

Background: Lysine acetylation, a prevalent post-translational modification, alters mitochondrial metabolism in response to nutrient changes.
Results: Quantitative proteomics distinguishes dynamic and static acetylation sites, highlighting 48 likely regulatory sites of thousands identified.
Conclusion: Acetylation of Acat1 lysine 260, a highly dynamic site, reversibly inhibits enzyme activity.
Significance: Quantitative, state-specific proteomic analyses accelerate the functional characterization of acetylation in mitochondrial remodeling.
Keywords: Energy Metabolism, Metabolic Regulation, Mitochondrial Metabolism, Protein Acylation, Proteomics, SIRT, Acat1
Abstract
Lysine acetylation is rapidly becoming established as a key post-translational modification for regulating mitochondrial metabolism. Nonetheless, distinguishing regulatory sites from among the thousands identified by mass spectrometry and elucidating how these modifications alter enzyme function remain primary challenges. Here, we performed multiplexed quantitative mass spectrometry to measure changes in the mouse liver mitochondrial acetylproteome in response to acute and chronic alterations in nutritional status, and integrated these data sets with our compendium of predicted Sirt3 targets. These analyses highlight a subset of mitochondrial proteins with dynamic acetylation sites, including acetyl-CoA acetyltransferase 1 (Acat1), an enzyme central to multiple metabolic pathways. We performed in vitro biochemistry and molecular modeling to demonstrate that acetylation of Acat1 decreases its activity by disrupting the binding of coenzyme A. Collectively, our data reveal an important new target of regulatory acetylation and provide a foundation for investigating the role of select mitochondrial protein acetylation sites in mediating acute and chronic metabolic transitions.
Introduction
Protein acetylation was first identified 50 years ago (1, 2) and has since become established as a key regulator of gene transcription through its role in chromatin remodeling. In recent years, fueled by technological advances in mass spectrometry-based proteomics, lysine acetylation has become recognized as a widespread post-translational modification (PTM)3 that rivals phosphorylation and ubiquitination in its prevalence (3–5). Metabolic enzymes, including many in mitochondria, are among the most heavily acetylated proteins (6–8). Mechanistic and physiological studies have begun to reveal that acetylation of these proteins plays a key role in regulating metabolic flux, and to suggest that aberrant levels of this PTM might contribute to disorders associated with metabolic inflexibility, including obesity, type 2 diabetes, and cancer (9–15).
Despite mounting evidence that acetylation is a prevalent protein modification, much work remains to establish it as a pervasive mitochondrial regulatory mechanism. This includes the need to distinguish bona fide functional acetylation sites from what may be widespread spurious or adventitious modifications (16). This is highlighted by the considerable gap between the large number of identified mitochondrial lysine acetylation sites (∼2,200) and the few with a validated regulatory function (∼12 proteins) (17). The reasons for this disparity stem in part from both the relative youth of the field and from technical limitations of most previous mass spectrometry-driven investigations, which were largely unable to produce accurate quantitative information on how acetylation levels change between contrasting biological states.
Here, we applied our newly developed quantitative acetyl-proteomics methods to measure changes in the mouse liver mitochondrial acetylproteome between two pairs of contrasting nutritional states (18). First, because PTMs are a primary mechanism of regulation at acute time scales, we assessed changes in the mitochondrial acetylproteome between fasted and refed mice. Second, because chronic dysregulation of PTMs can be an important factor in disease, we compared the mitochondrial acetylproteome of obese (Leptin-deficient, Leptinob/ob) and lean (Leptin+/+) C57/Bl6 (B6) mice. Our analyses reveal that, among a background of largely static acetylation sites, 237 mitochondrial proteins have a significant change (q ≤ 0.1, Welch's t test with Storey correction) in the relative occupancy of one or more acetylation sites during these transitions in nutritional status. These include a range of enzymes from core mitochondrial metabolic processes, such as oxidative phosphorylation, fatty acid β-oxidation, branched chain amino acid metabolism, and ketogenesis.
To further prioritize mitochondrial lysine acetylation sites that are the most likely to be important for metabolic flexibility, we integrated these large-scale data sets with our compendium of predicted Sirt3 targets (18). Together, these analyses highlighted a select set of mitochondrial proteins as being likely targets for regulatory acetylation, including acetyl-CoA acetyltransferase 1 (Acat1), which possessed several sites of highly dynamic acetylation. By performing site-specific acetyllysine incorporation and in vitro biochemical enzyme activity assays, we discovered that Sirt3-driven deacetylation of K260ac and K265ac significantly enhances Acat1 activity. Molecular modeling revealed that the inhibitory effects of acetylation on Acat1 activity are likely due to decreased affinity for CoA caused by a loss of a favorable electrostatic interaction between one or more positively charged lysines with the negatively charged 3′-phosphate of CoA. Collectively, our work reveals Acat1 as an important new metabolic target of reversible acetylation and provides a resource of prioritized acetylation sites for future investigations into the role of this PTM in regulating mitochondrial protein function (19).
EXPERIMENTAL PROCEDURES
Animal Models
Breeding, sacrificing, and tissue harvesting of mice were described previously for the fasting versus refeeding study (19) and the obese versus lean study (20). Briefly, male mice were bred and housed in an environmentally controlled facility with a 12-h light-dark cycle (6 am–6 pm light cycle) and provided water and standard rodent chow (Purina number 5008). For the fasting versus refeeding study, chow was removed for 16 h (5 pm–9 am), after which half of the animals were sacrificed, whereas the other half were allowed to feed ad libitum an additional 2 h before being sacrificed. Liver tissue was dissected and immediately enriched for mitochondria using differential centrifugation (19). For the obese versus lean study, chow was removed for 4 h (8 am–12 pm) before sacrifice, and the liver was flash-frozen in liquid nitrogen before being thawed and enriched for mitochondria.
Quantitative Acetylproteome Analysis
Crude mitochondria were subjected to quantitative proteomics/acetylomics using recently developed methods (18). Briefly, mitochondrial pellets were resolubilized, and the proteins were reduced and alkylated and digested enzymatically and labeled with isobaric tags (6-plex TMT or 8-plex iTRAQ). Samples were mixed and subjected to strong cation exchange chromatography, followed by immunoprecipitation with pan-acetyllysine antibody (Immunechem), and non-bound peptides were further fractionated by high pH reverse phase chromatography. All samples were subjected to data-dependent Nano-LC-MS/MS analysis on an ETD-enabled LTQ Orbitrap Velos (Thermo Fisher Scientific) using our previously described QuantMode instrumentation method (21) with higher energy collision-activated dissociation fragmentation. Our custom software COMPASS (22) was utilized to search the MS/MS spectra against a concatenated target-decoy mouse database (UniProt), to trim the identified peptides (1% false discovery rate), to normalize the intensities of isobaric tag reporter ions, to group all peptides to parsimonious protein groups (1% false discovery rate), to localize acetylation sites to specific residues (95% probability), and to sum reporter ion intensities for spectra identifying the same protein or acetyl-isoform. For more details, see the supplemental “Experimental Procedures”.
Measurement of Oxygen Consumption Rate
The oxygen consumption rate was measured using an XF-96 Extracellular Flux Analyzer (Seahorse Bioscience) with methods based on previous work (23). Briefly, isolated mitochondria were quantified by wet weight, and 40 μg were loaded per well and supplemented with 10 mm glutamate and 2 mm malate. Basal respiration was measured for 3 min, followed by ADP injection to a final concentration of 2 mm. ADP-stimulated oxygen consumption was calculated by subtracting basal respiration, after which the wells were averaged. Data analysis was performed using the XF96 software (version 1.4.1.4) and the “Level(Direct)Akos” algorithm. See the supplemental “Experimental Procedures” for further details.
Biochemical Assessment of Acat1 Activity
Recombinant Acat1 (amino acids 31–424) with a C-terminal hexahistidine tag was overexpressed in Escherichia coli and purified using metal affinity resin based on previous methods (24, 25). For site-specific acetyllysine incorporation, an acetyl-lysyl-tRNA synthetase/tRNACUA pair that recognizes an amber codon was co-expressed with Acat1 in the presence of 10 mm acetyllysine and 20 mm nicotinamide (26–28). Briefly, a plasmid encoding Acat1 was cotransformed with pBK AcKRS (acetyllysine tRNA synthase) and pCDF PyIT (tRNACUA) into BL21(DE3) E. coli. Cells were grown to an A600 of 0.5 to 0.7 and then supplemented with 10 mm acetyllysine and 20 mm nicotinamide for 30 min before the addition of 0.3 mm isopropyl 1-thio-β-d-galactopyranoside. Cells were purified using metal affinity resin as above with the inclusion of 20 mm nicotinamide in the lysis buffer. Incorporation was verified by mass spectrometry and immunoblots for Acat1 and acetyllysine. Sirt3 was purified as described previously (29).
Acat1 Activity Assays
Activity was assessed as described previously (24, 30). Briefly, the reaction mixture contained 50 mm Tris-HCl (pH 8.1), 20 mm MgCl2, 10 μm acetoacetyl-CoA, 40 mm KCl, and 8 ng of purified Acat1 enzyme (diluted into a buffer of 50 mm HEPES (pH 6.6), 9.5% glycerol, and 0.5 mg/ml gelatin). The reaction was initiated with the addition of CoA. The final volume of the reaction was 300 μl. We measured the change in absorbance at 303 nm over 30 min at room temperature (∼25 °C). For the determinations of Km and Vmax for CoA, the above procedure was followed, and the CoA concentration was varied between 15 and 300 μm. All absorbance measures were obtained using the BioTek Synergy 2 microplate spectrophotometer with Gen5 software. We analyzed the mean velocity over the first 5 min, whereas the reaction velocity curve remained linear over the first 10 min. Km and Vmax calculations were obtained using the Enzkin package in Matlab and SigmaPlot (version 12).
Sirt3 Treatments
Sirt3 treatments were performed at 37 °C for 30 min and contained 16 ng/μl Acat1, 0.375 μm Sirt3, 1 mm NAD+, 0.5 mm DTT in dilution buffer (50 mm HEPES, pH 8.23, 0.5 mg/ml gelatin, 9% glycerol). Deacetylation was verified with immunoblots for Acat1 and acetyllysine, and activity was measured as above.
Electrostatic Modeling
To assess the changes in charge complementarity for CoA in the active site of Acat1 caused by lysine acetylation, the tetrameric coordinates of Protein Data Bank code 2IBW (24) were prepared for electrostatic calculations. Water and other ligands were removed from the model. Lysine residues were either fully charged in the calculation or neutralized to simulate the effect of acetylation. An electrostatic map was calculated using the Adaptive Poisson-Boltzmann Solver (31) plugin in PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4, Schodinger, LLC) using default parameters. Local electrostatic environment was visualized as a continuous red-blue gradient running from −4 to +4 kT/e on the solvent accessible surface of the protein, or as an isosurface extending through space at +1 kT/e.
RESULTS
Quantitative Analysis of Mitochondrial Acetylation Dynamics during Fasting and Refeeding
To quantify the dynamics of mitochondrial acetylation with high resolution, we leveraged our recently developed strategy for efficient acetyl peptide enrichment (18) and our QuantMode method (21) for highly accurate quantitative proteomics with isobaric tags to profile mouse liver mitochondrial protein abundance and acetylation levels across the transition between fasting and refeeding. Eight mice were fasted overnight for 16 h, after which half were allowed to feed ad libitum for 2 h (Fig. 1A), a time point featuring marked PTM changes with limited protein abundance changes (19). We identified 1,915 unique acetylation isoforms, of which 1,796 were quantified (Fig. 1B and supplemental Fig. S1). Unsupervised average linkage hierarchical clustering of the data based on relative acetyl isoform occupancy (site-specific acetyl fold change divided by protein abundance fold change) grouped the refed and fasted mice separately (Fig. 2A), indicating that acetylation changes are a reproducible and distinguishing feature of the transition between fasting and refeeding (32, 33). Importantly, our quantitative approach reveals that ∼10% (188) of identified acetyl isoforms exhibited a statistically significant change (q ≤ 0.1) in acetyl occupancy in response to this acute perturbation of metabolic status.
FIGURE 1.
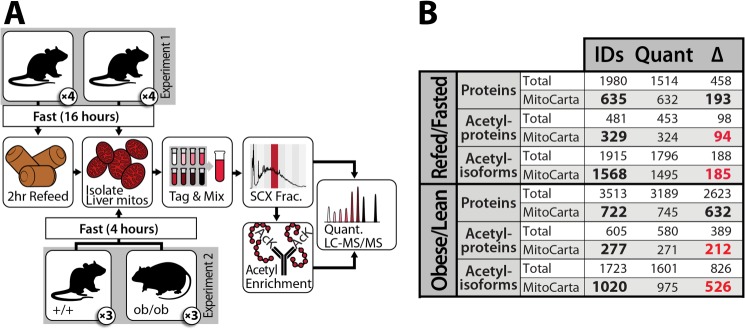
Schematic of workflow for quantitative proteomic and acetylomic analyses of mouse liver mitochondria. A, in experiment one, eight lean mice were fasted for 16 h and either sacrificed immediately or refed for 2 h before being sacrificed. In experiment two, three obese mice and three lean mice were fasted for 4 h before being sacrificed. In each case, liver mitochondria were enriched and analyzed by LC-MS/MS (see “Experimental Procedures”). SCX Frac., strong cation exchange fractionation. B, summary of protein and acetylation data. IDs, unique identifications at 1% false discovery rate; Quant, measurements quantified with isotopic reporter ions in all mice assessed; Δ, measurements significantly changing with q ≤ 0.1. See also supplemental Fig. S1.
FIGURE 2.
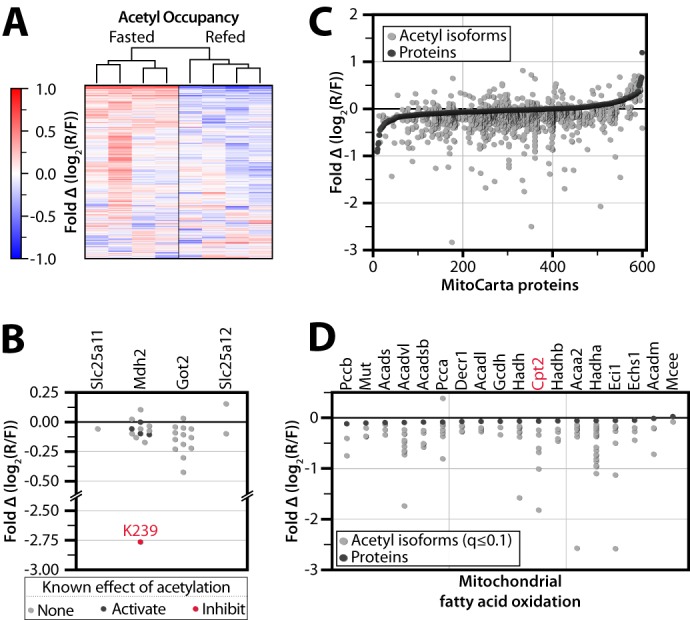
Global analysis of acute protein and acetylation changes during the transition from fasting to refeeding. A, heat map showing unsupervised average linkage hierarchical clustering of four fasted and four refed mice groups them according to nutrient status. Values are colored based on relative acetyl occupancy, normalized to the average of all eight mice, on a log2 scale from less than −1 to greater than 1. R/F, refed/fasted. B, relative acetyl occupancy fold change for proteins in the malate-aspartate shuttle. The acetylation site previously shown to inhibit Mdh2 activity is shown in red, whereas those previously shown to activate Mdh2 are shown in black. Other sites we detected as acetylated but that have no prior functional data are shown in gray. C, changes in acetylation are greater than changes in protein abundance. All quantified MitoCarta proteins are ranked on the x axis by protein abundance fold change, refed/fasted (black dots). Relative acetyl isoform changes are plotted in the same position on the x axis as the corresponding protein measurement (gray dots). D, acetyl and protein abundance changes in mitochondrial fatty acid oxidation. Proteins are marked in black, and significantly changing (q ≤ 0.1) acetyl isoforms are marked in gray.
We hypothesize that the subset of mitochondrial protein acetylation sites with changing occupancy upon refeeding represent those most likely to have a regulatory function during this transition. For example, we detect acetylation on all four mitochondrial members of the malate-aspartate shuttle (Mdh2, Got2, Slc25a11, and Slc25a12), which is responsible for relaying the reducing potential of NADH generated from glycolysis into the mitochondrial matrix (Fig. 2B) (34). Rat liver Mdh2 activity is increased upon feeding (35), and previous studies have reported that Mdh2 activity can be increased either by acetylation (on Lys-185, Lys-301, Lys-307, and Lys-314) (36) or by Sirt3-dependent deacetylation (of Lys-239) (18). Here, we detect a large decrease in acetylation occupancy of Mdh2 Lys-239 with little or no changes to the other sites. This indicates that the Sirt3-controlled acetylation site (Lys-239) is likely the dominant regulator of Mdh2 during this acute metabolic perturbation, consistent with an increased flux of reducing equivalents from the cytosol into the mitochondria for energy production upon feeding.
Quantifying the magnitude of acetylation changes across an acute time scale can provide insights into key acetyl sites without the confounding changes in protein levels that accompany longer perturbations (Fig. 2C). For example, despite unchanging protein levels among almost all enzymes involved in mitochondrial fatty acid oxidation (FAO), more than 90 distinct acetyl isoforms within this pathway are altered significantly upon refeeding (Fig. 2D). These include four acetylation sites on carnitine O-palmitoyltransferase 2 (Cpt2; Lys-104, Lys-544, Lys-537, and Lys-453), an enzyme involved in the transport of fatty acids from the cytosol into the mitochondrial matrix for β-oxidation (37). Similarly, energy production from amino acid degradation is robustly induced in the postprandial state (38), and several enzymes in amino acid catabolic processes exhibit large changes in relative acetylation occupancy without alteration to the underlying protein abundance levels. As one example, acetylation on Lys-329 of glutaminase (Gls2) decreases >2-fold following refeeding, which is consistent with this PTM serving an inhibitory function on this key enzyme of amino acid breakdown. Overall, the vast majority of dynamic acetylation sites decreases in relative acetyl occupancy upon refeeding, consistent with previous reports that fasting can lead to hyperacetylation of liver mitochondrial proteins (23).
Quantitative Analysis of Mitochondrial Acetylation Dynamics due to Chronic Obesity
Previous studies have noted general changes to the acetylproteome during the onset of obesity (13, 39); however, these studies were also limited by the semi-quantitative nature of their applied mass spectrometry methods. To evaluate and measure the key obesity-induced changes in acetylation of mitochondrial proteins with high accuracy and depth of coverage, we performed a large-scale quantitative comparison of the liver mitochondrial acetylproteome from three lean (Leptin+/+) and three obese (Leptinob/ob) mice at 10 weeks of age, each fed standard chow diet ad libitum (Fig. 1A). This approach provides a comparison that is similar in nature to the first perturbation (contrasting nutrient abundance) but markedly different in time scale (hours versus weeks). Leptin deficiency in B6 mice is known to cause hyperphagia-induced obesity, which is accompanied by a dramatic response to over-nutrition (e.g. hyperinsulinemia, hepatic steatosis) by 10 weeks (40). These obese mice, however, have only a mild, controlled elevation in blood glucose and model the prediabetic state, providing a model of obesity that is not confounded by diabetes. Here, we identified 1,723 unique acetyl isoforms, 1,601 of which were quantified, and 826 that were significantly changing (q ≤ 0.1; Fig. 1B, and supplemental Fig. S1).
Not surprisingly, given the relative magnitudes and durations of the perturbations involved, the changes in protein abundance and acetyl occupancy due to chronic obesity encompass a greater dynamic range than those seen following a mere 2-h refeeding (Fig. 1B and Fig. 3, A and B). This comparison could reveal key acetylation events potentially important for regulating mitochondrial function in states of chronic overnutrition. For instance, it is known that the protein abundance and the activity of complexes in the oxidative phosphorylation (OxPhos) pathway are increased in certain models of obesity (19, 41), and acetylation has been implicated in inhibiting all five complexes of OxPhos (39, 42–45); however, the specific regulatory sites are as yet unclear. To confirm the increase in OxPhos flux in our model of obesity, we isolated liver mitochondria from obese and lean livers and measured oxygen consumption using a Seahorse extracellular flux analyzer. As expected, mitochondria from obese mice exhibit an elevated state 3 oxygen consumption rate compared with their lean counterparts (Fig. 3D and supplemental Fig. S1). In our obese versus lean acetylome comparison, we identified 156 acetyl isoforms on OxPhos proteins (Fig. 3C). Ninety-seven of these sites change significantly (q ≤ 0.1) in acetyl occupancy, 91 of which decrease in the obese state. We therefore predict that a subset of these 91 sites (which we further prioritize below) plays an important role in the stimulation of OxPhos activity in obesity, irrespective of any protein abundance changes.
FIGURE 3.
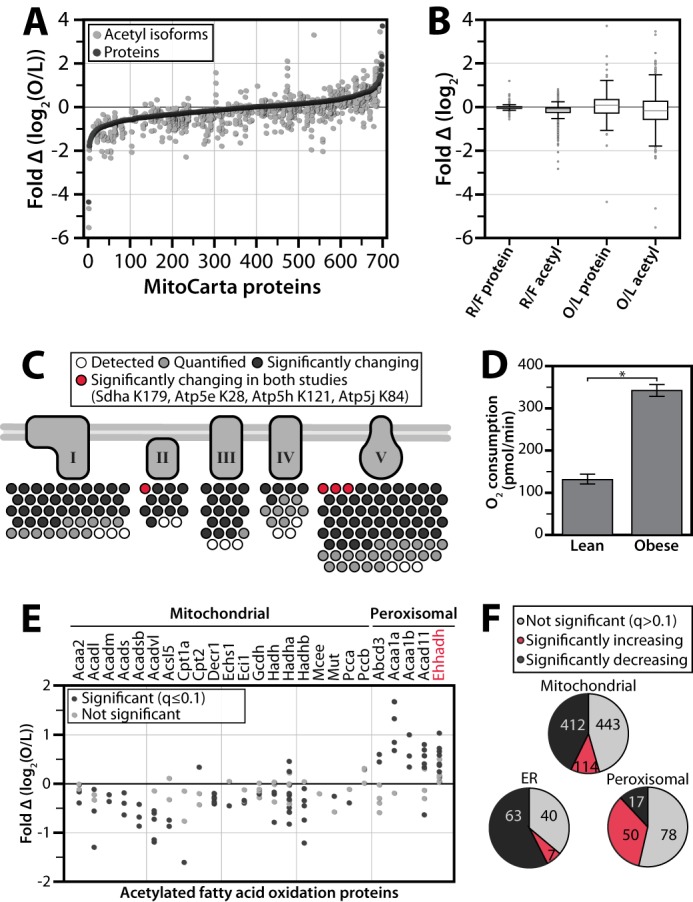
Comparison of of acetylproteome response to acute and chronic perturbations. A, all quantified MitoCarta proteins are ranked on the x axis by protein abundance fold change (black dots) for the chronic comparison of obese (O) and lean (L) mice. Relative acetyl isoform changes are plotted in the same position on the x axis as the corresponding protein measurement (gray dots). B, dynamic ranges of protein and acetyl abundance are greater in obese/lean study than in refed/fasted (R/F) study. A box plot comparing the ranges of protein abundance and acetyl isoform fold change in the acute (refed/fasted) study versus the chronic (obese/lean) study. Whiskers represent data within 50% of the range of the first and third quartiles, and remaining outlier data is represented as a gray dots. C, prioritization of acetylation sites in oxidative phosphorylation. Acetyl isoforms exhibiting significant changes (q ≤ 0.1) in both studies are in red; those significantly changing in the obese versus lean study only are in black; those isoforms that are not changing significantly are in gray; isoforms that were identified but not quantified are in white. D, ADP stimulated increase in oxygen consumption rate is greater in mitochondria isolated from obese livers than from lean livers. An asterisk indicates significance (p value = 3.9 × 10−14). E, relative acetyl occupancy changes in fatty acid oxidation. Sites with significantly changing (q ≤ 0.1) occupancy are marked in black, those that were quantified but not significantly changing in gray, and those that colocalize to peroxisomes are grouped. F, acetylation by organelle. Pie charts show the number of mitochondrial, peroxisomal, or endoplasmic reticular acetyl isoforms from the obese/lean study that are not significant (q > 0.1) (gray), significantly increasing (red), and significantly decreasing (black).
As with our fasting and refeeding study, we find that catabolic enzymes, including those of FAO and amino acid degradation, appear particularly susceptible to alterations in acetylation levels. For example, acetylation of Lys-42 on long chain acyl-CoA dehydrogenase (Acadl) is significantly decreased with obesity. This PTM has previously been shown to decrease enzymatic activity (46), consistent with elevated mitochondrial FAO in obese animals. Aminoadipate aminotransferase (Aadat), a protein involved in tryptophan degradation to kynurenine acid (47), has increased acetylation on Lys-263, which is involved in binding the pyridoxal phosphate cofactor (48, 49). We predict that acetylation of Lys-263 reduces aminoadipate aminotransferase activity by preventing this enzyme-cofactor interaction. This decrease in activity could shift tryptophan degradation from the kynurenine-kynurenic acid pathway toward the kynurenine-NAD+ pathway, which has previously been linked to hypertension, diabetes, atherosclerosis, obesity, and immunodeficiency (50).
Notably, for our proteomics analyses we purified mitochondria only to the extent required to achieve near comprehensive coverage of the liver MitoCarta protein list. (MitoCarta is a compendium of mitochondrial proteins (51).) As such, we were also able to profile the acetylproteomes of other co-purifying organelles, including peroxisomes and endoplasmic reticulum. Interestingly, FAO enzymes with evidence of mitochondrial/peroxisomal co-localization are generally increasing in relative occupancy with obesity (38 detected; 24 increase, one decreases, q ≤ 0.1) (Fig. 3E). In comparison, acetylation sites on mitochondria-specific FAO enzymes are mostly decreasing (89 detected; three increase, 49 decrease, q ≤ 0.1). Among the peroxisomal enzymes with increasing acetylation is Ehhadh (enoyl-CoA, hydratase/3-hydroxyacyl CoA dehydrogenase), which is activated by this PTM (36). We detect increasing acetylation on seven lysines, with the greatest change on Lys-344, consistent with the known increase peroxisomal fatty oxidation (i.e. α-oxidation) in obesity (35, 52). Overall, acetylation increases on most peroxisomal proteins in the obese state but decreases on most mitochondrial and endoplasmic reticulum proteins (Fig. 3F).
Integration of Disparate Data Sets Highlights Key Regulatory Acetylation Sites
The analyses above clarify the role of previously identified acetylation events and also quantify the acetyl occupancy of a wide range of uncharacterized acetylation sites. To further prioritize this extensive list for subsequent mechanistic studies, we compared the dynamic acetylation sites observed in both datasets. Of the thousands of mitochondrial sites we identify across the two studies, only 48 change significantly (q ≤ 0.1) due to both refeeding and obesity (Fig. 4C). We hypothesized that these isoforms likely represent key PTMs that serve as common mechanisms for mediating metabolic responses to a variety of different perturbations in nutrient status. Included in these are four of the OxPhos isoforms noted above (Sdha Lys-179 in complex II, and Atp5e Lys-28, Atp5h Lys-121, and Atp5j K84 in complex V), further highlighting them as high-likelihood regulatory sites from among the hundreds of OxPhos sites identified (Figs. 3C and 4C).
FIGURE 4.

Integration of different metabolic comparisons identifies candidate regulatory acetylation events. A, volcano plots of fold change in relative acetyl occupancy versus −log10 (p value). B and C, comparison of the relative acetyl occupancy fold change across the refed/fasted versus obese/lean comparisons. Only isoforms that were quantified in both studies are represented. B, putative Sirt3 targets are shown in red, other MitoCarta proteins are in black, and non-MitoCarta proteins are colored gray. Inset, Sirt3 protein abundance fold change by mass spectrometry. An asterisk indicates significance (p ≤ 0.05). C, acetylation sites that are significantly changing in both studies (q ≤ 0.1). Fold changes (log2) are shown in red (positive) and blue (negative) for refed/fasted (R/F) and obese/lean (O/L).
We reasoned that many bona fide regulatory acetylation sites would also be targets of Sirt3, the only well established mitochondrial deacetylase (17). Therefore, we further integrated our data sets with a compendium of predicted Sirt3 targets, which we recently developed from analysis of wild type and Sirt3−/− mouse liver mitochondria (18). Sirt3 target sites are among those with the largest decrease in relative acetyl occupancy due to acute refeeding or chronic obesity, indicative of an induction of Sirt3 activity in these states (Fig. 4, A and B). Strikingly, putative Sirt3 targets represent over half of the acetyl isoforms that exhibited a significant change (q ≤ 0.1) in relative acetyl occupancy in both studies despite accounting for <7% of all sites quantified. These results suggest a major, Sirt3-driven remodeling of the acetylproteome that is secondary to genetic obesity or to 2 h of refeeding after an overnight fast. Measurements for Sirt3 protein using both quantitative MS (Fig. 4B, inset) and immunoblotting (supplemental Fig. S2) indicate that Sirt3 abundance is increased in B6 10-week-old animals with obesity, consistent with our activity predictions. We note that this obesity-induced increase in Sirt3 abundance mirrors the induction of Sirt3 levels observed at early time points in the regimen of high fat diet feeding (13), as opposed to the decrease seen after many weeks of this diet. We did not observe a significant change in Sirt3 abundance following refeeding, despite the significant decrease in acetylation levels of predicted Sirt3 sites (Fig. 4, A–C, and supplemental Fig. S2). This is consistent with previous reports that alterations in Sirt3 activity are not always correlated with its protein abundance levels (39, 53, 54), suggesting that Sirt3 itself might be subject to post-translational regulation following acute perturbations. It is also consistent with indirect measures of mitochondrial redox state that suggest an increase in the NAD+/NADH ratio in the refed state (55).
Among the dynamic Sirt3 targets observed in both large-scale analyses are multiple sites on enzymes from core metabolic pathways involved in responding to changing nutrient status. Three of these processes, branched chain amino acid metabolism, fatty acid catabolism, and ketogenesis, exhibit acetylation changes in many pathway members and have in common a single enzyme: acetyl-coenzyme A acetyltransferase 1 (Acat1, NCBI gene ID 110446) (56, 57). Acat1 catalyzes two major reversible reactions: the cleavage of acetoacetyl-CoA into two acetyl-CoA, and the cleavage of 2-methyl acetoacetyl-CoA into propionyl-CoA and acetyl-CoA. We identified 18 total acetylation sites on this protein, nine of which are changing significantly (q ≤ 0.1) in at least one comparison. Relative acetyl occupancy at three of these sites changed significantly in both analyses (Lys-187, Lys-260, and Lys-265) (Fig. 5A), the latter two of which are predicted Sirt3 targets. Another recent Sirt3 knock-out study also identifies Lys-260 and Lys-265, but not Lys-187, as Sirt3 targets (15). In the present study, the acetylation levels of both putative Sirt3-target sites on Acat1 decrease markedly with obesity (relative to lean mice) and with refeeding (relative to fasted mice), whereas acetylation on Lys-187 increases due to obesity. These data led us to hypothesize that acetylation on Lys-187, Lys-260, and/or Lys-265 alters the activity of Acat1. Moreover, as Sirt3 most often activates its target enzymes (17), we further hypothesized that deacetylation of Lys-260 and Lys-265 increases Acat1 activity in response to nutrient excess.
FIGURE 5.
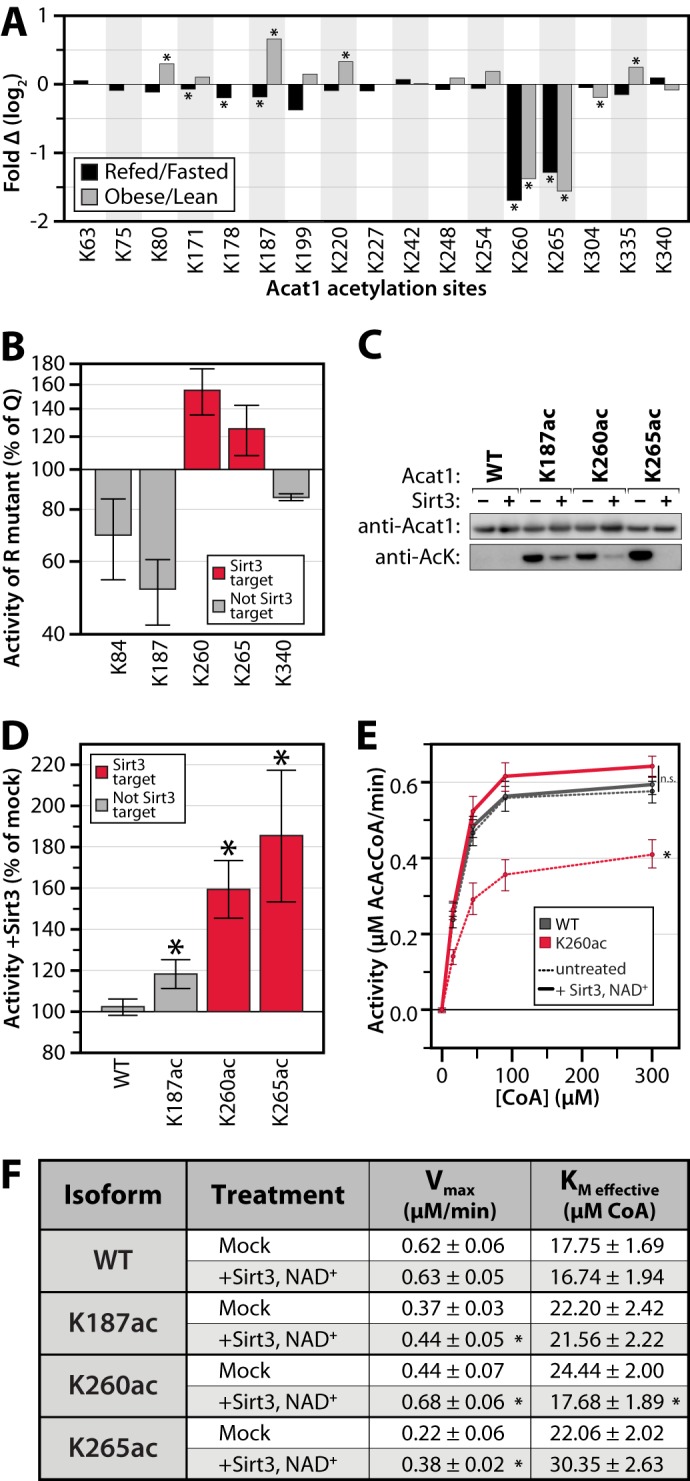
Reversible, Sirt3-regulated acetylation of Acat1 Lys-260 inhibits activity. A, relative acetyl occupancy fold change for 17 quantified sites on Acat1. An asterisk indicates q ≤ 0.1. B, in vitro enzyme activity assays of Acat1 mutants generated by mutating lysine to glutamine (Q, acetyl mimic) or arginine (R, deacetyl mimic). Data are expressed as the average activity of the Arg mutant as a percentage of that for the Gln mutant, at 300 μm CoA (± S.E., n ≥ 2). C, immunoblot for Acat1 and acetyllysine on samples of purified Acat1 protein used in kinetic assays. Proteins were treated with or without NAD+ and Sirt3. AcK, acetyllysine. C, representative of samples used in D. D–F, in vitro enzyme activity assays of Acat1 with site-specific incorporation of acetyllysine at the indicated residues, with or without deacetylase (Sirt3) treatment. D, data are expressed as the average activity after Sirt3 treatment as a percent of mock-treated enzyme, at 300 μm CoA (± S.E., n ≥ 3; an asterisk indicates a significant difference from mock treatment, p < 0.05). E, aggregated enzyme kinetic analysis (n = 4; an asterisk indicates a significant difference from all other treatments, p < 0.05; n.s. indicates no significant difference between the highlighted treatments). F, table of kinetic parameters of Acat1 isoforms ± Sirt3 treatment (± S.E., n ≥ 3; an asterisk indicates a significant difference from mock treatment, p < 0.05).
Acetylation of Lysine 260 and 265 on Acat1 Inhibits Enzymatic Activity
Our analyses suggest that acetylation of select Acat1 lysines is important for regulating enzyme activity. To begin testing this possibility, we individually mutated five acetylated lysine residues (Lys-84, Lys-187, Lys-260, Lys-265, and Lys-340) to glutamine to mimic the charge state of an acetylated lysine or mutated to arginine to mimic a deacetylated state (58). These sites were selected based on the structure of human ACAT1 (24) and on the dynamic changes seen in both data sets. Acat1 variants and wild type protein were purified from E. coli and tested by an in vitro activity assay. Mutation of lysines 260 and 265 to arginine (K260R, K265R) resulted in increased activity compared with the respective glutamine mutant (K260Q, K265Q), suggesting that acetylation would decrease activity (Fig. 5B). In contrast, K187R led to a loss of activity compared with K187Q. Interestingly, mutation of the other acetylation sites did not significantly change the activity of Acat1, further suggesting that Lys-187, Lys-260, and Lys-265 may be the most important acetylation sites for regulating the activity of this enzyme.
Although mutation of lysine to glutamine or arginine can approximate the effect of acetylation on protein function, these are imperfect analogs. To validate the ability of acetylation to regulate Acat1 enzymatic activity, we used site-specific acetyllysine incorporation, a technique that allows for the production of a protein with nearly full acetylation occupancy on a specific lysine (26, 27). Acat1 variants were generated and purified that each possessed a single acetylated lysine at position 187, 260, or 265 (K187ac, K260ac, K265ac). Acetyllysine incorporation was verified by immunoblot and MS analyses, and in vitro treatment of these proteins with recombinant Sirt3 effectively removed this acetylation in an NAD+-dependent manner (Fig. 5C and supplemental Fig. S3). Interestingly, in vitro Sirt3 treatment more readily removed the acetyl groups on Lys-260 and Lys-265 than that on Lys-187, consistent with the prediction that K260ac and K265ac are Sirt3 targets in vivo.
In vitro Acat1 activity assays of each isoform revealed that Sirt3-mediated deacetylation of K260ac and K265ac dramatically increased enzyme activity in an NAD+-dependent manner (Fig. 5, D–F and supplemental Fig. S4). In contrast, we observed minimal change in activity with identically treated wild type or K187ac proteins. We further analyzed the kinetic properties of the Acat1 K260 in its acetylated and deacetylated forms across several concentrations of substrate and determined that acetylation decreases the Vmax of the enzyme while increasing its Km for CoA (Fig. 5F). As physiological levels of CoA in liver are markedly higher than the Acat1 Km (59), these alterations in Vmax will likely alter metabolic flux in vivo.
Consistent with the hypothesis that regulatory residues are likely to be conserved throughout evolution (16, 60), Acat1 Lys-260 and Lys-265 are highly invariant and correspond to lysines 263 and 268 in the human ACAT1 ortholog (K263_Hs and K268_Hs). Recently, Haapalainen and colleagues (24) solved the crystal structure of human ACAT1 in complex with its CoA substrate. This structure reveals that positively charged K263_Hs is in close proximity to the negatively charged 3′-phosphate of CoA, which suggests that charge complementarity for CoA in the active site is important for substrate binding. Using this structure (Protein Data Bank code 2IBW) (Fig. 6A), we modeled a neutralized lysine residue at K263_Hs to simulate the effect of acetylation (Fig. 6B). We then calculated electrostatic isosurfaces extending through space at +1 kT/e with all lysines fully charged (Fig. 6C), or with K263_Hs neutralized (Fig. 6D). This analysis clearly reveals that the negatively charged 3′-phosphate of CoA is present within the positively charged space enclosed by this electrostatic isosurface in the fully charged enzyme, but not when K263_Hs is neutralized. These data suggest that acetylation of Acat1 Lys-260 (ACAT1 K263_Hs) likely reduces the favorable electrostatic interactions between CoA and the Acat1 binding pocket. This is similar to reports of the Sirt3-dependent regulation of other enzymes by an electrostatic repulsion mechanism, including Mn-SOD (61, 62). Together, in conjunction with our in vitro activity data, we propose that Sirt3 activates Acat1 by removing lysine acetylation that reduces the affinity of Acat1 for CoA.
FIGURE 6.
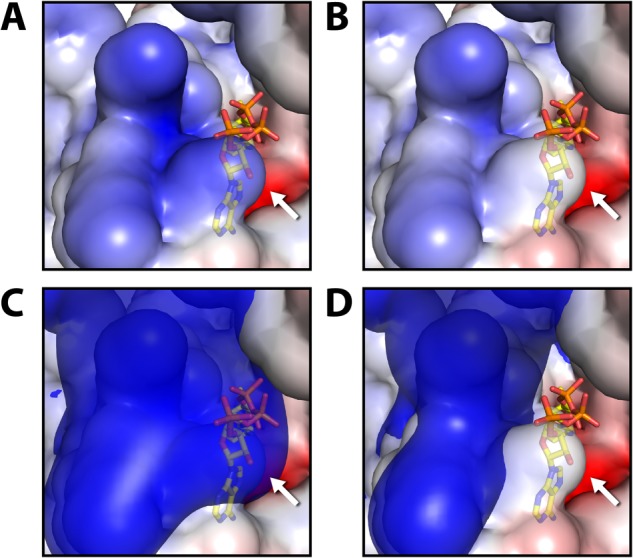
Electrostatic modeling of acetylation effects using the crystal structure of Homo sapiens ACAT1 (Protein Data Bank code 2IBW). Local electrostatic environment of ACAT1 active site surrounding K263_Hs (K260_Mm, identified by the white arrow). A and C represent wild-type ACAT1, whereas B and D mimic acetylation on K263_Hs by neutralizing its charge. The electrostatic environment is visualized as a continuous red-blue gradient from −4 to +4 kT/e on the solvent accessible surface of the protein. In C and D, it is overlaid with an electrostatic isosurface extending through space at +1 kT/e. The negatively charged 3′-phosphate of CoA is located within the space enclosed by the positive isosurface when K263_Hs is fully charged, but it is located outside when this lysine is neutralized.
DISCUSSION
Reversible lysine acetylation is rapidly becoming recognized as a pervasive PTM that regulates cellular processes ranging from gene transcription to intermediary metabolism. The impact of this modification extends to mitochondria, where a strikingly high percentage of proteins are acetylated on one or more lysines. A few of these sites are now known to modify metabolic flux by altering enzymatic activity or substrate availability, adding to the growing notion that PTMs are key to regulating mitochondrial function.
However, although thousands of mitochondrial acetylation sites have now been identified, little is known about the enzymes that control acetylation in mitochondria. There is only a single well established mitochondrial protein deacetylase, Sirt3 (17). Likewise, there is no known acetyltransferase within this organelle, although emerging evidence suggests that GCN5-L1 may be a regulator of a yet to be characterized protein acetyltransferase complex (63). Further confounding this issue is the fact that even at low concentrations of acetyl-CoA, acetylation can occur nonenzymatically (64, 65). Coupled with the knowledge that mitochondrial enzymes tend to become acetylated following nutritional changes that produce an increase in acetyl-CoA (66) and that there are a very limited number of validated regulatory sites, this has led to the speculation that many of the numerous mitochondrial acetylation events may be nonenzymatic and, thus, likely nonregulatory. As such, rigorous efforts are needed to spotlight and validate bona fide regulatory acetylation sites from a potentially noisy background of non-regulatory PTM “decorations” (67).
To prioritize candidate acetyl-regulated sites on mitochondrial proteins, we performed large-scale quantitative acetyl-proteomics across multiple contrasting biological states. Our data reveal that only a select subset of sites significantly change in occupancy during drastic acute and chronic alterations in nutritional status. From our recent study (18), we predict that many of these dynamic acetylation sites are targets of Sirt3, consistent with the role of this enzyme as a primary regulator of mitochondrial acetylation. Together, our integrated analyses highlight a collection of proteins with high-confidence candidate regulatory sites (Fig. 4C). Although other acetylation sites may prove to be regulatory under distinct metabolic transitions, our data nonetheless caution that many others might represent unregulated and likely non-regulatory targets of acetylation.
A second priority in the field is to rigorously validate and define the functions of this modification in regulating protein activity (16, 68). In this study, we investigated the importance of acetylation on Acat1. Our quantitative analyses nominated three of the 18 acetylated lysines on this protein as likely regulatory sites. Using site-specific acetyllysine incorporation, in vitro enzymatic analyses and molecular modeling, we demonstrated that acetylation of Lys-260 and Lys-265 inhibits Acat1 activity by disrupting CoA binding.
Acat1 is found at the cross-roads of branched chain amino acid degradation, ketogenesis, and fatty acid catabolism. Interestingly, more than one-third (19/48) of our high-confidence candidate regulatory sites are on proteins involved in these three pathways. As such, we hypothesize that the role of regulatory acetylation is not limited to isolated control points, but rather that it impacts multiple enzymes of a pathway to collectively alter net flux, often alongside other PTMs. Indeed, other key regulators are known for each of these pathways (e.g. Bckdha phosphorylation (69), Hmgcs2 acetylation (70) and phosphorylation (19), and Acadl acetylation (46)). Interestingly, Acat1 Lys-265 was also recently identified as a prominent site of reversible succinylation, further suggesting that this is an unusually important site of post-translational regulation (71). Based on our work here, we predict that this modification would likewise dampen Acat1 activity.
Although each of the three pathways mentioned above occurs in the liver, they are not simultaneously induced. For example, branched chain amino acid degradation is robustly induced by refeeding, as assessed by Bckdha phosphorylation status (19, 38) but fatty acid oxidation and ketogenesis are inhibited in that same state (56). Conversely, fatty acid oxidation and ketogenesis are highly active in the obese state (72). Thus, it seems reasonable to speculate that the overabundance of PTMs on ketogenesis, FAO, and branched chain amino acid enzymes noted above, including those on Acat1, operate combinatorially to help properly calibrate the individual fluxes through these interrelated but distinct pathways. Furthermore, although our biochemical data support a role for deacetylation of Acat1 in enhancing flux through any these pathways, its acetylation alone would likely not be sufficient for complete inactivation of any one of them. Further integration of our data sets with other protein PTM data and quantitative metabolic flux analyses will be necessary to better define the physiological ramifications of these modifications in collectively modifying metabolic output.
The results of our analyses both mirror current trends in the acetylation field and also raise new issues. Consistent with previous work, we find that acetylation is widespread in mitochondria and that its levels generally tend to track with predicted acetyl-CoA abundance (66). Our data also suggest that Sirt3 is more active in the refed state and early during the onset of obesity and that it tends to enhance the activity of its target enzymes. However, our data reveal that acetyl occupancy at several putative Sirt3-regulated sites (e.g. Eci2 Lys-60, Gstk1 Lys-93, Cs Lys-52, and Tst Lys-175) was significantly decreased due to refeeding but significantly increased due to obesity. This suggests that Sirt3 may possess an adjustable target preference under different nutritional states. Moreover, our data indicate that Sirt3 activity cannot necessarily be predicted from its abundance, implying that other regulatory post-transcriptional or post-translational features might be important for titrating its activity. Last, our data reveal that many probable non-Sirt3 sites also change considerably in acetylation occupancy, supporting the notion that other mitochondrial regulatory acetylation machinery awaits discovery.
Collectively, our work provides a quantitative map of the changing mitochondrial protein acetylation landscape during acute and chronic metabolic transitions, defines the role of deacetylation in stimulating Acat1 activity, and highlights many other high-priority acetylation sites for mechanistic follow-up work (19).
Acknowledgment
We thank A. J. Bureta for assistance with figure illustrations.
This work was supported by a Searle Scholars Award, a Shaw Scientist Award, a Glenn Foundation Award, and National Institutes of Health Grants RC1DK086410, U01GM094622, and R01DK098672 (to D. J. P.); National Science Foundation Graduate Fellowship DGE-1256259 and National Institutes of Health Training Grant 5T32GM007215-37 (to A. J. S.); National Institutes of Health Grants R01GM080148 (to J. J. Coon), DK066369 and DK058037 (to A. D. A.), AG038679 and GM065386 (to J. M. D.), T32DK007665 and T32GM008692 MSTP (to B. J. F.), and F32DK091049 (to P. A. G.); American Heart Association Grant 12PRE839 (to J. J. Carson); and a National Science Foundation graduate fellowship and National Institutes of Health Training Grant 5T32GM08349 (to K. E. D.).

This article contains supplemental “Experimental Procedures,” Figs. S1–S4, and additional references.
- PTM
- post-translational modification
- Acat1
- acetyl-CoA acetyltransferase 1
- FAO
- fatty acid oxidation
- OxPhos
- oxidative phosphorylation.
REFERENCES
- 1. Allfrey V. G., Faulkner R., Mirsky A. E. (1964) Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 51, 786–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Phillips D. M. (1963) The presence of acetyl groups of histones. Biochem. J. 87, 258–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gnad F., Forner F., Zielinska D. F., Birney E., Gunawardena J., Mann M. (2010) Evolutionary constraints of phosphorylation in eukaryotes, prokaryotes, and mitochondria. Mol. Cell. Proteomics 9, 2642–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Norvell A., McMahon S. B. (2010) Cell biology. Rise of the rival. Science 327, 964–965 [DOI] [PubMed] [Google Scholar]
- 5. Guan K. L., Xiong Y. (2011) Regulation of intermediary metabolism by protein acetylation. Trends Biochem. Sci. 36, 108–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 7. Lundby A., Lage K., Weinert B. T., Bekker-Jensen D. B., Secher A., Skovgaard T., Kelstrup C. D., Dmytriyev A., Choudhary C., Lundby C., Olsen J. V. (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Anderson K. A., Hirschey M. D. (2012) Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 52, 23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vadvalkar S. S., Baily C. N., Matsuzaki S., West M., Tesiram Y. A., Humphries K. M. (2013) Metabolic inflexibility and protein lysine acetylation in heart mitochondria of a chronic model of Type 1 diabetes. Biochem. J. 449, 253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lombard D. B., Tishkoff D. X., Bao J. (2011) Mitochondrial sirtuins in the regulation of mitochondrial activity and metabolic adaptation. Handb. Exp. Pharmacol. 206, 163–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vaitheesvaran B., Yang L., Hartil K., Glaser S., Yazulla S., Bruce J. E., Kurland I. J. (2012) Peripheral effects of FAAH deficiency on fuel and energy homeostasis: role of dysregulated lysine acetylation. PLoS One 7, e33717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim H. S., Patel K., Muldoon-Jacobs K., Bisht K. S., Aykin-Burns N., Pennington J. D., van der Meer R., Nguyen P., Savage J., Owens K. M., Vassilopoulos A., Ozden O., Park S. H., Singh K. K., Abdulkadir S. A., Spitz D. R., Deng C. X., Gius D. (2010) SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17, 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirschey M. D., Shimazu T., Jing E., Grueter C. A., Collins A. M., Aouizerat B., Stančáková A., Goetzman E., Lam M. M., Schwer B., Stevens R. D., Muehlbauer M. J., Kakar S., Bass N. M., Kuusisto J., Laakso M., Alt F. W., Newgard C. B., Farese R. V., Jr., Kahn C. R., Verdin E. (2011) SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 44, 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pereira C. V., Lebiedzinska M., Wieckowski M. R., Oliveira P. J. (2012) Regulation and protection of mitochondrial physiology by sirtuins. Mitochondrion 12, 66–76 [DOI] [PubMed] [Google Scholar]
- 15. Rardin M. J., Newman J. C., Held J. M., Cusack M. P., Sorensen D. J., Li B., Schilling B., Mooney S. D., Kahn C. R., Verdin E., Gibson B. W. (2013) Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. U.S.A. 110, 6601–6606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beltrao P., Albanèse V., Kenner L. R., Swaney D. L., Burlingame A., Villén J., Lim W. A., Fraser J. S., Frydman J., Krogan N. J. (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. He W., Newman J. C., Wang M. Z., Ho L., Verdin E. (2012) Mitochondrial sirtuins: regulators of protein acylation and metabolism. Trends Endocrinol. Metab. 23, 467–476 [DOI] [PubMed] [Google Scholar]
- 18. Hebert A. S., Dittenhafer-Reed K. E., Yu W., Bailey D. J., Selen E. S., Boersma M. D., Carson J. J., Tonelli M., Balloon A. J., Higbee A. J., Westphall M. S., Pagliarini D. J., Prolla T. A., Assadi-Porter F., Roy S., Denu J. M., Coon J. J. (2013) Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grimsrud P. A., Carson J. J., Hebert A. S., Hubler S. L., Niemi N. M., Bailey D. J., Jochem A., Stapleton D. S., Keller M. P., Westphall M. S., Yandell B. S., Attie A. D., Coon J. J., Pagliarini D. J. (2012) A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 16, 672–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao E., Keller M. P., Rabaglia M. E., Oler A. T., Stapleton D. S., Schueler K. L., Neto E. C., Moon J. Y., Wang P., Wang I. M., Lum P. Y., Ivanovska I., Cleary M., Greenawalt D., Tsang J., Choi Y. J., Kleinhanz R., Shang J., Zhou Y. P., Howard A. D., Zhang B. B., Kendziorski C., Thornberry N. A., Yandell B. S., Schadt E. E., Attie A. D. (2009) Obesity and genetics regulate microRNAs in islets, liver, and adipose of diabetic mice. Mamm. Genome 20, 476–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wenger C. D., Lee M. V., Hebert A. S., McAlister G. C., Phanstiel D. H., Westphall M. S., Coon J. J. (2011) Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat. Methods 8, 933–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wenger C. D., Phanstiel D. H., Lee M. V., Bailey D. J., Coon J. J. (2011) COMPASS: A suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 11, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rogers G. W., Brand M. D., Petrosyan S., Ashok D., Elorza A. A., Ferrick D. A., Murphy A. N. (2011) High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One 6, e21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haapalainen A. M., Meriläinen G., Pirilä P. L., Kondo N., Fukao T., Wierenga R. K. (2007) Crystallographic and kinetic studies of human mitochondrial acetoacetyl-CoA thiolase: the importance of potassium and chloride ions for its structure and function. Biochemistry 46, 4305–4321 [DOI] [PubMed] [Google Scholar]
- 25. Blommel P. G., Becker K. J., Duvnjak P., Fox B. G. (2007) Enhanced bacterial protein expression during auto-induction obtained by alteration of lac repressor dosage and medium composition. Biotechnol. Prog. 23, 585–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neumann H., Peak-Chew S. Y., Chin J. W. (2008) Genetically encoding Nϵ-acetyllysine in recombinant proteins. Nat. Chem. Biol. 4, 232–234 [DOI] [PubMed] [Google Scholar]
- 27. Neumann H., Hancock S. M., Buning R., Routh A., Chapman L., Somers J., Owen-Hughes T., van Noort J., Rhodes D., Chin J. W. (2009) A method for genetically installing site-Specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell 36, 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu W., Dittenhafer-Reed K. E., Denu J. M. (2012) SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 287, 14078–14086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hallows W. C., Lee S., Denu J. M. (2006) Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. U.S.A. 103, 10230–10235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Middleton B. (1973) The acetoacetyl-coenzyme A thiolases of rat brain and their relative activities during postnatal development. Biochem. J. 132, 731–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Hoon M. J., Imoto S., Nolan J., Miyano S. (2004) Open source clustering software. Bioinformatics 20, 1453–1454 [DOI] [PubMed] [Google Scholar]
- 33. Saldanha A. J. (2004) Java Treeview–extensible visualization of microarray data. Bioinformatics 20, 3246–3248 [DOI] [PubMed] [Google Scholar]
- 34. Locasale J. W., Cantley L. C. (2011) Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 14, 443–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Báez-Ruiz A., Escobar C., Aguilar-Roblero R., Vázquez-Martínez O., Díaz-Muñoz M. (2005) Metabolic adaptations of liver mitochondria during restricted feeding schedules. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G1015–G1023 [DOI] [PubMed] [Google Scholar]
- 36. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., Qin L., Chin J., Yang P., Chen X., Lei Q., Xiong Y., Guan K. L. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Longo N., Amat di San Filippo C., Pasquali M. (2006) Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. C Semin. Med. Genet. 142C, 77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adeva M. M., Calviño J., Souto G., Donapetry C. (2012) Insulin resistance and the metabolism of branched-chain amino acids in humans. Amino Acids 43, 171–181 [DOI] [PubMed] [Google Scholar]
- 39. Kendrick A. A., Choudhury M., Rahman S. M., McCurdy C. E., Friederich M., Van Hove J. L., Watson P. A., Birdsey N., Bao J., Gius D., Sack M. N., Jing E., Kahn C. R., Friedman J. E., Jonscher K. R. (2011) Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 433, 505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Keller M. P., Choi Y., Wang P., Davis D. B., Rabaglia M. E., Oler A. T., Stapleton D. S., Argmann C., Schueler K. L., Edwards S., Steinberg H. A., Chaibub Neto E., Kleinhanz R., Turner S., Hellerstein M. K., Schadt E. E., Yandell B. S., Kendziorski C., Attie A. D. (2008) A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 18, 706–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buchner D. A., Yazbek S. N., Solinas P., Burrage L. C., Morgan M. G., Hoppel C. L., Nadeau J. H. (2011) Increased mitochondrial oxidative phosphorylation in the liver is associated with obesity and insulin resistance. Obesity 19, 917–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Finley L. W., Haas W., Desquiret-Dumas V., Wallace D. C., Procaccio V., Gygi S. P., Haigis M. C. (2011) Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One 6, e23295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ahn B. H., Kim H. S., Song S., Lee I. H., Liu J., Vassilopoulos A., Deng C. X., Finkel T. (2008) A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 14447–14452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bao J., Scott I., Lu Z., Pang L., Dimond C. C., Gius D., Sack M. N. (2010) SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Rad. Biol. Med. 49, 1230–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cimen H., Han M. J., Yang Y., Tong Q., Koc H., Koc E. C. (2010) Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 49, 304–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hirschey M. D., Shimazu T., Goetzman E., Jing E., Schwer B., Lombard D. B., Grueter C. A., Harris C., Biddinger S., Ilkayeva O. R., Stevens R. D., Li Y., Saha A. K., Ruderman N. B., Bain J. R., Newgard C. B., Farese R. V., Jr., Alt F. W., Kahn C. R., Verdin E. (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Han Q., Cai T., Tagle D. A., Robinson H., Li J. (2008) Substrate specificity and structure of human aminoadipate aminotransferase/kynurenine aminotransferase II. Biosci. Rep. 28, 205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rossi F., Garavaglia S., Montalbano V., Walsh M. A., Rizzi M. (2008) Crystal structure of human kynurenine aminotransferase II, a drug target for the treatment of schizophrenia. J. Biol. Chem. 283, 3559–3566 [DOI] [PubMed] [Google Scholar]
- 49. Han Q., Robinson H., Li J. (2008) Crystal structure of human kynurenine aminotransferase II. J. Biol. Chem. 283, 3567–3573 [DOI] [PubMed] [Google Scholar]
- 50. Oxenkrug G. F. (2010) Metabolic syndrome, age-associated neuroendocrine disorders, and dysregulation of tryptophan-kynurenine metabolism. Ann. N.Y. Acad. Sci. 1199, 1–14 [DOI] [PubMed] [Google Scholar]
- 51. Pagliarini D. J., Calvo S. E., Chang B., Sheth S. A., Vafai S. B., Ong S. E., Walford G. A., Sugiana C., Boneh A., Chen W. K., Hill D. E., Vidal M., Evans J. G., Thorburn D. R., Carr S. A., Mootha V. K. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Noland R. C., Woodlief T. L., Whitfield B. R., Manning S. M., Evans J. R., Dudek R. W., Lust R. M., Cortright R. N. (2007) Peroxisomal-mitochondrial oxidation in a rodent model of obesity-associated insulin resistance. Am. J. Physiol. Endocrinol. Metab. 293, E986–E1001 [DOI] [PubMed] [Google Scholar]
- 53. Stein L. R., Imai S. (2012) The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 23, 420–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Feldman J. L., Dittenhafer-Reed K. E., Denu J. M. (2012) Sirtuin Catalysis and Regulation. J. Biol. Chem. 287, 42419–42427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Williamson D. H., Lund P., Krebs H. A. (1967) The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J. 103, 514–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McGarry J. D., Foster D. W. (1980) Regulation of hepatic fatty acid oxidation and ketone body production. Ann. Rev. Biochem. 49, 395–420 [DOI] [PubMed] [Google Scholar]
- 57. Korman S. H. (2006) Inborn errors of isoleucine degradation: a review. Mol. Genet. Metab. 89, 289–299 [DOI] [PubMed] [Google Scholar]
- 58. Megee P. C., Morgan B. A., Mittman B. A., Smith M. M. (1990) Genetic analysis of histone H4: essential role of lysines subject to reversible acetylation. Science (New York, NY) 247, 841–845 [DOI] [PubMed] [Google Scholar]
- 59. Herrera E., Freinkel N. (1967) Internal standards in the estimation of acetyl-CoA in liver extracts. J. Lipid Res. 8, 515–518 [PubMed] [Google Scholar]
- 60. Boekhorst J., van Breukelen B., Heck A., Jr., Snel B. (2008) Comparative phosphoproteomics reveals evolutionary and functional conservation of phosphorylation across eukaryotes. Genome Biol. 9, R144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Benovic J., Tillman T., Cudd A., Fridovich I. (1983) Electrostatic facilitation of the reaction catalyzed by the manganese-containing and the iron-containing superoxide dismutases. Arch. Biochem. Biophys. 221, 329–332 [DOI] [PubMed] [Google Scholar]
- 62. Tao R., Coleman M. C., Pennington J. D., Ozden O., Park S. H., Jiang H., Kim H. S., Flynn C. R., Hill S., Hayes McDonald W., Olivier A. K., Spitz D. R., Gius D. (2010) Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 40, 893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Scott I., Webster B. R., Li J. H., Sack M. N. (2012) Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem. J. 443, 655–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Paik W. K., Pearson D., Lee H. W., Kim S. (1970) Nonenzymatic acetylation of histones with acetyl-CoA. Biochim. Biophys. Acta 213, 513–522 [DOI] [PubMed] [Google Scholar]
- 65. Tanner K. G., Trievel R. C., Kuo M. H., Howard R. M., Berger S. L., Allis C. D., Marmorstein R., Denu J. M. (1999) Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J. Biol. Chem. 274, 18157–18160 [DOI] [PubMed] [Google Scholar]
- 66. Newman J. C., He W., Verdin E. (2012) Mitochondrial protein acylation and intermediary metabolism: regulation by sirtuins and implications for metabolic disease. J. Biol. Chem. 287, 42436–42443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Covian R., Balaban R. S. (2012) Cardiac mitochondrial matrix and respiratory complex protein phosphorylation. Am. J. Physiol. Heart Circ. Physiol. 303, H940–H966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xiong Y., Guan K. L. (2012) Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 198, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Popov K. M., Zhao Y., Shimomura Y., Kuntz M. J., Harris R. A. (1992) Branched-chain α-ketoacid dehydrogenase kinase. Molecular cloning, expression, and sequence similarity with histidine protein kinases. J. Biol. Chem. 267, 13127–13130 [PubMed] [Google Scholar]
- 70. Shimazu T., Hirschey M. D., Hua L., Dittenhafer-Reed K. E., Schwer B., Lombard D. B., Li Y., Bunkenborg J., Alt F. W., Denu J. M., Jacobson M. P., Verdin E. (2010) SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 12, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Park J., Chen Y., Tishkoff D. X., Peng C., Tan M., Dai L., Xie Z., Zhang Y., Zwaans B. M., Skinner M. E., Lombard D. B., Zhao Y. (2013) SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 50, 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Newgard C. B. (2012) Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 15, 606–614 [DOI] [PMC free article] [PubMed] [Google Scholar]