

Background: Stabilization of progesterone receptors contributes to mammary tumorigenesis in Brca1-deficient mice.
Results: Deficiency in phosphorylation on serine 390 of progesterone receptor A by GSK-3β enhances the receptor stability.
Conclusion: Stabilization of progesterone receptor A is mediated by GSK-3β kinase in the Brca1-deficient mammary gland.
Significance: This finding provides a novel insight of how tumor suppression of Brca1 is mediated by PR-A.
Keywords: BRCA1, Breast Cancer, Mammary Gland, Phosphorylation, Protein Degradation, Ubiquitination, Bard1, GSK-3, PR-A
Abstract
Germ line mutations of the BRCA1 gene increase the risk of breast and ovarian cancer, but the basis of this tissue-specific tumor predisposition is not fully understood. Previously, we reported that the progesterone receptors are stabilized in Brca1-deficient mammary epithelial cells, and treating with anti-progesterone delays mammary tumorigenesis in Brca1/p53 conditional knock-out mice, suggesting that the progesterone has a critical role in breast carcinogenesis. To further explore how the stability of progesterone receptor is modulated, here, we have found that glycogen synthase kinase (GSK)-3β phosphorylation of progesterone receptor-A (PR-A) facilitates its ubiquitination. GSK-3β-mediated phosphorylation of serine 390 in PR-A regulates its subsequent ubiquitination and protein stability. Expression of PR-AS390A mutant in the human breast epithelial cells, MCF-10A, results in enhanced proliferation and formation of aberrant acini structure in the three-dimensional culture. Consistently, reduction of phosphorylation of serine 390 of PR-A and GSK-3β activity is observed in the Brca1-deficient mammary gland. Taken together, these results provide important aspects of tissue specificity of BRCA1-mediated suppression of breast carcinogenesis.
Introduction
Progesterone receptors (PRs)2 play important roles in mammary gland development (reviewed in Ref. 1). PR activities have been linked to the expansion of mammary stem cells in a RANKL-dependent manner (2, 3). In hormone replacement randomized studies, postmenopausal women who received estrogen plus progestin have significantly increased breast cancer risk, by contrast, estrogen alone reduced the risk of breast cancer (4, 5). In mice, only the long isoform, PR-B but not PR-A, is required for mammary gland development; however, overexpression of PR-A leads to abnormal mammary gland development and ductal hyperplasia (6–8). These two isoforms differ in their amino terminus because of differential promoter usage that results in a PR-B-specific exon (9). Upon binding with progesterone, PR-B and PR-A are phosphorylated, ubiquitinated, and subsequently degraded through the proteasome pathway (10, 11). Several PR-B phosphorylation sites have been identified; serine 294, a MAPK site, is required for its ubiquitination and degradation (10). On the other hand, phosphorylation of serine 400, a CDK2 site, contributes to its ligand-independent transcription activities (12). These results suggest that post-translational modification of PR may be critical for fine tuning its biological activities.
Glycogen synthase kinase-3β (GSK-3β) is a serine/threonine kinase that phosphorylates a broad range of substrates including glycogen synthase, β-catenin, Axin (13), Cdc25A (14), glucocorticoid receptor, and elF-2B (15). Most GSK-3β substrates have been shown to contain the (pS/T)XXX(pS/T) motif; the GSK-3β-mediated S/T phosphorylation follows phosphorylation of the primary P+4 position (13). Unlike most kinases, GSK-3β is active in resting cells, and stimulation of cells by mitogens or growth factors leads to its inactivation (15). Interestingly, it is noted that GSK-3 modulates the progesterone responsiveness during Xenopus oocyte maturation, because treating with GSK-3 inhibitor 7-azaindolyl-pyrazinyl-maleimide increases progesterone response in Xenopus oocytes (16). This result suggests that PR may also be subjected to GSK-3 regulation.
It is noted that BRCA1 interacts with estrogen receptor α and two PR isoforms directly to modulate ligand-dependent and -independent transcription activities of estrogen receptor α and PRs. Both PR-B and PR-A become stabilized in Brca1-deficient mammary epithelial cells, correlating with the deregulated proliferation of mammary cells. Consistently, treatment with the progesterone receptor antagonist RU 486 delayed/prevented tumor development. These observations indicated that BRCA1 serves as a negative regulator for PRs (11). Similarly, increased PR expression and proliferation were reported in the normal breast epithelium of BRCA1 mutation carriers (17). BRCA1 contains E3 ubiquitin ligase at its amino-terminal region, which may directly dictate the stability of PR-A. However, direct evidence to support this observation remains obscure. It is likely that an additional factor such as phosphorylation by kinase may be needed.
In this study, we show that phosphorylation of PR-A at serine 390 by GSK-3β kinase is required for its ubiquitination and proteasome-mediated degradation. Expression of PR-A mutant with S390A substitution, but not wild-type PR-A, in nontumorigenic mammary epithelial cells, MCF-10A, resulted in increased proliferation and formation of abnormal acini structure. Consistently, in the Brca1/p53-mutated mammary gland, GSK-3β kinase activity was down-regulated, explaining the longer stability of PR-A. These results provide important aspects of tissue specificity of BRCA1-mediated suppression of breast carcinogenesis.
MATERIALS AND METHODS
Plasmids and siRNAs
PCR3.1 wild-type PR-A plasmid was a generous gift from Dr. Bert O'Malley. The PCR3.1 PR-AS390A construct was generated by site mutagenesis. To generate the His6-tagged PR-A, full-length PR-A cDNA was used as a template for PCR with a primer coding a His6 tag; the PCR fragment was subsequently subcloned into the pcDNA3.1 hygro vector. Full-length PR-A cDNA and PR-AS390A mutant were also subcloned into PQCXIP (Clontech). Wild-type GSK-3β, kinase dead GSK-3β, and HA-ubiquitin were as described in previous publication (18). GSK-3β siRNA was purchased from Santa Cruz Biotechnology, and control luciferase siRNA was customized at Dharmacon.
Antibodies
PR, cyclin D1, and GSK-3β antibodies were from Santa Cruz; GSK-3β serine 9 phosphospecific antibodies were from Abcam; HA antibody was from Sigma; and RANKL antibody was from R&D Systems. Vimentin, CK14, CD10, Muc1, phosphor-EGFR, and Bcl-2 antibodies were from GeneTex. Phosphospecific antibodies recognizing phosphoserine 390 of PR-A was raised against a synthetic phosphopeptide SEASQ(pS)PQYSFES and was made by GeneTex. Phosphospecific antibodies were obtained using a two-step affinity purification process; first by passing through a nonphosphospecific column followed by binding and elution from a phospho-peptide column.
Cell Culture
HeLa cells and 293T cells (ATCC) were cultured in DMEM supplemented with 10% FBS. MCF-10A (ATCC) were cultured in DMEM/F-12 supplemented with 5% horse serum (Hyclone), 10 μg/ml insulin (Sigma), 20 ng/ml EGF (Millipore), 100 ng/ml choleratoxin (Sigma), and 0.5 μg/ml hydrocortisone.
Pulse-Chase Studies for PR-A Stability
293T cells were grown in 100-mm culture dishes and transfected with the indicated plasmids or siRNA. Twenty-four hours later, cells were incubated in a methionine- and cysteine-free medium, metabolically labeled with a [35S]methionine/[35S]cysteine mixture (PerkinElmer Life Sciences), and then chased with complete medium supplemented with 20 nm R5020 (PerkinElmer Life Sciences). Cells from one-third of each plate were harvested at each indicated time point and lysed in Nonidet P-40 buffer. PR was immunoprecipitated with PR antibody, separated on SDS-PAGE, and analyzed by autoradiography.
Immunohistochemistry
Mammary glands were removed from different strains of mice as indicated, fixed in 4% paraformaldehyde, and embedded in paraffin. Sections were sliced at 5-μm thickness and subjected to immunohistochemical analyses. Immunostaining was performed following the instructions in the Vectastain Elite ABC kit (Vector Laboratories).
In Vivo Ubiquitination Assay
293T cells were transfected with the indicated plasmids or siRNAs for 48 h. Prior to harvest, cells were treated with 10 μm MG132 for 2 h, followed by 3 h of incubation with 20 nm R5020. The cells were lysed in a boiling solution containing SDS (1% in Tris-buffered saline) and then sonicated. Lysates were diluted 10-fold with Triton X-100 solution (1% in Tris-buffered saline). PR in the supernatants was immunoprecipitated using PR antibody, separated on SDS-PAGE, and visualized by immunoblotting using both HA and PR antibodies.
In Vitro Ubiquitination Assay
As previously reported, briefly, 1 μg of purified PR-A was preincubated with 2 units GSK3β (Sigma G1663) in the presence of the kinase buffer (50 mm Tris-HCl, pH 7.4, 5 mm NaF, 10 nm okadaic acid, 0.06 mm DTT, 2 mm Mg-ATP) at 30 °C for 30 min. The reaction products were added to the BRCA1-BARD1 complex that was immobilized to protein A beads with anti-FLAG antibody (15 μl in dried volume). The mixture was incubated at 4 °C for 60 min. The beads were then washed with buffer (100 mm NaCl, 0.5% Nonidet P-40, and phosphatase inhibitors) and used for the ubiquitination reactions. The ubiquitination reactions (30 μl) contained 1 μg of ubiquitin, 20 ng of E1, and 250 ng of E2, as well as 2 mm Mg-ATP in the ubiquitination buffer (50 mm Tris-HCl, pH 7.4, 2 mm NaF, 10 nm okadaic acid, 0.06 mm DTT) at 37 °C for 60 min. The reactions were terminated by boiling the samples in SDS buffer. The products were separated by SDS-PAGE and analyzed by Western blotting using anti-PR antibody.
Mammosphere Culture
Cells derived from MCF-10A were cultured in an ultra low attachment 24-well plate at a density of 2000 cells/well. The conditional medium consisted of 1× DMEM/F-12 medium, 0.4% BSA, 2% B27 supplement (v/v), 5 μg/ml insulin, 20 ng/ml basic FGF, 4 μg/ml heparin, 0.5 μg/ml hydrocortisone, and 10 nm R5020. The number of mammospheres was counted on day 8 using a Nikon Eclipse TE 2000-S microscope.
Mammary Acini Culture and Staining
The cells were seeded on a precoated Matrigel chamber slide (2000 cells/well), cultured in DMEM/F-12 supplemented with 2 ng/ml EGF, 2% horse serum, 10 μg/ml insulin, 100 ng/ml choleratoxin, 0.5 μg/ml hydrocortisone, 10 nm R5020, and 2% Matrigel (BD Biosciences; catalog no. 356231). Acini image was captured on day 16, and acini sizes were determined and analyzed using National Institutes of Health ImageJ software. For immunostaining, the acini were fixed with 4% paraformaldehyde for 20 min at room temperature, washed with 1× PBS twice, permeabilized with PBS containing 0.5% Triton X-100 for 10 min at 4 °C, washed with PBS/glycine (1× PBS, 100 mm glycine) three times followed by two washes with 1× PBS, and then stained with the indicated antibody overnight. Images were captured using a fluorescence microscope (Zeiss Axiovert 200M).
Nano-LC-MS/MS
Nano-LC-MS/MS experiments were performed on a LTQ-FT mass spectrometer (Thermo Fisher Scientific) equipped with a nano-electrospray ion source (New Objective) in positive ion mode. The liquid chromatography system was the Agilent 1100 Series binary high performance liquid chromatography pump (Agilent Technologies) with the Famos autosampler (LC Packings). The enzyme-digested protein samples were injected onto a self-packed precolumn (150-μm inner diameter × 20 mm, 5 μm, 200 Å). Chromatographic separation was performed on a self-packed reversed phase C18 nano-column (75-μm inner diameter × 300 mm, 5 μm, 100 Å) by using 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in 80% acetonitrile (mobile phase B). A linear gradient from 5 to 45% mobile phase B for 40 min at a flow rate of 300 nl/min was applied. Electrospray voltage was applied at 2 kV, and capillary temperature was set at 200 °C. A scan cycle was initiated with a full scan survey MS spectrum (m/z 300–2000) performed on the FT-ICR mass spectrometer with resolution of 100,000 at 400 Da. The ten most abundant ions detected in this scan were subjected to a MS/MS experiment performed in the linear quadrupole ion trap (LTQ) mass spectrometer. Ion accumulation (Auto Gain Control target number) and maximal ion accumulation time for full scan and MS/MS were set at 1 × 106 ions, 1000 ms and 5 × 104 ions, 200 ms. Ions were fragmented by use of collision-induced dissociation; the normalized collision energy was set to 35%, activation Q was 0.3, and activation time was 30 ms. For data analysis, all MS/MS spectra were converted as DTA format from experiment RAW file by Bioworks (Thermo Fisher Scientific) and then merged into a single file for Mascot (version 2.2, Matrix Science) MS/MS ion search. The search parameters in Mascot including the error tolerance of precursor ions, the MS/MS fragment ions in spectra were 10 ppm and 0.8 Da, and the enzyme miss cleavage number was 5.
RESULTS
GSK-3β Kinase Enhances PR-A Ubiquitination and Degradation
Upon ligand binding, progesterone receptors become phosphorylated at multiple sites; the receptors are subsequently polyubiquitinated and degraded by the proteasome. To test whether GSK-3β has any effect on PR-A polyubiquitination, we performed in vitro ubiquitination assay in the presence of E1, E2, and BRCA1-BARD1 complexes as previously described (11). As shown in Fig. 1A, PR-A was polyubiquitinated upon phosphorylation by GSK-3β (Fig. 1A, lane 4). To affirm this observation, we performed an in vivo polyubiquitination assay by expressing either wild-type, kinase dead GSK-3β (GSK-3β KD), or depleting GSK-3β with siRNA in 293T cells (SV40 T antigen transformed human embryonic kidney 293). As shown in Fig. 1B, ubiquitination of PR-A was the highest in cells overexpressing wild-type GSK-3β, PR-A, and HA-ubiquitin (Fig. 1B, lane 2) but not in GSK-3β KD-overexpressing (Fig. 1B, lane 1) or in GSK-3β-depleted cells (Fig. 1B, lane 4), indicating that GSK-3β activity enhances PR-A ubiquitination.
FIGURE 1.

Effects of GSK-3β on PR-A ubiquitination and stability. A, phosphorylation of PR-A by GSK-3β enhances ubiquitination of PR-A by BRCA1/BARD1 in vitro. PR-A protein was phosphorylated by GSK-3 first, and in vitro ubiquitination assay was carried out as indicated. Lane 1, control without GSK-3; lane 2, control without E1/E2; lane 3, control without E3 (BRCA1/BARD1 complex); lane 4, reaction with all components. B, effects of GSK-3β on PR-A ubiquitination. 293T cells were transfected with PR-A, HA-ubiquitin, and either GSK-3β KD (lane 1), GSK-3β WT (lane 2), control siRNA (lane 3), or GSK-3β siRNA (lane 4). Lysates were immunoprecipitated with PR antibody, and immunoblotting was with HA and PR antibodies; the HA immunoblot identified ubiquitinated PR. C, effects of GSK-3β on PR-A protein stability. Human kidney 293T cells were transfected with PR-A and GSK3β plasmids (WT or KD), subsequently labeled with a [35S]methionine/[35S]cysteine mixture, chased with complete medium supplemented with R5020(20 nm), and harvested at the indicated time points. Immunoprecipitation and SDS-PAGE followed by autoradiography were used to examine PR stability. Lanes 1–3, GSK-3β WT 0, 3, and 6 h, respectively; lanes 4–6, GSK-3β KD, 0, 3, and 6 h, respectively. D, effects of GSK-3β knockdown on PR-A expression. 293T cells were transfected with PR-A and GSK-3β siRNA or control siRNA, and PR-A expression was examined as described in C. Lanes 1–3, GSK-3β siRNA, 0, 3, and 6 h, respectively; lanes 4–6, control siRNA, 0, 3, and 6 h, respectively. Ubi-PR-A, ubiquitination of PR-A; siCtrl, control siRNA; siGSK-3, GSK-3β siRNA.
Consistently, expressing GSK-3β KD inhibited PR-A degradation (Fig. 1C, lanes 1–3) compared with that of expressing wild-type GSK-3β (Fig. 1C, lanes 4–6). On average, the half-life of PR-A was approximately 3 and 6 h in wild-type GSK-3β- and GSK-3β KD-expressing cells, respectively. Similarly, the half-life of PR-A was ∼4.5 h in GSK-3β-depleted cells (Fig. 1D, lanes 1–3) but was ∼3 h in control cells (Fig. 1D, lanes 4–6). Taken together, these results suggest that GSK-3β promotes ligand-induced ubiquitination and degradation of PR-A.
GSK-3β Binds to and Phosphorylate PR-A
To test whether PR-A is a direct substrate of GSK-3β, we performed co-immunoprecipitation using cells expressing FLAG-tagged PR-A and HA-tagged GSK-3β. HA-GSK-3β was presented in the anti-FLAG immunoprecipitates only when FLAG-PR-A was co-expressed (Fig. 2A); conversely, FLAG-PR-A was presented in the anti-HA only when HA-GSK-3β was co-expressed (Fig. 2B), indicating that GSK-3β and PR-A interact with each other.
FIGURE 2.

Interactions between GSK-3β and PR-A and mapping of GSK-3β phosphorylation sites in PR-A by mass spectroscopy. A and B, interaction between PR-A and GSK-3β. 293T cells were transfected with the indicated plasmids. Lane 1, no plasmid; lane 2, FLAG-PR-A; lane 3, HA-GSK-3β; lane 4, FLAG-PRA and HA-GSK-3β. 24 h later, cells were treated with R5020 (20 nm) for 1 h. Immunoprecipitation was carried out using either anti-FLAG (A) or anti-HA antibodies (B). Tagged GSK-3β or PR-A were visualized by immunoblotting using either anti-HA or anti-FLAG antibodies as indicated. C, the MS2 spectrum showing phosphorylation of serine 390 in PR-A. D, the MS2 spectrum showing phosphorylation of serine 512 in PR-A. E, schematic representation of PR-A phosphorylation sites revealed by mass spectroscopy. PR-A phosphorylation sites in insect Sf-9 cells or mammalian cells are indicated. Green dots, phosphorylation sites before kinase reaction; purple stars, phosphorylation sites after kinase reaction; red stars, phosphorylation sites in 293T cells expressing His6-tagged PR-A and treated with 20 nm R5020. AF1, transcription activation function 1; DBD, DNA binding domain; H, hinge region; LBD, ligand binding domain.
Next, we performed in vitro kinase assay using purified PR-A and GSK-3β followed with mass spectrometry analyses. Purified His6-tagged PR-A from Sf-9 cells in the presence of R5020 had phosphorylation sites at serines 26, 63, 112, 236, and 394. Upon incubation with GSK-3β in vitro, two additional phosphorylated sites, serines 164 and 390, were identified (Fig. 2, C and D). Mass spectroscopic analyses revealed that serine 390 but not serine 164 was phosphorylated when His6-tagged PR-A was purified from 293T cells treated with R5020 (Fig. 2, C and D). In addition to serine 390 phosphorylation, we also confirmed the previously reported phosphorylation sites, including serines 26, 63, 130, 236, and 512 in the latter setting (Fig. 2, C and E).
Characterization of Antibodies Specifically Recognize Serine 390 of PR-A
To confirm whether phosphorylation of serine 390 is GSK-3β-dependent, we generated phosphospecific antibodies against a synthetic phospho-peptide, SEASQ(pS)PQYSFESL, which corresponds to PR-A amino acids 385–398. These antibodies detected PR-A in R5020-treated human cervical cancer HeLa cells expressing wild-type PR-A, but not at the zero time point of treatment (Fig. 3A, lanes 1–3). No signals were detected in cells expressing PR-A mutant with serine 390 changed to alanine PR-A mutant (PR-AS390A; Fig. 3A, lane 4), suggesting that the antibodies are specific in recognizing PR-A with phosphorylated serine 390. These phospho-peptide antibodies also gave an immunostaining signal that co-localized with PR in cells treated with R5020 (data not shown).
FIGURE 3.

Serine 390 of PR-A is a GSK-3β phosphorylation site. A, the specificity of antibodies against phosphoserine 390 in the PR-A. Immunoblotting was performed using lysates from HeLa cells transfected with either wild-type PR-A (lanes 1 and 2) or PR-AS390A mutant (lanes 3 and 4) and treated with R5020 for 2 h (lane 2 and 4) or left untreated (lanes 1 and 3). Total PR and phosphorylated PR levels are indicated. B, phosphorylation of serine 390 in PR-A in cells with GSK-3β knockdown. Immunoblotting was performed using lysates from HeLa cell transfected with PR-A and control siRNA (lanes 1–3) or GSK-3β siRNA (lanes 4–6) treated with R5020. C, phosphorylation of serine 390 in PR-A in cells overexpressing either wild-type or GSK-3β KD mutant. Immunoblotting was performed using lysates from HeLa cell transfected with PR-A and GSK-3β WT plasmid (lanes 1 and 2) or GSK-3β KD (lanes 3 and 4) treated with R5020 for 2 h (lane 2 and 4) or untreated (lanes 1 and 3). D, the effects of GSK-3 inhibitor, BIO, on phosphorylation of serine 390 in PR-A. Phosphorylation was examined in HeLa cells transfected with PR-A, followed by either control dimethyl sulfoxide (lanes 1 and 2) or BIO (lanes 3 and 4) treatment. The cells were treated with R5020 for 2 h (lanes 2 and 4). p-PR, phosphorylated PR; siLuc, luciferase siRNA; siGSK-3, GSK-3β siRNA; DMSO, dimethyl sulfoxide.
Phosphorylation of Serine 390 in PR-A Is GSK-3β-dependent
Next, we assessed whether phosphorylation of serine 390 in PR-A is GSK-3β-dependent using these antibodies. Phosphorylation of serine 390 in PR-A became prominent upon R5020 treatment in HeLa cells expressing wild-type PR-A (Fig. 3B, lanes 2 and 3). In contrast, significantly reduced serine 390 phosphorylation was found in HeLa cells with GSK-3β depletion, whereas total PR-A levels were compatible in GSK-3β knockdown or control cells (Fig. 3B, lanes 4–6). Serine 390 phosphorylation was significantly reduced in HeLa cells expressing GSK-3β KD mutant compared with cells expressing GSK-3β WT upon ligand treatment (Fig. 3C, lanes 2 and 4). Similarly, phosphorylation of serine 390 was also reduced when cells were treated with the GSK-3 inhibitor BIO (Fig. 3D, lane 4 versus lane 2). Taken together, these results suggest that ligand-induced phosphorylation of serine 390 in PR-A is mediated by GSK-3β.
PR-AS390A Has Less Ubiquitination and Longer Half-life upon Ligand Treatment
If GSK-3β-mediated phosphorylation of PR-A serves as a signal for subsequent ubiquitination and proteasomal degradation, the PR-AS390A mutant would have a longer half-life than the wild-type PR-A. To explore this possibility, we introduced either wild-type PR-A or PR-AS390A and HA-ubiquitin into 293T cells that were treated with R5020 and MG132 for 3 h. Cell lysates were immunoprecipitated using PR antibodies followed by immunoblotting with HA antibody to detect ubiquitinated PR-A. As shown in Fig. 4A, PR-AS390A was less polyubiquitinated compared with the wild type (Fig. 4A, lane 1 versus lane 3). Furthermore, the stability of wild-type PR-A and PR-AS390A was compared in HeLa cells and immortalized human breast epithelial MCF-10A cells treated with R5020 and cycloheximide. Based on the amount of PR-A at various time points, the half-life of PR-A was ∼3.8 h in HeLa, whereas PR-AS390A was greater than 8.0 h (Fig. 4B). In MCF-10A cells, the half-life for the wild-type PR-A was 4.4 h, and that for PR-AS390A was ∼7.0 h (Fig. 4C). These results suggest that phosphorylation of serine 390 of PR-A by GSK-3β determines its stability.
FIGURE 4.

Serine 390 to alanine mutation in PR-A decreased PR-A ubiquitination and led to its stabilization. A, ubiquitination of wild-type PR-A and PR-AS390A. Immunoprecipitation followed by immunoblotting analyses of anti-PR immunoprecipitates from 293 cells co-transfected with HA-ubiquitin, and wild-type PR-A, PR-AS390A or control vector, treated with R5020 and MG132 for 3 h. Immunoblotting was performed with antibodies against PR or HA as indicated. Lane 1, PR-AS390A; lane 2, control; lane 3, PR-A. B and C, stability of PR-A and PR-AS390A in HeLa cells (B) or immortalized human mammary MCF-10A cells (C) upon ligand treatment. HeLa or MCF-10A cells expressing either wild-type or mutant PR-A were treated with R5020 and cycloheximide. Cells were harvested at the indicated time point, and PR levels were visualized by Western blotting. B, lanes 1–5, wild-type PR-A, 0, 2, 4, 6, and 8 h, respectively; lanes 6–10, PR-AS390A, 0, 2, 4, 6, and 8 h, respectively. C, lanes 1–5, WT PR, 0, 1.5, 3, 5, and 7 h, respectively; lanes 6–10, PR-AS390A, 0, 1.5, 3, 5, and 7 h, respectively.
Expression of PR-AS390A Promotes MCF-10A Cell Mammosphere Formation and Alters Its Growth Behavior in Three-dimensional Culture
To access the biological consequences of phosphorylation of serine 390 of PR-A, we expressed wild-type PR-A, PR-AS390A, or control vector pQCXIP in MCF-10A via retrovirus-mediated gene transfer. As shown in Fig. 5A, the expression of wild-type and mutant PR-A was comparable.
FIGURE 5.

PR-AS390A expression promoted proliferation of MCF-10A cells. A, effects of PR-A and PR-AS390A expression in MCF-10A cells infected with control viral vector (lane 1), WT PR-A (lane 2), or PR-AS390A retroviruses (lane 3). PR-A levels were visualized by immunoblotting. B and C, mammosphere formation. The number of mammospheres formed by MCF-10A cells after transduction with different retroviruses in a conditional medium supplemented with 10 nm R5020 and without EGF in the ultra low attachment plates was counted at 8 days. A representative figure of the mammospheres formed (B) and statistical analyses (C) are shown. WT PR-A+BIO was treated with the GSK-3 inhibitor BIO. D, acini formation. Acini formation of PR-A-expressing MCF-10A cells in a Matrigel-coated chamber slide and in medium containing 2% Matrigel were examined after 16 days culture. E, sizes of acini formed from WT PR-A and PR-AS390A. Sizes of acini were analyzed using National Institutes of Health ImageJ software (more than 100 acini were analyzed from each group). The average size of the control group was set to 1, WT PR-A group was 1.27, and PR-AS390A average size was 2.2. F, Ki-67 staining. Acini cultures were fixed and stained with Ki-67 antibody at day 16. Images were taken using a fluorescence microscopy. Vec, vector.
Using these cells, we compared their ability to form mammosphere because progesterone has been shown to promote mammary stem cell self-renewal (2, 3). Under a modified culture condition that contained R5020, instead of EGF, PR-AS390A-expressing cells formed ∼18 spheres, wild-type PR-A formed ∼9 spheres, but the vector only did not form any mammosphere (Fig. 5, B and C). The mammosphere formed under these conditions possessed multiple cell types as evidenced by the expression of vimentin, CK14, CD10, and MUC1 (data not shown). Interestingly, wild-type PR-A cells treated with the GSK-3β inhibitor BIO formed more mammospheres than mutant PR-A cells (Fig. 5C). These results suggest that the failure of phosphorylation of serine 390 of PR-A promotes stem cell renewal.
Next, we cultured these cells in Matrigel in low EGF-containing medium for 16 days to assess the effects of wild-type PR-A and PR-AS390A on their growth behavior. All three groups formed mammary acini (Fig. 5D), which consist of CK-14-positive and -negative cells (data not shown); interestingly, PR-AS390A-expressing cells had the largest acini. Using National Institutes of Health ImageJ software and statistical analyses of more than 100 acini in each group, we found that acini formed by PR-AS390A-expressing cells were 2.20-fold larger than the vector control group, whereas acini formed by the wild-type PR-A-expressing cells were 1.27-fold larger than the control group (Fig. 5E). Immunostaining of Ki-67 at day 16 revealed strong patched Ki-67 staining in acini formed by PR-AS390A-expressing cells, whereas control and wild-type PR-A-expressing cells had weak and even staining (Fig. 5F). These data suggest that PR-AS390A promotes proliferation. Although the PR-AS390A cells formed larger acini, there were fewer acini compared with the wild-type and control groups. Judging from stronger immunostaining of anti-cleaved caspase-3 antibodies in PR-AS390A-expressing cells compared with wild-type PR-A and control cells, an elevated apoptosis in PR-AS390A-expressing cells was observed (Fig. 6, A and B). In addition, we used antibodies recognizing PR target genes to compare their expressions among these three groups. RANKL expression was increased in PR-AS390A-expressing cells (Fig. 6C), whereas no changes in cyclin D1 expression were detected (Fig. 6D). Interestingly, Bcl-2 expression was decreased in cells expressing the mutant PR-A, which correlated with the increased cell death seen in these cells (Fig. 6E). Taken together, these results indicate that failure of the phosphorylation of serine 390 of PR-A promotes proliferation and apoptosis of MCF-10A cells in the three-dimensional culture.
FIGURE 6.

PR-AS390A expression altered growth behavior of MCF-10A cells in three-dimensional culture and expression of PR target genes. PR-A WT, PR-A S390A, or control cells were cultured in a Matrigel-coated chamber slide with medium containing 2% Matrigel. At the indicated time, acini were fixed and stained by antibody against cleaved caspase-3. A and B, acini after 6 (A) and 16 (B) days of culture. C, RANKL expression in MCF-10A cells stably expressing control vector, wild-type PR-A, or S390A mutant PR-A. The cells were fixed after 8 days culture in the three-dimensional Matrigel. D, cyclin D1 expression. See C for details. E, Bcl-2 expression. See C for details.
Phosphorylation of PR-A at Serine 390 Is Diminished in Brca1-deficient Mammary Epithelial Cells
Because PR-A has a longer half-life in Brca1-deficient cells, it is likely that phosphorylation of serine 390 of PR-A is attenuated in these cells. To test this possibility, mammary glands were harvested from mice at the estrous or diestrous stage, and PR-A and phospho-PR-A levels were analyzed by Western blot. As shown in Fig. 7A, phosphorylation of serine 390 was significantly reduced in the Brca1f/f p53f/fWapCrec mice both at estrous and diestrous compared with that of the wild-type mammary gland (Fig. 7A, lanes 3 and 4 versus lanes 1 and 2). Higher levels of total PR-A were found in Brca1f/f p53f/fWapCrec mice as previously reported (11). These results further suggest that phosphorylation of serine 390 of PR-A by GSK-3β plays an important role in BRCA1-mediated breast carcinogenesis.
FIGURE 7.
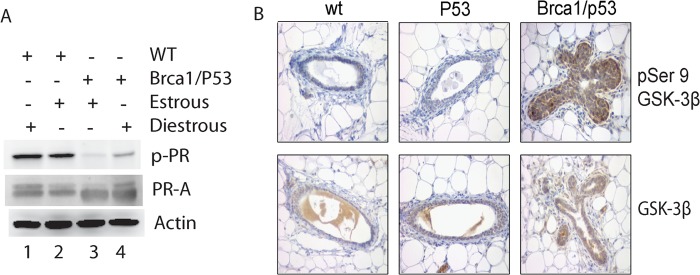
Diminished phosphorylation of serine 390 of PR-A and inactive GSK-3β in the Brca1-deficient mammary gland. A, phosphorylation of serine 390 in PR-A and total PR-A levels in the mammary gland of Brca1/p53-conditional knock-out mice. Immunoblotting was performed using lysates prepared from mammary glands of mice at either estrous and diestrous cycle as indicated. Lane 1, WT diestrous; lane 2; WT estrous; lane 3, Brca1/p53 estrous; lane 4, Brca1/p53 diestrous. B, GSK-3β activity in the Brca1-deficient mammary gland. Immunohistochemistry was performed using paraffin-fixed mammary gland sections from wild-type, p53f/f WapCrec (p53), and Brca1f/f p53f/fWapCrec (Brca1/p53) mice with antibodies against GSK-3β phosphoserine 9 (pSer 9) (top panel) and GSK-3β (bottom panel). p-PR, phosphorylated PR; wt, wild-type.
It was reported that BRCA1 negatively regulated AKT activity, which phosphorylates serine 9 of the GSK-3β and inhibits GSK-3β kinase activity (21). Thus, it is likely that GSK-3β activity is down-regulated in Brca1-deficient mammary epithelial cells. To test this possibility, we performed and compared immunostaining using mammary tissue sections from wild-type and Brca1f/f p53f/fWapCrec mice. As shown in Fig. 7B, the levels of GSK-3β serine 9 phosphorylation were significantly higher in Brca1f/f p53f/fWapCrec mice, compared with the mammary gland from both wild-type and p53f/f WapCrec mice (Fig. 7B, upper panels), whereas the total GSK-3β levels were compatible in these three groups (Fig. 7B, lower panels), indicating that GSK-3β activity was inhibited in the mammary epithelial cells of the Brca1f/f p53f/fWapCrec mice.
DISCUSSION
In this study, we found that progesterone receptor-A is a physiological substrate of GSK-3β kinase. Phosphorylation of PR-A on serine 390 by GSK-3β enhances its ubiquitination and degradation. Expression of PR-AS390A mutant in the human breast epithelial cells, MCF-10A, results in increased proliferation and formation of abnormal acini structure. Consistently, in the Brca1/p53-deficient mammary gland, phosphorylation on serine 390 of PR-A is down-regulated. These results suggest the importance of phosphorylation of PR-A by GSK-3β in BRCA1-mediated suppression of breast carcinogenesis.
Progesterone receptors, especially the dominant short form PR-A, accumulate in the mammary gland of Brca1 knock-out mice because of stabilization of the receptor (11). The PR activity is tightly modulated by phosphorylation. Both the long and short forms of PR become phosphorylated upon ligand binding, and several phosphorylation sites have been identified. For example, PR serine 400 (e.g., serine 236 in PR-A) is phosphorylated to mediate the ligand-independent activity of PR (12). Phosphorylation of PR serine 345 (e.g., serine 181 in PR-A) by p38 MAPK is required for tethering PR to specificity protein 1 to promote cell proliferation (19). Phosphorylation of PR serine 294 (e.g., serine 130 in PR-A) by p42/44 MAPK coupled with sumoylation of PR-B is essential for derepression of PR action (20). Consistently, phosphorylation of PR-A on serine 390 by GSK-3β, as described here, is essential for modulating PR-A stability.
GSK-3β is a key regulator of sex steroid receptors (reviewed in Ref. 21) and phosphorylates different steroid receptors including estrogen receptor α (22), glucocorticoid receptor (23), and androgen receptor (24). GSK-3β regulates the progesterone response in Xenopus oocyte meiotic entry, and treating with GSK-3β inhibitor enhances the progesterone sensitivity of Xenopus oocytes (16). Consistently, GSK-3β is the key kinase to modulate PR-A stability as described here.
Interestingly, many of GSK-3β substrates including glycogen synthase (13), CDC25A (14), and SRC-3 (steroid receptor coactivator 3) contain a (S/T)XXX(pS/T) motif for phosphorylation. However, phosphorylation of these substrates by GSK-3β requires a prior phosphorylation on priming site (the n + 4 S/T) by other kinases. On the other hand, many GSK-3β substrates such as Axin and Tau (15), steroid receptors (21, 22, 24), do not need priming phosphorylation. It was noted that phosphorylation of glucocorticoid receptor serine 404 by GSK-3β is also essential for modulating its stability (23). Interestingly, the surrounding amino acid sequences of serine 390 of PR-A share high homology with that of the glucocorticoid receptor serine 404, suggesting a potential recognition motif for GSK-3β for this class of proteins. This possibility warrants further exploration.
It has been shown that progesterone triggers two waves of proliferation in the mammary epithelial cells: the first short wave is dependent on cyclin D1 and acts in an autocrine fashion; the second long wave is dependent on RANKL and acts in a paracrine fashion (25). Progesterone induces mammary stem cell expansion (3). Interestingly, Hilton et al. (26) showed that the presence of PR- and basal markers-positive bi-potent progenitor cells in normal human breast. In contrast to the luminal epithelial cells, these PR-positive bi-potent progenitor cells are refractory to estrogen. We show that Brca1/p53-deficient mammary cells are uniquely sensitive to progesterone, in part because of stabilization of PR. Because PR degradation by the proteasome is the major pathway for termination of progesterone signaling, the degradation needs to be tightly regulated because prolonged progesterone response can trigger abnormal proliferation in the mammary gland (3). Consistently, the expression of PR-AS390A mutant triggered abnormal proliferation of MCF-10A cells in three-dimensional culture as indicated by larger acini and extensive Ki-67 staining (Fig. 6). In addition, increasing apoptosis was seen in the mutant PR-A group, which correlated with decreased levels of Bcl-2 expression. Bcl-2 is a known PR target (27). Although an extended PR-A half-life could lead to more Bcl-2 expression, the reduced Bcl-2 expression in these cells suggests that phosphorylation of PR-A S390A may have an opposite role in Bcl-2 transcription regulation. Further investigation is needed to clarify this issue.
Interestingly, Akt activity is up-regulated in the mammary gland of Brca1 knock-out mice (28); Brca1-deficient cells accumulate phosphorylated nuclear Akt, which increases phosphorylation of the Akt substrates including GSK-3β (29). Phosphorylation of GSK-3β by Akt inhibits GSK-3β kinase activity (30). Connecting these observations together, the longer half-life of PR-A in the BRCA1-deficient cells may be resulted from higher Akt activity, which inhibits GSK-3β kinase activity and reduces phosphorylation of serine 390 of PR-A. Thus, these results provide a plausible explanation for the tissue specificity of BRCA1-mediated suppression of breast carcinogenesis through progesterone receptors.
Acknowledgments
We thank Dr. Bert O'Malley for the His-PR-A overexpressed in the insect cells, Dr. Carol A Lange for PR reporter plasmid, Dr. Wen-Hwa Lee for critical reading, and Connie Tat and Serena Abbondante for comments.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA137102 and BCRF-35127 (to E. Y.-H. P. L.).
- PR
- progesterone receptor
- GSK
- glycogen synthase kinase
- KD
- kinase dead.
REFERENCES
- 1. Brisken C., O'Malley B. (2010) Hormone action in the mammary gland. Cold Spring Harb. Perspect. Biol. 2, a003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schramek D., Leibbrandt A., Sigl V., Kenner L., Pospisilik J. A., Lee H. J., Hanada R., Joshi P. A., Aliprantis A., Glimcher L., Pasparakis M., Khokha R., Ormandy C. J., Widschwendter M., Schett G., Penninger J. M. (2010) Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 468, 98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Joshi P. A., Jackson H. W., Beristain A. G., Di Grappa M. A., Mote P. A., Clarke C. L., Stingl J., Waterhouse P. D., Khokha R. (2010) Progesterone induces adult mammary stem cell expansion. Nature 465, 803–807 [DOI] [PubMed] [Google Scholar]
- 4. Chlebowski R. T., Anderson G. L. (2012) Changing concepts. Menopausal hormone therapy and breast cancer. J. Natl. Cancer Inst. 104, 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chlebowski R. T., Hendrix S. L., Langer R. D., Stefanick M. L., Gass M., Lane D., Rodabough R. J., Gilligan M. A., Cyr M. G., Thomson C. A., Khandekar J., Petrovitch H., McTiernan A. (2003) Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women. The Women's Health Initiative Randomized Trial. JAMA 289, 3243–3253 [DOI] [PubMed] [Google Scholar]
- 6. Shyamala G., Yang X., Silberstein G., Barcellos-Hoff M. H., Dale E. (1998) Transgenic mice carrying an imbalance in the native ratio of A to B forms of progesterone receptor exhibit developmental abnormalities in mammary glands. Proc. Natl. Acad. Sci. U.S.A. 95, 696–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mulac-Jericevic B., Lydon J. P., DeMayo F. J., Conneely O. M. (2003) Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc. Natl. Acad. Sci. U.S.A. 100, 9744–9749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lydon J. P., DeMayo F. J., Funk C. R., Mani S. K., Hughes A. R., Montgomery C. A., Jr., Shyamala G., Conneely O. M., O'Malley B. W. (1995) Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 9, 2266–2278 [DOI] [PubMed] [Google Scholar]
- 9. Kastner P., Krust A., Turcotte B., Stropp U., Tora L., Gronemeyer H., Chambon P. (1990) Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 9, 1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lange C. A., Shen T., Horwitz K. B. (2000) Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc. Natl. Acad. Sci. U.S.A. 97, 1032–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Poole A. J., Li Y., Kim Y., Lin S. C., Lee W. H., Lee E. Y. (2006) Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science 314, 1467–1470 [DOI] [PubMed] [Google Scholar]
- 12. Pierson-Mullany L. K., Lange C. A. (2004) Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol. Cell Biol. 24, 10542–10557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cohen P., Frame S. (2001) The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2, 769–776 [DOI] [PubMed] [Google Scholar]
- 14. Kang T., Wei Y., Honaker Y., Yamaguchi H., Appella E., Hung M. C., Piwnica-Worms H. (2008) GSK-3β targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3β inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell 13, 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harwood A. J. (2001) Regulation of GSK-3. A cellular multiprocessor. Cell 105, 821–824 [DOI] [PubMed] [Google Scholar]
- 16. Justman Q. A., Serber Z., Ferrell J. E., Jr., El-Samad H., Shokat K. M. (2009) Tuning the activation threshold of a kinase network by nested feedback loops. Science 324, 509–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martins F. C., De S., Almendro V., Gönen M., Park S. Y., Blum J. L., Herlihy W., Ethington G., Schnitt S. J., Tung N., Garber J. E., Fetten K., Michor F., Polyak K. (2012) Evolutionary pathways in BRCA1-associated breast tumors. Cancer Discov. 2, 503–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Plotnikov A., Li Y., Tran T. H., Tang W., Palazzo J. P., Rui H., Fuchs S. Y. (2008) Oncogene-mediated inhibition of glycogen synthase kinase 3β impairs degradation of prolactin receptor. Cancer Res. 68, 1354–1361 [DOI] [PubMed] [Google Scholar]
- 19. Faivre E. J., Daniel A. R., Hillard C. J., Lange C. A. (2008) Progesterone receptor rapid signaling mediates serine 345 phosphorylation and tethering to specificity protein 1 transcription factors. Mol. Endocrinol. 22, 823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Daniel A. R., Faivre E. J., Lange C. A. (2007) Phosphorylation-dependent antagonism of sumoylation derepresses progesterone receptor action in breast cancer cells. Mol. Endocrinol. 21, 2890–2906 [DOI] [PubMed] [Google Scholar]
- 21. Grisouard J., Mayer D. (2009) Specific involvement of glycogen synthase kinase-3 in the function and activity of sex steroid hormone receptors reveals the complexity of their regulation. J. Steroid Biochem. Mol. Biol. 117, 87–92 [DOI] [PubMed] [Google Scholar]
- 22. Medunjanin S., Hermani A., De Servi B., Grisouard J., Rincke G., Mayer D. (2005) Glycogen synthase kinase-3 interacts with and phosphorylates estrogen receptor α and is involved in the regulation of receptor activity. J. Biol. Chem. 280, 33006–33014 [DOI] [PubMed] [Google Scholar]
- 23. Spokoini R., Kfir-Erenfeld S., Yefenof E., Sionov R. V. (2010) Glycogen synthase kinase-3 plays a central role in mediating glucocorticoid-induced apoptosis. Mol. Endocrinol. 24, 1136–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salas T. R., Kim J., Vakar-Lopez F., Sabichi A. L., Troncoso P., Jenster G., Kikuchi A., Chen S. Y., Shemshedini L., Suraokar M., Logothetis C. J., DiGiovanni J., Lippman S. M., Menter D. G. (2004) Glycogen synthase kinase-3β is involved in the phosphorylation and suppression of androgen receptor activity. J. Biol. Chem. 279, 19191–19200 [DOI] [PubMed] [Google Scholar]
- 25. Beleut M., Rajaram R. D., Caikovski M., Ayyanan A., Germano D., Choi Y., Schneider P., Brisken C. (2010) Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc. Natl. Acad. Sci. U.S.A. 107, 2989–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hilton H. N., Graham J. D., Kantimm S., Santucci N., Cloosterman D., Huschtscha L. I., Mote P. A., Clarke C. L. (2012) Progesterone and estrogen receptors segregate into different cell subpopulations in the normal human breast. Mol. Cell Endocrinol. 361, 191–201 [DOI] [PubMed] [Google Scholar]
- 27. Yin P., Lin Z., Cheng Y. H., Marsh E. E., Utsunomiya H., Ishikawa H., Xue Q., Reierstad S., Innes J., Thung S., Kim J. J., Xu E., Bulun S. E. (2007) Progesterone receptor regulates Bcl-2 gene expression through direct binding to its promoter region in uterine leiomyoma cells. J. Clin. Endocrinol. Metab. 92, 4459–4466 [DOI] [PubMed] [Google Scholar]
- 28. Ma Y., Hu C., Riegel A. T., Fan S., Rosen E. M. (2007) Growth factor signaling pathways modulate BRCA1 repression of estrogen receptor-α activity. Mol. Endocrinol. 21, 1905–1923 [DOI] [PubMed] [Google Scholar]
- 29. Xiang T., Ohashi A., Huang Y., Pandita T. K., Ludwig T., Powell S. N., Yang Q. (2008) Negative Regulation of AKT Activation by BRCA1. Cancer Res. 68, 10040–10044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cross D. A., Alessi D. R., Cohen P., Andjelkovich M., Hemmings B. A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 [DOI] [PubMed] [Google Scholar]