

Background: H2O2 mediates signal transduction in response to TCR activation.
Results: Overexpressing mitochondrial Mn-SOD increases H2O2, enhancing intensity and duration of tyrosine phosphorylation after TCR activation, whereas overexpressing cytosolic Cu,Zn-SOD has no effect.
Conclusion: Mitochondrial H2O2 enhances TCR-mediated signal transduction selectively through JNK/cJun.
Significance: Mitochondrial translocation to the immunological synapse provides necessary proximity to raise effective H2O2 concentration to modulate TCR signaling.
Keywords: Hydrogen Peroxide; Jun N-terminal Kinase (JNK); Jun Transcription Factor; Mitochondria; Signal Transduction; T Cell Receptor; T Lymphocyte; Cytosolic SOD (Cu,Zn-SOD); Mitochondrial SOD (Mn-SOD)
Abstract
T cell receptor (TCR)-initiated signal transduction is reported to increase production of intracellular reactive oxygen species, such as superoxide (O2⨪) and hydrogen peroxide (H2O2), as second messengers. Although H2O2 can modulate signal transduction by inactivating protein phosphatases, the mechanism and the subcellular localization of intracellular H2O2 as a second messenger of the TCR are not known. The antioxidant enzyme superoxide dismutase (SOD) catalyzes the dismutation of highly reactive O2⨪ into H2O2 and thus acts as an intracellular generator of H2O2. As charged O2⨪ is unable to diffuse through intracellular membranes, cells express distinct SOD isoforms in the cytosol (Cu,Zn-SOD) and mitochondria (Mn-SOD), where they locally scavenge O2⨪ leading to production of H2O2. A 2-fold organelle-specific overexpression of either SOD in Jurkat T cell lines increases intracellular production of H2O2 but does not alter the levels of intracellular H2O2 scavenging enzymes such as catalase, membrane-bound peroxiredoxin1 (Prx1), and cytosolic Prx2. We report that overexpression of Mn-SOD enhances tyrosine phosphorylation of TCR-associated membrane proximal signal transduction molecules Lck, LAT, ZAP70, PLCγ1, and SLP76 within 1 min of TCR cross-linking. This increase in mitochondrial H2O2 specifically modulates MAPK signaling through the JNK/cJun pathway, whereas overexpressing Cu,Zn-SOD had no effect on any of these TCR-mediated signaling molecules. As mitochondria translocate to the immunological synapse during TCR activation, we hypothesize this translocation provides the effective concentration of H2O2 required to selectively modulate downstream signal transduction pathways.
Introduction
Reactive oxygen species (ROS),2 by-products of oxygen metabolism, consist of highly reactive charged oxygen free radicals such as superoxide anion (O2⨪, half-life of 10−6 s) and hydroxyl radical (OH−, half-life of 10−9 s) as well as non-radical oxidants such as hydrogen peroxide (H2O2) (1, 2). Until recently, ROS were considered damaging to DNA (3), lipids (4), and proteins (5), and generated as metabolic by-products of cellular respiration. More recently, there is increased recognition of ROS as effector molecules in various cellular processes (6), including host defense (7), hormone synthesis, and redox signaling involved in mitogenesis, apoptosis (8), and oxygen sensing (9). In particular, H2O2 acts as a mediator of biological processes, such as signal transduction (10). Many signaling pathways are propagated by protein phosphorylation; at low concentrations, H2O2 plays a modulatory role by reversible oxidation of cysteines in the active sites of phosphatases such as protein tyrosine phosphatases (11) and serine/threonine phosphatases (12). Thus, H2O2 can inhibit the dephosphorylation of signaling proteins and thereby modulate the intensity, kinetics, or branch points within a pathway (13). This is particularly evident in the T lymphocyte, where H2O2 mimics the stimulatory effects of growth factors and environmental agonists, including the tyrosine phosphorylation pattern unique to the T cell receptor (TCR) (14).
H2O2 is a stable diffusible oxidant in the cell, and can be generated or degraded rapidly in response to external stimuli (15). Recent studies reviewed by Gough and Cotter (16) reveal that the diverse roles of H2O2 as an oxidant and signaling molecule depend upon the subcellular source, location, and duration of its production. Dismutation of O2⨪ either spontaneously or by a family of cellular antioxidant enzymes known as superoxide dismutases (SODs (17)) leads to production of H2O2. The mitochondrial isoform of SOD (Mn-SOD) uses manganese as a cofactor (18), whereas the cytosolic isoform (Cu,Zn-SOD) uses both copper and zinc as cofactors (19). The physiological importance of SODs is illustrated by the severe pathologies evident in mice genetically engineered to lack these enzymes. Mn-SOD-deficient mice die within 10 days after birth due to dilated cardiomyopathy, accumulation of lipid in liver and skeletal muscle, and metabolic acidosis (20). Mice deficient in Cu,Zn-SOD exhibit a reduced life span, increased chances to hepatocellular carcinoma (21), and acceleration of age-related loss of skeletal muscle mass (22).
The concept of the mitochondria as crucial oxidative signaling organelle is an emerging field (23). Mitochondria in TCR-activated cells translocate to the immunological synapse (IS) (24). Intracellular generation of O2⨪ and H2O2 has been detected in T cells during receptor-mediated activation (25, 26). O2⨪ is converted to H2O2 through Mn-SOD and Cu,Zn-SOD, leading to increased production of H2O2 in mitochondria and cytosol of the T cell, respectively. Although it is widely accepted that T cell activation is a tightly controlled process to ensure sufficient immune protection and prevent autoimmunity, the intracellular source of H2O2 and the proximal downstream H2O2 sensitive targets required for modulation of TCR-mediated signaling are not characterized.
This work defines the intracellular source of H2O2 and its effect on the TCR-mediated activation in T lymphocytes. We hypothesized that overexpressing Mn-SOD and Cu,Zn-SOD in their defined physiological compartments will increase the H2O2 in their respective subcellular compartments specifically and define the role of H2O2 localization in TCR signal transduction. Jurkat T lymphoma cell lines, engineered to overexpress Mn-SOD and Cu,Zn-SOD, were generated and activated through the TCR. Our results identify H2O2 generated in mitochondria as a key modulator of the TCR-mediated signaling pathway. We demonstrate that mitochondrial H2O2 regulates TCR activation specifically through the JNK/cJun pathway and propose a crucial role for mitochondrial translocation toward the IS and the subsequent H2O2 generation in regulation of a T cell response.
EXPERIMENTAL PROCEDURES
Reagents
Antibodies (Abs) were obtained from Abcam (rabbit polyclonal to Mn-SOD, Cu,Zn-SOD, Prx1, Prx2, catalase, p-LAT (Tyr132), p-LAT (Tyr191), SLP-76 and rabbit monoclonal to p-SLP76), Cell Signaling (rabbit polyclonal to p-PLCγ1, p-ZAP70, p-Lck (Tyr505), p-LAT (Tyr171), p-ERK, p-p38, ERK1/2, PLCγ1, IκB, and rabbit monoclonal to ZAP70, p-cJun, p-JNK, JNK, p38, COX IV, mouse monoclonal to pIκB), Upstate (rabbit polyclonal to LAT, Lck), and Trevigen (rabbit polyclonal to GAPDH). HRP-conjugated pTyr was procured from Santa Cruz Biotechnology, and HRP-conjugated goat anti-rabbit and goat anti-mouse were purchased from Thermo Scientific. Anti-CD3 (OKT3) was procured from Imgenex, and anti-CD28 was obtained from Ancell. For molecular cloning purposes and where otherwise not mentioned, the reagents were purchased from Sigma Aldrich or Invitrogen. The chloromethyl derivative of dichlorodihydrofluorescein diacetate (CM-H2DCFDA) was purchased from Invitrogen.
Jurkat Cell Culture and Activation
The Jurkat T cell line (subclone E6) was cultured in Roswell Park Memorial Institute medium (RPMI 1640) supplemented with 10% FBS, 2 mm l-glutamine, 10 mm HEPES buffer, 0.1 mm non-essential amino acids, 1 mm sodium pyruvate (Invitrogen), and 0.1% 2-mercaptoethanol (Sigma Aldrich). For stimulation through the TCR, WT and transduced Jurkat cells were resuspended at 2 × 106/40 μl in RPMI 1640 and 25 mm HEPES buffer, and incubated at 37 °C for 5 min. Sheep anti-mouse F(ab′)2 (10 μg/ml) was added and incubated with the cells for 2 min. Cells were then stimulated via the CD3 and CD28 receptors with OKT3 Ab (10 μg/ml) and anti-CD28 Ab (10 μg/ml) at 37 °C for the times indicated in the figures. The reaction was stopped by the addition of an equal volume of Laemmli buffer (4% SDS, 120 mm Tris-HCl (pH 6.8) 0.02% bromphenol blue, and 0.04% 2-mercaptoethanol), and the sample was boiled for 10 min. The cleared lysate was stored at −20 °C until use. Unstimulated cells received only the sheep anti-mouse F(ab′)2.
Gene Cloning, Transduction, and Overexpression
Plasmids having Mn-SOD (coding sequence NM_000636.2, accession no. BC012423, pCMV-SPORT6) or Cu,Zn-SOD (coding sequence NM_000454.4, accession no. BC001034, pINCY) were purified from bacterial glycerol stocks (Open Biosystems). Gene specific primers for Mn- and Cu,Zn-SOD cDNA were designed with engineered restriction sites (EcoRI and BglII; specific to the pMSCV vector) and used for cloning and PCR confirmation of the transgenes (5′-ACTGACTAGATCTGCCACCATGTTGAGCCGGGCAGTGTG-3′ (sense) and 5′-ACTGACTGAATTCTTACTTTTTGCAAGCCATGTATC-3′ (antisense) for Mn-SOD, and 5′-ACTGACTAGATCTGCCACCATGGCGACGAAGGCCGTGTG-3′ (sense) and 5′-ACTGACTGAATTCTTATTGGGCGATCCCAATTACACC-3′ (antisense) for Cu,Zn-SOD). All vector construction was done using standard cloning and PCR techniques (27). All constructs were verified by dideoxynucleotide sequencing at the Case Western Reserve University Department of Genetics Genomics Core. Retrovirus was assembled in the human embryonic kidney cell line (HEK293T cells), using the three-plasmid retroviral packaging system pMSCV-Puro vector (Addgene; a kind gift of Dr. J. Karn, Case Western Reserve University, Cleveland, OH) constitutively driving either Mn- or Cu,Zn-SOD under the CMV immediate early promoter. The three plasmids were (i) pMSCV expressing the targeted gene, (ii) the group antigen polyprotein gene that forms the viral core structure, RNA genome-binding proteins, and nucleoprotein core particle, and reverse transcriptase gene that encodes reverse transcriptase, integrase, and RNase H activity, and (iii) the vesicular stomatitis virus G envelope protein. HEK293T cells were transfected using Lipofectamine (Invitrogen Life Technologies) in Opti-MEM (Invitrogen Life Technologies) media for 9–12 h, after which cells were incubated in RPMI 1640 containing 10% FBS for 48 h. Retroviruses were harvested from the conditioned medium and concentrated by ultracentrifugation (28,000 rpm for 30 min) and used to transduce Jurkat cells. After infection (48 h), the cells were selected in a medium containing puromycin (4 μg/ml) for 3–4 weeks. Overexpression of the transgenes was verified by immunoblot analysis and confocal imaging.
Isolation of Total Genomic DNA and PCR Confirmation of the Transgenes
Integration of the transgene cDNA for both Mn-SOD and Cu,Zn-SOD was confirmed by PCR with the same transgene specific primers used for cloning. DNA was isolated from WT and transduced Jurkat cells using the PureLinkTM Genomic DNA mini kit (Invitrogen).
Immunoblot Analysis
Protein isolation was performed on equal numbers of Jurkat cells and fractionated by size on a reducing 10% SDS-PAGE. Proteins were electrotransferred to nitrocellulose membrane (Invitrogen). Membranes were incubated at room temperature for 1 h in blocking buffer (3% BSA or 5% nonfat milk, 10 mm Tris, pH 7.6, 100 mm NaCl, and 0.1% Tween 20). Primary and secondary Abs were diluted, as recommended by the manufacturer, in blocking buffer and incubated overnight at 4 °C for primary and 1 h at room temperature for secondary antibody with three washes in between incubations. Detection of HRP-conjugated Abs was performed using Supersignal (Pierce) and Kodak Biomax Film MR.
Confocal Microscopy
Actively growing WT and transduced Jurkat cells were adhered at 37 °C for 2 h to slides coated with 0.01% poly-l-lysine. These cells were subsequently fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.2% Triton X-100 for 20 min. The cells were then blocked with 10% goat serum in 1× PBS for 1 h. After blocking, cells were incubated with primary Ab diluted in 10% goat serum overnight at 4 °C. After three washes with 1× PBS, Alexa Fluor 488-conjugated secondary Ab was added in 10% goat serum for 40 min. Cells were incubated with DRAQ5 (1/250) for 3 min in the dark. Following staining, coverslips were mounted on glass slides using Fluoromount (Vector Labs, Inc.), allowed to dry, and visualized on a confocal microscope (Perkin Elmer UltraVIEW® 3D Live Cell Imaging System connected to a Leica DMI 6000B microscope). For co-localization studies, live cells were stained with MitoTracker Red dye (100 nm, Invitrogen) for 30 min at 37 °C. After fixation and permeabilization, as described above, cells were stained with anti-Mn-SOD or anti-Cu,Zn-SOD Ab. Nuclei were stained with Hoechst (10 μm). The 488- and 579-nm lines of a krypton/argon laser were used for measuring the fluorescence excitation of green Mn-SOD or Cu,Zn-SOD and MitoTracker Red, respectively.
Subcellular Fractionation
To isolate cytosol and mitochondria, Jurkat cells were fractionated using Qproteome mitochondria isolation kit (Qiagen). Protein from cytosolic, mitochondrial, nuclear, and microsomal fractions was probed with Cu,Zn-SOD- and Mn-SOD-specific Abs to confirm the cytosolic and mitochondrial localization of the enzymes, respectively. GAPDH was used as a cytosolic positive control, and COX IV was used as a mitochondrial positive control.
Determination of H2O2 Generation
Intracellular H2O2 production was determined by staining with CM-H2DCFDA. Jurkat cells were washed with 1× PBS and incubated with 0.5 μm CM-H2DCFDA in phenol red-free RPMI 1640 for 10 min at 37 °C. After the incubation, cells were washed in 1X PBS and resuspended in RPMI 1640 and 5% FBS for 10 min at 37 °C. The cells were analyzed by flow cytometry (excitation, 488 nm; emission, 585 nm; LSRII, BD Biosciences). Autofluorescence of Jurkat cells was measured using unstained WT cells. Data from 10,000 events were collected and analyzed with FloJo software (BD Biosciences).
RESULTS
Targeted Overexpression of Mn-SOD and Cu,Zn-SOD to Mitochondria and Cytosol, Respectively
Jurkat cell lines transduced with either Mn-SOD or Cu,Zn-SOD were selected by continuous growth in 4 μg/ml puromycin-containing medium for 2 weeks. Successful transduction was confirmed after amplifying genomic DNA isolated from those cells by PCR, using the same gene-specific cDNA primers used for cloning (data not shown). The endogenous genomic amplicons were multiple kilobases in size and were not amplified. Protein expression for each SOD transductant was validated by immunoblot, using Mn-SOD and Cu,Zn-SOD gene-specific antibodies (Fig. 1, A and B). Serial 2-fold dilutions of transduced whole cell extracts were electrophoresed in parallel to an undiluted extract from untransformed Jurkat cells (WT). Both proteins (Mn-SOD and Cu,Zn-SOD) exhibited similar levels of overexpression (∼2-fold, using Quantity One software). Overexpression was confirmed by confocal microcopy (Fig. 1, C and D), in which Jurkat cell lines expressing Mn-SOD or Cu,Zn-SOD exhibited a notable increase in green fluorescence due to the FITC fluorochrome conjugated to the appropriate antibody. Mn-SOD staining displayed a punctate pattern, consistent with mitochondrial localization not observed in staining for Cu,Zn-SOD. Mitochondrial localization of overexpressed Mn-SOD was confirmed with confocal microscopy by identifying the mitochondria with the staining dye MitoTracker Red (Fig. 1E). The green-labeled Mn-SOD Ab colocalized with MitoTracker Red, yielding yellow fluorescence in the merged images. Because mitochondria are known to associate closely with the endoplasmic reticulum, this causes aggregates to form in the cell for Mn-SOD staining (28, 29). Parallel staining with MitoTracker Red and Cu,Zn-SOD Ab in WT and Cu,Zn-SOD overexpressing cells did not exhibit any yellow fluorescence in the merged images (Fig. 1F), confirming that Mn-SOD and Cu,Zn-SOD were overexpressed in different subcellular compartments. Overexpression and subcellular localization of cytosolic Cu,Zn-SOD and mitochondrial Mn-SOD were confirmed by cell fractionation using density medium centrifugation, followed by extraction of cytosolic, mitochondrial, nuclear/membrane, and microsomal proteins and analysis by immunoblot. Cytosolic protein levels of Cu,Zn-SOD were considerably higher in overexpression lines than levels of the WT, whereas mitochondrial, nuclear/membrane, and microsomal fractions did not show any bands (Fig. 1G). GAPDH, another cytosolic protein, was used as a loading control. Similarly, the mitochondrial fraction of Mn-SOD overexpression lines exhibit increased levels of Mn-SOD signal as compared with that seen in WT cells (Fig. 1H). Another mitochondrial protein COX IV was used as a loading control. With the Qproteome kit, during cell lysis, some of the mitochondria also lyse and the resulting fragments sediment with the nuclear/membrane fraction, thus explaining Mn-SOD and COX IV bands in the nuclear fraction. The presence of mitochondrial protein in other fractions is most likely due to minor contamination during fractionation.
FIGURE 1.

Increased protein expression of Mn-SOD and Cu,Zn-SOD in transduced Jurkat cells. Immunoblot analysis was performed to determine the level of Mn-SOD (A) and Cu,Zn-SOD (B) protein expression in non-transduced Jurkat cells (WT) and Jurkat cells transduced by Mn-SOD and Cu,Zn-SOD, respectively. Serial dilutions of cell extracts isolated from transduced cells are shown in comparison to undiluted WT cell extract. WT and Jurkat cells transduced with Mn-SOD (C) and Cu,Zn-SOD (D) were stained with their respective antibodies and imaged by confocal microscopy. The relative florescence intensity of each overexpression line is greater than that of WT. Nuclei were stained with DRAQ. WT (upper panel) and Mn-SOD (E) or Cu,Zn-SOD (F) overexpressing (lower panel) Jurkat cells were stained with MitoTracker Red dye and Mn-SOD Ab or Cu,Zn-SOD Ab, respectively, followed by appropriate secondary Ab. The nuclei were stained with Hoechst dye, and the cells were imaged by confocal microscopy. Merging the images demonstrates mitochondrial specific localization of Mn-SOD, and that Cu,Zn-SOD does not localize to the mitochondria. Cytosolic, mitochondrial, nuclear, and microsomal fractions of WT and Cu,Zn-SOD (G) or Mn-SOD (H) transductants were isolated, fractionated by PAGE, and probed with Cu,Zn-SOD or Mn-SOD specific Abs, respectively. GAPDH, a cytosolic protein, or COX IV, a mitochondrial protein, were used both as localization and loading controls.
Overexpression of both Mn-SOD and Cu,Zn-SOD Increases Intracellular H2O2 but Does Not Change Levels of Endogenous Peroxide Scavengers
Jurkat cell lines overexpressing either Mn-SOD or Cu,Zn-SOD demonstrated increased basal levels of intracellular H2O2, as compared with those in WT controls (Fig. 2A). CM-H2DCFDA, a fluorophore that measures intracellular H2O2, was used to stain cells for flow cytometric analysis. To confirm that elevated H2O2 levels in resting T cells was due to dismutation of superoxide by the overexpressed SOD enzymes and not attributable to a change in peroxide scavenging, protein levels of three intracellular antioxidant enzymes from different subcellular compartments were analyzed by immunoblot (Fig. 2B). The expression level of other endogenous peroxide scavengers such as peroxisomal catalase, membrane-bound peroxiredoxin (Prx)1, and cytosolic Prx2 was not altered. Similarly, the levels of the alternate intracellular SOD in each transductant remained unchanged, enabling us to attribute any change in the T cell response directly to the overexpressed SOD.
FIGURE 2.
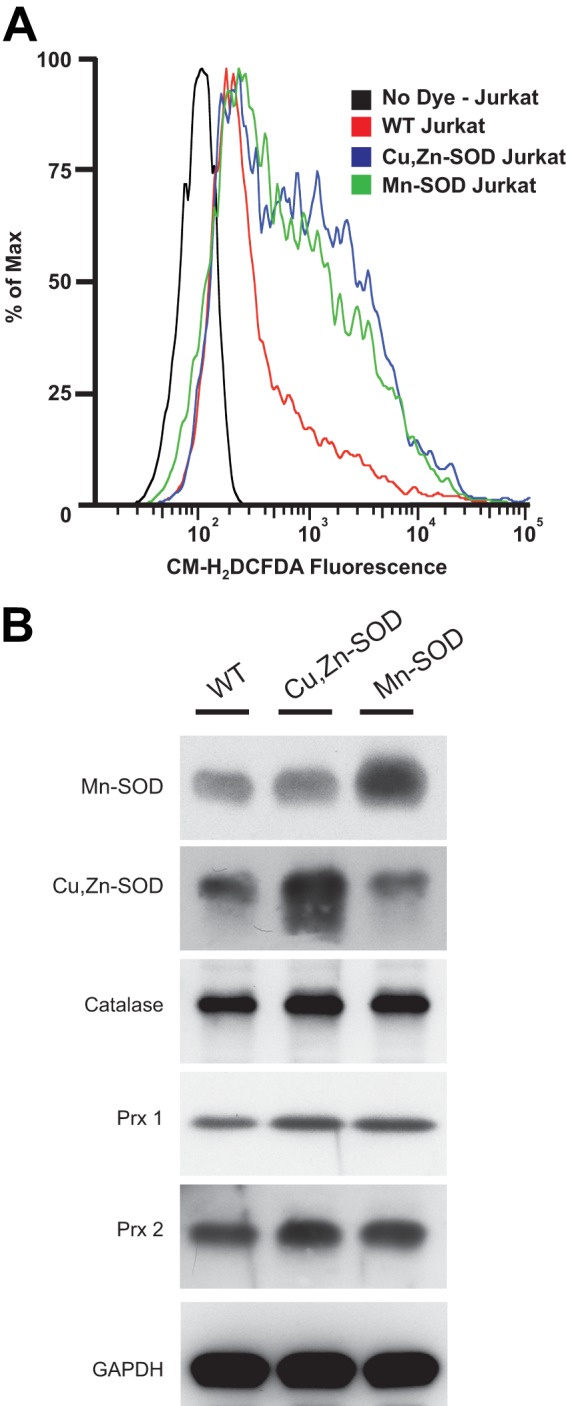
Mn-SOD and Cu,Zn-SOD overexpression increases intracellular levels of H2O2, without altering the level of endogenous antioxidant enzymes. A, WT, Mn-SOD, and Cu,Zn-SOD overexpressing Jurkat cell lines were loaded with 0.5 μm CM-H2DCFDA, and intracellular levels of H2O2 were analyzed using flow cytometry. Unstained WT were used as an autofluorescence control. B, immunoblots of WT, Cu,Zn-SOD, and Mn-SOD overexpression cell lysates were probed with Abs specific for Mn-SOD, Cu,Zn-SOD, catalase, Prx1, and Prx 2. Anti-GAPDH Ab was used as a loading control.
Overexpression of Mn-SOD and Not Cu,Zn-SOD Increases Global Tyrosine Phosphorylation and Membrane Proximal Signaling after TCR Cross-linking
H2O2 induces tyrosine phosphorylation of many proteins, often attributed to its ability to oxidize the cysteine in the active site of protein tyrosine phosphatase (30). To determine the effect of increased H2O2 produced in either the mitochondria or cytosol on TCR-mediated signaling, Jurkat cell lines overexpressing either Mn-SOD or Cu,Zn-SOD and WT controls were activated with anti-CD3 cross-linking and anti-CD28 for 1, 5, and 10 min. The reaction was stopped, and whole cell lysates were prepared by adding Laemmli buffer. The global tyrosine phosphorylation response was analyzed by immunoblotting (Fig. 3A). The intensity of phosphotyrosine proteins was increased in Jurkat cell lines overexpressing Mn-SOD (lanes 10–12), whereas Cu,Zn-SOD transduced cells exhibited only a minor change in protein phosphorylation (lanes 6–8) when compared with that seen in WT cells (lanes 2–4). Interestingly, phosphorylation of TCRζ increased with Mn-SOD overexpression (lanes 10–12), whereas it decreased in Jurkat cells overexpressing Cu,Zn-SOD (lanes 6–8) in comparison with WT (lanes 2–4) at all time points after stimulation (Fig. 3A). This finding underscores a critical role for mitochondrial generation of H2O2 to enhance global tyrosine phosphorylation after TCR activation. The minimal effect of overexpressing cytosolic Cu,Zn-SOD in global tyrosine phosphorylation is surprising in that signaling events take place in the cytosol. To characterize the early events (1, 2, 4 and 6 min) in redox modulation of TCR activation, we analyzed the tyrosine phosphorylation of specific membrane proximal TCR signaling proteins (p-Lck394, p-ZAP70, p-LAT132, p-LAT171, p-LAT191, p-PLCγ1, and p-SLP76) in Jurkat cell lines overexpressing Mn-SOD and Cu,Zn-SOD, in comparison with WT. Mn-SOD-transduced cells exhibited greater levels of protein tyrosine phosphorylation for membrane proximal signaling molecules within 1 min after TCR activation and remained elevated for 6 min (Fig. 3B), whereas Cu,Zn-SOD overexpression did not increase protein tyrosine phosphorylation after TCR cross-linking (Fig. 3C). The observed increase in tyrosine phosphorylation of each of these proteins in Mn-SOD overexpressing Jurkat cell lines returns to basal levels within 15 min (data not shown).
FIGURE 3.

Overexpression of Mn-SOD increases global tyrosine phosphorylation of membrane proximal signaling proteins early after TCR engagement. WT, Mn-SOD, and Cu,Zn-SOD overexpressing Jurkat cells were left unstimulated or stimulated by anti-CD3 cross-linking and anti-CD28 for 1, 2, 4 and 6 min. A, cell extracts were analyzed by phosphotyrosine immunoblotting. Immunoblot for GAPDH was used as a control for total protein loading. Protein extracts from Jurkat cells overexpressing Mn-SOD (B) and Cu,Zn-SOD (C) in comparison with WT controls were analyzed by immunoblotting with p-Lck, p-ZAP70, p-LAT, p-PLCγ1, and p-SLP76 Abs. Immunoblots for respective total proteins were used as a loading control.
Mitochondrial Generation of H2O2 Selectively Enhances Phosphorylation of the JNK/cJun Pathway
To evaluate the effect of increased phosphorylation of membrane proximal proteins in Mn-SOD overexpression cell lines on second messenger pathways, we activated both Mn-SOD and Cu,Zn-SOD overexpressing Jurkat cell lines along with WT controls for 15, 30, 60, and 120 min and probed with antibodies specific for MAPKs (p-JNK, p-ERK, and p-p38; Fig. 4, A and B, respectively). After TCR activation, Mn-SOD overexpression specifically increased phosphorylation of JNK, whereas phosphorylation of ERK was not enhanced in comparison with the WT controls (Fig. 4A), suggesting the selectivity of mitochondrial H2O2 in inducing the JNK pathway. We also observed a minimal increase in phosphorylation of p38 with Mn-SOD overexpression (Fig. 4A). This result defeats the notion of H2O2 as a nonspecific oxidant and brings to light a highly selective nature of this redox modulator. The Cu,Zn-SOD line did not exhibit an increase in protein phosphorylation for any MAPK over the WT (Fig. 4B). JNK phosphorylates Ser63 on cJun, which upon phosphorylation translocates to the nucleus and up-regulates gene expression for inflammatory cytokines such as interleukin-2 (IL-2). Extending this pathway in Mn-SOD overexpressing cell lines, there is also increased phosphorylation of cJun (Fig. 4C), which intensifies with time, remaining stable for 120 min. In contrast, Cu,Zn-SOD overexpression has a minimal effect on cJun phosphorylation, which is maximal at 30 min and then declines (Fig. 4D). To characterize the specificity of mitochondrial generation of H2O2 on signal transduction, phosphorylation of the inhibitor of kappa B (IκB) was studied as a negative control (Fig. 4, C and D). In resting cells, NFκB is present in the cytosol bound to IκB. After TCR activation, IκB is phosphorylated facilitating its ubiquitination and subsequent proteosomal degradation releasing NFκB, which then translocates to the nucleus and binds to the promoter region of its target genes. Overexpressing either Mn-SOD (Fig. 4C) or Cu,Zn-SOD (Fig. 4D) had no effect on the phosphorylation of IκB, suggesting that TCR-mediated redox regulation selectively activates genes targeted by the JNK/cJun pathway.
FIGURE 4.

Overexpression of Mn-SOD specifically increases threonine/tyrosine phosphorylation of JNK and serine phosphorylation of cJun after TCR engagement. Mn-SOD and Cu,Zn-SOD overexpressing Jurkat cells with WT controls were left unstimulated or stimulated by anti-CD3 cross-linking and anti-CD28 for 15, 30, 60, and 120 min, after which JNK, ERK, p38, (A and B) cJun, and IκB (C and D) phosphorylation was measured by immunoblot. Immunoblot for respective total proteins (ERK, p38, IκB) or GAPDH was used as a loading control.
DISCUSSION
Modulation of the TCR signaling pathway, by both exogenous mediators (e.g. costimulation, adhesion, and cytokines) and endogenous factors (e.g. phosphatases, assembly of the IS, changes in the cytoskeleton), is critical for orchestrating a coordinated, focused immune response. In this report, we reveal that increased generation of H2O2 due to overexpression of Mn-SOD augments membrane proximal tyrosine phosphorylation emanating from the TCR and selectively enhances the JNK/cJun second messenger pathway. These results are consistent with the growing field of evidence that mitochondria, a major generator of physiological H2O2 (31), are critical in T cell activation. A recent report demonstrates that mitochondrial metabolism, producing ROS through complex III, is required for activation of nuclear factor of activated T cells and IL-2 induction (32). Shortly after TCR engagement and formation of the IS, the large interconnected mitochondrial network undergoes fragmentation thereby facilitating transport (33) to the IS (34). These mitochondria are strategically localized beneath the IS and modulate intracellular calcium signals following IS formation (35). We propose a model (Fig. 5) that upon TCR activation, mitochondria translocate toward the IS. It is their proximity to the synapse that provides a biologically effective concentration of H2O2, which modulates tyrosine phosphorylation-mediated signaling of membrane proximal proteins, which, in turn, transduces downstream through the JNK/cJun pathway, and subsequent transcription of inflammatory cytokine genes. Our hypothesis is consistent with other evidence that changes in the microenvironment within a T cell function to restrict its cellular and metabolic choices (36).
FIGURE 5.

Model depicting mitochondrial generation of H2O2 as a key modulator of TCR-mediated signal transduction. Mitochondrial translocation to the IS during TCR activation brings mitochondria in proximity to the TCR signaling complex. After TCR engagement, mitochondria produce an H2O2 burst that inhibits phosphatases, thereby enhancing and prolonging protein phosphorylation. Enhanced signaling from these membrane proximal proteins specifically up-regulates second messenger signal transduction through the JNK/cJun pathway. Phosphorylated cJun translocates into the nucleus and activates transcription factors for cytokine and chemokine production. Circles indicate protein phosphorylation (green, tyrosine; blue, threonine/tyrosine; red, serine).
It is well known that exogenous H2O2 is produced by granulocytes and macrophages during inflammation (37). H2O2 acts as a chemoattractant to direct leukocytes to injury sites, demonstrating that immune cells are able to respond to exogenous H2O2 in addition to producing it (38). H2O2 also plays a mitogenic role in lymphocyte proliferation (39), as it mimics the effect of growth factors and antigenic stimulation (40, 41). Our initial report on redox regulation of T cell activation showed that addition of exogenous H2O2 to primary cultures of human blood-derived T cells leads to an increase in global phosphotyrosine (42). In addition, we demonstrated that the strength of the signal that emanates from TCR cross-linking is directly correlated with endogenous H2O2 production, and when H2O2 levels are depleted, signal transduction from the TCR is similarly reduced. Unfortunately, evaluation of endogenous H2O2 production and localization on T cell activation was not possible, due to the inability of T cells to remain viable after long term exposure to ROS or TCR cross-linking in culture. Therefore, we developed transfected Jurkat T cell lines overexpressing mitochondrial Mn-SOD and cytosolic Cu,Zn-SOD to define the localization and understand the effects of H2O2 on TCR-mediated signal transduction. Consistent with existing data, we observed in this current study increased intracellular H2O2 in T cells in response to exogenous H2O2 (data not shown). It is becoming more widely accepted that endogenous H2O2 plays an important role as a second messenger, inhibiting protein phosphatases and thereby amplifying and modulating signals emanating from the TCR (43) and affecting immune regulation (44). H2O2 has also been implicated in formation of lipid rafts and downstream intracellular signaling after T cell activation (45).
SODs are major antioxidative enzymes in the cell, providing a first line of defense against O2⨪ by efficiently dismutating it to H2O2 and O2 (reaction rate constant at pH 7.8, K = 2 × 109 m−1 s−1) (46). Mn-SOD is present exclusively in the mitochondrial matrix, whereas Cu,Zn-SOD is present in the cytosol. Our results confirmed increased intracellular levels of H2O2 by overexpressing either Mn-SOD or Cu,Zn-SOD in their respective organelles, which did not lead to the up-regulation of H2O2 catabolizing enzymes. We attribute these results to the moderate levels of overexpression. Although initially unexpected, we show that increased production of H2O2 in the cytosol had a minimal effect on TCR-mediated immune signaling. Our results suggest that even during Cu,Zn-SOD overexpression cytosolic H2O2 did not reach the effective local concentration required for immune modulation at the IS. Thus, we propose that it is the location and duration of H2O2 production that selectively modulates T cell signaling.
We characterized the effects of mitochondria-derived H2O2 on TCR-mediated signal transduction by studying the tyrosine phosphorylation levels of the membrane proximal signaling complex. Co-stimulation of Jurkat cells with cross-linked OKT3 and anti-CD28 led to the simultaneous phosphorylation of both inhibitory (Tyr505, data not shown) and catalytic (Tyr394) sites of Lck, leading to downstream activation of the cell. A recent study also confirmed the simultaneous phosphorylation of regulatory and enzymatic sites in Lck upon activation by exogenous H2O2 and through TCR activation (47). Downstream of p-Lck, we found that increased mitochondrial H2O2 enhanced the level and longevity of tyrosine phosphorylation of ZAP70, LAT at Tyr134, Tyr171, and Tyr191, SLP76, and PLCγ1, whereas increased cytosolic H2O2 had minimal if any effect on the phosphorylation of these molecules. In agreement, a report from other investigators (48) indicates that an increase in intracellular steady state production of H2O2 by Mn-SOD blocks TNF-α-induced apoptosis. Another study also found mitochondrial ROS as an important regulator of LPS-mediated production of proinflammatory cytokines (49).
To study the possible selectivity of mitochondrial-mediated H2O2 generation, we found that increased mitochondrial H2O2 specifically enhances the phosphorylation of JNK, whereas an increase in cytosolic H2O2 has no effect on MAPK phosphorylation. Activated JNK phosphorylates its major substrate cJun as well as several other transcription factors required for cell survival, proliferation, transformation, and death (50). The dual role of JNK in both apoptotic and survival (51) signaling pathways indicates that the functional role of JNK is complex. Throughout the study, we did not observe any increase in cell death in either Mn-SOD or Cu,Zn-SOD transduced Jurkat cell lines in comparison with WT cells, although Jurkat cells overexpressing Mn-SOD showed increased proliferation (data not shown). This finding is consistent with a report that insulin-like growth factor-1-induced protection from apoptotic death in CD28 co-stimulated Jurkat cells is mediated by JNK activation (52). To further underscore the selective role of H2O2 in modulating TCR-mediated signaling, the activation of NFκB was studied through phosphorylation of IκB. Neither a mitochondrial nor cytosolic increase in H2O2 had any effect on the activation of NFκB, as shown by the lack of a change in phosphorylation of IκB in the SOD overexpression cell lines. Because Jurkat cells do not produce IL-2 or other cytokines upon TCR activation, further studies in primary T cells to understand the changes in T cell biology after modulating intracellular H2O2 are warranted.
In summary, we describe a novel mechanism for the proinflammatory effects of mitochondrial generation of H2O2, which causes the selective phosphorylation of cJun in activated Jurkat cells through JNK-mediated phosphorylation. Our findings that implicate mitochondrial and not cytosolic H2O2 in modulating TCR-mediated activation are consistent with an emerging theme of mitochondrial translocation to the IS during T lymphocyte activation.
Acknowledgments
We thank Drs. Scott Howell and Minh Lam and Charlotte Chung for help with confocal microscopy, Drs. Jonathan Karn and Uri Mbonye for providing the pMSCV vector, and Jeffrey Meisch for help with flow cytometry.
This work was supported by National Institutes of Health Grants R21 AI-083609 (to A. D. L.) and P30 AR-039750 to the Case Western Reserve University Skin Diseases Research Center and the Ohio Department of Development Center for Innovative Immunosuppressive Therapeutics (Prime Award, TECH 09-023).
- ROS
- reactive oxygen species
- O2⨪
- superoxide free radical
- TCR
- T cell receptor
- Mn-SOD
- manganese superoxide dismutase
- Cu,Zn-SOD
- copper zinc superoxide dismutase
- IS
- immunological synapse
- Ab
- antibody
- LAT
- linker for activation of T cells
- SLP76
- Src homology 2 (SH2) domain-containing leukocyte protein of 76 kDa
- Lck
- lymphocyte-specific protein tyrosine kinase
- PLCγ1
- phospholipase C γ1
- ZAP70
- γ-chain-associated protein kinase 70
- COX IV
- cytochrome c oxidase IV
- CD28
- cluster of differentiation 28
- CM-H2DCFDA
- chloromethyl derivative of dichlorodihydrofluorescein diacetate.
REFERENCES
- 1. Loschen G., Azzi A., Richter C., Flohé L. (1974) Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 42, 68–72 [DOI] [PubMed] [Google Scholar]
- 2. Sies H. (1993) Strategies of antioxidant defense. Eur. J. Biochem. 215, 213–219 [DOI] [PubMed] [Google Scholar]
- 3. Cooke M. S., Evans M. D., Dizdaroglu M., Lunec J. (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 17, 1195–1214 [DOI] [PubMed] [Google Scholar]
- 4. Morel I., Lescoat G., Cillard J., Pasdeloup N., Brissot P., Cillard P. (1990) Kinetic evaluation of free malondialdehyde and enzyme leakage as indices of iron damage in rat hepatocyte cultures. Involvement of free radicals. Biochem. Pharmacol. 39, 1647–1655 [DOI] [PubMed] [Google Scholar]
- 5. Mallis R. J., Buss J. E., Thomas J. A. (2001) Oxidative modification of H-ras: S-thiolation and S-nitrosylation of reactive cysteines. Biochem. J. 355, 145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dröge W. (2002) Free radicals in the physiological control of cell function. Physiol. Rev. 82, 47–95 [DOI] [PubMed] [Google Scholar]
- 7. Leto T. L., Geiszt M. (2006) Role of Nox family NADPH oxidases in host defense. Antioxid. Redox. Signal 8, 1549–1561 [DOI] [PubMed] [Google Scholar]
- 8. Simon H. U., Haj-Yehia A., Levi-Schaffer F. (2000) Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 5, 415–418 [DOI] [PubMed] [Google Scholar]
- 9. Williams M. S., Kwon J. (2004) T cell receptor stimulation, reactive oxygen species, and cell signaling. Free Radic. Biol. Med. 37, 1144–1151 [DOI] [PubMed] [Google Scholar]
- 10. Groeger G., Quiney C., Cotter T. G. (2009) Hydrogen peroxide as a cell-survival signaling molecule. Antioxid. Redox. Signal. 11, 2655–2671 [DOI] [PubMed] [Google Scholar]
- 11. Denu J. M., Tanner K. G. (1998) Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37, 5633–5642 [DOI] [PubMed] [Google Scholar]
- 12. O'Loghlen A., Pérez-Morgado M. I., Salinas M., Martín M. E. (2003) Reversible inhibition of the protein phosphatase 1 by hydrogen peroxide. Potential regulation of eIF2 alpha phosphorylation in differentiated PC12 cells. Arch. Biochem. Biophys. 417, 194–202 [DOI] [PubMed] [Google Scholar]
- 13. Rhee S. G. (2006) Cell signaling. H2O2, a necessary evil for cell signaling. Science 312, 1882–1883 [DOI] [PubMed] [Google Scholar]
- 14. Jin Y. J., Friedman J., Burakoff S. J. (1998) Regulation of tyrosine phosphorylation in isolated T cell membrane by inhibition of protein tyrosine phosphatases. J. Immunol. 161, 1743–1750 [PubMed] [Google Scholar]
- 15. Rhee S. G. (1999) Redox signaling: hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 31, 53–59 [DOI] [PubMed] [Google Scholar]
- 16. Gough D. R., Cotter T. G. (2011) Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2, e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fridovich I. (1995) Superoxide radical and superoxide dismutases. Ann. Rev. Biochem. 64, 97–112 [DOI] [PubMed] [Google Scholar]
- 18. Weisiger R. A., Fridovich I. (1973) Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J. Biol. Chem. 248, 4793–4796 [PubMed] [Google Scholar]
- 19. Harris E. D. (1992) Copper as a cofactor and regulator of copper,zinc superoxide dismutase. J. Nutr. 122, 636–640 [DOI] [PubMed] [Google Scholar]
- 20. Li Y., Huang T. T., Carlson E. J., Melov S., Ursell P. C., Olson J. L., Noble L. J., Yoshimura M. P., Berger C., Chan P. H., Wallace D. C., Epstein C. J. (1995) Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 11, 376–381 [DOI] [PubMed] [Google Scholar]
- 21. Elchuri S., Oberley T. D., Qi W., Eisenstein R. S., Jackson Roberts L., Van Remmen H., Epstein C. J., Huang T. T. (2005) CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 24, 367–380 [DOI] [PubMed] [Google Scholar]
- 22. Muller F. L., Song W., Liu Y., Chaudhuri A., Pieke-Dahl S., Strong R., Huang T. T., Epstein C. J., Roberts L. J., 2nd, Csete M., Faulkner J. A., Van Remmen H. (2006) Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 40, 1993–2004 [DOI] [PubMed] [Google Scholar]
- 23. Quintana A., Schwindling C., Wenning A. S., Becherer U., Rettig J., Schwarz E. C., Hoth M. (2007) T cell activation requires mitochondrial translocation to the immunological synapse. Proc. Natl. Acad. Sci. U.S.A. 104, 14418–14423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baixauli F., Martín-Cófreces N. B., Morlino G., Carrasco Y. R., Calabia-Linares C., Veiga E., Serrador J. M., Sánchez-Madrid F. (2011) The mitochondrial fission factor dynamin-related protein 1 modulates T-cell receptor signalling at the immune synapse. EMBO J. 30, 1238–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rhee S. G., Bae Y. S., Lee S. R., Kwon J. (2000) Sci STKE 2000, pe1. [DOI] [PubMed] [Google Scholar]
- 26. Devadas S., Zaritskaya L., Rhee S. G., Oberley L., Williams M. S. (2002) Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J. Exp. Med. 195, 59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, Third Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 28. Levine T. (2004) Short-range intracellular trafficking of small molecules across endoplasmic reticulum junctions. Trends Cell Biol. 14, 483–490 [DOI] [PubMed] [Google Scholar]
- 29. Levine T., Rabouille C. (2005) Endoplasmic reticulum: one continuous network compartmentalized by extrinsic cues. Curr. Opin. Cell Biol. 17, 362–368 [DOI] [PubMed] [Google Scholar]
- 30. Meng T. C., Fukada T., Tonks N. K. (2002) Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Molecular cell 9, 387–399 [DOI] [PubMed] [Google Scholar]
- 31. Balaban R. S., Nemoto S., Finkel T. (2005) Mitochondria, oxidants, and aging. Cell 120, 483–495 [DOI] [PubMed] [Google Scholar]
- 32. Sena L. A., Li S., Jairaman A., Prakriya M., Ezponda T., Hildeman D. A., Wang C. R., Schumacker P. T., Licht J. D., Perlman H., Bryce P. J., Chandel N. S. (2013) Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kamiński M. M., Röth D., Sass S., Sauer S. W., Krammer P. H., Gülow K. (2012) Manganese superoxide dismutase: a regulator of T cell activation-induced oxidative signaling and cell death. Biochim. Biophys. Acta 1823, 1041–1052 [DOI] [PubMed] [Google Scholar]
- 34. Quintana A., Hoth M. (2012) Mitochondrial dynamics and their impact on T cell function. Cell Calcium 52, 57–63 [DOI] [PubMed] [Google Scholar]
- 35. Quintana A., Kummerow C., Junker C., Becherer U., Hoth M. (2009) Morphological changes of T cells following formation of the immunological synapse modulate intracellular calcium signals. Cell Calcium 45, 109–122 [DOI] [PubMed] [Google Scholar]
- 36. Pearce E. L., Pearce E. J. (2013) Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nathan C. F., Brukner L. H., Silverstein S. C., Cohn Z. A. (1979) Extracellular cytolysis by activated macrophages and granulocytes. I. Pharmacologic triggering of effector cells and the release of hydrogen peroxide. J. Exp. Med. 149, 84–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Niethammer P., Grabher C., Look A. T., Mitchison T. J. (2009) A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 459, 996–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Staal F. J., Anderson M. T., Staal G. E., Herzenberg L. A., Gitler C. (1994) Redox regulation of signal transduction: tyrosine phosphorylation and calcium influx. Proc. Natl. Acad. Sci. U.S.A. 91, 3619–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Davicino R., Manuele M. G., Ferraro G., Micalizzi B., Anesini C. (2009) Modulatory effect of hydrogen peroxide on tumoral lymphocytes proliferation. Immunopharmacol. Immunotoxicol. 31, 130–139 [DOI] [PubMed] [Google Scholar]
- 41. Lee S. R., Kwon K. S., Kim S. R., Rhee S. G. (1998) Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273, 15366–15372 [DOI] [PubMed] [Google Scholar]
- 42. Reyes B. M., Danese S., Sans M., Fiocchi C., Levine A. D. (2005) Redox equilibrium in mucosal T cells tunes the intestinal TCR signaling threshold. J. Immunol. 175, 2158–2166 [DOI] [PubMed] [Google Scholar]
- 43. Reth M. (2002) Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 3, 1129–1134 [DOI] [PubMed] [Google Scholar]
- 44. Kesarwani P., Murali A. K., Al-Khami A. A., Mehrotra S. (2013) Redox regulation of T-cell function: from molecular mechanisms to significance in human health and disease. Antioxid. Redox. Signal. 18, 1497–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lu S. P., Lin Feng M. H., Huang H. L., Huang Y. C., Tsou W. I., Lai M. Z. (2007) Reactive oxygen species promote raft formation in T lymphocytes. Free Radic. Biol. Med. 42, 936–944 [DOI] [PubMed] [Google Scholar]
- 46. Fridovich I. (1983) Superoxide radical: an endogenous toxicant. Annu. Rev. Pharmacol. Toxicol. 23, 239–257 [DOI] [PubMed] [Google Scholar]
- 47. Nyakeriga A. M., Garg H., Joshi A. (2012) TCR-induced T cell activation leads to simultaneous phosphorylation at Y505 and Y394 of p56(lck) residues. Cytometry 81, 797–805 [DOI] [PubMed] [Google Scholar]
- 48. Dasgupta J., Subbaram S., Connor K. M., Rodriguez A. M., Tirosh O., Beckman J. S., Jourd'Heuil D., Melendez J. A. (2006) Manganese superoxide dismutase protects from TNF-α-induced apoptosis by increasing the steady-state production of H2O2. Antioxid. Redox. Signal. 8, 1295–1305 [DOI] [PubMed] [Google Scholar]
- 49. Bulua A. C., Simon A., Maddipati R., Pelletier M., Park H., Kim K. Y., Sack M. N., Kastner D. L., Siegel R. M. (2011) Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208, 519–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shaulian E., Karin M. (2002) AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–136 [DOI] [PubMed] [Google Scholar]
- 51. Yu C., Minemoto Y., Zhang J., Liu J., Tang F., Bui T. N., Xiang J., Lin A. (2004) JNK suppresses apoptosis via phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol. Cell 13, 329–340 [DOI] [PubMed] [Google Scholar]
- 52. Walsh P. T., Smith L. M., O'Connor R. (2002) Insulin-like growth factor-1 activates Akt and Jun N-terminal kinases (JNKs) in promoting the survival of T lymphocytes. Immunology 107, 461–471 [DOI] [PMC free article] [PubMed] [Google Scholar]