

Background: Glioma cancer stem cells (CSCs) are believed to drive tumorigenesis.
Results: Glioma CSCs show constitutive activation of the STAT3/NF-κB signaling pathway and the Notch pathway.
Conclusion: A novel relationship between glioma CSCs and the Notch pathway is defined, involving the constitutive activation of STAT3 and NF-κB signaling.
Significance: The STAT3, NF-κB, and Notch pathways provide novel therapeutic targets to treat glioma.
Keywords: Cancer Stem Cells, Glioblastoma, NF-κB (NF-κB), Notch Pathway, STAT3, Glioma
Abstract
Malignant gliomas are locally aggressive, highly vascular tumors that have a dismal prognosis, and present therapies provide little improvement in the disease course and outcome. Many types of malignancies, including glioblastoma, originate from a population of cancer stem cells (CSCs) that are able to initiate and maintain tumors. Although CSCs only represent a small fraction of cells within a tumor, their high tumor-initiating capacity and therapeutic resistance drives tumorigenesis. Therefore, it is imperative to identify pathways associated with CSCs to devise strategies to selectively target them. In this study, we describe a novel relationship between glioblastoma CSCs and the Notch pathway, which involves the constitutive activation of STAT3 and NF-κB signaling. Glioma CSCs were isolated and maintained in vitro using an adherent culture system, and the biological properties were compared with the traditional cultures of CSCs grown as multicellular spheres under nonadherent culture conditions. Interestingly, both adherent and spheroid glioma CSCs show constitutive activation of the STAT3/NF-κB signaling pathway and up-regulation of STAT3- and NF-κB-dependent genes. Gene expression profiling also identified components of the Notch pathway as being deregulated in glioma CSCs, and the deregulated expression of these genes was sensitive to treatment with STAT3 and NF-κB inhibitors. This finding is particularly important because Notch signaling appears to play a key role in CSCs in a variety of cancers and controls cell fate determination, survival, proliferation, and the maintenance of stem cells. The constitutive activation of STAT3 and NF-κB signaling pathways that leads to the regulation of Notch pathway genes in glioma CSCs identifies novel therapeutic targets for the treatment of glioma.
Introduction
Primary brain tumors occur in approximately one of 5000 Americans and represent an important cause of cancer-related morbidity and mortality in the United States. About half of all brain tumors are gliomas. Malignant gliomas are locally aggressive, highly vascular tumors that are very difficult to treat. The median survival for patients with glioblastoma multiforme (GBM),2 the most common histological subtype of glioma in adults, has remained at less than one year for several decades (1). Surgical resection of GBM is the primary treatment modality because present adjuvant therapies provide little improvement in the disease course and outcome.
For many cancers the tumorigenic process may be initiated and sustained by a rare, stem cell-like subpopulation, denoted cancer stem cells (CSCs) (2). These cells behave similarly to normal stem cells in that they can self-renew, but they form tumors upon serial transplantation into host mice and recapitulate the tumor phenotype (3). CSCs are often generated on the basis of their ability to grow as multicellular, nonadherent spheres from single cell suspensions (4, 5), which are denoted as spheroid CSCs. However, expansion of spheroid CSCs is technically challenging and expensive, and as spheres enlarge, they become hypoxic, and dying cells accumulate in the core of the sphere. As an alternative approach, adherent CSCs grown in laminin-coated tissue culture flasks have been found to display stem cell properties and initiate high-grade gliomas following xenotransplantation (6). The ineffectiveness of current cancer therapies is believed to reflect their lack of activity against the CSCs, which remain viable despite therapy and, thus, retain their capacity to regenerate tumors. Therefore, it is critical to define the pathways selectively activated in CSCs to specifically target them.
Transcription factors found constitutively activated in CSCs include STAT3 and NF-κB (7, 8). STAT3 was originally identified as the transcription factor responsible for the induction of acute-phase response genes, but STAT3 is activated through tyrosine phosphorylation (pSTAT3) by a wide variety of cytokines, suggesting that it integrates diverse signals into common transcriptional responses (9–11). High persistent activation of STAT3 is found in diverse human tumors, including gliomas (12, 13). Moreover, considerable evidence suggests that STAT3 actively participates in tumor formation and progression (13). The family of NF-κB transcription factors, which includes NFκB1 (constitutively processed to p50), NFκB2 (stimulus-induced processing to p52), RelA (p65), RelB, and cRel, has also been observed to be persistently activated in many human tumors, including glioma, and they are believed to contribute to tumor cell survival, proliferation, migration, and therapeutic resistance and to regulating genes that play important roles in immunity and inflammation (14–18). Under most circumstances, NF-κB is inactivated in the cytoplasm through the binding of IκB inhibitory proteins. In general various stimuli promote the dissociation of the cytosolically inactive NF-κB/IκB complexes via IKK activation, which results in the serine phosphorylation and degradation of IκB and the translocation of p50:p65 dimers into the nucleus and DNA binding. Specifically, NF-κB blocks apoptosis by stimulating the induction of antiapoptotic genes as well as suppressing apoptosis-inducing genes (19).
In this study, we characterized the properties of glioblastoma CSCs grown as adherent cultures in neurobasal medium on laminin-coated plates. Glioma CSCs have markedly enhanced tumor-initiating activity when compared with the glioma monolayers from which they were derived. These adherent glioma CSCs share many properties with CSCs grown as traditional tumor spheres. Interestingly, adherent glioma CSCs show constitutive activation of the STAT3/NF-κB signaling pathway and up-regulation of STAT3- and NF-κB-dependent genes. Gene expression profiling also identified components of the Notch pathway as being deregulated in glioma CSCs, and the deregulated expression of these genes was sensitive to treatment with STAT3 inhibitors. This finding is particularly important because Notch signaling appears to play a key role in CSCs in a variety of cancers and controls cell fate determination, survival, proliferation, and the maintenance of stem cells (20).
EXPERIMENTAL PROCEDURES
Cell Culture
The GBM6 (provided by Dr. C. David James, Department of Neurological Surgery, University of California, San Francisco, CA) and MT330 (provided Dr. Christopher Duntsch, Department of Neurosurgery, UTHSC) human glioma cell lines were grown in monolayer culture in DMEM (Cellgro, Mediatech) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone), 100 units/ml penicillin, and 100 μg/ml streptomycin. GBM6 cells were continuously maintained as subcutaneous xenografts in NOD.Cg Prkdcscid Il2rgtm1Wjl/SzJ mice, and monolayer and CSC cultures were derived from freshly harvested tumor tissue. Adherent and spheroid glioma CSCs were maintained in NeuroBasal-A medium (Invitrogen) containing 2% B27 supplement, 2 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, EGF (20 ng/ml), and basic FGF (40 ng/ml). For isolation of adherent CSCs, culture flasks were coated with 100 μg/ml poly-D-lysine (Sigma) for 1 h, followed by coating with 10 μg/ml laminin (Invitrogen) for 2 h prior to use. Adherent CSCs were plated at 1 × 105 cells/75-cm2 flask, grown to confluence, dissociated with HyQTase (Thermo Scientific), and split 1:3. For isolation of spheroid CSCs, glioma cells were dissociated with HyQtase and plated at ∼1 × 105 cells/ml in ultra-low adhesion flasks.
Quantitative RT-PCR
Total RNA was extracted using the QIAshredder and RNeasy mini kits (Qiagen Inc.) according to the protocol of the manufacturer. RT-PCR was performed on an iCyclerIQ (Bio-Rad) using an iScript one-step RT-PCR kit with SYBR Green (Bio-Rad). Reaction parameters were as follows: cDNA synthesis at 50 °C for 20 min, transcriptase inactivation at 95 °C for 5 min, and PCR cycling at 95 °C for 10 s and 60 °C for 30 s for 40 cycles. The following primers were used for RT-PCR: β-actin, 5′-AGAAGGAGATCACTGCCCTG-3′ (forward) and 5′-CACATCTGCTGGAAGGTGGA-3′ (reverse); CD133, 5′-CATCCACAGATGCTCCTAAGG-3′ (forward) and 5′-AAGAGAATGCCAATGGGTCCA-3′ (reverse); SOX2, 5′-GCCGAGTGGAAACTTTTGTCG-3′ (forward) and 5′-GCAGCGTGTACTTATCCTTCTT-3′ (reverse); IL8, 5′-TAGCAAAATTGAGGCCAAGG-3′ (forward) and 5′-AGCAGACTAGGGTTGCCAGA-3′ (reverse); Trail, 5′-GAGCTGAAGCAGATGCAGGAC-3′ (forward) and 5′-TGACGGAGTTGCCACTTGACT-3′ (reverse); CXCL11, 5′-ATGAGTGTGAAGGGCATGGGC-3′ (forward) and 5′-TCACTGCTTTTACCCCAGGG-3′ (reverse); Bcl-2, 5′-CCGGAGGCGCTTTACTACC-3” (forward) and 5′-TAGGGGTGTAGGCAGGTTCAC-3′ (reverse); Bcl-X, 5′-GGTCGCATTGTGGCCTTTTTC-3′ (forward) and 5′-AGGGGCTTGGTTCTTACCCA-3′ (reverse); caspase 3, 5′-CATGGAAGCGAATCAATGGACT-3′ (forward) and 5′-CTGTACCAGACCGAGATGTCA-3′ (reverse); NOTCH1, 5′-GAGGCGTGGCAGACTATGC-3′ (forward) and 5′-CTTGTACTCCGTCAGCGTGA-3′ (reverse); HES5, 5′-AGTCCCAAGGAGAAAAACCGA-3′ (forward) and 5′-GCTGTGTTTCAGGTAGCTGAC-3′ (reverse); JAG1, 5′-GTCCATGCAGAACGTGAACG-3′ (forward) and 5′-GCGGGACTGATACTCCTTGA-3′ (reverse); NUMBL, 5′-TGGTGGACGACAAAACCAAGG-3′ (forward) and 5′-ACGACAGATATAGGAAGCCT-3′ (reverse); DTX3, 5′-TCGTTCGTCCTGTCCAGAATG-3′ (forward) and 5′-AAGTCTCGCCATCTATGAGGAT-3′ (reverse); DVL3, 5′-GACGCCGTACCTTGTGAAG-3′ (forward) and 5′-CGCTGCAAAACGCCCTTAAA-3′ (reverse); and RBPJ, 5′-CGGCCTCCACCTAAACGAC-3′ (forward) and 5′-TCCATCCACTGCCCATAAGAT-3′ (reverse).
Immunoblot Analysis
For preparation of whole cell extracts, cells were lysed in radioimmune precipitation assay buffer (Thermo Scientific) containing 1 mm NaF, 1 mm Na3VO4, 1 mm PMSF, and protease inhibitor mixture (Sigma) at 4 °C for 30 min and precleared by centrifugation (12,000 × g, 15 min). Nuclear extracts were prepared using the Active Motif nuclear extract kit as described previously (21). The amount of protein was determined using a BCA protein assay kit (Thermo Scientific, Pierce, Rockford, IL). Extracts (25 μg) were separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes (Millipore), and immunoblotted with antibodies against the following proteins: β-tubulin III and glial fibrillary acidic protein (Sigma), Nestin (Abcam), STAT3 and phospho-STAT3 (Cell Signaling Technology), and p65 and lamin (Santa Cruz Biotechnology). Following the addition of IRDye800CW goat anti-mouse IgG or IRDye680 goat anti-rabbit IgG (LICOR Biosciences), blots were visualized on an Odyssey infrared imaging system (LICOR Biosciences).
Immunofluorescence and Confocal Microscopy
Cells grown in 8-well chamber slides (Millipore) to ∼50% confluence were washed with PBS, fixed with 4% paraformaldehyde and methanol, and permeabilized with 1% Triton X-100. After blocking with 5% goat serum, cells were incubated with nestin, STAT3, pSTAT3, and p65 antibodies and subsequently stained with Alexa Fluor 488 (goat anti-mouse, Invitrogen, catalog no. A11029) and Alexa Fluor 555 (goat anti-rabbit and DNA counterstained with Vectashield mounting medium with DAPI (Vectra Laboratories) (Invitrogen, catalog no. A21429). Images were captured on a Zeiss LSM700 laser-scanning confocal microscope.
Tumor Xenografts in Mice
All animal experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center. Cells were dissociated with HyQTase, resuspended in PBS, enumerated in a Coulter counter analyzer, and resuspended in 100 μl of PBS at the desired cell number for subcutaneous injections. Tumor xenografts were established in five-week-old male NOD.Cg Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory) by direct injection of glioma cells into the flanks. Tumors were measured biweekly until reaching a volume of ∼400 mm3. Then, mice were sacrificed and tumors harvested.
Colony Formation Assay
Single-cell suspensions of 5000 cells in 1 ml of 0.4% agarose in tissue culture medium were added to triplicate wells of ultralow adhesion 6-well plates. Cells were fed twice a week with an additional 0.5 ml of medium. At day 14, plates were stained with MTT (10 μg/ml) for 3 h, and colonies were counted on a light microscope.
MTT Cell Viability Assay
Glioma monolayer cells or adherent CSCs in 96-well plates were treated with varying concentrations of WP1066 or S3I-201 (Selleckchem). After 72 h, 10 μl of MTT stock solution (10 mg/ml) was added to each well and incubated at 37 °C for 2–4 h. MTT was solubilized by adding 100 μl of 10% SDS in 0.01 m HCl, and plates were incubated at 37 °C for 4 h in a humidified chamber. Plates were read at 570 nm on a Bio-Rad plate reader.
Apoptosis Assay
The induction of apoptosis was monitored by flow cytometry (Accuri Model 6C) using the annexin V-FITC apoptosis detection kit (BD Biosciences) according to the instructions of the manufacturer.
p65-GST Pull-down Assay
The p65 cDNA was cloned into pGEX-KG expressed in Escherichia coli strain BL21 and affinity-purified in glutathione-Sepharose beads as described previously (22). For pull-down assays, nuclear extracts from glioma monolayer cells or adherent CSCs were incubated with the p65-GST fusion protein bound to glutathione-agarose beads at 4 °C overnight. The bound proteins were washed extensively and eluted with Laemmli buffer, resolved by SDS-PAGE (10%), blotted on PVDF membranes, and probed with anti-STAT3.
Microarray Analysis
Total cellular RNA was extracted from cells with TRIzol reagent (Invitrogen) and submitted to the UTHSC Center of Genomics and Bioinformatics (Memphis, TN) for labeling and hybridization to Human-HT12 BeadChips (Illumina Inc.). Microarray data analysis was then carried out using GenomeStudio 3.4.0 (Illumina Inc., CA) and GeneSpring software 7.0 (Silicon Genetics, Inc.), and expression values for each gene were normalized as described previously (23). The average fold change in gene expression from three independent sets of GeneChip data for glioma monolayers and adherent CSCs was subjected to non-parametric t testing. The microarray data were subjected to gene ontology modular enrichment analysis using the Database for Annotation, Visualization, and Integrated Discovery (24), and the protein-protein interaction network of the Notch pathway was determined by Search Tool for the Retrieval on Interacting Genes/Proteins analysis (25).
Chromatin Immunoprecipitation
ChIP was carried out using the ChIP-ITTM Express Enzymatic kit (Active Motif, Carlsbad, CA) according to the instructions of the manufacturer. In brief, chromatin from cells was cross-linked with 1% formaldehyde (10 min at 22 °C), sheared to an average size of ∼200 bp, and then immunoprecipitated with anti-p65 or STAT3 (Santa Cruz Biotechnology). ChIP-PCR primers were designed to amplify a proximal promoter region containing putative STAT3 (-1940 to −1919) and NF-κB (-2145 to −2135) binding sites in the Notch1 promoter. The primers used were as follows: for the STAT3 site, 5′-CACTGGCTGTTTCCAGAGTG-3′ (forward) and 5′-GGGAGGGACCTAGGACTGTG-3′ (reverse) and for NF-κB site, 5′CTACTTGCCAGGGGCCTAC-3′ (forward) and 5′-GCTCATAAGCCCGCGTTAC-3′ (reverse).
Statistical Analysis
At least three independent experiments were performed in duplicate, and data are presented as mean ± S.D. Analysis of variance and post hoc least significant difference analysis or Student's t tests were performed. p < 0.05 was considered to be statistically significant.
RESULTS
Expression of CSC Markers and Tumorigenicity of Adherent CSCs
CSCs are often generated on the basis of their ability to grow as multicellular, nonadherent spheres in low-adhesion flasks (4, 5), which we denoted spheroid CSCs. Alternatively, CSCs can also be grown in laminin-coated flasks, which we denote as adherent CSCs (6). To define the biological properties of adherent and spheroid CSCs derived from glioma lines, we examined the expression of two classical stem cell markers, CD133 and Sox2. In brief, total RNA was prepared from GBM6 and MT330 gliomas, which were grown in monolayer or under the two different CSC conditions, and the expression of CD133 and Sox2 was determined by qPCR. As shown in Fig. 1A, CD133 and Sox2 gene expression was greater in both adherent and spheroid glioma CSCs when compared with glioma cells grown in monolayer. Furthermore, as determined by immunostaining, nestin (a neural stem cell marker) was highly expressed in adherent and spheroid glioma CSCs when compared with glioma monolayers, whereas βIII-tubulin (a neural differentiation marker) and glial fibrillary acid protein (astrocyte differentiation marker) were more highly expressed in glioma monolayers (Fig. 1B).
FIGURE 1.

Expression of CSC markers and tumorigenicity of adherent CSCs. A, RNA was prepared from monolayer (mono) and adherent (Ad-C) and spheroid (Sp-C) CSC cultures of GBM6 and MT330 gliomas, and CD133 and SOX2 expression was quantified by qPCR and normalized to actin expression (n = 3). Error bars show S.D. **, p < 0.01; ***, p < 0.001. B, GBM6 monolayer cells and adherent CSCs (Ad-CSC) were fixed and immunostained for Nestin, tubulin, or glial fibrillary acidic protein (GFAP) (green), counterstained with DAPI (blue), and analyzed by confocal microscopy. C, mice were injected subcutaneously with varying concentrations of GBM6 cells grown in monolayer (GBM6-M) or adherent CSCs in laminin-coated plates (GBM6 Ad-C), and tumor volume was determined by caliper measurement three times per week (n = 10/group). D, GBM6 monolayer and adherent CSC cultures were plated in soft agar (10,000 cells/well of 6-well plates), and the colony formation potential was assessed.
Because CSC are believed to have enhanced tumorigenicity, we next compared the tumorigenicity of the GBM6 glioma cells grown in monolayer or as adherent CSCs. In brief, 1 × 106 cells were injected subcutaneously in the flanks of NOD.Cg Prkdcscid Il2rgtm1Wjl/SzJ mice, and tumor engraftment was determined by caliper measurement. As shown in Fig. 1C, adherent glioma CSCs formed tumors rapidly and grew to ∼400 mm3, whereas only small tumors developed in mice injected with monolayer cultures at 40 days. The tumorigenic potential of adherent glioma CSCs was further characterized by performing a limiting dilution analysis. We found that as few as 10,000 glioma CSCs were capable of consistently forming tumors and, most interestingly, that the adherent CSCs were nearly 100 times more potent in inducing tumors than the glioma monolayer cultures. Soft agar assays for anchorage-independent growth, which is used as an in vitro correlate for tumorigenic potential, showed that adherent CSCs have a 68-fold greater colony formation potential than glioma monolayer cells, 6.8% versus 0.1%, respectively. (Fig. 1D). Thus, the adherent glioma cells grown in laminin-coated plates exhibit characteristics ascribed previously to cancer stem cells, i.e. they self-renew, display neural stem cell markers, form tumors upon serial transplantation that recapitulate the tumor phenotype, and exhibit enhanced tumorigenicity.
Constitutive Activation of the STAT3 and NF-κB Signaling Pathways in Glioma CSCs
The STAT3 and NF-κB signaling pathways have been found to be constitutively active in various human cancers, including glioma, and their activation is believed to play a critical role in the stem cell phenotype. To characterize the NF-κB and STAT3 signaling pathways in glioma CSCs, we examined the intracellular localization of STAT3 and the p65 subunit of NF-κB by confocal microscopy. GBM6 glioma cells were grown as monolayers or adherent glioma CSCs on glass slides and immunostained with antibodies specific for STAT3, pSTAT3 (as a measure of transcriptionally active STAT3), and p65. Cells were counterstained with DAPI to define nuclear localization of proteins. As shown in Fig. 2A, although STAT3 and p65 are present in the cytoplasm and nucleus of both GBM6 glioma monolayers and adherent CSCs, their colocalization is only evident in the nucleus of adherent CSCs. Moreover, as shown in Fig. 2B, although tyrosine phosphorylated STAT3 is undetectable in glioma monolayer cultures, pSTAT3 is clearly present in glioma CSCs and is selectively colocalized in the nucleus with p65.
FIGURE 2.
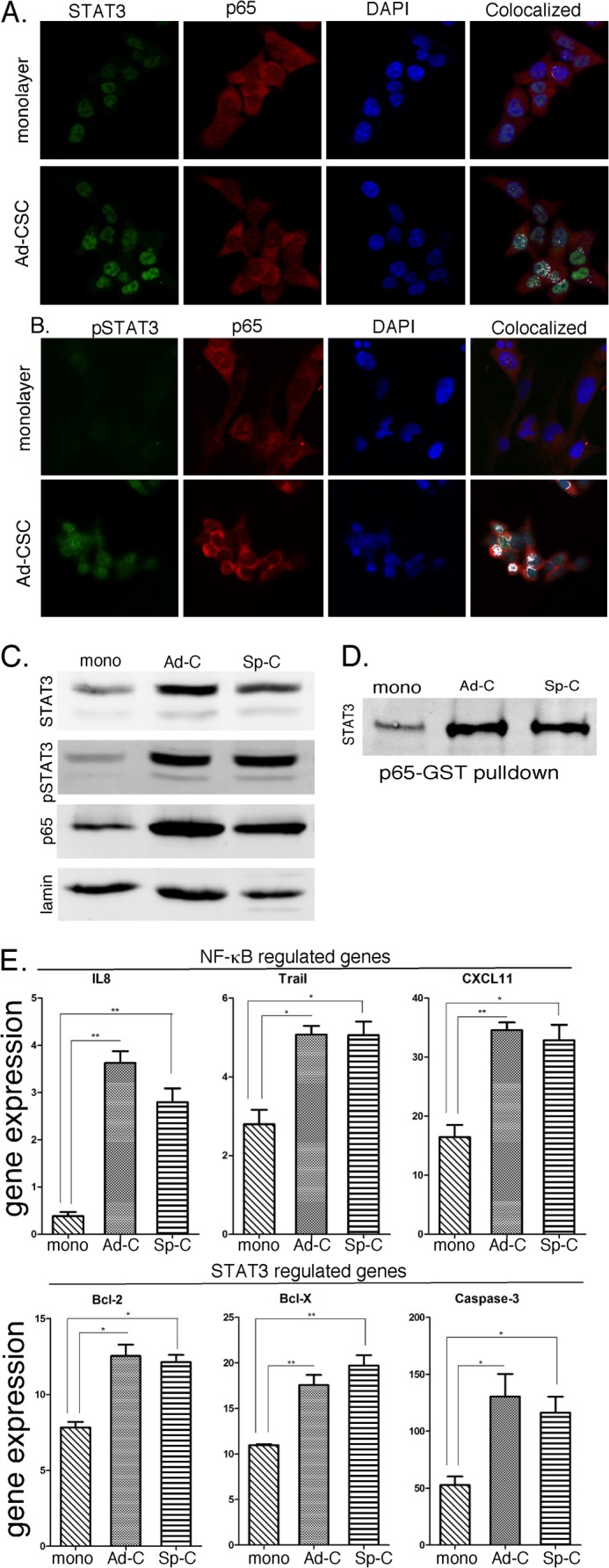
Constitutive activation of STAT3 and NF-κB signaling pathways in glioma adherent CSCs. GBM6 monolayer and adherent CSC (Ad-CSC) cultures were fixed and immunostained with antibodies as indicated and analyzed by confocal microscopy. STAT3 (A) and phospho-STAT3 (B) staining is represented in green, p65 is represented in red, and nuclear DAPI staining is shown in blue. White pixels represent the colocalization of STAT3 (A) or phospho-STAT3 (B) and p65 proteins within the cells. C, the expression of STAT3, pSTAT3, and p65 in nuclear extract-prepared glioma cells was determined by immunoblotting. mono, monolayer; Ad-C, adherent CSC; Sp-C, spheroid CSC. D, GST-p65 pull-down assays were performed to assess the interaction of STAT3 and p65 in glioma CSCs. E, RNA was prepared from GBM6 monolayer and adherent CSC cultures, and the expression of IL8, Trail, CXCL11, Bcl-2, Bcl-X and Caspase 3 was quantified by qPCR and normalized to actin expression (n = 3). Error bars show S.D. *, p < 0.05; **, p < 0.01.
To further characterize the activation of the STAT3 signaling pathway in gliomas, nuclear extracts were prepared from GBM6 cells grown under the different culture conditions and analyzed by immunoblotting for phospho-STAT3 and STAT3. As shown in Fig. 2C and consistent with immunostaining results, both STAT3 and pSTAT3 were clearly detectable in nuclear extracts of glioma cells irrespective of whether they were grown as monolayers or CSCs. However, basal activation of STAT3, as determined by the nuclear levels of tyrosine-phosphorylated STAT3, was markedly greater in CSCs. This finding is in contrast to normal (untransformed) cells, such as fibroblasts, where basal activation of STAT3 is undetectable. Moreover, nuclear expression of p65 is higher in both adherent and spheroid CSCs. Thus, we provide strong evidence that the STAT3 (as measured by nuclear STAT3 and phospho-STAT3 levels) and NF-κB (as measured by nuclear p65 levels) signaling pathways are constitutively activated in glioma cells and that activation is markedly greater in glioma CSCs. In addition, as shown in Fig. 2D, the interaction between STAT3 and p65 in the nucleus is further borne out by pull-down assays with p65-GST, which show a greater interaction of STAT3 with p65 in nuclear extracts from adherent and spheroid CSCs.
To characterize the functional significance of constitutive activation of these signaling pathways, we examined the expression of several STAT3- and NF-κB-regulated genes in glioma monolayer and CSCs. As shown in Fig. 2E, the expression of several NF-κB-regulated genes (IL8, CXCL11, and Trail) was significantly higher in both adherent and spheroid glioma CSCs when compared with monolayer cultures. Most notably, IL8 gene expression was 6- to 8-fold higher in CSCs. In addition, the expression of known STAT3-regulated genes (Bcl-2, Bcl-X, and Caspase 3) was also markedly elevated in both adherent and spheroid CSCs (Fig. 2F). These results suggest that because the transcriptionally active forms of p65 and STAT3 are colocalized in the nucleus of glioma CSCs, the constitutively activated forms of these important transcription factors may interact and drive the expression of critical STAT3- and NF-κB-regulated genes.
The Effects of Specific STAT3 Inhibitors on Glioma Monolayers and Glioma CSCs
We next sought to determine the efficacy of STAT3 inhibitors on the constitutively activated STAT3 pathway in CSCs. Adherent GBM6 CSCs were plated and treated with a pharmacological inhibitor of STAT3 (WP1066 or S3I-201) for 2 h at varying concentrations. Nuclear extracts were prepared, and STAT3 and pSTAT3 expression was determined by immunoblotting. As shown in Fig. 3A, both WP1066 and S3I-201 were extremely effective in inhibiting STAT3 tyrosine phosphorylation in CSCs, with nearly complete inhibition of pSTAT3 at 50 μm WP1066 and 300 μm S3I-201. However, both inhibitors had little or no effect on nuclear STAT3 levels. We then examined the effects of WP1066 (50 μm for 2 h) on intracellular localization of STAT3 and the p65 subunit of NF-κB in glioma CSCs by confocal microscopy. As shown in the top row of Fig. 3B and in Fig. 2A, STAT3 and p65 are colocalized in the nucleus of adherent CSCs. In contrast, treatment with WP1066 markedly inhibited the nuclear colocalization of STAT3 with p65 in GBM6 glioma CSCs (Fig. 3A, bottom row). Similar results were obtained when cells were immunostained for pSTAT3 and p65, i.e. the STAT3 inhibitor ablated the nuclear colocalization of pSTAT3 with p65.
FIGURE 3.
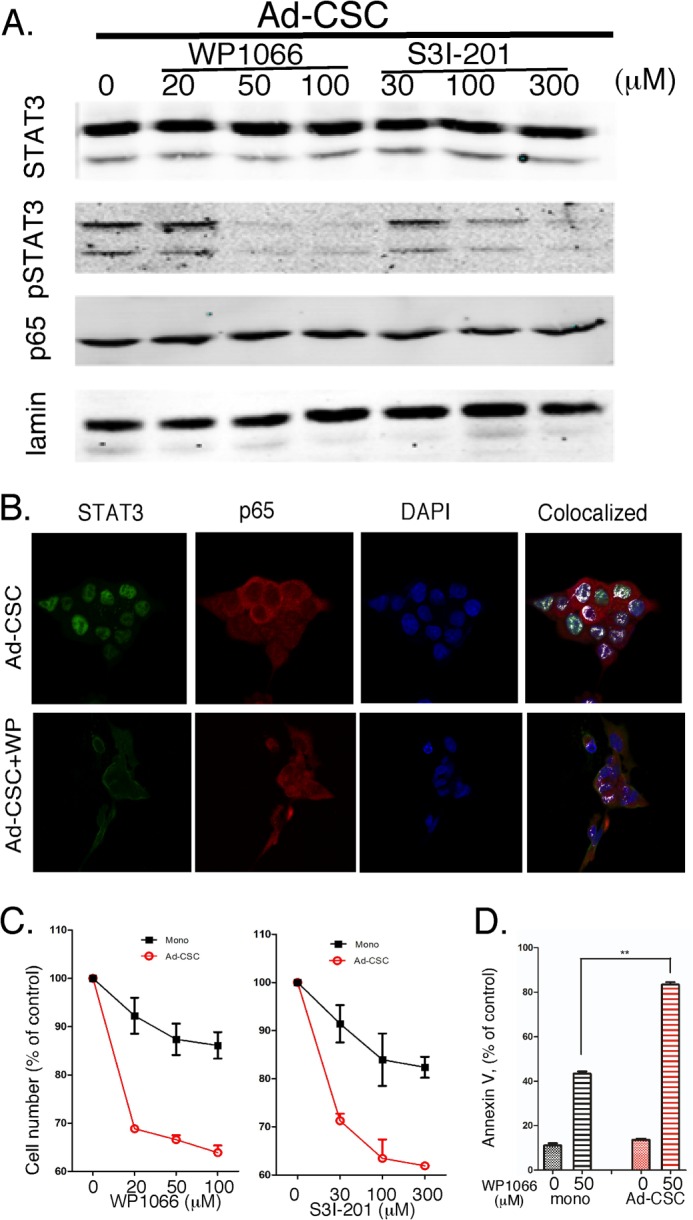
Effects of selective STAT3 inhibitors on adherent glioma CSCs. A, GBM6 cells were treated with WP1066 and S3I-201 at the indicated concentrations, and the expression of STAT3, pSTAT3, and p65 proteins was determined by immunoblotting. Ad-CSC, adherent CSC. B, cells were treated with WP1066 (50 μm for 2 h) or vehicle, and colocalization of STAT3 and p65 was determined by immunostaining as in Fig. 2. C, proliferation of GBM6 monolayer and adherent CSC cultures was measured by MTT assay after treatment (72 h) with WP1066 or S3I-201 at the indicated concentrations. D, 48 h after treatment with 50 μm WP1066, apoptosis was determined by annexin V staining and quantified by flow cytometry.
We then examined the effects of WP1066 or S3I-201 on the cell viability/proliferation of glioma monolayers and adherent CSCs. In brief, cells in 96-well plates were treated with varying concentrations of the pharmacological STAT3 inhibitors for 72 h, and cell proliferation was determined by MTT assays. As shown in Fig. 3B, although both inhibitors induced a dose-dependent reduction in cell number in both glioma monolayers and adherent CSCs, there was a greater effect of both inhibitors on adherent CSCs. Moreover, flow cytometric analysis of annexin V-stained cells demonstrated that treatment with the STAT3 inhibitor WP1066 had a greater effect on the induction of apoptosis in adherent CSCs than in glioma monolayers (Fig. 3C). Taken together, these results show that STAT3 activation is greater in adherent CSCs.
Microarray Analysis Identifies Up-regulation of the Notch Pathway in Glioma CSCs
To identify genes differentially expressed in glioma CSCs, we performed a preliminary microarray analysis. In brief, whole genome expression profiling was performed on RNA prepared from three independent biological replicates of MT330 and GBM6 grown as monolayers and spheroid glioma CSCs. The samples were submitted to the UTHSC Center of Genomics and Bioinformatics (Memphis, TN) for labeling and hybridization to HT-12 expression BeadChips (Illumina Inc.). RNA integrity was validated on an Agilent bioanalyzer, and all samples showed distinct peaks corresponding to intact 28 S and 18 S ribosomal RNA. Hybridization signals were processed using Illumina GenomeStudio software (annotation, background subtraction, quantile normalization, and presence call filtering). GeneSpring GX software (Agilent Technologies) was used for statistical computing. Functional annotation of the genes differentially expressed in glioma CSCs revealed that genes in the Notch signaling pathway were enriched significantly (p < 0.05, Database for Annotation, Visualization, and Integrated Discovery Bioinformatics Resources 6.7). The expression of these Notch pathway genes is shown in Fig. 4A, and their interaction with the STAT3 and NF-κB signaling pathways is shown schematically in B. Most notably although Notch1, Notch3, Notch4, Hes5, Hey1, and Jag1, known positive regulators of this pathway, were up-regulated in glioma CSCs, the negative regulators of this pathway, CTBP1 and RBPJ, were down-regulated.
FIGURE 4.
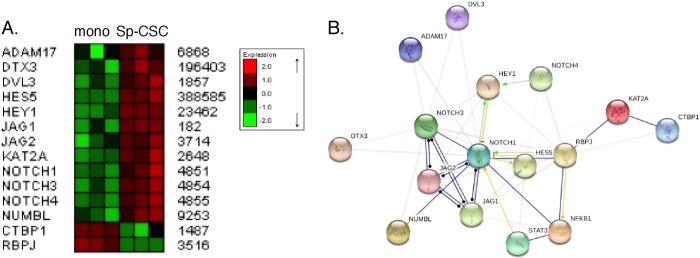
Enrichment of the Notch signaling pathway in glioma CSCs. A, RNA from MT330 and GBM6 monolayer (mono) and spheroid CSC (Sp-CSC) cultures was prepared, and microarray analysis was performed. B, schematic of the interaction of the Notch pathway with the STAT3 and NF-κB signaling pathways using Search Tool for the Retrieval on Interacting Genes/Proteins analysis.
The Roles of STAT3 and p65 in the Up-regulation of the Notch Pathway in Glioma CSCs
To confirm that these genes were indeed differentially regulated in CSCs, qPCR was performed on RNA extracted from GBM6 monolayers and adherent glioma CSCs. As shown in Fig. 5, expression of Notch1, Hes5, Jag1, Numbl, Dtx3, and Dvl3 was up-regulated in adherent glioma CSCs, whereas RBPJ was down-regulated. Most interestingly, pretreatment with either STAT3 inhibitor or the NF-κB inhibitor Velcade reduced the expression level of the genes up-regulated in the CSCs to levels observed in monolayer cultures, whereas these inhibitors increased the level of RBPJ expression in glioma CSCs. Thus, these genes in the Notch pathway are regulated both by STAT3 and NF-κB.
FIGURE 5.

Effects of STAT3 and NF-κB inhibitors on the expression of components in the Notch signaling cascade in vitro. GBM6 cells were treated with STAT3 (50 μm WP1066 (WP) and 300 μm S3I-201 (S3I)), NF-κB (10 nm Velcade (Vel)), or γ-secretase (10 μm N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT)) inhibitors. RNA was prepared, and the gene expression of Notch1, Hes5, JAG1, NUMBL, DTX3, DVL3, and RBPJ was quantified by qPCR and normalized to actin expression (n = 3). Error bars show S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001. mono, monolayer; con, control; Ad-CSC, adherent CSC.
In addition, treatment with N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (a γ-secretase inhibitor), which blocks Notch activation, also reduced the expression level of the genes up-regulated in CSCs, whereas it increased the level of expression of RBPJ in glioma CSCs. These results are consistent with the critical role the Notch receptor plays in regulating multiple genes in both normal neural stem cells and glioma CSCs (26, 27). Because p65 and STAT3 appear to be involved in Notch1 expression, we next examined whether they directly bind to the Notch1 promoter. Examination of putative transcription binding sites revealed a STAT3 and a NF-κB binding site proximal to the Notch1 promoter. Specific primers were designed and synthesized for each of these potential binding sites in the Notch1 promoter, and the binding of STAT3 and p65 was demonstrated by ChIP analysis. In brief, protein-DNA complexes were cross-linked with formaldehyde, chromatin was sheared to an average of 200 bp and immunoprecipitated with anti-STAT3 or anti-p65, cross-linking was reversed, and the resulting DNA sequences were detected by PCR and qPCR. As shown in Fig. 6A, significantly increased binding of STAT3 to the Notch1 promoter was observed in adherent CSCs as compared with glioma monolayer cultures. We then examined whether p65 bound to the NF-κB binding site that was near the STAT3 site in the Notch1 promoter. As shown in Fig. 6B, significantly increased binding of p65 to the Notch1 promoter was also observed in adherent CSCs. Taken together, these results show that STAT3 and p65 regulate Notch1 expression in adherent CSCs by directly binding to the Notch1 promoter.
FIGURE 6.

The binding of STAT3 and the p65 subunit of NF-κB to the Notch1 promoter. ChIP analyses of STAT3 (A) and p65 (B) binding to the Notch1 promoter. The ChIP-enriched DNA levels analyzed by qPCR were normalized to input DNA, followed by subtraction of nonspecific binding determined by control IgG. mono, monolayer; Ad-CSC, adherent CSC.
DISCUSSION
Numerous studies support the concept that many types of malignancies, including glioblastoma, originate from a CSC population that is able to initiate and maintain tumors (28, 29). Although CSCs only represent a small fraction of cells within a tumor, their high tumor-initiating capacity and therapeutic resistance drives tumorigenesis. Therefore, it is imperative to identify pathways associated with CSCs to devise strategies to selectively target them. In this study, we describe a novel relationship between glioblastoma CSCs and the Notch pathway, which involves the constitutive activation of STAT3 and NF-κB signaling. Glioma CSCs were isolated and maintained in vitro using the adherent culture system described previously (6), and the biological properties were compared with the traditional cultures of CSCs grown as multicellular spheres under nonadherent culture conditions (4, 5). Under the different CSC growth conditions, the expression of CD133, Sox2, and Nestin, which are markers of neural and brain cancer stem cells (30), were similar but increased when compared with glioma cells grown in monolayer. Examination of the tumorigenicity of the adherent glioma CSCs in vitro by soft agar colony formation assays and in vivo by limiting dilution analysis showed that these cells were ∼100 times more tumorigenic than monolayer-cultured glioma cells. These findings are consistent with previous studies showing that CD133 expression correlated with chemoresistance of short-term cultures of glioma cells isolated from patients with primary or recurrent tumors, and the percentage of tumor cells expressing CD133 increased in all recurrent patient tumors compared with the primary tumor (31). Furthermore, 100-fold fewer CD133-positive cells of the murine GL261 glioma line were sufficient to initiate tumors in the brains of mice when compared with CD133-negative cells (32). Taken together, these findings demonstrate that adherent glioma CSCs exhibit characteristics described previously for CSCs grown in suspension culture and, thus, provide a valuable model for studying glioma CSC behavior.
The STAT3 and NF-κB pathways have been linked to cancer, and they trigger critical target genes regulating cell proliferation and survival. Both pathways have been found to be constitutively active in a number of human cancers (33), including glioma, but their role in the glioma CSC subpopulation is not well understood. For example, aberrant nuclear expression of NF-κB was found in a panel of GBM cell lines, whereas untransformed glial cells did not display NF-κB activity (34). In addition, constitutively high STAT3 activity has been observed in a number of glioma cell lines and correlated with poor prognosis (35). In this study, although the STAT3 and NF-κB signaling pathways are constitutively activated in glioma lines, we found that these pathways are dramatically activated in glioma CSCs. For example, nuclear STAT3 and phospho-STAT3 levels found in glioma CSCs were similar to the high cytokine-induced levels found in glioma monolayers that, at base line, were low but detectable. Moreover, NF-κB activation in glioma CSCs was demonstrated by high levels of the p65 subunit of NF-κB present in nuclear extracts. Furthermore, constitutive activation of the STAT3 and NF-κB pathways and their direct interaction in glioma CSCs was evidenced by confocal microscopy of glioma CSCs stained for STAT3, pSTAT3, and p65. These STAT3 and NF-κB proteins were colocalized in the nuclei of glioma CSCs, with the transcriptionally active form of STAT3 (i.e. p-STAT3) and p65 exclusively found in the nucleus. Evidence of the functional significance of STAT3 and NF-κB activation in glioma CSCs was provided by the finding that some of their known target genes (Bcl-2, Bcl-X, IL8, CXCL11, Trail, and Caspase3) were overexpressed in adherent and spheroid glioma CSCs relative to glioma monolayers. Because targeting these signaling pathways in glioma CSCs would be a novel approach in glioma treatment, we examined the effects of two STAT3 inhibitors, WP1066 and S3I-201, in glioma CSCs. The inhibitors blocked STAT3 tyrosine phosphorylation, thereby preventing its nuclear translocation (36), which was confirmed by its ability to reduce the high basal nuclear levels of pSTAT3 in glioma CSCs. Treatment with the STAT3 inhibitor led to loss of nuclear colocalization of these proteins as well as their interaction. In addition, treatment with either STAT3 inhibitor resulted in a growth-suppressive effect on the monolayer and adherent CSC cultures, but there was a markedly greater growth-suppressive effect on glioma CSCs, suggesting that targeted therapy of these key pathways in glioma CSCs may be possible.
To further investigate potential biomarkers in glioma CSCs, microarray analysis was performed and revealed deregulation of the Notch signaling pathway. Notch is involved in cell fate decisions throughout normal brain development and in stem cell proliferation and maintenance, and its role in glioma is firmly established (20). Although the expression of Notch1, Hes5, Jag1, Numbl, Dtx3, and Dvl3 was up-regulated in glioma CSCs, the expression of CTBP1 and RBPJ, negative regulators of Notch signaling, was down-regulated in glioma CSCs. The differential expression of these genes was validated by qPCR. The novel identification of Hes5 as a potential glioma CSC biomarker is of great interest because Hes5 appears to play an important role in neural development (37).
In addition, we defined molecular cross-talk between the STAT3, NF-κB, and Notch signaling pathways in glioma CSCs. Although STAT3 and NF-κB inhibitors reduced expression of Notch-related genes in glioma CSCs, they increased expression of the negative regulator of Notch, RBPJ. Moreover, ChIP assays showed that STAT3 and the p65 subunit of NF-κB bind to adjacent sites in the Notch1 promoter. It has been reported previously that in the developing central nervous system, there is cross-talk between the Notch and STAT3 pathways. For example, the activation and phosphorylation of STAT3 is mediated by the direct binding of several Hes family members (Notch effectors) to STAT3 (38). Interestingly, activation of the Notch pathway leads to serine phosphorylation (Ser-727) but not tyrosine phosphorylation (Tyr-705) of STAT3 (27), suggesting that the constitutive activation of STAT3 in glioma CSCs, as determined by its tyrosine phosphorylation, lies upstream of Notch pathway activation. These results on STAT3 activation are consistent with previous studies on the Notch pathway in neural stem cells (26). In addition, the Notch and NF-κB signaling pathways apparently collaborate throughout normal brain development and function and may regulate stem cell renewal and differentiation (39). We hypothesize that the interactions of the STAT3, NF-κB, and Notch signaling pathways that occur during normal brain development are deregulated in glioma CSCs. As shown in this study, there is cross-talk between these signaling pathways in the glioma CSC subpopulation that drives gliomagenesis. The constitutive activation of STAT3 and NF-κB signaling pathways and the up-regulation of the Notch pathway in glioma CSCs identifies novel therapeutic targets for the treatment of glioma. Future studies will be required to validate these findings in vivo and decipher the underlying molecular mechanisms.
Acknowledgments
We thank Dr. Christopher Nosrat for input during the initial stages of this research.
This work was supported, in whole or in part, by National Institutes of Health Grant CA133322 (to L. M. P. and A. M. D.). This work was also supported by Department of Defense Grant W81XWH-11-1-0533 (to L. M. P.), by Cancer Center Support Grant 21766 from the National Cancer Institute and the Assisi Foundation of Memphis (to A. M. D.), by the UTHSC Muirhead Chair Endowment (to L. M. P.), and by the American Lebanese Syrian Associated Charities (to A. M. D.).
The amino acid sequence of this protein can be accessed through the NCBI Protein Database under NCBI accession number GSE48079.
- GBM
- glioblastoma multiforme
- CSC
- cancer stem cell
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- qPCR
- quantitative RT-PCR
- UTHSC
- University of Tennessee Health Science Center.
REFERENCES
- 1. Stupp R., Mason W. P., van den Bent M. J., Weller M., Fisher B., Taphoorn M. J., Belanger K., Brandes A. A., Marosi C., Bogdahn U., Curschmann J., Janzer R. C., Ludwin S. K., Gorlia T., Allgeier A., Lacombe D., Cairncross J. G., Eisenhauer E., Mirimanoff R. O., European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups, and National Cancer Institute of Canada Clinical Trials Group (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 [DOI] [PubMed] [Google Scholar]
- 2. Clarke M. F., Dick J. E., Dirks P. B., Eaves C. J., Jamieson C. H., Jones D. L., Visvader J., Weissman I. L., Wahl G. M. (2006) Cancer stem cells. Perspectives on current status and future directions. AACR Workshop on cancer stem cells. Cancer Res. 66, 9339–9344 [DOI] [PubMed] [Google Scholar]
- 3. Huntly B. J., Gilliland D. G. (2005) Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat. Rev. Cancer 5, 311–321 [DOI] [PubMed] [Google Scholar]
- 4. Dontu G., Abdallah W. M., Foley J. M., Jackson K. W., Clarke M. F., Kawamura M. J., Wicha M. S. (2003) In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17, 1253–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee A., Kessler J. D., Read T. A., Kaiser C., Corbeil D., Huttner W. B., Johnson J. E., Wechsler-Reya R. J. (2005) Isolation of neural stem cells from the postnatal cerebellum. Nat. Neurosci. 8, 723–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pollard S. M., Yoshikawa K., Clarke I. D., Danovi D., Stricker S., Russell R., Bayani J., Head R., Lee M., Bernstein M., Squire J. A., Smith A., Dirks P. (2009) Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4, 568–580 [DOI] [PubMed] [Google Scholar]
- 7. Marotta L. L., Almendro V., Marusyk A., Shipitsin M., Schemme J., Walker S. R., Bloushtain-Qimron N., Kim J. J., Choudhury S. A., Maruyama R., Wu Z., Gönen M., Mulvey L. A., Bessarabova M. O., Huh S. J., Silver S. J., Kim S. Y., Park S. Y., Lee H. E., Anderson K. S., Richardson A. L., Nikolskaya T., Nikolsky Y., Liu X. S., Root D. E., Hahn W. C., Frank D. A., Polyak K. (2011) The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Invest. 121, 2723–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shostak K., Chariot A. (2011) NF-κB, stem cells and breast cancer. The links get stronger. Breast Cancer Res. 13, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang C. H., Shi W., Basu L., Murti A., Constantinescu S. N., Blatt L., Croze E., Mullersman J. E., Pfeffer L. M. (1996) Direct association of STAT3 with the IFNAR1 signal transducing chain of the type I IFN receptor. J. Biol. Chem. 271, 8057–8061 [DOI] [PubMed] [Google Scholar]
- 10. Akira S., Nishio Y., Inoue M., Wang X.-J., Wei S., Matsusaka T., Yoshida K., Sudo T., Naruto M., Kishimoto T. (1994) Molecular Cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77, 63–71 [DOI] [PubMed] [Google Scholar]
- 11. Zhong Z., Wen Z., Darnell J. E., Jr. (1994) Stat3. A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98 [DOI] [PubMed] [Google Scholar]
- 12. Turkson J., Jove R. (2000) STAT proteins. Novel molecular targets for cancer drug discovery. Oncogene 19, 6613–6626 [DOI] [PubMed] [Google Scholar]
- 13. Bowman T., Garcia R., Turkson J., Jove R. (2000) STATs in oncogenesis. Oncogene 19, 2474–2488 [DOI] [PubMed] [Google Scholar]
- 14. Aggarwal B. B. (2004) Nuclear factor κB. The enemy within. Cancer Cell 6, 203–238 [DOI] [PubMed] [Google Scholar]
- 15. Beg A. A., Baltimore D. (1996) An essential role for NF-κB in preventing TNF-α-induced cell death. Science 274, 782–784 [DOI] [PubMed] [Google Scholar]
- 16. Beg A. A., Sha W. C., Bronson R. T., Ghosh S., Baltimore D. (1995) Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 376, 167–170 [DOI] [PubMed] [Google Scholar]
- 17. Van Antwerp D. J., Martin S. J., Kafri T., Green D. R., Verma I. M. (1996) Suppression of TNF-α-induced apoptosis by NF-κB. Science 274, 787–789 [DOI] [PubMed] [Google Scholar]
- 18. Wang C.-Y., Mayo M. W., Baldwin A. S., Jr. (1996) TNF- and cancer therapy-induced apoptosis. Potentiation by inhibition of NF-κB. Science 274, 784–787 [DOI] [PubMed] [Google Scholar]
- 19. Du Z., Wei L., Murti A., Pfeffer S. R., Fan M., Yang C. H., Pfeffer L. M. (2007) Non-conventional signal transduction by type 1 interferons: the NF-κB pathway. J. Cell Biochem. 102, 1087–1094 [DOI] [PubMed] [Google Scholar]
- 20. Lino M. M., Merlo A., Boulay J. L. (2010) Notch signaling in glioblastoma. A developmental drug target? BMC Med. 8, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Du Z., Fan M., Kim J. G., Eckerle D., Lothstein L., Wei L., Pfeffer L. M. (2009) Interferon-resistant Daudi cell line with a Stat2 defect is resistant to apoptosis induced by chemotherapeutic agents. J. Biol. Chem. 284, 27808–27815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang C. H., Murti A., Pfeffer L. M. (2005) IFN induces NIK/TRAF-dependent NF-κB activation to promote cell survival. J. Biol. Chem. 280, 31530–31536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wei L., Sandbulte M. R., Thomas P. G., Webby R. J., Homayouni R., Pfeffer L. M. (2006) NF κB negatively regulates interferon-induced gene expression and anti-influenza activity. J. Biol. Chem. 281, 11678–11684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 25. Jensen L. J., Kuhn M., Stark M., Chaffron S., Creevey C., Muller J., Doerks T., Julien P., Roth A., Simonovic M., Bork P., von Mering C. (2009) STRING 8. A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Androutsellis-Theotokis A., Leker R. R., Soldner F., Hoeppner D. J., Ravin R., Poser S. W., Rueger M. A., Bae S. K., Kittappa R., McKay R. D. (2006) Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442, 823–826 [DOI] [PubMed] [Google Scholar]
- 27. Fan X., Khaki L., Zhu T. S., Soules M. E., Talsma C. E., Gul N., Koh C., Zhang J., Li Y. M., Maciaczyk J., Nikkhah G., Dimeco F., Piccirillo S., Vescovi A. L., Eberhart C. G. (2010) NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28, 5–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mani S. A., Guo W., Liao M. J., Eaton E. N., Ayyanan A., Zhou A. Y., Brooks M., Reinhard F., Zhang C. C., Shipitsin M., Campbell L. L., Polyak K., Brisken C., Yang J., Weinberg R. A. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marotta L. L., Polyak K. (2009) Cancer stem cells. A model in the making. Curr. Opin. Genet. Dev. 19, 44–50 [DOI] [PubMed] [Google Scholar]
- 30. Ma Y. H., Mentlein R., Knerlich F., Kruse M. L., Mehdorn H. M., Held-Feindt J. (2008) Expression of stem cell markers in human astrocytomas of different WHO grades. J. Neurooncol. 86, 31–45 [DOI] [PubMed] [Google Scholar]
- 31. Liu G., Yuan X., Zeng Z., Tunici P., Ng H., Abdulkadir I. R., Lu L., Irvin D., Black K. L., Yu J. S. (2006) Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 5, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu A., Oh S., Wiesner S. M., Ericson K., Chen L., Hall W. A., Champoux P. E., Low W. C., Ohlfest J. R. (2008) Persistence of CD133+ cells in human and mouse glioma cell lines. Detailed characterization of GL261 glioma cells with cancer stem cell-like properties. Stem Cells Dev. 17, 173–184 [DOI] [PubMed] [Google Scholar]
- 33. Grivennikov S. I., Karin M. (2010) Dangerous liaisons. STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 21, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gill J. S., Zhu X., Moore M. J., Lu L., Yaszemski M. J., Windebank A. J. (2002) Effects of NFκB decoy oligonucleotides released from biodegradable polymer microparticles on a glioblastoma cell line. Biomaterials 23, 2773–2781 [DOI] [PubMed] [Google Scholar]
- 35. Mizoguchi M., Betensky R. A., Batchelor T. T., Bernay D. C., Louis D. N., Nutt C. L. (2006) Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas. Correlation with EGFR status, tumor grade, and survival. J. Neuropathol. Exp. Neurol. 65, 1181–1188 [DOI] [PubMed] [Google Scholar]
- 36. Hussain S. F., Kong L. Y., Jordan J., Conrad C., Madden T., Fokt I., Priebe W., Heimberger A. B. (2007) A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 67, 9630–9636 [DOI] [PubMed] [Google Scholar]
- 37. Hitoshi S., Ishino Y., Kumar A., Jasmine S., Tanaka K. F., Kondo T., Kato S., Hosoya T., Hotta Y., Ikenaka K. (2011) Mammalian Gcm genes induce Hes5 expression by active DNA demethylation and induce neural stem cells. Nat. Neurosci. 14, 957–964 [DOI] [PubMed] [Google Scholar]
- 38. Kamakura S., Oishi K., Yoshimatsu T., Nakafuku M., Masuyama N., Gotoh Y. (2004) Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat. Cell Biol. 6, 547–554 [DOI] [PubMed] [Google Scholar]
- 39. Ang H. L., Tergaonkar V. (2007) Notch and NFκB signaling pathways: Do they collaborate in normal vertebrate brain development and function? BioEssays 29, 1039–1047 [DOI] [PubMed] [Google Scholar]