were treated for 72 h with or without 30 nm FK866 in the presence or absence of 10 μm of NMN (added twice a day). Cells were harvested and lysed in 0.6 m PCA, and NAD+ content was measured in neutralized extracts. NAD+ values were normalized to protein content. Data are expressed as mean ± S.D. (error bars) (n = 3). *, p < 0.05 compared with untreated siRNA control cells; #, p < 0.01 compared with FK866-treated siRNA control cells and with FK866 + NMN-treated siRNA CD73 cells. Inset, 24 h after transfection; a, qPCR analysis was performed, and expression of CD73 was normalized to that of the housekeeping genes GAPDH and HPRT1 and compared with negative control; b, Western blot analysis of CD73 protein level was performed (results from one representative experiment are shown). B, U87 cells were transfected in parallel with pcDNA3.1/V5-HisTOPO® (empty plasmid) or with CD73-pcDNA3.1/V5-His TOPO® (CD73 plasmid) using the Nucleofector system and seeded in 12-well plates (2 × 105 cells/well). Cells were then treated for 72 h with or without 30 nm FK866 in the presence or absence of 10 μm of NMN (added twice a day). Cells were harvested and lysed in 0.6 m PCA, and NAD content was measured in neutralized extracts. NAD+ values were normalized to protein content. Data are expressed as mean ± S.D. (n = 3). *, p < 0.05 compared with untreated empty plasmid cells; †, p < 0.05 compared with untreated CD73 plasmid cells; §, p < 0.0001 compared with FK866-treated empty plasmid cells; #, p < 0.01 compared with FK866 + NMN-treated empty plasmid cells. Inset, 24 h after transfection; a, transfected cells were subjected to total RNA extraction, and qPCR analysis for CD73 was performed. Results are normalized on the reference genes GAPDH and HPRT1 and compared with empty plasmid; b, Western blot analysis of cell lysates was performed, using a monoclonal antibody against the V5 epitope (one representative experiment of three is shown).
CD73 Inhibition Potentiates FK866-induced Cell Death in the Presence of Ectocellular NMN
In an attempt to confirm that NMN conversion to NR by CD73 was responsible for the rescue from cell death in FK866-treated cells, CD73 expression was silenced in A549 cells, and cells were treated (or not) with FK866 in the presence or absence of extracellular NMN or NR (added twice a day for 72 h, at 10 μm final concentration). Indeed, whereas CD73 silencing did not affect the viability rescue by extracellular NR, it significantly decreased (by ∼70%) the rescue by NMN (Fig. 5A).
FIGURE 5.
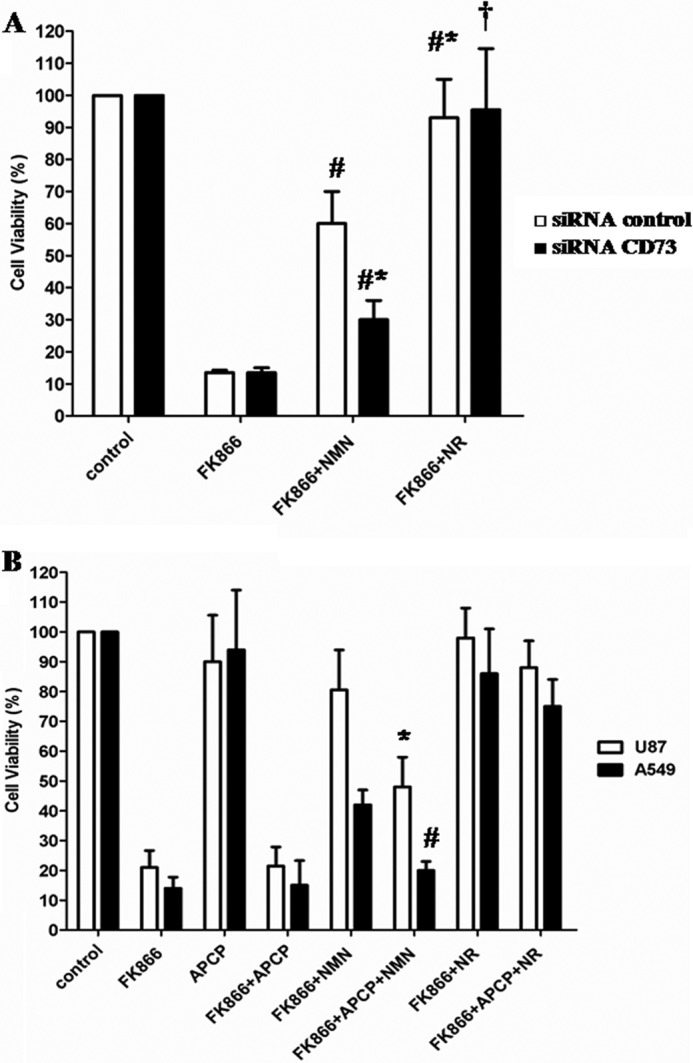
CD73 mediates the NMN-triggered rescue from FK866-induced cell death. A, 24 h after transfection, negative control and CD73-silenced A549 cells (5 × 103 cells/well in 96-well plates) were incubated for 72 h in the presence or absence of 30 nm FK866, with or without 10 μm NMN or NR (added twice a day). Cell viability was evaluated by SBC. Results are expressed as a percentage of cell growth relative to untreated, control cells. Data are expressed as the mean ± S.D. (error bars) (n = 3). #, p < 0.01 compared with FK866-treated siRNA control cells; *, p < 0.05 compared with FK866 + NMN-treated siRNA control cells; †, p < 0.01 compared with FK866- and FK866 + NMN-treated siRNA CD73 cells. B, A549 and U87 cell viability (5 × 103/well in 96-well plates) was evaluated by SBC after a 72-h incubation of the cells in complete medium with or without 30 nm FK866 in the presence or absence of 1 μm APCP or of 10 μm NMN or NR (added twice a day). Results are expressed as a percentage of cell growth relative to untreated, control cells. Data are expressed as mean ± S.D. (n = 3). *, p < 0.05; #, p < 0.01 compared with the corresponding FK866 + NMN-treated cells.
In the presence of a specific CD73 inhibitor (APCP) (51), which per se did not affect viability of control or of FK866-treated cells (Fig. 5B), NMN was considerably less effective than NR at counteracting toxicity of FK866 in A549 and U87 cells; as shown in Fig. 5B, cell viability in FK866-treated cells in the presence of extracellular NMN was reduced by ∼55 and 81% in U87 and A549 cells, respectively, upon the addition of APCP. Conversely, the NR-induced salvaging effect on FK866-treated cells was not modified by the presence of APCP. The lack of sensitivity to APCP in FK866- and NR-treated cells is probably due to the fact that NAD+ synthesis from NR bypasses the reactions inhibited by both NAMPT and CD73.
DISCUSSION
In this study, we addressed the role of two ectoenzymes, CD38 and CD73, in the ectocellular metabolism of precursors for intracellular NAD+ biosynthesis. Starting from the observation that a higher expression of CD38 in A549 cells was accompanied by a reduced efficacy of NAD+ in rescuing from cell death due to NAMPT inhibition, we down- or up-regulated CD38 levels by specific CD38 silencing or overexpression, respectively. By degrading NAD+ and NMN to NAM, CD38 impairs NAD+- and NMN-mediated rescue from FK866-induced intracellular NAD+ depletion and from consequent cell death because NAM utilization by these cells is blocked by NAMPT inhibition. Thus, CD38 expression by tumor cells could account for their inability to overcome NAMPT inhibition, when they are supplemented with NAM-containing precursors. Interestingly, preclinical and clinical studies revealed a peculiar sensitivity of normal lymphocytes to FK866, as inferred from the lymphopenia consistently observed in response to this drug (25). This fact is consonant with a high CD38 expression in lymphocytes (52).
On the other hand, our results demonstrate that CD73 expression leads to the degradation of extracellular NAD+ and NMN to NR, which can enter cells (27) and be used by FK866-treated cells to fuel NAD+ synthesis, thereby bypassing NAMPT inhibition. This is the first study demonstrating a clear role for endogenous CD73 in mediating intracellular NAD+ synthesis from appropriate extracellular precursors. A potential role of CD73 in NMN dephosphorylation had been postulated (3), and recently we demonstrated that purified recombinant human CD73 is active on both NAD+ and NMN, in addition to its canonical substrate AMP, whereas it does not degrade NR (40).
From a clinical perspective, double targeting of NAMPT and CD73 seems to hold promise for cancer treatments aimed at inhibiting NAD+ biosynthetic pathways starting from NAM and NMN. This view is supported by the present results, showing that NMN-mediated salvage of FK866-treated cells is reduced when CD73 is either silenced or pharmacologically inhibited (Fig. 5). Comparison of results obtained with two different tumor cell lines, expressing different endogenous levels of CD38 and CD73, suggests that CD73 inhibition would prove advantageous in tumor cells expressing low levels of CD38 (in our experimental settings, U87 cells). Indeed, constitutively high levels of CD38 can undermine the ability of a cell to exploit extracellular NMN for intracellular NAD+ synthesis. This view is supported by the fact that, in cells expressing high levels of CD38, such as A549 cells, NAD+ was mainly converted to ADPR and NAM (Table 1), and the production of adenosine by endogenous CD73 was not detectable (not shown).
Extracellular NR rescues cells from FK866-induced cell death (Fig. 5). Thus, in principle, the presence of extracellular NR could impair antitumor activity of CD73-targeting inhibitors. However, no NR has been detected in animal plasma (33). Conversely, NMN is present in human plasma (33, 34).
Increased expression of CD73 has been observed in several types of cancer, and the tumor microenvironment contains factors modulating CD73 expression (53). High CD73 expression is currently believed to confer on cancer cells a survival advantage during therapies with antimetabolites (54), and CD73 expression and activity in tumor cells are associated with poor prognosis and may promote metastasis (55). The mechanisms that were proposed to account for these effects include the production by CD73 of adenosine, which would act on A2A and A2B receptors to promote tumor cell motility and metastases (56). However, the existence of additional protumorigenic effects of CD73 has been postulated (reviewed by Salmi and Jalkanen (55)). Indeed, despite some concern that CD73 inhibition in humans may have consequences that are somehow different from what is observed in mice (55), CD73 is currently considered an appealing therapeutic target for treating cancer (53, 57–60). The CD73 inhibitor APCP, a non-hydrolyzable structural mimic of ADP, appears to be well tolerated in vivo, although its half-life in vivo and biodistribution are not well characterized (57), and it shows promising anticancer activity (57, 60).
In conclusion, our study delineates an enzymatic pathway through which ectocellular NAD+ precursors appear to contribute to intracellular NAD+ biosynthesis and to possibly affect the ultimate efficacy of NAMPT inhibitors as anticancer agents. We also devise a metabolic strategy (CD73 inhibition) that could prove useful to increase the efficacy of these agents.
Acknowledgment
We are indebted to Prof. Elena Zocchi for advice and critical review of the manuscript.
This work was supported by the European Seventh Framework Program (project number 256986, PANACREAS), by the Ministero della Salute (GR-2008-1135635), by the Ministero dell'Istruzione, Università e Ricerca Scientifica (MIUR, FIRB 2003, RBLA039LSF_002), by the Associazione Italiana per la Ricerca sul Cancro (AIRC, Code 6108) (to A. N.), by the Compagnia di San Paolo, and by the University of Genova.
- ADPR
- ADP-ribose
- NAD+e
- extracellular NAD+
- [NAD+]i
- intracellular NAD+ concentration
- NAM
- nicotinamide
- NR
- nicotinamide riboside
- NAMPT
- NAM phosphoribosyltransferase
- NMN
- nicotinamide mononucleotide
- APCP
- adenosine 5′-(α,β-methylene)diphosphate
- PCA
- perchloric acid
- qPCR
- quantitative PCR
- SBC
- sulforhodamine B colorimetric assay.
REFERENCES
- 1. Lee H. C. (2012) Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 287, 31633–31640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee H. C. (2012) The cyclic ADP-ribose/NAADP/CD38-signaling pathway. Past and present. Messenger 1, 16–33 [Google Scholar]
- 3. Belenky P., Bogan K. L., Brenner C. (2007) NAD+ metabolism in health and disease. Trends Biochem. Sci. 32, 12–19 [DOI] [PubMed] [Google Scholar]
- 4. Koch-Nolte F., Haag F., Guse A. H., Lund F., Ziegler M. (2009) Emerging roles of NAD+ and its metabolites in cell signaling. Sci. Signal. 2, mr1. [DOI] [PubMed] [Google Scholar]
- 5. Chiarugi A., Dölle C., Felici R., Ziegler M. (2012) The NAD metabolome. A key determinant of cancer cell biology. Nat. Rev. Cancer 12, 741–752 [DOI] [PubMed] [Google Scholar]
- 6. Magni G., Amici A., Emanuelli M., Raffaelli N., Ruggieri S. (1999) Enzymology of NAD+ synthesis. Adv. Enzymol. Relat. Areas Mol. Biol. 73, 135–182, xi [DOI] [PubMed] [Google Scholar]
- 7. Houtkooper R. H., Auwerx J. (2012) Exploring the therapeutic space around NAD+. J. Cell Biol. 199, 205–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rongvaux A., Shea R. J., Mulks M. H., Gigot D., Urbain J., Leo O., Andris F. (2002) Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur. J. Immunol. 32, 3225–3234 [DOI] [PubMed] [Google Scholar]
- 9. Lau C., Niere M., Ziegler M. (2009) The NMN/NaMN adenylyltransferase (NMNAT) protein family. Front. Biosci. 14, 410–431 [DOI] [PubMed] [Google Scholar]
- 10. Stein L. R., Imai S. (2012) The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 23, 420–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hasmann M., Schemainda I. (2003) FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 63, 7436–7442 [PubMed] [Google Scholar]
- 12. Bruzzone S., Fruscione F., Morando S., Ferrando T., Poggi A., Garuti A., D'Urso A., Selmo M., Benvenuto F., Cea M., Zoppoli G., Moran E., Soncini D., Ballestrero A., Sordat B., Patrone F., Mostoslavsky R., Uccelli A., Nencioni A. (2009) Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS One 4, e7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Montecucco F., Bauer I., Braunersreuther V., Bruzzone S., Akhmedov A., Lüscher T. F., Speer T., Poggi A., Mannino E., Pelli G., Galan K., Bertolotto M., Lenglet S., Garuti A., Montessuit C., Lerch R., Pellieux C., Vuilleumier N., Dallegri F., Mage J., Sebastian C., Mostoslavsky R., Gayet-Ageron A., Patrone F., Mach F., Nencioni A. (2013) Inhibition of nicotinamide phosphoribosyltransferase reduces neutrophil-mediated injury in myocardial infarction. Antioxid. Redox Signal. 18, 630–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watson M., Roulston A., Bélec L., Billot X., Marcellus R., Bédard D., Bernier C., Branchaud S., Chan H., Dairi K., Gilbert K., Goulet D., Gratton M. O., Isakau H., Jang A., Khadir A., Koch E., Lavoie M., Lawless M., Nguyen M., Paquette D., Turcotte E., Berger A., Mitchell M., Shore G. C., Beauparlant P. (2009) The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis. Strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell Biol. 29, 5872–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hjarnaa P. J., Jonsson E., Latini S., Dhar S., Larsson R., Bramm E., Skov T., Binderup L. (1999) CHS 828, a novel pyridyl cyanoguanidine with potent antitumor activity in vitro and in vivo. Cancer Res. 59, 5751–5757 [PubMed] [Google Scholar]
- 16. Fleischer T. C., Murphy B. R., Flick J. S., Terry-Lorenzo R. T., Gao Z. H., Davis T., McKinnon R., Ostanin K., Willardsen J. A., Boniface J. J. (2010) Chemical proteomics identifies Nampt as the target of CB30865, an orphan cytotoxic compound. Chem. Biol. 17, 659–664 [DOI] [PubMed] [Google Scholar]
- 17. Lockman J. W., Murphy B. R., Zigar D. F., Judd W. R., Slattum P. M., Gao Z. H., Ostanin K., Green J., McKinnon R., Terry-Lorenzo R. T., Fleischer T. C., Boniface J. J., Shenderovich M., Willardsen J. A. (2010) Analogues of 4-[(7-bromo-2-methyl-4-oxo-3H-quinazolin-6-yl)methylprop-2-ynylamino]-N-(3-pyridy lmethyl)benzamide (CB-30865) as potent inhibitors of nicotinamide phosphoribosyltransferase (Nampt). J. Med. Chem. 53, 8734–8746 [DOI] [PubMed] [Google Scholar]
- 18. Cea M., Cagnetta A., Fulciniti M., Tai Y. T., Hideshima T., Chauhan D., Roccaro A., Sacco A., Calimeri T., Cottini F., Jakubikova J., Kong S. Y., Patrone F., Nencioni A., Gobbi M., Richardson P., Munshi N., Anderson K. C. (2012) Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 120, 3519–3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fuchs D., Rodriguez A., Eriksson S., Christofferson R., Sundberg C., Azarbayjani F. (2010) Metronomic administration of the drug GMX1777, a cellular NAD synthesis inhibitor, results in neuroblastoma regression and vessel maturation without inducing drug resistance. Int. J. Cancer 126, 2773–2789 [DOI] [PubMed] [Google Scholar]
- 20. Beauparlant P., Bédard D., Bernier C., Chan H., Gilbert K., Goulet D., Gratton M. O., Lavoie M., Roulston A., Turcotte E., Watson M. (2009) Preclinical development of the nicotinamide phosphoribosyl transferase inhibitor prodrug GMX1777. Anti-cancer drugs 20, 346–354 [DOI] [PubMed] [Google Scholar]
- 21. Zoppoli G., Cea M., Soncini D., Fruscione F., Rudner J., Moran E., Caffa I., Bedognetti D., Motta G., Ghio R., Ferrando F., Ballestrero A., Parodi S., Belka C., Patrone F., Bruzzone S., Nencioni A. (2010) Potent synergistic interaction between the Nampt inhibitor APO866 and the apoptosis activator TRAIL in human leukemia cells. Exp. Hematol. 38, 979–988 [DOI] [PubMed] [Google Scholar]
- 22. Pogrebniak A., Schemainda I., Azzam K., Pelka-Fleischer R., Nüssler V., Hasmann M. (2006) Chemopotentiating effects of a novel NAD biosynthesis inhibitor, FK866, in combination with antineoplastic agents. Eur. J. Med. Res. 11, 313–321 [PubMed] [Google Scholar]
- 23. Cea M., Soncini D., Fruscione F., Raffaghello L., Garuti A., Emionite L., Moran E., Magnone M., Zoppoli G., Reverberi D., Caffa I., Salis A., Cagnetta A., Bergamaschi M., Casciaro S., Pierri I., Damonte G., Ansaldi F., Gobbi M., Pistoia V., Ballestrero A., Patrone F., Bruzzone S., Nencioni A. (2011) Synergistic interactions between HDAC and sirtuin inhibitors in human leukemia cells. PloS One 6, e22739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bajrami I., Kigozi A., Van Weverwijk A., Brough R., Frankum J., Lord C. J., Ashworth A. (2012) Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol. Med. 4, 1087–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Holen K., Saltz L. B., Hollywood E., Burk K., Hanauske A. R. (2008) The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest. New Drugs 26, 45–51 [DOI] [PubMed] [Google Scholar]
- 26. von Heideman A., Berglund A., Larsson R., Nygren P. (2010) Safety and efficacy of NAD depleting cancer drugs. Results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother. Pharmacol. 65, 1165–1172 [DOI] [PubMed] [Google Scholar]
- 27. Nikiforov A., Dölle C., Niere M., Ziegler M. (2011) Pathways and subcellular compartmentation of NAD biosynthesis in human cells. From entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 286, 21767–21778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang P., Xu T. Y., Guan Y. F., Tian W. W., Viollet B., Rui Y. C., Zhai Q. W., Su D. F., Miao C. Y. (2011) Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann. Neurol. 69, 360–374 [DOI] [PubMed] [Google Scholar]
- 29. De Flora A., Zocchi E., Guida L., Franco L., Bruzzone S. (2004) Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann. N.Y. Acad. Sci. 1028, 176–191 [DOI] [PubMed] [Google Scholar]
- 30. Billington R. A., Bruzzone S., De Flora A., Genazzani A. A., Koch-Nolte F., Ziegler M., Zocchi E. (2006) Emerging functions of extracellular pyridine nucleotides. Mol. Med. 12, 324–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruzzone S., Guida L., Sturla L., Usai C., Zocchi E., De Flora A. (2012) Subcellular and intercellular traffic of NAD+, NAD+ precursors and NAD+-derived signal metabolites and second messengers. Old and new topological paradoxes. Messenger 1, 34–52 [Google Scholar]
- 32. Bieganowski P., Brenner C. (2004) Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117, 495–502 [DOI] [PubMed] [Google Scholar]
- 33. Yoshino J., Mills K. F., Yoon M. J., Imai S. (2011) Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 14, 528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Revollo J. R., Körner A., Mills K. F., Satoh A., Wang T., Garten A., Dasgupta B., Sasaki Y., Wolberger C., Townsend R. R., Milbrandt J., Kiess W., Imai S. (2007) Nampt/PBEF/visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 6, 363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hara N., Yamada K., Shibata T., Osago H., Tsuchiya M. (2011) Nicotinamide phosphoribosyltransferase/visfatin does not catalyze nicotinamide mononucleotide formation in blood plasma. PLoS One 6, e22781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Imai S. (2009) The NAD world. A new systemic regulatory network for metabolism and aging. Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem. Biophys. 53, 65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang P., Xu T. Y., Guan Y. F., Su D. F., Fan G. R., Miao C. Y. (2009) Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor. Role of nicotinamide mononucleotide. Cardiovasc. Res. 81, 370–380 [DOI] [PubMed] [Google Scholar]
- 38. Sauve A. A., Munshi C., Lee H. C., Schramm V. L. (1998) The reaction mechanism for CD38. A single intermediate is responsible for cyclization, hydrolysis, and base-exchange chemistries. Biochemistry 37, 13239–13249 [DOI] [PubMed] [Google Scholar]
- 39. Bogan K. L., Brenner C. (2008) Nicotinic acid, nicotinamide, and nicotinamide riboside. A molecular evaluation of NAD precursor vitamins in human nutrition. Annu. Rev. Nutr. 28, 115–130 [DOI] [PubMed] [Google Scholar]
- 40. Garavaglia S., Bruzzone S., Cassani C., Canella L., Allegrone G., Sturla L., Mannino E., Millo E., De Flora A., Rizzi M. (2012) The high-resolution crystal structure of periplasmic Haemophilus influenzae NAD nucleotidase reveals a novel enzymatic function of human CD73 related to NAD+ metabolism. Biochem. J. 441, 131–141 [DOI] [PubMed] [Google Scholar]
- 41. Wellings D. A., Atherton E. (1997) Standard Fmoc protocols. Methods Enzymol. 289, 44–67 [DOI] [PubMed] [Google Scholar]
- 42. Bruzzone S., De Flora A., Usai C., Graeff R., Lee H. C. (2003) Cyclic ADP-ribose is a second messenger in the lipopolysaccharide-stimulated proliferation of human peripheral blood mononuclear cells. Biochem. J. 375, 395–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zocchi E., Daga A., Usai C., Franco L., Guida L., Bruzzone S., Costa A., Marchetti C., De Flora A. (1998) Expression of CD38 increases intracellular calcium concentration and reduces doubling time in HeLa and 3T3 cells. J. Biol. Chem. 273, 8017–8024 [DOI] [PubMed] [Google Scholar]
- 44. Vichai V., Kirtikara K. (2006) Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 1, 1112–1116 [DOI] [PubMed] [Google Scholar]
- 45. Bruzzone S., Guida L., Zocchi E., Franco L., De Flora A. (2001) Connexin 43 hemichannels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 15, 10–12 [DOI] [PubMed] [Google Scholar]
- 46. Loewenstein W. R. (1981) Junctional intercellular communication. The cell-to-cell membrane channel. Physiol. Rev. 61, 829–913 [DOI] [PubMed] [Google Scholar]
- 47. Braet K., Vandamme W., Martin P. E., Evans W. H., Leybaert L. (2003) Photoliberating inositol-1,4,5-trisphosphate triggers ATP release that is blocked by the connexin mimetic peptide gap 26. Cell Calcium 33, 37–48 [DOI] [PubMed] [Google Scholar]
- 48. Zocchi E., Franco L., Guida L., Benatti U., Bargellesi A., Malavasi F., Lee H. C., De Flora A. (1993) A single protein immunologically identified as CD38 displays NAD/glycohydrolase, ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities at the outer surface of human erythrocytes. Biochem. Biophys. Res. Commun. 196, 1459–1465 [DOI] [PubMed] [Google Scholar]
- 49. Goding J. W., Terkeltaub R., Maurice M., Deterre P., Sali A., Belli S. I. (1998) Ecto-phosphodiesterase/pyrophosphatase of lymphocytes and non-lymphoid cells. Structure and function of the PC-1 family. Immunol. Rev. 161, 11–26 [DOI] [PubMed] [Google Scholar]
- 50. Resta R., Yamashita Y., Thompson L. F. (1998) Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol. Rev. 161, 95–109 [DOI] [PubMed] [Google Scholar]
- 51. Colgan S. P., Eltzschig H. K., Eckle T., Thompson L. F. (2006) Physiological roles for ecto-5′-nucleotidase (CD73). Purinergic Signal. 2, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ferrero E., Malavasi F. (1999) The metamorphosis of a molecule. From soluble enzyme to the leukocyte receptor CD38. J. Leukoc. Biol. 65, 151–161 [DOI] [PubMed] [Google Scholar]
- 53. Beavis P. A., Stagg J., Darcy P. K., Smyth M. J. (2012) CD73. A potent suppressor of antitumor immune responses. Trends Immunol. 33, 231–237 [DOI] [PubMed] [Google Scholar]
- 54. Ujházy P., Klobusická M., Babusíková O., Strausbauch P., Mihich E., Ehrke M. J. (1994) Ecto-5′-nucleotidase (CD73) in multidrug-resistant cell lines generated by doxorubicin. Int. J. Cancer 59, 83–93 [DOI] [PubMed] [Google Scholar]
- 55. Salmi M., Jalkanen S. (2011) Homing-associated molecules CD73 and VAP-1 as targets to prevent harmful inflammations and cancer spread. FEBS Lett. 585, 1543–1550 [DOI] [PubMed] [Google Scholar]
- 56. Stagg J., Divisekera U., McLaughlin N., Sharkey J., Pommey S., Denoyer D., Dwyer K. M., Smith M. J. (2010) Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. U.S.A. 107, 1547–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang B. (2010) CD73. A novel target for cancer immunotherapy. Cancer Res. 70, 6407–6411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang B. (2012) CD73 promotes tumor growth and metastasis. Oncoimmunology 1, 67–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou X., Zhi X., Zhou P., Chen S., Zhao F., Shao Z., Ou Z., Yin L. (2007) Effects of ecto-5′-nucleotidase on human breast cancer cell growth in vitro and in vivo. Oncol. Rep. 17, 1341–1346 [PubMed] [Google Scholar]
- 60. Forte G., Sorrentino R., Montinaro A., Luciano A., Adcock I. M., Maiolino P., Arra C., Cicala C., Pinto A., Morello S. (2012) Inhibition of CD73 improves B cell-mediated anti-tumor immunity in a mouse model of melanoma. J. Immunol. 189, 2226–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]