

Background: Acetylcholinesterase (AChE) and amyloid precursor protein (APP) are implicated in Alzheimer's disease, but their specific biological roles remain unclear.
Results: Overexpression or knockdown of neuronal APP modulates AChE mRNA, protein levels, and enzyme activity.
Conclusion: APP can act as a transcriptional regulator through a novel mechanism independently of γ-secretase.
Significance: Understanding the physiological functions of APP will lead to a greater understanding of Alzheimer's disease etiology.
Keywords: Acetylcholinesterase, Alzheimer's Disease, Amyloid Precursor Protein, Cell Culture, Transfection, γ-Secretase Inhibitor, AICD
Abstract
The toxic role of amyloid β peptides in Alzheimer's disease is well documented. Their generation is via sequential β- and γ-secretase cleavage of the membrane-bound amyloid precursor protein (APP). Other APP metabolites include the soluble ectodomains sAPPα and sAPPβ and also the amyloid precursor protein intracellular domain (AICD). In this study, we examined whether APP is involved in the regulation of acetylcholinesterase (AChE), which is a key protein of the cholinergic system and has been shown to accelerate amyloid fibril formation and increase their toxicity. Overexpression of the neuronal specific isoform, APP695, in the neuronal cell lines SN56 and SH-SY5Y substantially decreased levels of AChE mRNA, protein, and catalytic activity. Although similar decreases in mRNA levels were observed of the proline-rich anchor of AChE, PRiMA, no changes were seen in mRNA levels of the related enzyme, butyryl-cholinesterase, nor of the high-affinity choline transporter. A γ-secretase inhibitor did not affect AChE transcript levels or enzyme activity in SN56 (APP695) or SH-SY5Y (APP695) cells, showing that regulation of AChE by APP does not require the generation of AICD or amyloid β peptide. Treatment of wild-type SN56 cells with siRNA targeting APP resulted in a significant up-regulation in AChE mRNA levels. Mutagenesis studies suggest that the observed transcriptional repression of AChE is mediated by the E1 region of APP, specifically its copper-binding domain, but not the C-terminal YENTPY motif. In conclusion, AChE is regulated in two neuronal cell lines by APP in a manner independent of the generation of sAPPα, sAPPβ, and AICD.
Introduction
The amyloid precursor protein (APP)2 and acetylcholinesterase (AChE) are both multifaceted proteins with a wide range of functions. Although they are both linked with growth and developmental processes, especially in the brain (1–3), they are also crucially linked with the pathogenesis of Alzheimer's disease (AD). APP is the precursor of the Aβ peptide, which is suggested to be the pathological agent in AD (4), with its oligomers considered the most toxic (5, 6). AChE has also been linked to the disease pathogenesis by exacerbating amyloid fibril formation and toxicity (7–10) and is the main target of clinically available AD drugs (11).
AChE is a key protein in cholinergic signaling, of which there are several main systems in the brain. Among these, the basal forebrain cholinergic system (12) is strongly linked to AD (13). There are reports specifically and consistently linking cholinergic hypofunction and cognitive decline. For reasons unknown, cholinergic neurons of the basal forebrain are specifically affected in AD (14). The predominant neurotransmitter in this system is acetylcholine (ACh), which, like AChE, has been suggested to have trophic functions (15).
The individual subunits of AChE can associate with each other, forming both dimers and tetramers. The proline-rich membrane anchor (PRiMA) is a 20-kDa protein responsible for both AChE tetramerization and its anchorage to the membrane in neuronal cells. This is a crucial role because tetramers of AChE form the functional units at cholinergic synapses (16–19). PRiMA has a proline-rich attachment domain like its counterpart, ColQ, which serves as a membrane anchor for AChE at neuromuscular junctions. The AChE-PRiMA association occurs between the C-terminal t peptides of AChE and the proline-rich attachment domain of PRiMA. It has also been suggested that disulfide bonds form between four Cys residues at the N terminus of PRiMA and the C-terminal Cys in the AChE t peptide (17). Immunofluorescence studies have shown strong colocalization between AChE and PRiMA in cholinergic neurons but no localization of PRiMA in either dopaminergic or GABAergic neurons (18).
APP is a type I integral membrane protein (see Fig. 1A) (20). It exists in three isoforms (APP695, APP751, and APP770) generated by differential splicing of exons 7 and 8 (21, 22). In terms of distribution, APP mRNA is expressed in almost every tissue, where only the isoform ratio differs (23). It is APP695 that predominates in neurons (2), and APP mRNA represents 0.2% of the total mRNA in these cells (24). There are two proteolytic pathways of APP processing. Amyloidogenic processing is the minor pathway (∼5%) and involves sequential cleavage of APP by β-secretase and the γ-secretase complex, both aspartic proteases. These proteolytic events release Aβ and sAPPβ, with the former, although linked to AD, having physiological roles, such as ion channel regulation (25). The second, non-amyloidogenic pathway involves α-secretase cleavage of APP, which occurs within the Aβ region (26, 27), precluding formation of Aβ, while generating a large, soluble ectodomain, the neuroprotective sAPPα (27–29). Both pathways result in the generation of the amyloid precursor protein intracellular domain (AICD), which can act as a transcriptional regulator (30–34), although there remains some controversy about the precise cohort of genes involved in AICD-mediated transcriptional regulation (35). Important residues in APP695 for transcriptional regulation are Tyr-682 and Tyr-687, which represent the N- and C-terminal residues of the YENPTY motif in the cytoplasmic domain of APP (Fig. 1A) (36). Functional AICD is mostly formed in the amyloidogenic pathway, predominantly from the APP695 isoform (37–39). Much of the transcriptional regulation by AICD is dependent on binding of the Fe65 protein, which may facilitate its nuclear translocation (40, 41). Although the APP C terminus is the predominant region for protein-protein interactions, other regions are also involved, e.g. via the extracellular E1 region with reelin (42), fibulin-1 (43), and integrin β1 (44, 45) and also in dimerization of APP (46). Within the E1 domain, there are subdomains, including the His-rich copper-binding domain (CuBD) (36, 47), which has an important role in mediating APP dimerization (48).
FIGURE 1.
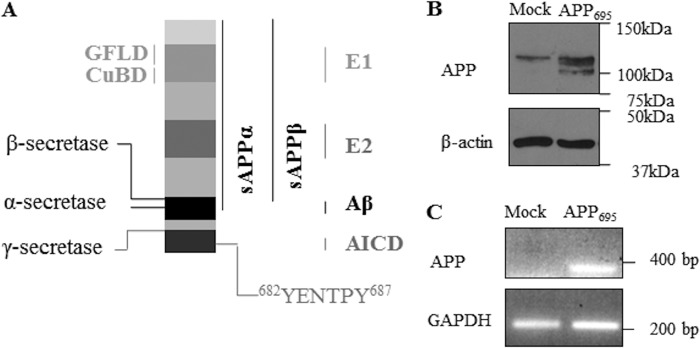
Schematic representation of APP695 and data of overexpression of APP695 in cholinergic SN56 cells. A, the amyloid precursor protein is a multidomain protein. The key domains growth factor-like domain (GFLD), CuBD, E1, E2, Aβ, and AICD are indicated. The YENTPY motif in the C-terminal region encompasses residues 682–687 in APP695, which are crucial in the interaction of APP with intracellular binding partners. The full-length protein can be cleaved sequentially by α- and γ-secretases at the indicated sites, yielding the sAPPα fragment and AICD. Alternatively, APP can be cleaved sequentially by β- and γ-secretases, yielding sAPPβ, Aβ, and functional AICD. B, Western blot analysis for APP (top panel) and β-actin (bottom panel) in lysates of mock-transfected SN56 cells and SN56 cells overexpressing APP695. C, RT-PCR of DNA extracted from the same cell lines, assessing levels of APP (top panel) and GAPDH (bottom panel).
The main goal of this study was to investigate whether APP695 regulates AChE expression in neuronal cells. The concept of the cholinergic system regulating APP has been well documented (49, 50), but data about APP regulation of the cholinergic system are sparse. Here we show that overexpression or, conversely, knockdown of APP695 can modulate mRNA, protein, and activity levels of AChE. In addition, PRiMA appears to be regulated via the same mechanism, whereas BChE and CHT remain unaffected. This novel mechanism is γ-secretase-independent and is likely mediated via the E1 domain of APP, specifically the CuBD therein.
EXPERIMENTAL PROCEDURES
Cell Culture
Cholinergic SN56 cells (provided by Prof. A. Szutowicz, Medical University of Gdańsk, Poland) and GD25/GD25β1 cells (a gift from Prof. S. Johansson, Uppsala University, Sweden) were cultured in DMEM (Lonza, Basel, Switzerland), and SH-SY5Y cells were cultured in DMEM F-12 (Lonza). All media were supplemented with 10% fetal bovine serum and 1% penicillin, streptomycin, and l-glutamine. For SH-SY5Y cells, 1% non-essential amino acids were added. For transfected cells, either 150 μg/ml hygromycin B (wild-type APP695 or APP695 with tyrosine/histidine mutations) (Invitrogen) or 10 μg/ml blasticidin S (APPΔE1) (Invitrogen) was added. All cells were incubated at 37 °C and 5% CO2 in a humidified atmosphere.
Constructs
The APP695 (pIRESHyg) construct was provided by Dr. A. R. Whyteside (University of Leeds). APP695 constructs expressing tyrosine mutations (all pIRESHyg, Y682G, Y687G and Y682G + Y687G double mutant) were prepared as follows. These APP Y682G and Y687G constructs were generated using site-directed mutagenesis to convert tyrosine codons (TAC) to glycine (GGC) according to the instructions of the manufacturer (Agilent Technologies UK Ltd, Stockport, UK) using pIREShyg-wt-APP695 as a template. The APP Y682G + Y687G double mutant was then generated in the same manner but using previously mutated APPY682G as a template. The APPΔCuBD (H147A, H149A, H151A triple mutant) was a gift from Dr. Edward Neale (Lancaster University, UK). The APPΔE1 (pLBCX, c-myc, 6× His tag) was provided by Prof. C. U. Pietrzik (Department of Pathobiochemistry, University Medical Center of the Johannes Gutenberg University, Mainz, Germany).
Transformation
Where appropriate, JM109-competent cells (Agilent Technologies) were transformed and cultured on ampicillin agar plates overnight. Plasmid DNA was extracted according to the instructions of the manufacturer using the Plasmid Maxi kit (Qiagen, Crawley, UK), checked for APP expression by polymerase chain reaction, and then further verified by sequencing (GATC Biotech, Konstanz, Germany).
Transfection
SH-SY5Y-APP695 cells were provided by Dr. I. J. Whitehouse (University of Leeds). SN56 cells were grown to 50–80% confluence and incubated for 3–4 h in 5 ml of OptiMEM (Invitrogen) containing 2 μl of Lipofectamine (Invitrogen) and 5 μg of DNA (absent in mock-transfected cells) encoding APP695 (or mutants thereof). Standard culture medium was added after 3–4 h, and the medium was changed after 24 h. At 48 h, cells were either assayed (for transient transfection) or split 1:50 and grown in 300 μg/ml hygromycin B or 20 μg/ml blasticidin S to select transfected cells (for stable transfection).
Cell Treatment
SN56-APP695 or SH-SY5Y-APP695 cells at ≥ 80% confluence were incubated for 24 h in OptiMEM containing either vehicle (dimethyl sulfoxide, control), 25 μm GM6001 (InSolutionTM, Calbiochem, Merck Chemicals, Nottingham, UK), 1 μm β-IV (Calbiochem), or 10 μm N-(N-(3,5-difluorophenacetyl)-l-alanyl)-S-phenylglycine t-butyl ester (DAPT; InSolutionTM γ-secretase inhibitor IX, Calbiochem). The latter treatment was also performed in wild-type cells. After incubation, media samples were taken and concentrated, and cell lysates were prepared as described below. Alternatively, RNA was extracted from the cells for PCR analysis.
Preparation of Cell Lysates for Analysis of Cellular Proteins
Cells at ≥ 80% confluence or after pharmacological treatment, as indicated above, were washed twice in ice-cold PBS and harvested in 10 ml PBS. Cells were pelleted at 2700 × g for 5 min (4 °C) and resuspended in 6× volume of lysis buffer (50 mm Tris-HCl (pH 7.4) with 1% Triton X-100 and 0.5% sodium deoxycholate) with a 21-gauge needle and syringe. Lysis was performed for 30 min on ice, followed by centrifugation at 2700 × g for 5 min to clarify the lysates. Supernatants were collected for assays.
Preparation of Cell Media for the Analysis of Secreted Proteins
Cells were washed with OptiMEM and incubated for 24 h in OptiMEM. The cell medium was then collected, and 5 ml was centrifuged (2400 × g, 5 min, 4 °C) to remove cell debris. Media samples were then added to a 6-ml, 10-kDa molecular weight cutoff Spin X-UF 20 concentrator (Corning Life Sciences, Amsterdam, The Netherlands). This was followed by centrifugation (2400 × g, 4 °C) until the volume reached 0.75–1.0 ml. At this point, the medium samples were centrifuged (10,000 × g, 10 min) in a new Eppendorf tube to clear cell debris.
Cholinesterase Assay
This followed the classical method (51), modified as described before (52). Absorbance changes were measured using a plate reader (412 nm) (Fluostar Omega, BMG LabTech, Aylesbury, Buckinghamshire, UK). Type IV AChE from Electrophorus electricus (diluted 1:12500) (Sigma-Aldrich) was used as a positive control in the assays.
Determination of Protein Concentration
The BCA assay method was used for determining protein concentration. Both the bicinchoninic acid and 4% copper (II) pentahydrate solutions were supplied by Sigma-Aldrich.
SDS-PAGE and Western blotting
An 8% gel was used unless stated otherwise. Protein samples (20–50 μg) were run for 90 min (30 mA and 300 V) using a Bio-Rad gel rig and Invitrogen PowerEase 500 power supply. Western blotting was performed as described previously (37). Primary antibodies used were for AChE (AChE (C-16) catalog no. sc-6430, goat, 1:250, Santa Cruz Biotechnology), APP (22C11, mouse, 1:2000, Millipore, Billerica, MA or anti-C-terminal fragment, rabbit, 1:1000, Sigma-Aldrich), sAPPβ (rabbit, 1:250, Signet Laboratories), and β-actin (1:10000, mouse, Sigma-Aldrich).
RT-PCR
RNA was isolated using the RNeasy kit (Qiagen) according to the instructions of the manufacturer. cDNA was prepared using the iScript cDNA synthesis kit (Bio-Rad) and amplified using conventional PCR with TaqDNA polymerase (New England Biolabs, Hitchin, UK). Conditions were as follows: 94 °C (5 min), 60 °C (30 s), and 68 °C (50 s) for 35 cycles and then 68 °C (10 min) using a PTC-200 Peltier thermal cycler (MJ Research). Amplified DNA was resolved on 1% agarose gels with 50 μg of ethidium bromide and visualized on the Molecular Imager Gel Doc XR system with the Quantity One 4.6.1 program (Bio-Rad).
Primers (Sigma-Aldrich) were as follows: APP, AAGAAGCCGATGATGACGAG (forward) and TTCTCATCCCCAGGTGTCTC (reverse) and GAPDH, AACTTTGGCATTGTGGAAGG (forward) and CACATTGGGGGTAGGAACAC (reverse).
Quantitative PCR (qPCR)
RNA was isolated and cDNA synthesized as above. Transcript levels were assessed using SensiMix SYBR Green (Bioline Reagents, London, UK) on a Rotor-Gene 6000 (Corbett Life Sciences, Cambridge, UK).
Primers used were for human genes as follows: AChE, TTCCTCCCCAAATTGCTCAG (forward) and TCCAGTGCACCATGTAGGAG (reverse); PRiMA, TGATCATCATTGCCGTATGC (forward) and GGTGCCATTTTCGTCTTTTC (reverse); neprilysin, CCTGGAGATTCATAATGGATCTTG (forward) and AAAGGGCCTTGCGCAAAG (reverse); and GAPDH, CAATGACCCCTTCATTGACC (forward) and GACAAGCTTCCCGTTCTCAG (reverse).
Primers used were for mouse genes as follows: PRiMA, ATCATTGTCGCTGTGGTCTG (forward) and GGTGCCATTCTCATCCTTTC (reverse); BChE, TTACAACCAAGACCGGAAGG (forward) and GTTGTGCATAGGGGATACCG (reverse); CHT- F, ATATGGGCTGCATGGAAAAC (forward) and CACCAACCAACAAACCAATG (reverse); and U6- F, CTCGCTTCGGCAGCACA (forward) and AACGCTTCACGAATTTGCGT (reverse). The AChE primers were used in both cell lines because they bind in a conserved area of the transcript.
siRNA Knockdown
Wild-type SN56 cells were transfected with either 25 nm siRNA targeting APP (SMARTpool, Dharmacon, Thermo Scientific) or a scrambled sequence (siRNA negative control, Ambion, Invitrogen) at an equivalent concentration using Lipofectamine (Invitrogen). The transfection medium was replaced with OptiMEM after 6–8 h, and cells were harvested after 24 h.
Statistical Analysis
All statistical analyses were performed using an unpaired, two-tailed Student's t test (Microsoft Excel 2007). All error bars displayed are ± S.E. Statistical significance is defined as follows: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
RESULTS
Overexpression of APP695 in Neuronal Cell Lines
To investigate the possible regulation of AChE by APP, two neuronal cell lines, the human SH-SY5Y neuroblastoma cell line and the mouse cholinergic SN56 cell line, were used. The SN56 cell line is a neuronal cell line derived from murine septal neurons of the basal forebrain (53). A stable SH-SY5Y (APP695) cell line has been generated and characterized previously (37). We also generated a stable SN56 (APP695) cell line that showed substantial increases in APP695 protein (mature and immature) and also in mRNA transcripts as compared with mock-transfected cells (Fig. 1, B and C).
Analysis of AChE Activity, Protein, and mRNA in Transfected Cell Lines
AChE activity was assessed in cell lysates from both the SN56 and SH-SY5Y cell lines (Fig. 2). The activity was significantly reduced in cell lines stably overexpressing APP695 compared with mock-transfected cells, by 75% in SN56 and by 40% in SH-SY5Y cells (Fig. 2, A and B). This correlated with protein level changes caused by APP695 overexpression, as shown by Western blot analysis (Fig. 2, A and B, top panels). To assess the effects of APP695 overexpression on AChE transcript levels, quantitative RT-PCR was used (Fig. 2, C and D). The results of mRNA analyses correlate with AChE activity and protein levels, with a 95% reduction in AChE mRNA in SN56 (APP695) cells and a 50% mRNA reduction in SH-SY5Y (APP695) cells. Furthermore, AChE activity was decreased in the medium of SN56 (APP695) cells in proportion to the decrease seen in the lysates (data not shown).
FIGURE 2.

Effect of APP695 overexpression on AChE activity, protein, and mRNA levels and mRNA levels of other cholinergic genes. A, AChE activity in SN56 (Mock) and SN56 (APP695) cell lysates (n = 6, p < 0. 001) and Western blot analysis for AChE (top panel) and β-actin (bottom panel) in lysates of SN56 cells (using 10% gel). B, AChE activity assays and Western blot analysis for AChE in SH-SY5Y cells (n = 9, p = 0.01) The vertical dashed line on the blot indicates alignment of samples from distal lanes run on the same immunoblot analysis. C, qPCR analysis of AChE mRNA transcripts (n = 6, p = 0.001) in SN56 cells and in SH-SY5Y cells (n = 6, p = 0.02) (D). *, p ≤ 0.05; **, p ≤ 0.01; or ***, p {ltequ] 0.001.
Regulation of Other Cholinergic Genes by APP695
Because catalytically active AChE is PRiMA-bound (17), we investigated whether PRiMA expression also changed with APP695 overexpression, possibly either through coregulation or in response to the significant decrease in AChE expression (Fig. 3A). Quantitative PCR showed a significant 50% decrease in PRiMA transcripts in SN56 (APP695) cells (p < 0.001). We further investigated the transcript levels of another cholinergic protein, namely BChE, which, however, showed no change with overexpression of APP695 (Fig. 3B, p = 0.51). To further expand our assessment of a more global regulatory effect by APP on the cholinergic system, we again utilized qPCR to assay mRNA levels of the high-affinity choline transporter (CHT). We confirmed expression of this protein in SH-SY5Y and SN56 cells by Western blot analysis (data not shown). In SN56 cells overexpressing APP695, there were no changes in mRNA levels of CHT (Fig. 3C, p = 0.99).
FIGURE 3.

QPCR analysis of mRNA levels of cholinergic genes upon stable overexpression of APP695 in SN56 cells. A, qPCR analysis of PRiMA mRNA transcripts (n = 7, p < 0.001) (A), BChE mRNA transcripts (n = 6, p = 0.51) (B), and CHT transcripts (n = 9, p = 0.99) (C). ***, p ≤ 0.001.
Assessment of AChE mRNA Levels after siRNA-mediated Knockdown of Endogenous APP
After demonstrating the effects of APP overexpression on AChE mRNA levels and activity, we next examined the effects of knockdown of endogenous APP using siRNA. Transfection of wild-type SN56 cells with a scrambled sequence oligonucleotide showed no effect on APP expression, but the APP-specific siRNA caused large reductions in APP protein levels (Fig. 4, inset). We then assessed AChE mRNA levels after siRNA knockdown of APP. Again, the scrambled sequence had no effect, but siRNA knockdown of APP resulted in a significant increase in transcription of AChE (Fig. 4).
FIGURE 4.
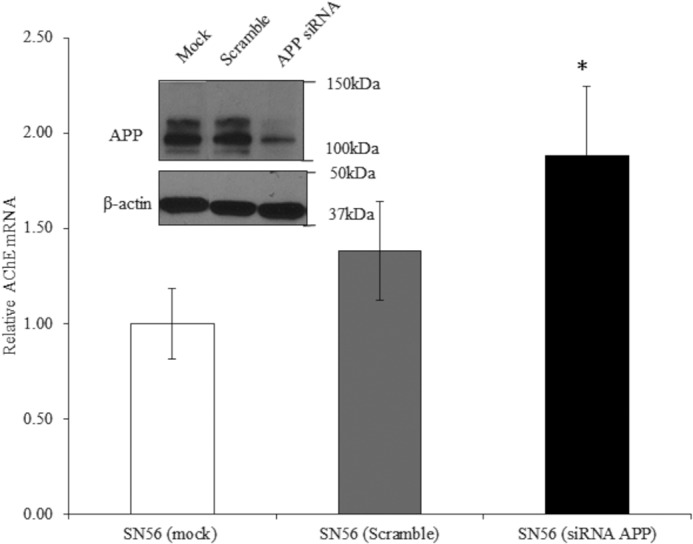
Effect of APP knockdown on AChE mRNA. qPCR analysis of AChE mRNA transcripts (scramble, n = 7, p = 0.43; siRNA APP, n = 9, p = 0.03) and (inset) Western blot analysis for APP (top panel) and β-actin (bottom panel) in lysates of SN56 cells treated with buffer (mock), scrambled oligonucleotide, or siRNA targeting APP. *, p ≤ 0.05.
Investigation of the Role of APP Metabolites in AChE Regulation
We next examined the potential role of AICD in the APP-mediated regulation of AChE expression by using the γ-secretase inhibitor DAPT to block AICD production (Fig. 5). Inhibition of the γ-secretase complex precludes generation of AICD and up-regulation of neprilysin (33, 38). Inhibition of AICD generation had no effect on total AChE activity in SH-SY5Y (APP695) or SN56 (APP695) cells (Fig. 5, A and B). Furthermore, analysis of AChE mRNA levels by quantitative RT-PCR revealed no changes in these cell lines (Fig. 5, C and D). Similarly, DAPT had no effect on AChE activity in wild-type SN56 cells (data not shown). As with AChE, PRiMA mRNA transcripts showed no change on treatment with DAPT (data not shown). Successful inhibition of γ-secretase was confirmed by a significant reduction in transcript levels of the AICD-regulated gene neprilysin (Fig. 5E).
FIGURE 5.

Effects of γ-secretase inhibition on AChE activity and mRNA levels. A, AChE activity in cell lysates from SN56 (APP695) and SN56 (APP695) cells treated with 10 μm DAPT for 24 h (n = 6, p = 0. 41) and in SH-SY5Y cells (n = 6, p = 0.26) (B). C, qPCR analysis of AChE mRNA transcripts with same treatment in SN56 cells (n = 6, p = 0.54) and SH-SY5Y cells (n = 6, p = 0.55) (D). E, qPCR for neprilysin in SH-SY5Y (APP695) cells, either control or 24 h of 10 μm DAPT (n = 5, p = 0.001). **, p ≤ 0.01
To assess the potential roles of the soluble ectodomains sAPPα and sAPPβ in the transcriptional repression of AChE, we blocked their formation in SH-SY5Y (APP695) and SN56 (APP695) cells using the α-secretase inhibitor GM6001 and the β-secretase inhibitor β-IV. Neither of these inhibitors was able to alter cellular AChE activity in SH-SY5Y (APP695) or SN56 (APP695) cells (Fig. 6, A and B). Confirmation that the inhibitors were functional at the concentrations used was demonstrated by Western blot analysis for the APP ectodomains sAPPα and sAPPβ, the production of which is dependent on α-secretase and β-secretase, respectively (Fig. 6C).
FIGURE 6.

Effects of α- and β-secretase inhibition on AChE activity. A, AChE activity in cell lysates from SN56 (APP695) treated with 25 μm GM6001 for 24 h (n = 6, p = 0.78) or 1 μm β-IV for 24 h (n = 6, p = 0.52). B, AChE activity in cell lysates from SH-SY5Y (APP695) cells (n = 7; p = 0.83 (GM6001); p = 0.51 (β-IV)). C, Western blot analyses for sAPPα in cell culture medium from SH-SY5Y (APP695) cells, either control or after 24 h of 25 μm GM6001 treatment or for sAPPβ in medium from SH-SY5Y (APP695) cells after 24 h of 1 μm β-IV treatment.
Effects of Mutagenesis of Key Residues and Domains in APP695 on AChE Regulation.
To elucidate the regions of APP that may be responsible for mediating transcriptional repression of AChE, we overexpressed three APP695 mutant constructs in SN56 cells (Fig. 7, A and B). Numerous interactions of proteins with APP are mediated through its C-terminal region (36), the majority of which involve the YENTPY motif. The key residues therein, Tyr-682 and Tyr-687, were mutated both individually and together. These mutations did not change the ability of APP to exert its repressive effects on AChE. When assessing both AChE activity and mRNA in these cell lines, we have found significant reductions after overexpression of the APP mutants (Fig. 7, C and D). Although these APP constructs can undergo proteolysis in a slightly different manner compared with the wild-type protein, we have already shown that proteolytically derived APP metabolites have no effect on AChE expression (Figs. 5 and 6), which would suggest that differential proteolysis would not have any effect on AChE.
FIGURE 7.

Effect of APP695 mutagenesis and overexpression on AChE activity and mRNA. Western blot analysis for APP (top panel) and β-actin (bottom panel) in lysates of mock-transfected SN56 cells SN56 cells overexpressing mutant APP695, specifically Y682G and Y687G (A) and the Y682G, Y687G double mutant (YYGG) (B). C, AChE activity in SN56 (mock) and SN56 (mutant APP695) cell lysates (Y682G, n = 6, p < 0.001; Y687G, n = 6, p = 0.02; YYGG, n = 6, p < 0.001). D, qPCR analysis of AChE mRNA transcripts (Y682G, n = 6, p = 0.01; Y687G, n = 6, p = 0.005; YYGG, n = 6, p = 0.009). *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. E, Western blot analysis for APP (1:1000 anti-C-terminal fragment) (top panel) and β-actin (bottom panel) in lysates of mock-transfected SN56 cells or SN56 cells overexpressing APPΔE1. The vertical dashed line on the blot indicates alignment of samples from distal lanes run on the same immunoblot analysis. F, AChE activity in SN56 (mock) and SN56 (APPΔE1) cell lysates (n = 27, p = 0.51). G, qPCR analysis of AChE mRNA transcripts (n = 9, p = 0.33). H, qPCR analysis of PRiMA mRNA transcripts (n = 6, p = 0.4).
Another region of APP that has several interacting partners is the extracellular E1 domain. To assess a possible role of this APP domain in transcriptional repression of APP, we expressed a deletion construct (APPΔE1) in SN56 cells (Fig. 7E). Assessment of AChE activity and mRNA levels in these cells showed, in contrast to wild-type APP695, no AChE-repressing effect of the APPΔE1 construct (Fig. 7, F and G). Similarly, expression of this construct did not result in any significant change in PRiMA mRNA levels (Fig. 7H).
A significant reduction in AChE activity and mRNA levels was seen after transient transfection with the APP695 construct (Fig. 8, B and C). However, transient transfection of wild-type SN56 cells with the APPΔE1 construct showed no change in AChE activity or mRNA levels compared with mock-transfected cells (Fig. 8, E and F), further supporting a role for the E1 domain in mediating AChE repression.
FIGURE 8.

Effects of transient APP695 overexpression on AChE activity and mRNA levels. A, Western blot analysis for APP (1:2000, 22C11) (top panel) and β-actin (bottom panel) in lysates of mock-transfected SN56 cells and SN56 cells transiently transfected with APP695. The vertical dashed line on the blot indicates alignment of samples from distal lanes run on the same immunoblot analysis. B, AChE activity in SN56 (mock) and SN56 (transient APP695) cell lysates (n = 3, p = 0.005). C, qPCR analysis of AChE mRNA transcripts in mock SN56 cells and cells transiently transfected with APP695 (n = 5, p = 0.01). D, Western blot analysis for APP (1:1000, anti- C-terminal fragment) (top panel) and β-actin (bottom panel) in lysates of mock-transfected SN56 cells and cells transiently transfected with APPΔE1. E, AChE activity in SN56 (mock) and SN56 (transient APPΔE1) cell lysates (n = 9, p = 1.0). F, qPCR analysis of AChE mRNA transcripts in SN56 (mock) and SN56 (transient APPΔE1) cells (n = 6, p = 0.96). **, p ≤ 0.01.
Effects of Overexpression of APP695 (ΔCuBD)
The E1 domain has been linked to a number of APP-interacting proteins and also as containing critical residues in mediating APP dimerization (36, 45, 46). Indeed, studies of the APP interactome frequently list APP/APLP (APP-like protein) as an interacting partner for APP itself (54). Therefore, we mutated a key motif in APP dimerization and protein-protein interactions, the CuBD, mutating three critical His residues to Ala, generating APP695ΔCuBD. We overexpressed this APP construct in SN56 cells (Fig. 9A) to assess whether mutation of the CuBD would compromise the ability of APP to repress the transcription of AChE. These data showed that the SN56 (APPΔCuBD) cells were unable to repress AChE activity in cell lysates (Fig. 9B). These findings were recapitulated at the mRNA level (Fig. 9C).
FIGURE 9.

Analysis of AChE activity and mRNA levels after overexpression of APP695 (ΔCuBD). A, Western blot analysis for APP (1:1000, 22C11) (top panel) and β-actin (bottom panel) in lysates of mock-transfected SN56 cells and SN56 cells stably overexpressing APP695 (ΔCuBD). B, AChE activity in lysates of mock-transfected SN56 cells and those overexpressing either of the aforementioned APP construct (n = 12, p = 0.53) followed by qPCR analysis for AChE in these cell lines (n = 6, p = 0.32) (C).
Integrin β1 Is Not Involved in Mediating the Transcriptional Repression of AChE by APP695
Integrins are transmembrane proteins with an intracellular NPXY motif, downstream of which intracellular signaling is better defined than any other candidate APP N-terminal ligands (44, 45). Therefore, we investigated whether ITGB1 may be the transmembrane interacting partner of APP through the C-terminal region of which intracellular signals are transduced. We used GD25 cells that are null for ITGB1 (55) and their ITGB1-expressing counterparts, GD25β1. Transient transfection of both cell lines with wild-type APP695 resulted in non-significant decreases in AChE mRNA levels, and there was no significant difference in the magnitude of this effect between GD25 and GD25β1 cells (data not shown).
DISCUSSION
Although full-length APP has a number of putative functions, a detailed investigation of these has been diverted by substantial focus on the amyloid hypothesis, dominated by Aβ (56). Similarly, with AChE, attention has been diverted from the investigation of its non-catalytic physiological roles. Although a protein with significant links with AD, AChE research has mostly focused on inhibition of its catalytic activity for therapeutic benefits rather than understanding its physiology and metabolism in relation to the AD pathology.
To elucidate any possible direct relationship between APP and AChE, the possible regulation of AChE expression by APP was investigated. Our data show that, in two different neuronal cell lines, SN56 and SH-SY5Y, APP695 overexpression repressed expression of AChE. This negative regulation is occurring on a transcriptional level with consequent reductions in AChE protein levels and enzyme activity. Although the data from our overexpression studies showed a clear relationship between the levels of APP and AChE expression and activity, we deemed it important to also investigate the effect of endogenous APP. Using siRNA, we were able to show that endogenous APP exerts a repressive effect on AChE transcription, showing that APP is a physiologically relevant modulator of AChE transcription. Furthermore, it clearly demonstrates that our data are not artifacts of an overexpression system, with the relationship induced by large increases in protein translation. Some studies in vivo have reported changes in AChE activity when APP is overexpressed. A study by Van Dam et al. (57) using the APP23 mouse model showed a significant reduction in AChE activity in the basal forebrain but not other regions examined. A further study in APP23 mice observed moderate decreases in AChE activity but also reported reductions in cholinergic fiber density (58), and Boncristiano et al. (59) reported reduced neuronal volume in the basal forebrain of APP23 mice. These data show that the degenerative phenotype in these AD model mice may be a confounding factor in the interpretation of AChE activity differences between wild-type and transgenic mice. We explored the possibility that APP had some pan-regulatory role in the cholinergic system, perhaps regulating a cohort of cholinergic genes rather than one in particular.
Apart from AChE, we also found a regulatory relationship between APP and another cholinergic system protein, namely PRiMA, which, like AChE, was down-regulated after APP695 overexpression. This shows that, at least in this pathway, AChE and PRiMA levels are closely coupled. However, it is still unclear whether PRiMA is directly regulated by APP or whether its down-regulation is a consequence of modifications in AChE levels.
On the contrary, APP had no effect on another mammalian cholinesterase, BChE, because its levels were unchanged after APP695 overexpression. Similarly, we were unable to find any effect on mRNA levels of the high-affinity choline transporter CHT. This suggests that the regulatory effects downstream of APP695 overexpression exhibit some selectivity and specificity. Our data, then, argue against regulation of a subset of cholinergic genes by APP, with only AChE and its membrane anchor directly modulated.
In addition to APP itself, its metabolites have also been shown to exert biological effects (60, 61). These metabolites include AICD, which regulates expression of numerous genes (61, 62), as well as sAPPα and sAPPβ, whose mechanisms of action are still poorly understood (60). Of the APP fragments, AICD is by far the best characterized as a transcriptional regulator (31, 61–63). A large number of genes have been linked to AICD (64), with some consistently shown to be up-regulated, such as neprilysin (33, 34), and a much smaller number to be down-regulated, such as the EGF receptor (65).
However, the failure of a γ-secretase inhibitor to affect AChE mRNA or activity levels demonstrates that AICD does not regulate AChE in the mammalian neuronal cell lines used, with the repression of AChE expression unchanged by γ-secretase inhibition and, hence, ablation of AICD production. Because the effects of APP on AChE expression did not involve AICD, the possible involvement of the soluble APP ectodomains was also examined because they may have some roles in gene expression (60). sAPPα has well reported neuroprotective and neurotrophic effects (23, 66), and there are data reporting an increase in neuronal survival and neurite outgrowth promoted by sAPPα in rat cortical neurons (23). Further works have implicated enhancement of mitogenesis and synapse formation and stability as features of sAPPα action (67, 68). Neuroprotective roles of sAPPα against glucose deprivation, glutamate toxicity, and ischemia have also been reported (66, 69, 70). Although there is scant evidence linking sAPPα to any specific genes, (60), sAPPβ has been linked to transcriptional regulation of transthyretin and Klotho (71). However, despite such a range of physiological properties, we failed to observe any effect of inhibition of sAPPα or sAPPβ production on AChE activity.
The elimination of the major secretase-derived APP fragments (AICD, sAPPα, and sAPPβ) as having a contributory role in repression of AChE transcription leaves full-length APP as the most likely responsible species. It is not surprising that full-length APP is involved in gene regulation because it is linked to numerous signaling pathways and other processes (1, 2). In terms of interactions of APP, the intracellular C-terminal region has the most documented interacting proteins, with key residues demonstrated to be Tyr-682 and Tyr-687 within the YENPTY motif (36). However, our data demonstrate that mutation of these residues did not reduce the repressive effect exerted by APP and suggest that the observed transcriptional repression is unlikely to be mediated by the C-terminal region.
For further investigation of the role of APP in AChE repression, we analyzed the effect of deletion of the E1 region in APP. One possibility is that deletion of this domain prevents binding of an APP-interacting protein. Several N-terminal APP binding partners have been reported, namely fibulin-1, reelin, F-spondin, Lingo-1, contactin 2, pancortins 1 and 3, and integrin β1 (ITGB1) (36, 44, 45). Of these, ITGB1 is capable of binding copper-binding domains (72), although this has not been directly shown for APP. A necessary requirement of a binding partner is that the interacting protein must be able to transduce intracellular signals, which ultimately results in transcriptional repression of AChE. Most of the APP binding partners listed are adhesion proteins, and many do not have any defined intracellular signaling activity. However, integrins do have defined intracellular signaling activity, modulating such proteins as focal adhesion kinase and Akt (73). However, our studies have shown that, although ITGB1 is an interacting partner of APP, it is unlikely to be the N-terminal binding partner through which repression of AChE is transduced.
The data obtained in our work have clearly indicated that deletion of the E1 domain ablates the repressive activity of APP695, which suggests that this domain can mediate transcriptional repression of AChE. Previous studies have shown that the APPΔE1 construct traffics and localizes in the same manner as wild-type APP695 (74). We were able to characterize the CuBD within E1 as the region responsible for this transcriptional repression of AChE. This APPΔCuBD construct has been shown to traffic and undergo proteolysis in a manner very similar wild-type APP695, although sAPPβ is reduced relative to wild-type.3 Copper binding has been linked to promoting APP dimerization (48), but binding of interacting proteins to this region has also been suggested (75), and previous work has implicated integrins as being capable of binding CuBDs (72). Although ITGB1 does not appear to be the relevant interacting partner in the transduction mechanism, other interacting partners or even perturbation of dimerization may be factors.
The data provide an insight into the relationship between APP and AChE and highlight a novel function for APP in the regulation of gene expression independent of its intracellular domain. Although AD-linked genes have been shown to be regulated by APP, none of these regulatory pathways have been shown, so far, to involve the E1 domain of full-length APP, although the holoprotein has been recently linked to regulation of cholesterol metabolism at a transcriptional level (76). It is possible that repression of AChE serves to maintain cholinergic signaling by reducing ACh hydrolysis in a neurodegenerative environment. However, it is also possible that this relationship is independent of the catalytic function of AChE. AChE and APP both have roles in cell adhesion and synaptic integrity (3, 77), so perhaps this novel relationship serves to regulate these non-catalytic functions. Finally, AChE has been shown to have a role in apoptosis, being up-regulated by certain apoptotic stimuli and then participating in the process of apoptosis (78, 79), including in AD (80). As such, this down-regulation of AChE might be an example of a novel neuroprotective function of the APP holoprotein.
To elucidate any direct links between APP and the key cholinergic system proteins in mammalian cells, we have shown that APP695 represses transcription of AChE in SH-SY5Y and SN56 cells. The mechanism is not dependent on APP metabolites produced by α-, β-, or γ-secretases because their inhibition in cell culture does not ablate the effect of APP695 overexpression. Furthermore, mutagenesis studies show that the C-terminal region of APP is unlikely to have any involvement in this process. However, the E1 region of APP appears to play a role in the transcriptional repression of APP. Because current therapy focuses on inhibition of AChE, an understanding of the APP-AChE regulatory axis and the manipulation thereof may lead to novel therapeutic strategies.
Acknowledgments
We thank Dr. S. Isbert (University Medical Center of the Johannes Gutenberg University, Mainz, Germany) for assistance with the APPΔE1 construct and Dr. C. Kerridge (University of Leeds) for transformation of tyrosine mutant APP plasmids into competent cells and subsequent extraction. We also thank Prof. N. M. Hooper (University of Leeds) for helpful discussion of the manuscript.
This work was supported by the UK Biotechnology and Biological Sciences Research Council (studentship support to D. A. H.), the UK Medical Research Council, Alzheimer's Research UK, Russian Foundation for Basic Research Grant FBR 13-04-00388, and the Fundamental Sciences to Medicine Programme of the Russian Academy of Sciences.
E. T. Parkin and M. Gough, unpublished observations.
- APP
- amyloid precursor protein
- AChE
- acetylcholinesterase
- ACh
- acetylcholine
- AD
- Alzheimer's disease
- Aβ
- amyloid β peptide
- PRiMA
- proline rich membrane anchor
- AICD
- amyloid precursor protein intracellular domain
- CuBD
- copper-binding domain
- BChE
- butyrylcholinesterase
- qPCR
- quantitative PCR
- CHT
- choline transporter
- DAPT
- N-(N-(3,5-difluorophenacetyl)-l-alanyl)-S-phenylglycine t-butyl ester.
REFERENCES
- 1. Zheng H., Koo E. H. (2011) Biology and pathophysiology of the amyloid precursor protein. Mol. Neurodegener. 6, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gralle M., Ferreira S. T. (2007) Structure and functions of the human amyloid precursor protein. The whole is more than the sum of its parts. Prog. Neurobiol. 82, 11–32 [DOI] [PubMed] [Google Scholar]
- 3. Halliday A. C., Greenfield S. A. (2012) From protein to peptides. A spectrum of non-hydrolytic functions of acetylcholinesterase. Protein Pept. Lett. 19, 165–172 [DOI] [PubMed] [Google Scholar]
- 4. Hardy J. A., Higgins G. A. (1992) Alzheimer's disease. The amyloid cascade hypothesis. Science 256, 184–185 [DOI] [PubMed] [Google Scholar]
- 5. Walsh D. M., Selkoe D. J. (2007) Aβ oligomers. A decade of discovery. J. Neurochem. 101, 1172–1184 [DOI] [PubMed] [Google Scholar]
- 6. Haass C., Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration. Lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 7. Inestrosa N. C., Alvarez A., Pérez C. A., Moreno R. D., Vicente M., Linker C., Casanueva O. I., Soto C., Garrido J. (1996) Acetylcholinesterase accelerates assembly of amyloid-β-peptides into Alzheimer's fibrils. Possible role of the peripheral site of the enzyme. Neuron 16, 881–891 [DOI] [PubMed] [Google Scholar]
- 8. Alvarez A., Opazo C., Alarcón R., Garrido J., Inestrosa N. C. (1997) Acetylcholinesterase promotes the aggregation of amyloid-β-peptide fragments by forming a complex with the growing fibrils. J. Mol. Biol. 272, 348–361 [DOI] [PubMed] [Google Scholar]
- 9. Dinamarca M. C., Sagal J. P., Quintanilla R. A., Godoy J. A., Arrázola M. S., Inestrosa N. C. (2010) Amyloid-β-acetylcholinesterase complexes potentiate neurodegenerative changes induced by the Aβ peptide. Implications for the pathogenesis of Alzheimer's disease. Mol. Neurodegener. 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rees T., Hammond P. I., Soreq H., Younkin S., Brimijoin S. (2003) Acetylcholinesterase promotes β-amyloid plaques in cerebral cortex. Neurobiol. Aging 24, 777–787 [DOI] [PubMed] [Google Scholar]
- 11. Stahl N., Borchelt D. R., Hsiao K., Prusiner S. B. (1987) Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51, 229–240 [DOI] [PubMed] [Google Scholar]
- 12. Auld D. S., Kornecook T. J., Bastianetto S., Quirion R. (2002) Alzheimer's disease and the basal forebrain cholinergic system: relations to β-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 68, 209–245 [DOI] [PubMed] [Google Scholar]
- 13. Coyle J. T., Price D. L., DeLong M. R. (1983) Alzheimer's disease. A disorder of cortical cholinergic innervation. Science 219, 1184–1190 [DOI] [PubMed] [Google Scholar]
- 14. Madhusudan A., Sidler C., Knuesel I. (2009) Accumulation of reelin-positive plaques is accompanied by a decline in basal forebrain projection neurons during normal aging. Eur. J. Neurosci. 30, 1064–1076 [DOI] [PubMed] [Google Scholar]
- 15. Isacson O., Seo H., Lin L., Albeck D., Granholm A. C. (2002) Alzheimer's disease and Down's syndrome. Roles of APP, trophic factors and ACh. Trends Neurosci. 25, 79–84 [DOI] [PubMed] [Google Scholar]
- 16. Dvir H., Silman I., Harel M., Rosenberry T. L., Sussman J. L. (2010) Acetylcholinesterase. From 3D structure to function. Chem. Biol. Interact. 187, 10–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Perrier A. L., Massoulié J., Krejci E. (2002) PRiMA. The membrane anchor of acetylcholinesterase in the brain. Neuron 33, 275–285 [DOI] [PubMed] [Google Scholar]
- 18. Henderson Z., Matto N., John D., Nalivaeva N. N., Turner A. J. (2010) Co-localization of PRiMA with acetylcholinesterase in cholinergic neurons of rat brain. An immunocytochemical study. Brain Res. 1344, 34–42 [DOI] [PubMed] [Google Scholar]
- 19. Hicks D., John D., Makova N. Z., Henderson Z., Nalivaeva N. N., Turner A. J. (2011) Membrane targeting, shedding and protein interactions of brain acetylcholinesterase. J. Neurochem. 116, 742–746 [DOI] [PubMed] [Google Scholar]
- 20. Weidemann A., König G., Bunke D., Fischer P., Salbaum J. M., Masters C. L., Beyreuther K. (1989) Identification, biogenesis, and localization of precursors of Alzheimer's disease A4 amyloid protein. Cell 57, 115–126 [DOI] [PubMed] [Google Scholar]
- 21. Tanzi R. E., Gusella J. F., Watkins P. C., Bruns G. A., St George-Hyslop P., Van Keuren M. L., Patterson D., Pagan S., Kurnit D. M., Neve R. L. (1987) Amyloid β protein gene. cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 235, 880–884 [DOI] [PubMed] [Google Scholar]
- 22. Sandbrink R., Masters C. L., Beyreuther K. (1996) APP gene family. Alternative splicing generates functionally related isoforms. Ann. N.Y. Acad. Sci. 777, 281–287 [DOI] [PubMed] [Google Scholar]
- 23. Araki W., Kitaguchi N., Tokushimà Y., Ishii K., Aratake H., Shimohama S., Nakamura S., Kimura J. (1991) Trophic effect of β-amyloid precursor protein on cerebral cortical neurons in culture. Biochem. Biophys. Res. Commun. 181, 265–271 [DOI] [PubMed] [Google Scholar]
- 24. Beyreuther K., Multhaup G., Mönning U., Sandbrink R., Beher D., Hesse L., Small D. H., Masters C. L. (1996) Regulation of APP expression, biogenesis and metabolism by extracellular matrix and cytokines. Ann. N.Y. Acad. Sci. 777, 74–76 [DOI] [PubMed] [Google Scholar]
- 25. Pearson H. A., Peers C. (2006) Physiological roles for amyloid β peptides. J. Physiol. 575, 5–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuhn P. H., Wang H., Dislich B., Colombo A., Zeitschel U., Ellwart J. W., Kremmer E., Rossner S., Lichtenthaler S. F. (2010) ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 29, 3020–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lichtenthaler S. F. (2011) α-Secretase in Alzheimer's disease. Molecular identity, regulation and therapeutic potential. J. Neurochem. 116, 10–21 [DOI] [PubMed] [Google Scholar]
- 28. Lichtenthaler S. F., Haass C., Steiner H. (2011) Regulated intramembrane proteolysis. Lessons from amyloid precursor protein processing. J. Neurochem. 117, 779–796 [DOI] [PubMed] [Google Scholar]
- 29. Allinson T. M., Parkin E. T., Condon T. P., Schwager S. L., Sturrock E. D., Turner A. J., Hooper N. M. (2004) The role of ADAM10 and ADAM17 in the ectodomain shedding of angiotensin converting enzyme and the amyloid precursor protein. Eur. J. Biochem. 271, 2539–2547 [DOI] [PubMed] [Google Scholar]
- 30. Kim H. S., Kim E. M., Lee J. P., Park C. H., Kim S., Seo J. H., Chang K. A., Yu E., Jeong S. J., Chong Y. H., Suh Y. H. (2003) C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3β expression. FASEB J. 17, 1951–1953 [DOI] [PubMed] [Google Scholar]
- 31. Cao X., Südhof T. C. (2001) A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115–120 [DOI] [PubMed] [Google Scholar]
- 32. Schettini G., Govoni S., Racchi M., Rodriguez G. (2010) Phosphorylation of APP-CTF-AICD domains and interaction with adaptor proteins. Signal transduction and/or transcriptional role. Relevance for Alzheimer pathology. J. Neurochem. 115, 1299–1308 [DOI] [PubMed] [Google Scholar]
- 33. Belyaev N. D., Nalivaeva N. N., Makova N. Z., Turner A. J. (2009) Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter. Implications for Alzheimer disease. EMBO Rep. 10, 94–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pardossi-Piquard R., Petit A., Kawarai T., Sunyach C., Alves da Costa C., Vincent B., Ring S., D'Adamio L., Shen J., Müller U., St George Hyslop P., Checler F. (2005) Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of βAPP and APLP. Neuron 46, 541–554 [DOI] [PubMed] [Google Scholar]
- 35. Hébert S. S., Serneels L., Tolia A., Craessaerts K., Derks C., Filippov M. A., Müller U., De Strooper B. (2006) Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes. EMBO Rep 7, 739–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aydin D., Weyer S. W., Müller U. C. (2012) Functions of the APP gene family in the nervous system. Insights from mouse models. Exp. Brain Res. 217, 423–434 [DOI] [PubMed] [Google Scholar]
- 37. Belyaev N. D., Kellett K. A., Beckett C., Makova N. Z., Revett T. J., Nalivaeva N. N., Hooper N. M., Turner A. J. (2010) The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a β-secretase dependent pathway. J. Biol. Chem. 285, 41443–41454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goodger Z. V., Rajendran L., Trutzel A., Kohli B. M., Nitsch R. M., Konietzko U. (2009) Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. J. Cell Sci. 122, 3703–3714 [DOI] [PubMed] [Google Scholar]
- 39. Flammang B., Pardossi-Piquard R., Sevalle J., Debayle D., Dabert-Gay A. S., Thévenet A., Lauritzen I., Checler F. (2012) Evidence that the amyloid-β protein precursor intracellular domain, AICD, derives from β-secretase-generated C-terminal fragment. J. Alzheimer's Dis. 30, 145–153 [DOI] [PubMed] [Google Scholar]
- 40. Cao X., Südhof T. C. (2004) Dissection of amyloid-β precursor protein-dependent transcriptional transactivation. J. Biol. Chem. 279, 24601–24611 [DOI] [PubMed] [Google Scholar]
- 41. Kimberly W. T., Zheng J. B., Guénette S. Y., Selkoe D. J. (2001) The intracellular domain of the β-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J. Biol. Chem. 276, 40288–40292 [DOI] [PubMed] [Google Scholar]
- 42. Hoe H. S., Lee K. J., Carney R. S., Lee J., Markova A., Lee J. Y., Howell B. W., Hyman B. T., Pak D. T., Bu G., Rebeck G. W. (2009) Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J. Neurosci. 29, 7459–7473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ohsawa I., Takamura C., Kohsaka S. (2001) Fibulin-1 binds the amino-terminal head of β-amyloid precursor protein and modulates its physiological function. J. Neurochem. 76, 1411–1420 [DOI] [PubMed] [Google Scholar]
- 44. Young-Pearse T. L., Chen A. C., Chang R., Marquez C., Selkoe D. J. (2008) Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin β1. Neural Dev. 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rice H. C., Young-Pearse T. L., Selkoe D. J. (2013) Systematic evaluation of candidate ligands regulating ectodomain shedding of amyloid precursor protein. Biochemistry 52, 3264–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Isbert S., Wagner K., Eggert S., Schweitzer A., Multhaup G., Weggen S., Kins S., Pietrzik C. U. (2012) APP dimer formation is initiated in the endoplasmic reticulum and differs between APP isoforms. Cell. Mol. Life Sci. 69, 1353–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Barnham K. J., McKinstry W. J., Multhaup G., Galatis D., Morton C. J., Curtain C. C., Williamson N. A., White A. R., Hinds M. G., Norton R. S., Beyreuther K., Masters C. L., Parker M. W., Cappai R. (2003) Structure of the Alzheimer's disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J. Biol. Chem. 278, 17401–17407 [DOI] [PubMed] [Google Scholar]
- 48. Noda Y., Asada M., Kubota M., Maesako M., Watanabe K., Uemura M., Kihara T., Shimohama S., Takahashi R., Kinoshita A., Uemura K. (2013) Copper enhances APP dimerization and promotes Aβ production. Neurosci. Lett. 547, 10–15 [DOI] [PubMed] [Google Scholar]
- 49. Canet-Aviles R. M., Anderton M., Hooper N. M., Turner A. J., Vaughan P. F. (2002) Muscarine enhances soluble amyloid precursor protein secretion in human neuroblastoma SH-SY5Y by a pathway dependent on protein kinase C(α), src-tyrosine kinase and extracellular signal-regulated kinase but not phospholipase C. Brain Res. Mol. Brain Res. 102, 62–72 [DOI] [PubMed] [Google Scholar]
- 50. Nitsch R. M., Slack B. E., Wurtman R. J., Growdon J. H. (1992) Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science 258, 304–307 [DOI] [PubMed] [Google Scholar]
- 51. Ellman G. L., Courtney K. D., Andres V., Jr., Feather-Stone R. M. (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95 [DOI] [PubMed] [Google Scholar]
- 52. Hicks D. A., Makova N. Z., Nalivaeva N. N., Turner A. J. (2013) Characterisation of acetylcholinesterase release from neuronal cells. Chem. Biol. Interact. 203, 302–308 [DOI] [PubMed] [Google Scholar]
- 53. Hammond D. N., Lee H. J., Tonsgard J. H., Wainer B. H. (1990) Development and characterization of clonal cell lines derived from septal cholinergic neurons. Brain Res. 512, 190–200 [DOI] [PubMed] [Google Scholar]
- 54. Bai Y., Markham K., Chen F., Weerasekera R., Watts J., Horne P., Wakutani Y., Bagshaw R., Mathews P. M., Fraser P. E., Westaway D., St George-Hyslop P., Schmitt-Ulms G. (2008) The in vivo brain interactome of the amyloid precursor protein. Mol. Cell. Proteomics 7, 15–34 [DOI] [PubMed] [Google Scholar]
- 55. Fässler R., Pfaff M., Murphy J., Noegel A. A., Johansson S., Timpl R., Albrecht R. (1995) Lack of β 1 integrin gene in embryonic stem cells affects morphology, adhesion, and migration but not integration into the inner cell mass of blastocysts. J. Cell Biol. 128, 979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hardy J. (2009) The amyloid hypothesis for Alzheimer's disease. A critical reappraisal. J. Neurochem. 110, 1129–1134 [DOI] [PubMed] [Google Scholar]
- 57. Van Dam D., Marescau B., Engelborghs S., Cremers T., Mulder J., Staufenbiel M., De Deyn P. P. (2005) Analysis of cholinergic markers, biogenic amines, and amino acids in the CNS of two APP overexpression mouse models. Neurochem. Int. 46, 409–422 [DOI] [PubMed] [Google Scholar]
- 58. Diez M., Danner S., Frey P., Sommer B., Staufenbiel M., Wiederhold K. H., Hökfelt T. (2003) Neuropeptide alterations in the hippocampal formation and cortex of transgenic mice overexpressing β-amyloid precursor protein (APP) with the Swedish double mutation (APP23). Neurobiol. Dis. 14, 579–594 [DOI] [PubMed] [Google Scholar]
- 59. Boncristiano S., Calhoun M. E., Kelly P. H., Pfeifer M., Bondolfi L., Stalder M., Phinney A. L., Abramowski D., Sturchler-Pierrat C., Enz A., Sommer B., Staufenbiel M., Jucker M. (2002) Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J. Neurosci. 22, 3234–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chasseigneaux S., Allinquant B. (2012) Functions of Aβ, sAPPα and sAPPβ. Similarities and differences. J. Neurochem. 120, 99–108 [DOI] [PubMed] [Google Scholar]
- 61. Chang K. A., Suh Y. H. (2010) Possible roles of amyloid intracellular domain of amyloid precursor protein. BMB Rep. 43, 656–663 [DOI] [PubMed] [Google Scholar]
- 62. Beckett C., Nalivaeva N. N., Belyaev N. D., Turner A. J. (2012) Nuclear signalling by membrane protein intracellular domains. The AICD enigma. Cell. Signal. 24, 402–409 [DOI] [PubMed] [Google Scholar]
- 63. Pardossi-Piquard R., Checler F. (2012) The physiology of the β-amyloid precursor protein intracellular domain AICD. J. Neurochem. 120, 109–124 [DOI] [PubMed] [Google Scholar]
- 64. Ohkawara T., Nagase H., Koh C. S., Nakayama K. (2011) The amyloid precursor protein intracellular domain alters gene expression and induces neuron-specific apoptosis. Gene 475, 1–9 [DOI] [PubMed] [Google Scholar]
- 65. Zhang Y. W., Wang R., Liu Q., Zhang H., Liao F. F., Xu H. (2007) Presenilin/γ-secretase-dependent processing of β-amyloid precursor protein regulates EGF receptor expression. Proc. Natl. Acad. Sci. U.S.A. 104, 10613–10618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Smith-Swintosky V. L., Pettigrew L. C., Craddock S. D., Culwell A. R., Rydel R. E., Mattson M. P. (1994) Secreted forms of β-amyloid precursor protein protect against ischemic brain injury. J. Neurochem. 63, 781–784 [DOI] [PubMed] [Google Scholar]
- 67. Barger S. W., Mattson M. P. (1995) The secreted form of the Alzheimer's β-amyloid precursor protein stimulates a membrane-associated guanylate cyclase. Biochem. J. 311, 45–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Furukawa K., Barger S. W., Blalock E. M., Mattson M. P. (1996) Activation of K+ channels and suppression of neuronal activity by secreted β-amyloid-precursor protein. Nature 379, 74–78 [DOI] [PubMed] [Google Scholar]
- 69. Barger S. W., Fiscus R. R., Ruth P., Hofmann F., Mattson M. P. (1995) Role of cyclic GMP in the regulation of neuronal calcium and survival by secreted forms of β-amyloid precursor. J. Neurochem. 64, 2087–2096 [DOI] [PubMed] [Google Scholar]
- 70. Mattson M. P., Cheng B., Culwell A. R., Esch F. S., Lieberburg I., Rydel R. E. (1993) Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron 10, 243–254 [DOI] [PubMed] [Google Scholar]
- 71. Li H., Wang B., Wang Z., Guo Q., Tabuchi K., Hammer R. E., Südhof T. C., Zheng H. (2010) Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc. Natl. Acad. Sci. U.S.A. 107, 17362–17367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Weaver M. S., Workman G., Sage E. H. (2008) The copper binding domain of SPARC mediates cell survival in vitro via interaction with integrin β1 and activation of integrin-linked kinase. J. Biol. Chem. 283, 22826–22837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Clarke N. E., Fisher M. J., Porter K. E., Lambert D. W., Turner A. J. (2012) Angiotensin converting enzyme (ACE) and ACE2 bind integrins and ACE2 regulates integrin signalling. PLoS ONE 7, e34747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kaden D., Voigt P., Munter L. M., Bobowski K. D., Schaefer M., Multhaup G. (2009) Subcellular localization and dimerization of APLP1 are strikingly different from APP and APLP2. J. Cell Sci. 122, 368–377 [DOI] [PubMed] [Google Scholar]
- 75. Kong G. K., Miles L. A., Crespi G. A., Morton C. J., Ng H. L., Barnham K. J., McKinstry W. J., Cappai R., Parker M. W. (2008) Copper binding to the Alzheimer's disease amyloid precursor protein. Eur. Biophys. J. 37, 269–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pierrot N., Tyteca D., D'auria L., Dewachter I., Gailly P., Hendrickx A., Tasiaux B., Haylani L. E., Muls N., N'kuli F., Laquerrière A., Demoulin J. B., Campion D., Brion J. P., Courtoy P. J., Kienlen-Campard P., Octave J. N. (2013) Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol. Med. 5, 608–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Müller U. C., Zheng H. (2012) Physiological functions of APP family proteins. Cold Spring Harb. Perspect. Med. 2, a006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang X. J., Greenberg D. S. (2012) Acetylcholinesterase involvement in apoptosis. Front. Mol. Neurosci. 5, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Toiber D., Greenberg D. S., Soreq H. (2009) Pro-apoptotic protein-protein interactions of the extended N-AChE terminus. J. Neural Transm. 116, 1435–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Toiber D., Berson A., Greenberg D., Melamed-Book N., Diamant S., Soreq H. (2008) N-acetylcholinesterase-induced apoptosis in Alzheimer's disease. PLoS ONE 3, e3108. [DOI] [PMC free article] [PubMed] [Google Scholar]