

Background: KCNJ10 is a key component of the basolateral K channels in DCT.
Results: SFK phosphorylates KCNJ10 at Tyr9, and inhibition of SFK decreases basolateral K channel activity in DCT.
Conclusion: SFK stimulates the basolateral K channels in DCT by phosphorylating KCNJ10.
Significance: Tyrosine phosphorylation of KCNJ10 plays a role in regulating membrane transport in DCT.
Keywords: Ion Channels, Kidney, Membrane Transport, Phosphorylation, Potassium Channels
Abstract
The loss of function of the basolateral K channels in the distal nephron causes electrolyte imbalance. The aim of this study is to examine the role of Src family protein tyrosine kinase (SFK) in regulating K channels in the basolateral membrane of the mouse initial distal convoluted tubule (DCT1). Single-channel recordings confirmed that the 40-picosiemen (pS) K channel was the only type of K channel in the basolateral membrane of DCT1. The suppression of SFK reversibly inhibited the basolateral 40-pS K channel activity in cell-attached patches and decreased the Ba2+-sensitive whole-cell K currents in DCT1. Inhibition of SFK also shifted the K reversal potential from −65 to −43 mV, suggesting a role of SFK in determining the membrane potential in DCT1. Western blot analysis showed that KCNJ10 (Kir4.1), a key component of the basolateral 40-pS K channel in DCT1, was a tyrosine-phosphorylated protein. LC/MS analysis further confirmed that SFK phosphorylated KCNJ10 at Tyr8 and Tyr9. The single-channel recording detected the activity of a 19-pS K channel in KCNJ10-transfected HEK293T cells and a 40-pS K channel in the cells transfected with KCNJ10+KCNJ16 (Kir.5.1) that form a heterotetramer in the basolateral membrane of the DCT. Mutation of Tyr9 did not alter the channel conductance of the homotetramer and heterotetramer. However, it decreased the whole-cell K currents, the probability of finding K channels, and surface expression of KCNJ10 in comparison to WT KCNJ10. We conclude that SFK stimulates the basolateral K channel activity in DCT1, at least partially, by phosphorylating Tyr9 on KCNJ10. We speculate that the modulation of tyrosine phosphorylation of KCNJ10 should play a role in regulating membrane transport function in DCT1.
Introduction
The DCT3 is responsible for the reabsorption of 5–10% of the filtered NaCl load and is the target of thiazide diuretics (1, 2). It is composed of early (DCT1) and late DCT (DCT2), and the expression of ion transporters is also not identical between DCT1 and DCT2. Although the Na/Cl cotransporter (NCC) is expressed in the apical membrane throughout the DCT (3, 4), ROMK (renal outer medullary K) and ENaC (epithelial Na channel) have been shown to be highly expressed in the apical membrane of DCT2 but not of DCT1 (5, 6). The absorption of NaCl in DCT1 is a two-step process. Na and Cl enter the cells across the apical membrane through the NCC, and Na is then pumped out the cell through the basolateral Na-K-ATPase, whereas Cl exits the cell along its electrochemical gradient by basolateral Cl channels (7).
The basolateral K channel in DCT1 plays several important roles in regulating the transepithelial membrane transport in DCT1. First, the K channels participate in generating the cell membrane potential, which is the driving force for Cl exit across the basolateral membrane (8). Therefore, the activity of the basolateral K channel has a significant effect on the transepithelial Cl absorption in DCT1. Second, they are responsible for K recycling, which is essential for maintaining Na-K- ATPase activity (9). It has been suggested that the activity of Na-K-ATPase must be matched with the basolateral K conductance to sustain transepithelial Na transport (9). Finally, the basolateral K channel provides the electrochemical driving force for apical Mg2+ entering the DCT cells.
The patch clamp experiments have identified a 40-pS K channel as the only type of K channel in the basolateral membrane of DCT1 (10). It has been established generally that the basolateral 40-pS K channel is a heterotetramer and composed of KCNJ10 (Kir.4.1) and KCNJ16 (Kir5.1) (10–13). This is on the basis of the observations that expression of Kir.4.1 and Kir.5.1 formed a 40-pS K channel, whereas expression of Kir.4.1 alone formed a 20-pS K channel, and expression of Kir5.1 failed to produce a functional K channel (10, 14, 15). Although the molecular nature of the basolateral K channels is established, the regulatory mechanism of the basolateral K channels is poorly understood. It has been demonstrated that the 40-pS K channel in the DCT is inhibited by Mg2+ and pH-sensitive (10). Because SFKs such as c-Src are highly expressed in the distal nephron, including the DCT, and plays an important role in regulating ROMK (Kir1.1) (16, 17), we suspect that SFKs may also regulate the basolateral K channels. Therefore, the aim of this study is to explore the role of SFK in regulating the basolateral K channels in DCT1 and to determine the molecular mechanisms of the effect of SFKs.
EXPERIMENTAL PROCEDURES
Preparation of DCT1
C57BL/6 mice (either sex, 4–6 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME). The mice were fed with control diet and had free access to water. After the mice were sacrificed by cervical dislocation, we perfused the left kidney with 5 ml collagenase type 2 (1 mg/1 ml) containing L-15 medium (Life Technology). The collagenase-perfused kidney was removed, and the renal cortex was cut with a sharp razor. The renal cortex was further cut into small pieces that were then incubated in collagenase-containing L-15 medium for 45–60 min. After collagenase treatment, the tissue was washed three times with L-15 medium and transferred to an ice-cold chamber for dissection. Fig. 1A shows an isolated tubule in which DCT is included. The experiments were performed on DCT1, a nephron segment immediately adjacent to the glomerulus (Fig. 1, B and C). The isolated DCT was transferred onto a 5 × 5-mm coverglass coated with polylysine (Sigma) to immobilize the tubule, and the coverglass was placed in a chamber mounted on an inverted microscope (Nikon). The DCT was superfused with HEPES-buffered NaCl solution containing 140 mm NaCl, 5 mm KCl, 1.8 mm MgCl2, 1.8 mm CaCl2, and 10 mm HEPES (pH = 7.4) at room temperature.
FIGURE 1.

A, light microscope image showing an isolated DCT localized anatomically next to a thick ascending limb (TAL) and adjacent to the glomerulus. B, patch clamp pipette attached on the basolateral membrane of DCT1. C, the same tubule as shown in B, demonstrating that a pipette was placed on the basolateral membrane of DCT1.
Cell Culture and Transient Transfection
HEK293T cells (ATCC) were used for transient expression of FLAG-tagged or GFP-tagged KCNJ10 and GFP-tagged KCNJ16. The cells were grown in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen) in 5% CO2 and 95% air at 37 °C. Cells were grown to 50–70% confluence for transfection, and the corresponding cDNAs were applied simultaneously to the cells using TurboFect transfection reagent (Fermentas). Briefly, a cDNA mixture (0.5 μg of KCNJ10 for Kir4.1-transfected cells and 0.25 μg of KCNJ10/0.25 μg of KCNJ16 for Kir4.1/5.1-transfected cells) was diluted with 200 μl of serum-free DMEM and further mixed with 4 μl of TurboFect transfection reagent for the transfection of cells cultured in a 35-mm Petri dish. Cells transfected with the vector alone were used as a control, and their background currents were subtracted from that of the experimental groups. After 15 min of incubation at room temperature, the mixture of the transfection agents was applied to the cells, followed by an additional 24 h of incubation before use.
Liquid Chromatography and Mass Spectrometry
Proteins were prepared for LC/MS analysis, and phosphopeptides were enriched as described previously (10). Capillary LC/MS was performed on an Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) connected to a nanoAcquity UPLC (Waters, Milford, MA). Liquid chromatography was performed at 35 °C with a setup consisting of a commercially available 180 μm × 20 mm C18 nanoAcquity UPLC trap column and a BEH130C18 Waters symmetry 75-μm inner diameter × 250 mm capillary column packed with 5- and 1.7-μm particles, respectively. Mass spectrometry was performed with a spray voltage of 1.8 kV and a capillary temperature of 270 °C. A top 10 higher collisional energy dissociation method with one precursor survey scan (300–1750 Da) and up to 10 tandem MS spectra was performed with an isolation window of 2 Da and a normalized collision energy of 40 eV. The resolving power (at m/z = 400) of the Orbitrap was 30,000 for the precursor and 7500 for the fragment ion spectra, respectively. Raw files from the Orbitrap were processed with Mascot Distiller and searched in-house with Mascot (v. 2.3.02) against the NCBInr_20110627 protein database. The MASCOT search results were deposited in the Yale Protein Expression Database (18).
Purification of FLAG-KCNJ10
Cells cultured in 10- mm Petri dishes were transfected with 10 μg of pcDNA3-FLAG-KCNJ10 with TurboFect. After 24 h of incubation, the cells were treated with 1 ml of 1% PBST lysis buffer with protease and phosphatase inhibitors, followed by centrifugation at 13,000 rpm for 20 min at 4 °C. The supernatant was saved at −80 °C for the pull-down experiments in the next step. For harvesting FLAG-tagged KCNJ10 in the sample, 80 μl of anti-FLAG affinity gel (Sigma) was washed with PBS and added to the sample, and 4 μg of IgG conjugated beads were added to the sample as a control. The sample was gently rocked for 2 h at 4 °C and then subjected to centrifugation at 6000 rpm for 2 min to harvest KCNJ10 proteins.
Preparation of Protein Samples
The cells were placed in a lysis buffer containing 150 mm NaCl, 50 mm Tris HCl, 1% Nonidet P-40 (pH 8.0), and protease inhibitor mixture (1%) (Sigma). The prepared tissue was then homogenized and kept on ice for an additional 30 min. The sample was subjected to centrifugation at 13,000 rpm for 8 min at 4 °C, and protein concentrations were measured in duplicate using a Bio-Rad Dc protein assay kit.
Immunoprecipitation and Western Blot Analysis
The corresponding antibodies were added to the protein samples (500 μg) harvested from cell cultures with a ratio of 4:1 mg/1iter protein. The mixture was gently rotated at 4 °C overnight, followed by incubation with 25 μl of protein A/G plus agarose (Santa Cruz Biotechnology, Santa Cruz, CA) for an additional 2 h at 4 °C. The tube containing the mixture was centrifuged at 3000 rpm, and the agarose bead pellets were mixed with 25 μl of 2× SDS sample buffer containing 4% SDS, 100 mm Tris·HCl (pH 6.8), 20% glycerol, 200 mm DTT, and 0.2% bromphenol blue. After boiling the sample for 5 min, the proteins were resolved by electrophoresis on 8% SDS-polyacrylamide gels and then transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk in TBS and incubated overnight with the primary antibody at 4 °C. The membrane was then washed with 0.05% Tween 20-Tris-buffered saline three times followed by incubation for 30 min with the respective second antibody. After three washes, the membranes were scanned by an Odyssey infrared imaging system (LI-COR) at a wavelength of 680 or 800 nm.
Electrophysiology
Within 24 h after transfection, the cells were treated with trypsin-containing medium (TrypleEcpresscare) (Invitrogen) for 10 min to detach the cells. We followed the method described previously to prepare the cells for the patch clamp experiments (19). We carried out the perforated whole-cell patch clamp experiments at room temperature. The cells were incubated with a bath solution containing 140 mm KCl, 1.8 mm MgCl2, 1.8 mm CaCl2, and 10 mm HEPES (pH 7.4). Fluorescence signal (an indication of positive transfection) was detected with an intensified video imaging system, including a SIT 68 camera (Long Island Industries). Borosilicate glass (1.7-mm outer diameter) was used to make the patch clamp pipettes that were pulled with a Narishege electrode puller. The pipette had a resistance of 2–4 MΩ when filled with 140 mm KCl. The tip of the pipette was filled with pipette solution containing 140 mm KCl, 2 mm MgCl2, 1 mm EGTA, and 5 mm HEPES (pH 7.4). The pipette was then backfilled with amphotericin B (20 μg/0.1 ml) containing the pipette solution. After forming a high-resistance seal (> 2 GΩ), the membrane capacitance was monitored until the whole-cell patch configuration was formed. The cell membrane capacitance was measured and compensated. The K currents were measured by an Axon 200A patch clamp amplifier. The currents were low pass-filtered at 1 kHz, digitized by an Axon interface (Digidata 1200), and data were analyzed using the pClamp software system 9 (Axon).
The same patch clamp equipment described above was used for the single-channel recording, and the pipette solution contained 140 mm KCl, 1.8 mm MgCl2, 10 mm HEPES (pH = 7.4). The currents were low pass-filtered at 1 kHz and digitized by an Axon interface. Data were analyzed using the pClamp software system 9 (Axon). Channel activity defined as NPo (a product of channel number and open probability) was calculated from data samples of 60-s duration in the steady state as follows:
where ti is the fractional open time spent at each of the observed current levels. The channel conductance was determined by measuring the current amplitudes over several voltages. For the calculation of channel numbers, we selected a channel recording at least 10 min long. Because K current levels in HEK293T cells transfected with KCNJ10 and KCNJ16 were less than 6 and single-channel Po was close to 0.5, the channel numbers could be calculated by counting the highest K current levels in a 10-min single channel recording.
Biotinylation
The HEK293T cells were transfected with FLAG-tagged KCNJ10 and GFP-tagged KCNJ16 for 24 h. The cells were washed gently and incubated with 2 mg/ml EZ-Link Sulfo-NHS-SS-Biotin in 1× PBS for 1 h at 4 °C. The biotin-labeling was ended with 100 mm glycine in 1× PBS to absorb the unbound biotin. The cells were washed twice with 1× TBS and lysed with radioimmune precipitation assay buffer (100 mm Tris-HCl (pH7.4), 150 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate) in the presence of a protease inhibitor mixture and phosphatase inhibitor I and II (Sigma). After centrifugation, the protein sample (100 μg in 20 μl) was mixed with 20 μl of immobilized NeutrAvidinTM gel in 500 μl of radioimmune precipitation assay buffer and placed on a rocker overnight at 4 °C. The bead fraction was harvested, mixed with 2× sample buffer, boiled for 5 min, and resolved by electrophoresis. The biotin-labeled KCNJ10 was detected with FLAG or KCNJ10 antibody. The surface expression of KCNJ10 was normalized by calculating the ratio between the surface and the total expression of KCNJ10.
Experimental Materials and Statistics
Anti-tyrosine phosphorylation antibody (PY20), c-Src, and tubulin antibodies were purchased from Santa Cruz Biotechnology. Antibodies for KCNJ10, FLAG, GFP, and IgG were obtained from Sigma. All chemicals, including PP1 (4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]-pyrimidine) were purchased from Sigma. Collagenase type 2 was purchased from Worthington (Lakewood, NJ). The data are presented as mean ± S.E. We used Student t' test or one-way analysis of variance to determine the statistical significance. p < 0.05 was considered to be significant.
RESULTS
The patch clamp experiments were performed (140 mm NaCl in the bath and 140 mm KCl in the pipette) in the basolateral membrane of the isolated DCT1, which was determined by its unique location next to the glomerulus (Fig. 1B). In 350 patches with high seal resistance (> 1 GΩ), we identified a 39.5 ± 3 pS (n = 25) K channel in 178 patches. Moreover, this type of K channel was the only type of K channel in the basolateral membrane of the DCT1. Thus, we confirmed the previous report that the 40 pS K channel was the main type of K channels in the basolateral membrane of DCT1 (10). Fig. 2A is a typical channel recording showing that the K channel activity was not voltage-dependent between 0 and −60 mV and that the K channel current was typically inwardly rectifying. SFK has been shown to play a role in regulating K channels, such as ROMK, in the aldosterone-sensitive distal nephron (21, 22). To examine whether SFK also regulates the basolateral 40-pS K channels in the DCT1, we tested the effect of PP1, a specific inhibitor of SFK (23), on the 40-pS K channels in the DCT1. Fig. 3 is a recording showing that application of 100 nm PP1 inhibited the 40-pS K channel. From the inspection of Fig. 3, it is apparent that the inhibition of SFK not only reduced channel activity (NPo) from 2.9 ± 0.4 to 0.2 ± 0.1 (n = 10) but also decreased K current amplitude, suggesting that inhibition of the 40-pS K channel caused membrane depolarization. However, inhibition of SFK did not alter channel conductance (supplemental Fig. S1). Fig. 2B is an I (current)/V (voltage) curve demonstrating that the channel conductance was about 39.6 ± 3 pS in the presence of PP1. However, inhibition of SFK shifted the I/V curve to the left (depolarization). The effect of PP1 was reversible because washout restored the channel activity. Therefore, the results support the role of SFK in stimulating basolateral K channel activity.
FIGURE 2.

A, recording showing the activity of the basolateral 40-pS K channel in DCT1 at clamping potentials from 0 mV to −60 mV with a 20-mV step. The experiments were performed in a cell-attached patch with 140 mm KCl in the patch pipette and 140 mm NaCl/5 mm KCl in the bath. The channel closed level (c) is indicated by a dotted line. B, I/V curve obtained in the absence (Control, ●) and presence of PP1 (○).
FIGURE 3.

A channel recording demonstrates the effect of SFK inhibitor (PP1) on the basolateral K channel activity in the DCT1. The top trace shows the time course of the experiments, and three parts of the recording indicated by numbers are extended to show the fast time resolution. The channel closed level (c) is indicated by a dotted line. The experiments were performed in a cell-attached patch with 140 mm NaCl/5 mm KCl in the bath and 140 mm KCl in the pipette. The shift of the base line in the top trace was induced by changing the bath solution.
After demonstrating that suppression of SFK inhibited the 40-pS K channel, we used perforated whole-cell recordings to measure Ba2+-sensitive whole-cell K currents under control conditions (without PP1) and in the presence of PP1. Because we (data not shown) and others failed to detect any functional K channel activity in the apical membrane of DCT1 (8), the K currents measured with a perforated whole-cell recording in DCT1 cells should represent the whole population of basolateral 40-pS K channels. Fig. 4A shows a set of whole-cell recordings under control conditions (without PP1) and in the presence of 100 nm PP1 (lower panel). The whole-cell K currents in DCT1 show a typical inwardly rectifying characteristic. Moreover, inhibition of SFK decreased the Ba2+-sensitive inward K currents at −60 mV from 1296 ± 205pA (n = 10) under control conditions to 330 ± 144 pA (n = 8) (Fig. 4B). Therefore, the results obtained from the whole-cell recordings are consistent with the finding made in the single-channel recordings and support the role of SFK in regulating basolateral K channels in DCT1.
FIGURE 4.

A, whole-cell recording showing the Ba2+-sensitive K currents measured with perforated patches under control conditions and in the presence of 100 nm PP1, an SFK inhibitor. The bath solution and the pipette solution contained a symmetrical 140 mm KCl. B, bar graph summarizing the results of experiments in which K currents were measured with perforated whole-cell patches at −60 mV under control conditions and in the presence of an SFK inhibitor (100 nm PP1). #, significant difference. nA, nanoamperes. C, the Ba2+-sensitive K currents measured with the perforated whole-cell recording from −160 mV to 40 mV under control conditions and in the presence of 100 nm PP1. DCT1 was bathed in a solution containing 140 mm NaCl/5 mm KCl, whereas the pipette solution contained 140 mm KCl.
Because the 40-pS K channel is the only type of K channel expressed in the basolateral membrane of DCT1, inhibition of the 40-pS K channel activity is expected to depolarize the cell membrane potential in DCT1. This hypothesis was confirmed by measuring the K reversal potential in DCT1 with ramp voltage from −160 to 40 mV. Fig. 4C is a recording showing the I/V curve with 140 mm NaCl/5 mm KCl in the bath and 140 mm KCl in the pipette. Under control conditions, the K reversal potential was −65 ± 5 mV, whereas inhibition of SFK with 100 nm PP1 shifted the K reversal potential to −43 ± 8 mV (n = 6). This is consistent with the results reported in Fig. 2B in which inhibition of SFP shifted the I/V curve to the right (depolarization) about 20 mV. Hence, the results suggest that SFK plays an important role in determining the basolateral 40-pS K channel activity and the cell membrane potential in DCT1.
Immunostaining and electrophysiological studies have indicated that KCNJ10 is an important component of the basolateral K channels in the DCT (12, 15). Thus, we hypothesize that SFK may regulate the basolateral K channels in DCT1 by phosphorylating KCNJ10. Thus, we used a tyrosine phosphorylation antibody to examine whether c-Src phosphorylates KCNJ10. KCNJ10 proteins were harvested in HEK293T cells transfected with FLAG-tagged-KCNJ10 by immunoprecipitation of the cell lysates with FLAG antibody. Tyrosine-phosphorylated KCNJ10 was detected with anti-tyrosine phosphorylation antibody (PY20). Fig. 5 shows a Western blot analysis demonstrating that KCNJ10 is a tyrosine-phosphorylated protein and that the expression of c-Src increased tyrosine phosphorylation of KCNJ10. We followed the methods described previously and used mass spectrometry to identify phosphorylation sites of KCNJ10 (24). We isolated FLAG-tagged KCNJ10 from HEK293T cells, digested the protein with trypsin, and analyzed the peptides with LC/MS. We observed significant peptides that covered 25% of the protein sequence and unambiguously identified two phosphotyrosine residues at positions 8 (Tyr8) (supplemental Fig. S2) and 9 (Tyr9) in the amino terminus of KCNJ10 (Fig. 6).
FIGURE 5.
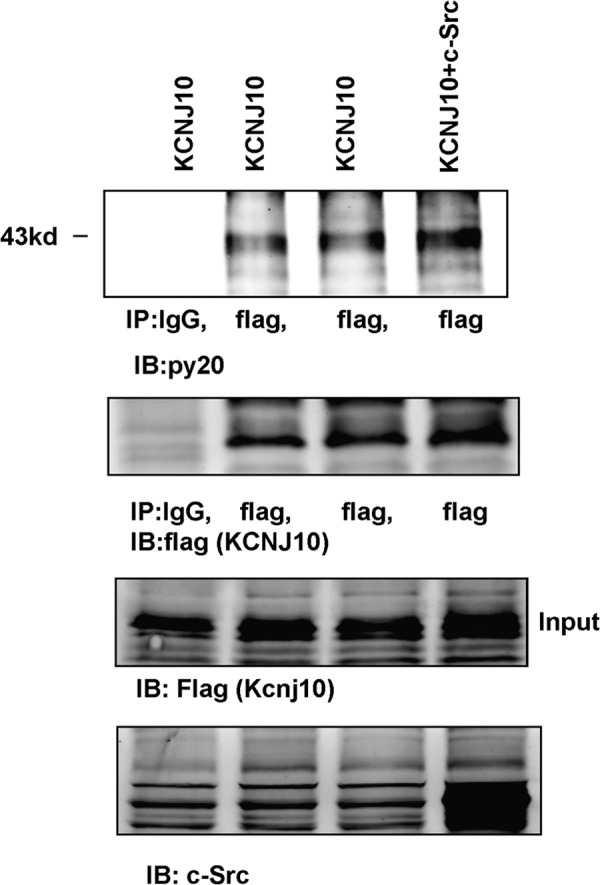
Shown is a Western blot analysis demonstrating the tyrosine phosphorylation of KCNJ10 in the absence or presence of c-Src. HEK293T cells were transfected with FLAG-tagged KCNJ10 or KCNJ10 + c-Src. KCNJ10 proteins were harvested with FLAG antibody, and tyrosine-phosphorylated KCNJ10 proteins were detected with PY20. The expression of KCNJ10 and c-Src is shown in the two lower panels. IP, immunoprecipitation; IB, immunoblot.
FIGURE 6.

LC/MS workflow for KCNJ10 phosphotyrosine identification. Immunoprecipitated KCNJ10 was digested with trypsin, and phosphopeptides were enriched with titanium dioxide (TiO2) and identified with LC/MS. A representative annotated tandem MS spectrum and sequence coverage (y and b ions) are shown for the KCNJ10 phosphopeptide VYYPSQTTQTESRPLMoxGPGIR (m/z = 794.03).
After demonstrating that KCNJ10 is a tyrosine-phosphorylated protein, we examined the role of tyrosine phosphorylation in regulating KCNJ10 activity in HEK293T cells transfected with WT or KCNJ10Y9F. We detected a 19 ± 1-pS K channel in the cells transfected with WT KCNJ10 but not in the vector-transfected cells (Fig. 7A). Thus, our finding is in agreement with the report that the conductance of the basolateral K channel in DCT is about 20 pS in KCNJ16 knockout mice in whom four KCNJ10 monomers presumably form a homotetramer (15). We also used perforated whole-cell recordings to measure the Ba2+-sensitive K currents in the cells transfected with WT or KCNJ10 mutants in which Tyr8 (KCNJ10Y8F), Tyr9 (KCNJ10Y9F), or Tyr8/9 (KCNJ10Y8/9F) were each mutated to phenylalanine. Fig. 8A shows a recording of a typical inwardly rectifying K current with symmetrical 140 mm KCl solution in the bath and pipette. It is apparent that the K currents in the cells transfected with either KCNJ10Y9F or KCNJ10Y8/9F were significantly lower than those with WT KCNJ10, whereas the protein expression of KCNJ10Y9F and KCNJ10Y8/9F was similar to those of WT KCNJ10. The results summarized in Fig. 8B demonstrate that K currents in cells transfected with KCNJ10Y9F and KCNJ10Y8/9F were 57 ± 14 pA/pF and 80 ± 20 pA/pF (n = 6), a value significantly lower than in those transfected with WT KCNJ10 (290 ± 40 pA/pF, n = 6). The mutation of the Tyr9-induced decrease in whole-cell K currents was specific because of the K currents measured in the cells transfected with KCNJ10Y8F (270 ± 40 pA/pF, n = 6). KCNJ10Y51F (295 ± 42 pA/pF, n = 6) and KCNJ10Y349F (310 ± 50 pA/PF, n = 6) were similar to those of WT K channels (supplemental Fig. S3 and Fig. 8B).
FIGURE 7.

A recording showing the representative K channel activity in HEK293T cells transfected with GFP-tagged KCNJ10 (A) or with KCNJ10Y9F (B). The experiments were performed in a cell-attached patch with 140 mm KCl in the patch pipette and 140 mm NaCl/5 mm KCl in the bath. The channel closed level is indicated by a dotted line, and the clamping potential is indicated at the top of each trace. C, bar graph showing the total number of patches with channel activity (gray bar) and without K channel activity (black bar) in cells transfected with WT or KCNJ10Y9F. D, bar graph showing the mean channel activity (NPo) in cells transfected with WT (n = 23) or KCNJ10Y9F (n = 14).
FIGURE 8.

A, a set of recordings showing K currents measured with the perforated whole-cell recording in HEK293T cells transfected with WT KCNJ10 and KCNJ10 mutants. The pipette solution and the bath solution contained a symmetrical 140 mm KCl, and the K currents were measured from −60 to 60 mV at 20-mV steps (the protocol of the voltage clamp is included). Also shown are Western blot analyses showing the expression of KCNJ10 and its mutants (bottom panel). A Western blot analysis of α-tubulin was used as a loading control. IB, immunoblot. B, bar graph summarizing the results of experiments in which Ba2+-sensitive K currents were measured at −60 mV with a perforated whole-cell recording in the cells transfected with KCNJ10 or the mutant.
To examine whether the mutation of the Tyr9-induced decrease in K currents was due to diminishing K channel conductance or channel activity (NPo), we examined the K channel activity in the cells transfected with WT or KCNJ10Y9F. The mutation of Tyr9 did not significantly affect K channel conductance (18.5 ± 1 pS) in comparison to that of the WT (Fig. 7B), but it significantly decreased the probability of finding the 19-pS K channels in the KCNJ10Y9F-transfected cells. We observed 19-pS K channels in WT KCNJ10-transfected cells in 23 of 41 total patches (56%), whereas this K channel was only observed in 14 of 45 total patches (31%) in cells transfected with KCNJ10Y9F (Fig. 7C). Moreover, the mean NPo per patch with the 19-pS K channel was also significantly lowered (1.0 ± 0.15) in cells transfected with KCNJ10Y9F than in those with the WT KCNJ10 ((1.59 ± 0.1) (Fig. 7D). Thus, the decrease in K currents of cells transfected with KCNJ10Y9F was possibly induced by decreasing the channel open probability and reducing the number of active K channels in the plasma membrane.
To test the role of Tyr9 phosphorylation in the regulation of the basolateral K channel in the DCT, we extended the study by examining the channel activity in cells transfected with KCNJ10 + KCNJ16 because the basolateral K channel in the native DCT is a heterotetramer of Kir4.1/Kir5.1 (10). Fig. 9 shows a single-channel recording in HEK293T cells transfected with KCNJ10/KCNJ16 or KCNJ10Y9F/KCNJ16. The experiments were performed in cell-attached patches with 140 mm NaCl/5 mm KCl in the bath and 140 mm KCl in the pipette. We confirmed a previous report that coexpression of KCNJ10 and KCNJ16 formed a 40-pS K channels (10). Fig. 10A shows an I/V curve of WT and the mutant heterotetramer (KCNJ10Y9F + KCNJ16). It is apparent that the mutation of Tyr9 did not affect single K channel conductance. Moreover, the analysis of the single-channel recording in those patches with K channel activity showed that the mean channel open probability (Po) in the cells transfected with KCNJ10Y9F + KCNJ16 (0.55 ± 0.1, n = 5) was not significantly lower than those with WT K channels (0.75 ± 0.1, n = 6) (Fig. 10B). However, as observed in the cells transfected with KCNJ10 alone, the probability of finding the 40-pS K channel in KCNJ10Y9F + KCNJ16-transfected cells was lower than in those with KCNJ10 + KCNJ16. We observed a 40-pS K channel in WT heterotetramer-transfected cells in 13 of 26 total patches (50%), whereas this K channel was only observed in 9 of 33 total patches (27%) in the cells transfected with KCNJ10Y9F + KCNJ16 (Fig. 10C). Thus, the mean number per patch in the cells transfected with KCNJ10 + KCNJ16 (1.8 ± 0.4, n = 26) was significantly higher than in those with the mutant (0.8 ± 0.25, n = 33). The whole-cell K currents in the cells transfected with KCNJ10Y9F + KCNJ16 (161 ± 12 pA/pF) were also significantly lower than in those with WT K channels (440 ± 30 pA/pF, n = 7) (Fig. 11A). The notion that the mutation of Tyr9 of KCNJ10 decreased the functional K channel number in the plasma membrane is also supported by biotin surface labeling. Fig. 11B shows a Western blot analysis showing the expression of KCNJ10 in the plasma membrane of the cells transfected with FLAG-tagged KCNJ10 and GFP-tagged KCNJ16. Although the total expression of KCNJ10 and KCNJ16 was similar in WT or in the mutant-transfected cells, the expression of KCNJ10 in the plasma membrane of the mutant-transfected cells was lower than those in WT heterotetramer-transfected cells. In four such experiments, the normalized surface expression of KCNJ10 in cells transfected with mutant Kir4.1/5.1 was only 40 ± 8% (p < 0.05) of those with WT K channels (data were normalized in comparison with tubulin). Thus, a Tyr9 mutation-induced decrease in K channel activity results, at least partially, from a decrease in K channel number in the plasma membrane.
FIGURE 9.

A recording showing the representative K channel activity in HEK293T cells transfected with FLAG-tagged KCNJ10 and GFP-tagged KCNJ16 (left panel) or with KCNJ10Y9F and KCNJ16 (right panel). The experiments were performed in a cell-attached patch with 140 mm KCl in the patch pipette and 140 mm NaCl/5 mm KCl in the bath. The channel closed level is indicated by a dotted line, and the clamping potential is indicated at the top of the each trace. A short bar indicates each K channel level.
FIGURE 10.

A, I/V curve obtained in the cells transfected with FLAG-tagged KCNJ10 and GFP-tagged KCNJ16 (●) or with KCNJ10Y9F and KCNJ16 (○). B, bar graph showing the mean channel open probability (Po) of the WT and the mutant heterotetramer, respectively. NS, not significant. C, bar graph showing the total number of the patches with channel activity (gray bar) and without K channel activity (black bar) in cells transfected with KCNJ10/KCNJ16 or KCNJ10Y9F/KCNJ16. D, mean channel number per patch in the cells transfected with KCNJ10/KCNJ16 or KCNJ10Y9F/KCNJ16.
FIGURE 11.

A, whole-cell recording showing the Ba2+-sensitive K currents measured from −60 to 60 mV in the cells transfected with KCNJ10/KCNJ16 or KCNJ10Y9F/KCNJ16. The results are summarized in a bar graph (bottom panel). B, Western blotting demonstrating the expression of biotin-labeled KCNJ10, total KCNJ10, KCNJ16, and α-tubulin in HEK293T cells transfected with FLAG-tagged KCNJ10/mutant and GFP-tagged KCNJ16. The number of experiments was four. IP, immunoprecipitation; IB, immunoblot.
DISCUSSION
This study confirmed the previous finding that the inwardly rectifying 40-pS K channel is the only type of K channel expressed in the basolateral membrane of DCT1(10). The basolateral K channels in DCT1 are responsible for maintaining K recycling across the basolateral membrane and participate in generating cell membrane potential, which provides the driving force for Cl exit across the basolateral membrane. Thus, it is conceivable that basolateral K channel activity should play a role in regulating transepithelial Cl movement in the DCT1 so that a high basolateral K channel activity stimulates Cl secretion, whereas a low basolateral K channel activity inhibits it. Relevant to this notion is the observation that inhibition of basolateral membrane K conductance decreased the short-circuit current and the net rate of transepithelial Cl movement in the airway epithelium (25). Because the intracellular Cl concentration has been shown to affect Na-coupled Cl transporters (26), it is possible that basolateral K channel activity may also regulate NCC activity through altering the intracellular Cl levels in the DCT. Hence, the basolateral K channels are critically involved in the maintenance of the trans-epithelial transport in the DCT.
Three lines of evidence indicate that SFK plays an important role in regulating the basolateral 40-pS K channels. First, the inhibition of SFK decreased the 40-pS K channel activity defined by NPo. Second, inhibition of SFK reduced the whole-cell Ba2+-sensitive K currents in DCT1. Because no functional apical K channel activity was detected in DCT1, a decrease in the whole-cell K currents induced by inhibiting SFK must be the result of inhibiting the basolateral K channels. Third, inhibiting SFK shifted the K reversal potential of DCT1 from −65 mV to −43 mV, an indication of membrane depolarization. This is consistent with the notion that the basolateral 40-pS K channel is the main K channel type in DCT1. Thus, SFK-mediated regulation of the 40-pS K channel plays an important role in determining membrane potential in DCT1. In contrast to the effect of SFK on the basolateral 40-pS K channels in DCT1, SFK has been shown to inhibit ROMK channels in the CCD (16, 27–29). This effect on ROMK is achieved through direct phosphorylation and by abolishing the stimulatory effect of serum glucocorticoid-inducible kinase 1 (SGK1) on with-no-lysine (K) kinase 4 (WNK4), which inhibits ROMK (16, 19, 30). We speculate that the different responses of ROMK and basolateral 40-pS K channels to SFK may play a role in regulating renal K secretion. For instance, a high K intake has been shown to suppress the expression of SFK in the kidney (31). A decrease in SFK activity in the DCT1 is expected to inhibit basolateral 40-pS K channels, thereby depolarizing cell membrane potential. A depolarization in the basolateral membrane should lead to a decrease in the driving force for Cl exit in the basolateral membrane, thereby inhibiting the interaction between WNK and Ste20-related proline/alanine-rich kinase (SPAK) (26), which is required for the activation of the NCC (32, 33). Hence, a decrease in basolateral K channel activity should inhibit Na absorption and increase Na delivery to the connecting tubule and the CCD. Accordingly, it stimulates K secretion in the connecting tubule/CCD. On the other hand, the down-regulation of SFK activity induced by high K intake is expected to stimulate ROMK channels (34). Thus, SFK may function to synchronize apical ROMK channel activity in the connecting tubule/CCD and basolateral 40-pS K channels in the DCT to stimulate renal K secretion.
A large body of evidence indicates that the basolateral 40-pS K channel is composed of KCNJ10 and KCNJ16, which form a heterotetramer in the basolateral membrane of the DCT (10, 11, 13). First, Kir4.1 and Kir5.1 proteins are highly expressed in the basolateral membrane of the DCT (10, 12). Second, coexpression of Kir4.1 and Kir5.1 in HEK293 cells (35) and in Xenopus oocytes (14) formed a K channel sharing the biophysical properties of the basolateral 40-pS K channel in the DCT. We also showed that coexpression of KCNJ10 and KCNJ16 formed a 40-pS K channel. Third, an immunostaining study has shown that both Kir4.1 and the calcium-sensing receptor were colocalized in the basolateral membrane of the DCT (36). The significance of Kir4.1 in regulating membrane transport in the DCT is demonstrated in patients with SeSAME or EAST (epilepsy, ataxia, sensorineural deafness, and tubulopathy) syndrome, characterized by seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (15,33). The renal phenotypes of SeSAME syndrome are hypokalemia, metabolic alkalosis and hypomagnesemia(37). Hypomagnesemia should be the result of the decrease in the driving force for Mg2+ entry across the apical membrane because of the depolarization of the DCT, whereas hypokalemia results from compromised NCC activity induced by the diminished Cl exit across the basolateral membrane. Consequently, delivery of NaCl to both the connecting tubule and the CCD is increased, and it leads to enhanced Na absorption at the expense of increased K secretion.
The observation that KCNJ10 is phosphorylated by SFK suggests that the stimulatory effect of SFK on the basolateral 40-pS K channel is achieved, at least in part, by phosphorylation of KCNJ10 at Tyr9. Although LS/MS analysis identified the phosphorylation of KCNJ10 at Tyr8 and Tyr9, two lines of evidence suggest that phosphorylation of Tyr9 is essential for the effect of SFK on the basolateral 40-pS K channels. First, mutation of Tyr9 decreased K currents in the cells transfected with KCNJ10 alone or KCNJ10 + KCNJ16. Second, the probability of finding KCNJ10 was significantly lower in cells transfected with KCNJ10Y9F than those with WT KCNJ10. Furthermore, the effect of SFK on the basolateral 40-pS K channel is, at least in part, achieved by increasing surface K channel expression. This conclusion is supported by the observation that the surface expression of KCNJ10 is significantly lower in cells transfected with KCNJ10Y9F than those with the WT. However, it is also possible that mutation of Tyr9 of KCNJ10 may also have a minor effect on the channel open probability.
Although the role of SFK in stimulating basolateral K channels in the DCT1 was established, the physiological stimuli that activate SFK and regulate the basolateral K channels are not known. Angiotensin II has been shown not only to play an important role in regulating the NCC in the DCT but also to stimulate SFK activity (20, 38). Therefore, it is possible that angiotensin II may stimulate the basolateral K channel activity by an SFK-dependent pathway. Further experiments are required to test this hypothesis. In conclusion, the basolateral 40-pS K channel is phosphorylated by SFK and Tyr9 of KCNJ10 and is one of the important sites that regulate the basolateral K channel activity in DCT1.
Acknowledgment
We thank Dr. Ute Scholl (laboratory of Dr. R. Lifton) for providing the KCNJ10/16 construct.
This work is supported, in whole or in part, by National Institutes of Health Grants DK54983 (to W. H. W.) and K01DK089006 (to J. R.). This work was also supported by Chinese National Natural Science Foundation Grant 31171109 (to C. Z.).

This article contains supplemental Figs. S1–S3.
- DCT
- distal convoluted tubule
- NCC
- NaCl cotransporter
- SFK
- Src family protein tyrosine kinase
- I/V
- voltage/current
- SeSAME
- seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance
- pS
- picosiemens
- pF
- picofarads
- CCD
- cortical collecting duct
- PP1
- 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]-pyrimidine.
REFERENCES
- 1. Ellison D. H., Valazquez H., Wright F. S. (1987) Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am. J. Physiol. Renal Physiol. 253–22:F546–FF554 [DOI] [PubMed] [Google Scholar]
- 2. Ellison D. H., Velázquez H., Wright F. S. (1989) Adaptation of the distal convoluted tubule of the rat. Structural and functional effects of dietary salt intake and chronic diuretic infusion. J. Clin. Invest. 83, 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Obermüller N., Bernstein P., Velázquez H., Reilly R., Moser D., Ellison D. H., Bachmann S. (1995) Expression of the thiazide-sensitive Na-Cl cotransporter in rat and human kidney. Am. J. Physiol. 269, F900–F910 [DOI] [PubMed] [Google Scholar]
- 4. Gamba G. (1999) Molecular biology of distal nephron sodium transport mechanisms. Kidney Int. 56, 1606–1622 [DOI] [PubMed] [Google Scholar]
- 5. Wade J. B., Fang L., Coleman R. A., Liu J., Grimm P. R., Wang T., Welling P. A. (2011) Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. Am. J. Physiol. Renal Physiol. 300, F1385–F1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schmitt R., Ellison D. H., Farman N., Rossier B. C., Reilly R. F., Reeves W. B., Oberb+ñumer I., Tapp R., Bachmann S. (1999) Developmental expression of sodium entry pathways in rat nephron. Am. J. Physiol. Renal Physiol. 276, F367–F381 [DOI] [PubMed] [Google Scholar]
- 7. Bernstein P. L., Ellison D. H. (2011) Diuretics and salt transport along the nephron. Semin. Nephrol. 31, 475–482 [DOI] [PubMed] [Google Scholar]
- 8. Hebert S. C., Desir G., Giebisch G., Wang W. (2005) Molecular diversity and regulation of renal potassium channels. Physiol. Rev. 85, 319–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schultz S. G. (1981) Homocellular regulatory mechanisms in sodium-transporting epithelia: avoidance of extinction by “flush-through”. Am. J. Physiol. 241, F579–F590 [DOI] [PubMed] [Google Scholar]
- 10. Lourdel S., Paulais M., Cluzeaud F., Bens M., Tanemoto M., Kurachi Y., Vandewalle A., Teulon J. (2002) An inward rectifier K+ channel at the basolateral membrane of the mouse distal convoluted tubule. Similarities with Kir4-Kir5.1 heteromeric channels. J. Physiol. 538, 391–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Williams D. M., Lopes C. M., Rosenhouse-Dantsker A., Connelly H. L., Matavel A., O-Uchi J., McBeath E., Gray D. A. (2010) Molecular basis of decreased Kir4.1 function in SeSAME/EAST syndrome. J. Am. Soc. Nephrol. 21, 2117–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lachheb S., Cluzeaud F., Bens M., Genete M., Hibino H., Lourdel S., Kurachi Y., Vandewalle A., Teulon J., Paulais M. (2008) Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am. J. Physiol. Renal Physiol. 294, F1398–F1407 [DOI] [PubMed] [Google Scholar]
- 13. Cha S. K., Huang C., Ding Y., Qi X., Huang C. L., Miller R. T. (2011) Calcium-sensing receptor decreases cell surface expression of the inwardly rectifying K+ channel Kir4.1. J. Biol. Chem. 286, 1828–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pessia M., Tucker S. J., Lee K., Bond C. T., Adelman J. P. (1996) Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J. 15, 2980–2987 [PMC free article] [PubMed] [Google Scholar]
- 15. Paulais M., Bloch-Faure M., Picard N., Jacques T., Ramakrishnan S. K., Keck M., Sohet F., Eladari D., Houillier P., Lourdel S., Teulon J., Tucker S. J. (2011) Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 10361–10366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin D. H., Yue P., Rinehart J., Sun P., Wang Z., Lifton R., Wang W. H. (2012) Protein phosphatase 1 modulates the inhibitory effect of with-no-lysine kinase 4 on ROMK channels. Am. J. Physiol. Renal Physiol. 303, F110–F119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin D. H., Sterling H., Yang B., Hebert S. C., Giebisch G., Wang W. H. (2004) Protein tyrosine kinase is expressed and regulates ROMK1 location in the cortical collecting duct. Am. J. Physiol. Renal Physiol. 286, F881–F892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shifman M. A., Li Y., Colangelo C. M., Stone K. L., Wu T. L., Cheung K. H., Miller P. L., Williams K. R. (2007) YPED. A web-accessible database system for protein expression analysis. J. Proteome Res. 6, 4019–4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yue P., Lin D. H., Pan C. Y., Leng Q., Giebisch G., Lifton R. P., Wang W. H. (2009) Src family protein tyrosine kinase (PTK) modulates the effect of SGK1 and WNK4 on ROMK channels. Proc. Natl. Acad. Sci. U.S.A. 106, 15061–15066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei Y., Zavilowitz B., Satlin L. M., Wang W. H. (2007) Angiotensin II inhibits the ROMK-like small-conductance K channel in renal cortical collecting duct during dietary K restriction. J. Biol. Chem. 282, 6455–6462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moral Z., Dong K., Wei Y., Sterling H., Deng H., Ali S., Gu R., Huang X. Y., Hebert S. C., Giebisch G., Wang W. H. (2001) Regulation of ROMK1 channels by protein-tyrosine kinase and -tyrosine phosphatase. J. Biol. Chem. 276, 7156–7163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin D. H., Sterling H., Yang B., Hebert S. C., Giebisch G., Wang W. H. (2004) Protein tyrosine kinase is expressed and regulates ROMK1 location in the cortical collecting duct. Am. J. Physiol. Renal Physiol. 286, F881–F892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hanke J. H., Gardner J. P., Dow R. L., Changelian P. S., Brissette W. H., Weringer E. J., Pollok B. A., Connelly P. A. (1996) Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem. 271, 695–701 [DOI] [PubMed] [Google Scholar]
- 24. Rinehart J., Maksimova Y. D., Tanis J. E., Stone K. L., Hodson C. A., Zhang J., Risinger M., Pan W., Wu D., Colangelo C. M., Forbush B., Joiner C. H., Gulcicek E. E., Gallagher P. G., Lifton R. P. (2009) Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138, 525–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Welsh M. J. (1983) Barium inhibition of basolateral membrane potassium conductance in tracheal epithelium. Am. J. Physiol. Renal Physiol. 244, F639–F645 [DOI] [PubMed] [Google Scholar]
- 26. Ponce-Coria J., San-Cristobal P., Kahle K. T., Vazquez N., Pacheco-Alvarez D., de Los Heros P., Juárez P., Muñoz E., Michel G., Bobadilla N. A., Gimenez I., Lifton R. P., Hebert S. C., Gamba G. (2008) Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc. Natl. Acad. Sci. U.S.A. 105, 8458–8463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin D., Kamsteeg E. J., Zhang Y., Jin Y., Sterling H., Yue P., Roos M., Duffield A., Spencer J., Caplan M., Wang W. H. (2008) Expression of tetraspan protein CD63 activates protein-tyrosine kinase (PTK) and enhances the PTK-induced inhibition of ROMK channels. J. Biol. Chem. 283, 7674–7681 [DOI] [PubMed] [Google Scholar]
- 28. Sterling H., Lin D. H., Gu R. M., Dong K., Hebert S. C., Wang W. H. (2002) Inhibition of protein-tyrosine phosphatase stimulates the dynamin-dependent endocytosis of ROMK1. J. Biol. Chem. 277, 4317–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang W., Lerea K. M., Chan M., Giebisch G. (2000) Protein tyrosine kinase regulates the number of renal secretory K channels. Am. J. Physiol. Renal Physiol. 278, F165–F171 [DOI] [PubMed] [Google Scholar]
- 30. Lin D. H., Sterling H., Lerea K. M., Welling P., Jin L., Giebisch G., Wang W. H. (2002) K depletion increases the protein tyrosine-mediated phosphorylation of ROMK. Am. J. Physiol. Renal Physiol. 283, F671–F677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wei Y., Bloom P., Lin D., Gu R., Wang W. H. (2001) Effect of dietary K intake on the apical small-conductance K channel in the CCD. Role of protein tyrosine kinase. Am. J. Physiol. Renal Physiol. 281, F206–F212 [DOI] [PubMed] [Google Scholar]
- 32. San-Cristobal P., Pacheco-Alvarez D., Richardson C., Ring A. M., Vazquez N., Rafiqi F. H., Chari D., Kahle K. T., Leng Q., Bobadilla N. A., Hebert S. C., Alessi D. R., Lifton R. P., Gamba G. (2009) Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc. Natl. Acad. Sci. U.S.A. 106, 4384–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reichold M., Zdebik A. A., Lieberer E., Rapedius M., Schmidt K., Bandulik S., Sterner C., Tegtmeier I., Penton D., Baukrowitz T., Hulton S. A., Witzgall R., Ben-Zeev B., Howie A. J., Kleta R., Bockenhauer D., Warth R. (2010) KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc. Natl. Acad. Sci. U.S.A. 107, 14490–14495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang W., Lerea K. M., Chan M., Giebisch G. (2000) Protein tyrosine kinase regulates the number of renal secretory K channel. Am. J. Physiol. Renal Physiol. 278, F165–F171 [DOI] [PubMed] [Google Scholar]
- 35. Tanemoto M., Kittaka N., Inanobe A., Kurachi Y. (2000) In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J. Physiol. 525, 587–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang C., Sindic A., Hill C. E., Hujer K. M., Chan K. W., Sassen M., Wu Z., Kurachi Y., Nielsen S., Romero M. F., Miller R. T. (2007) Interaction of the Ca2+-sensing receptor with the inwardly rectifying potassium channels Kir4.1 and Kir4.2 results in inhibition of channel function. Am. J. Physiol. Renal Physiol. 292, F1073–F1081 [DOI] [PubMed] [Google Scholar]
- 37. Scholl U. I., Choi M., Liu T., Ramaekers V. T., Häusler M. G., Grimmer J., Tobe S. W., Farhi A., Nelson-Williams C., Lifton R. P. (2009) Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. U.S.A. 106, 5842–5847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Castañeda-Bueno M., Cervantes-Pérez L. G., Vázquez N., Uribe N., Kantesaria S., Morla L., Bobadilla N. A., Doucet A., Alessi D. R., Gamba G. (2012) Activation of the renal Na:Cl cotransporter by angiotensin II is a WNK4-dependent process. Proc. Natl. Acad. Sci. U.S.A. 109, 7929–7934 [DOI] [PMC free article] [PubMed] [Google Scholar]