

Abstract
Background and Purpose
Chemogenomics focuses on the discovery of new connections between chemical and biological space leading to the discovery of new protein targets and biologically active molecules. G-protein coupled receptors (GPCRs) are a particularly interesting protein family for chemogenomics studies because there is an overwhelming amount of ligand binding affinity data available. The increasing number of aminergic GPCR crystal structures now for the first time allows the integration of chemogenomics studies with high-resolution structural analyses of GPCR-ligand complexes.
Experimental Approach
In this study, we have combined ligand affinity data, receptor mutagenesis studies, and amino acid sequence analyses to high-resolution structural analyses of (hist)aminergic GPCR-ligand interactions. This integrated structural chemogenomics analysis is used to more accurately describe the molecular and structural determinants of ligand affinity and selectivity in different key binding regions of the crystallized aminergic GPCRs, and histamine receptors in particular.
Key Results
Our investigations highlight interesting correlations and differences between ligand similarity and ligand binding site similarity of different aminergic receptors. Apparent discrepancies can be explained by combining detailed analysis of crystallized or predicted protein-ligand binding modes, receptor mutation studies, and ligand structure-selectivity relationships that identify local differences in essential pharmacophore features in the ligand binding sites of different receptors.
Conclusions and Implications
We have performed structural chemogenomics studies that identify links between (hist)aminergic receptor ligands and their binding sites and binding modes. This knowledge can be used to identify structure-selectivity relationships that increase our understanding of ligand binding to (hist)aminergic receptors and hence can be used in future GPCR ligand discovery and design.
Linked Articles
This article is part of a themed issue on Histamine Pharmacology Update. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2013.170.issue-1
Keywords: aminergic, chemogenomics, chemical similarity, GPCR, histamine receptors, site-directed mutagenesis, structure-affinity relationship, protein-ligand interactions, crystal structures, transmembrane proteins
Introduction
The aim of chemogenomics is to derive predictive links between the chemical structures of biologically active molecules and the protein targets with which these molecules interact (Klabunde, 2007; Rognan, 2007). Based on the assumption that similar proteins bind similar ligands (and vice versa) knowledge on the molecular determinants of protein-ligand interactions can be used to identify novel ligands for a given target or a novel target for a given ligand. Generally, chemogenomic analyses are based on the comparison of the molecular and structural properties of ligands, protein targets or ligand-protein complexes (Keiser et al., 2007; Klabunde, 2007; Rognan, 2007; Jacoby, 2009; Garland and Gloriam, 2011b). The family of GPCRs is a particularly interesting system for chemogenomic analyses for several reasons: (i) GPCRs are targeted by ∼30% of the currently marketed drugs (Overington et al., 2006); (ii) large experimental GPCR ligand binding data sets are available (Knox et al., 2011; Gaulton et al., 2012); (iii) GPCRs share ligands within and between receptor subfamilies [Jacoby et al., 2006; Brianso et al., 2011; Besnard et al., 2012; Sanders et al., 2012) (and with other protein families (Morphy and Rankovic, 2005; Keiser et al., 2009; de Graaf et al., 2013)], (iv) GPCRs consist of seven transmembrane helices (7TM) that share a similar fold (Katritch et al., 2012) (Figure 1A), which allows the definition of a generic GPCR ligand binding site consisting of a small set of residues at conserved locations in the TM helices (Figure 1B,C) (Surgand et al., 2006; Gloriam et al., 2009). Until the past few years, chemogenomic analyses of GPCR-ligand interactions have been limited to protein information derived from sequence alignments (Attwood and Findlay, 1994; Kolakowski, 1994) or GPCR homology models based on the bovine rhodopsin crystal structure, the first (and for long time only) solved GPCR crystal structure (Palczewski et al., 2000). These studies have been used to identify privileged GPCR ligand scaffolds (Jacoby et al., 2006; Johansson et al., 2013) and complementary structural or sequence motifs in GPCR binding sites (Bondensgaard et al., 2004; Garland and Gloriam, 2011a; Surgand et al., 2006) that offer insight in GPCR ligand selectivity profiles (Besnard et al., 2012; de Graaf et al., 2013), the construction of ligand- and protein-based virtual screening models for GPCR ligands (Klabunde et al., 2009; Weill and Rognan, 2009) and receptor deorphanization (Gloriam et al., 2011; Weill, 2011). The increased number of GPCR crystal structures in the past 5 years (Jacobson and Costanzi, 2012), in particular of the aminergic GPCR subfamily (27 structures for 6 of the 42 aminergic receptors) (Surgand et al., 2006; Katritch et al., 2012), now for the first time allow to combine chemogenomic studies with high-resolution structural analyses of GPCR-ligand complexes. In addition to the emerging information on GPCR structures, also more complete GPCR ligand data sets are becoming more and more accessible to further push the limits of (structural) chemogenomic investigation of GPCR-ligand interaction space. The binding affinities of large numbers of small molecule ligands against many individual GPCRs have been determined over the past decades, but only in the past few years these data can be systematically analysed in publically accessible libraries of protein-target annotated ligands [e.g. ChEMBL (Gaulton et al., 2012), DrugBank (Knox et al., 2011), BindingDB (Chen et al., 2001)]. Secondly, the first consistent and complete experimental screening data of (small fragment-like) ligand libraries against multiple GPCR targets are being reported (Besnard et al., 2012; de Graaf et al., 2013). Our structural chemogenomics study combines these emerging data and insights on the aminergic GPCR family, a receptor family that has been extensively investigated by ligand structure-activity/affinity relationships (SAR) and site-directed protein mutagenesis studies (Table 1, Supporting Information Table S1) (Paolini et al., 2006; Shi and Javitch, 2002; Surgand et al., 2006). We will show how this analysis can be used to elucidate the molecular determinants of ligand binding to a particular subfamily of aminergic GPCRs, namely the histamine receptor family, important players in allergy, acid secretion, inflammation and CNS disorders (Engelhardt et al., 2009; Kiss and Keseru, 2012; Kuhne et al., 2011; Parsons and Ganellin, 2006; Simons and Simons, 2011).
Figure 1.

(A) Conserved TM–fold of crystallized aminergic GPCRs (one crystal structure per receptor). The co-crystallized ligand doxepin (1) in H1R is depicted using black carbon atoms. (B) Top view of the H1R with doxepin (1, black carbon atoms). Magenta spheres depict C-alpha atoms from the binding pocket residues. The side chain of the key ionic anchor D3.32 is displayed in black. Both the major and minor binding pocket are highlighted. (C) Sequence alignment of putative binding site residues of the human H1R, human H2R, human H3R, human H4R, human M2R, rat and human M3R (the pocket residues for M3R rat and human are identical); human α1AR, turkey α1AR, human α2AR and human D3R. The lower case character preceding the receptor abbreviation indicates the species, h (human), r (rat) and m (turkey). Binding site residues are assigned based on the basis of 30 residues proposed by (Surgand et al., 2006) plus an additional 24 residues based on the six aminergic crystal structures and SDM studies (Table 1, Supporting Information Table S1) (Vroling et al., 2011). Residues in contact with the ligand in the crystal structure are coloured cyan. Magenta highlights residue W7.40, which is an aminergic family specific conserved residue. The conserved residue D3.32 is coloured red. Capital letters at the bottom indicate a partially conserved residue (>75%) and * indicates a fully conserved residue. All cysteines that form a disulphide bridge are highlighted in yellow.
Table 1.
Binding affinities from site directed mutagenesis studies on histamine receptors and crystallized aminergic GPCRs
| Ligand | Reference | WT | 2.57 | 2.61 | 2.64 | 3.24 | 3.28 | 3.32 | 3.33 | 3.36 | 3.37 | 3.40 | 4.57 | 45.49 | 45.50 | 45.51 | 5.39 | 5.42 | 5.43 | 5.46 | 6.48 | 6.51 | 6.52 | 6.55 | 7.31 | 7.39 | 7.42 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Histamine H1 receptor | V | N | Y | L | W | D | Y | S | T | I | V | K | C | E | K | T | A | N | W | Y | F | F | N | I | G | ||
| 8 | Moguilevsky et al. (1995) | 5.9 | 5.2 (A) | 3.6 (A) | |||||||||||||||||||||||
| 19 | Moguilevsky et al. (1995) | 7.0 | 8.0 (A) | ||||||||||||||||||||||||
| 8 | Gillard et al. (2002) | 5.9 | 4.7 (A) | 5.2 (A) | |||||||||||||||||||||||
| 18 | Gillard et al. (2002) | 8.5 | 7.9 (A) | 8.7 (A) | |||||||||||||||||||||||
| 19 | Gillard et al. (2002) | 7.1 | 6.3 (A) | 8.2 (A) | |||||||||||||||||||||||
| 16 | Nonaka et al. (1998) | 8.6 | 7.3 (A) | 8.4 (A) | 8.4 (A) | ||||||||||||||||||||||
| 1 | Nonaka et al. (1998) | 9.2 | 6.7 (A) | ||||||||||||||||||||||||
| 8 | Ohta et al. (1994) | 4.8 | N.D. (A) | 4.2 (A) | <2.0 (A) | ||||||||||||||||||||||
| 8 | Bruysters et al. (2004) | 4.3 | N.D. (A) | 3.9 (A) | 2.6 (A) | N.D. (A) | 2.7 (A) | ||||||||||||||||||||
| 8 | Wieland et al. (1999) a | 4.1 | 3.5 (A) | 3.7 (M) | 4.5 (M) | ||||||||||||||||||||||
| 17 | Wieland et al. (1999) a | 8.0 | 6.3 (A) | ||||||||||||||||||||||||
| 19 | Wieland et al. (1999) a | 7.2 | 6.3 (A) | ||||||||||||||||||||||||
| 8 | Leurs et al. (1995) a | 4.5 | 3.8 (A) | ||||||||||||||||||||||||
| 9 | Leurs et al. (1994) a | 9.1 | 9.3 (A) | 8.3 (A) | |||||||||||||||||||||||
| 12 | Leurs et al. (1994) a | 7.2 | 7.4 (A) | 6.5 (A) | |||||||||||||||||||||||
| 8 | Bakker et al. (2004) | 4.2 | <2.2 (A) | 3.7 (A) | 3.0 (A) | ||||||||||||||||||||||
| Histamine H2 receptor | V | S | Y | F | Y | D | V | C | T | I | S | K | C | K | G | D | G | T | W | Y | F | F | N | L | G | ||
| 8 | Gantz et al. (1992) b | - | ↓ (N) | ↓ (A) | ↓ (A) | ||||||||||||||||||||||
| Histamine H3 receptor | C | Y | Y | L | W | D | Y | C | T | A | Y | H | C | Y | L | A | S | E | W | Y | T | M | P | F | L | ||
| 8 | Uveges et al. (2002) | 7.8 | 7.7 (A) | 7.2 (Q) | 4.5 (A) | ||||||||||||||||||||||
| 29 | Yao et al. (2003) | 9.4 | 9.1 (A) | 10.5 (V) | |||||||||||||||||||||||
| 21 | Yao et al. (2003) | 7.2 | 8.2 (A) | 8.4 (V) | |||||||||||||||||||||||
| 22 | Yao et al. (2003) | 6.1 | 7.6 (A) | 7.4 (V) | |||||||||||||||||||||||
| Histamine H4 receptor | S | Y | H | I | W | D | Y | C | T | V | N | E | C | E | L | T | S | E | W | Y | S | T | K | F | Q | ||
| 31 | Schultes et al. (2012) | 7.1 | 6.5 (V) | 6.7 (Q) | |||||||||||||||||||||||
| 32 | Schultes et al. (2012) | 7.7 | 6.4 (V) | 7.5 (Q) | |||||||||||||||||||||||
| 33 | Schultes et al. (2012) | 8.3 | 7.7 (V) | 7.2 (Q) | |||||||||||||||||||||||
| 34 | Schultes et al. (2012) | 8.7 | 7.2 (V) | 6.7 (Q) | |||||||||||||||||||||||
| 29 | Istyastono et al. (2011b) | 8.1 | 8.0 (A) | 7.8 (Y) | 8.6 (A) | 7.3 (Q) | 8.8 (M) | 9.0 (L) | |||||||||||||||||||
| 30 | Istyastono et al. (2011b) | 7.5 | 7.2 (A) | 6.5 (Y) | 7.9 (A) | 6.8 (Q) | 7.9 (M) | 7.9 (L) | |||||||||||||||||||
| 8 | Jongejan et al. (2008) | 7.7 | N.D. (A) | 5.0 (Q) | |||||||||||||||||||||||
| 48 | Jongejan et al. (2008) | 6.3 | N.D. (A) | 6,2 (Q) | |||||||||||||||||||||||
| 48 | Lim et al. (2010) | 6.4 | 4.7 (H) | 7.0 (V) | 6.6 (L) | ||||||||||||||||||||||
| 32 | Lim et al. (2010) | 8.3 | 7.7 (H) | 7.1 (V) | 7.8 (L) | ||||||||||||||||||||||
| 8 | Shin et al. (2002) | 7.8 | N.D. (A) | 7.5 (T) | 7.3 (A) | 7.5 (A) | N.D. (A) | 7.1 (F) | |||||||||||||||||||
| 8 | Shin et al. (2002) | 7.8 | N.D. (E) | 7.2 (A) | 6.8 (D) | 7.5 (A) | |||||||||||||||||||||
| 8 | Shin et al. (2002) | 7.8 | N.D. (N) | N.D. (N) | |||||||||||||||||||||||
| Dopamine D3 receptor | V | V | L | C | F | D | V | C | T | I | S | V | C | S | V | S | S | S | W | F | F | H | S | T | G | ||
| 35 | Dorfler et al. (2008) | 7.1 | 6.0 (E) | <4.1 (W) | |||||||||||||||||||||||
| 37 | Dorfler et al. (2008) | 8.4 | 5.9 (E) | 6.6 (W) | |||||||||||||||||||||||
| 38 | Dorfler et al. (2008) | 8.6 | 5.8 (E) | 7.5 (W) | |||||||||||||||||||||||
| 39 | Ehrlich et al. (2009) | 6.1 | 5.6 (F) | 6.1 (F) | 6.2 (E) | 7.2 (A) | |||||||||||||||||||||
| 40 | Ehrlich et al. (2009) | 7.6 | 6.1 (F) | 6.4 (F) | 6.2 (E) | 7.9 (A) | |||||||||||||||||||||
| 35 | Alberts et al. (1998) | 8.4 | 7.0 (S) | ||||||||||||||||||||||||
| 36 | Lundstrom et al. (1998) | 8.6 | 8.8 (H) | 7.6 (L) | 8.7 (A) | 9.1 (V) | |||||||||||||||||||||
| 37 | Lundstrom et al. (1998) | 8.3 | 8.9 (H) | 8.4 (L) | 8.6 (A) | 9.3 (V) | |||||||||||||||||||||
| 35 | Lundstrom et al. (1998) | 7.2 | 7.1 (H) | 6.0 (L) | 7.1 (A) | 7.9 (V) | |||||||||||||||||||||
| 41 | Lundstrom et al. (1998) | 8.6 | 9.1 (H) | 7.9 (L) | 9.1 (A) | 9.4 (V) | |||||||||||||||||||||
| 35 | Sartania and Strange (1999) | 7.4 | 5.7 (A) | 7.1 (A) | 7.5 (A) | ||||||||||||||||||||||
| Muscarinic M2 receptor | S | Y | Y | V | W | D | Y | S | N | V | W | E | C | Y | T | T | A | A | W | Y | N | V | P | Y | C | ||
| 44 | Gregory et al. (2010) | 10.0 | 9.6 (A) | 9.6 (A) | 8.4 (A) | 9.7 (A) | 10.0 (A) | 9.49 (A) | |||||||||||||||||||
| 42 | Gregory et al. (2010) | 5.1 | 4.9 (A) | 4.5 (A) | 2.5 (A) | 3.8 (A) | 5.0 (A) | 3.06 (A) | |||||||||||||||||||
| 49 | Gregory et al. (2010) | 5.7 | 7.1 (A) | 8.8 (A) | 5.2 (A) | 5.7 (A) | 6.2 (A) | 6.27 (A) | |||||||||||||||||||
| 47 | Gregory et al. (2010) | 6.2 | 5.9 (A) | 6.3 (A) | 5.8 (A) | 5.7 (A) | 6.2 (A) | 5.85 (A) | |||||||||||||||||||
| 45 | Heitz et al. (1999) | 9.6 | 9.6 (A) | N.D. (A) | N.D. (A) | 9.1 (A) | 9.3 (A) | N.D. (A) | N.D. (A) | N.D. (A) | |||||||||||||||||
| 44 | Heitz et al. (1999) | 9.7 | 9.4 (A) | N.D. (A) | 8.5 (A) | 9.0 (A) | 9.8 (A) | 9.1 (A) | N.D. (A) | N.D. (A) | |||||||||||||||||
| 42 | Hulme et al. (1995) | 5.6 | 3.6 (E) | ||||||||||||||||||||||||
| Muscarinic M3 receptor | S | F | Y | A | W | D | Y | S | N | V | W | E | C | F | T | T | A | A | W | Y | N | V | P | Y | C | ||
| 45 | Schmidt et al. (2003) c | 9.3 | 10.0 (A) | 8.5 (T) | 9.6 (I) | ||||||||||||||||||||||
| 42 | Wess et al. (1993) c | 6.1 | 4.8 (F) | ||||||||||||||||||||||||
| 45 | Wess et al. (1991) c | 10.3 | 9.3 (A) | 9.9 (F) | 10.4 (A) | 10.3 (A) | 10.1 (F) | 9.7 (F) | |||||||||||||||||||
| 42 | Wess et al. (1991) c | 5.8 | 5.4 (A) | 4.7 (F) | 4.7 (A) | 4.2 (A) | 4.1 (F) | 4.4 (F) | |||||||||||||||||||
| 42 | Bluml et al. (1994) c | 5.7 | 5.0 (A) | ||||||||||||||||||||||||
| 43 | Bluml et al. (1994) c | 10.5 | 9.5 (D) | ||||||||||||||||||||||||
| 46 | Bluml et al. (1994) c | 11.0 | 6.5 (D) | ||||||||||||||||||||||||
| β2 adrenoceptor | V | G | H | W | W | D | V | V | T | I | S | C | C | D | A | S | S | S | W | F | F | N | R | N | G | ||
| 7 | Dohlman et al. (1990) | 9.9 | 8.5 (V) | 8.5 (V) | |||||||||||||||||||||||
| 50 | Dohlman et al. (1990) | 8.4 | 6.8 (V) | 6.2 (V) | |||||||||||||||||||||||
| 52 | Dohlman et al. (1990) | 8.2 | 6.9 (V) | 6.9 (V) | |||||||||||||||||||||||
| 53 | Dohlman et al. (1990) | 9.4 | 7.3 (V) | 7.2 (V) | |||||||||||||||||||||||
| 7 | Fraser (1989) | 8.0 | 7.2 (S) | 5.2 (S) | |||||||||||||||||||||||
| 50 | Fraser (1989) | 6.7 | 4.7 (S) | 4.3 (S) | |||||||||||||||||||||||
| 56 | Fraser (1989) | 9.7 | 7.7 (S) | 6.2 (S) | |||||||||||||||||||||||
| 55 | Fraser (1989) | 7.7 | 5.5 (S) | 5.0 (S) | |||||||||||||||||||||||
| 53 | Liapakis et al. (2000) | 6.9 | 6.5 (T) | 5.4 (A) | |||||||||||||||||||||||
| 7 | Suryanarayana and Kobilka (1993) | 6.3 | 4.2 (T) | ||||||||||||||||||||||||
| 54 | Suryanarayana and Kobilka (1993) | 5.7 | 4.0 (T) | ||||||||||||||||||||||||
| 51 | Ambrosio et al. (2000) | 6.7 | 5.3 (A) | 5.5 (A) | |||||||||||||||||||||||
| 7 | Wieland et al. (1996) | 6.6 | 6.7 (A) | 5.6 (L) | |||||||||||||||||||||||
| 50 | Wieland et al. (1996) | 5.0 | 5.1 (A) | 4.8 (L) | |||||||||||||||||||||||
| 54 | Wieland et al. (1996) | 6.8 | 5.2 (L) |
The annotated binding affinities (pIC50/pKi) for the mutants are followed by their respective mutation in brackets. Human (unless otherwise specified) SDM binding affinity data was collected from GPCRdb (Vroling et al., 2011) and an exhaustive literature search. The complete overview is available in the supporting information (Table S1) from which a subselection of single-point mutations within the predefined binding pocket is presented here.
SDM study on guinea pig H1R.
Only functional SDM data available for histamine (8) in H2R (Gantz et al., 1992).
SDM study on rat M3R.
Conserved bioaminergic GPCR ligand binding site
While the crystal structures of aminergic, adenosine, chemokine, lipid, opioid, opsin and peptide GPCR subfamilies show differences in helical bends [e.g. TM2 in CXCR4 (Wu et al., 2010)] and relative orientations of helices [e.g. TM5 in β2AR (Katritch and Abagyan, 2011)], the overall protein fold around the TM binding pocket is well conserved, in particular when comparing GPCRs of the same family (Figure 1A) (Katritch et al., 2012). Generally, two subpockets in which ligands can bind are defined within the GPCR TM bundle, i.e. a minor pocket consisting of TMs 1, 2, 3, and 7 and a major pocket consisting of TMs 3, 4, 5, and 6 (Figure 1B) (Surgand et al., 2006).
Throughout this manuscript, we use the Ballesteros-Weinstein residue numbering scheme (Ballesteros and Weinstein, 1995) that is based on the presence of several highly conserved residues among class A GPCRs: N1.50 in TM 1, D2.50 in TM2, R3.50 in TM3, W4.50 in TM4, P5.50 in TM5, P6.50 in TM6 and P7.50 in TM7. D3.32, for example, is part of TM3 and is located 18 residues before the highly conserved R3.50 (Supporting Information Figures S1 and S3). We have defined the ligand binding pocket in the TM domain of aminergic GPCRs by considering 54 positions: 30 residues defined by Surgand et al. (2006) based on the bovine rhodopsin crystal structure and 24 additional residues that are accessible from the ligand binding pocket in six aminergic GPCR crystal structures (Figure 1C) and have been investigated in site-directed mutagenesis studies (Table 1, Supporting Information Table S1).
GPCRs have been divided in seven classes (Kolakowski, 1994) (class A-F and O), five families (Fredriksson et al., 2003) (rhodopsin, glutamate, secretin, adhesion and frizzled/taste), and many subfamilies (Surgand et al., 2006). Up to now crystal structures have been published for 15 different class A GPCRs, including six GPCRs targeted by biogenic amines (i.e. aminergic GPCRs): human histamine H1 receptor, human dopamine D3 receptor, human muscarinic M2 receptor, rat muscarinic M3 receptor, turkey β1 adrenoceptor and human β2 adrenoceptor (Jacobson and Costanzi, 2012; Katritch et al., 2012). These GPCRs are abbreviated to H1R, D3R, M2R, M3R, β1AR and β2AR, respectively and will be used throughout this paper (Alexander et al., 2011). For β1AR and β2AR multiple crystal structures with different ligand types are available [i.e. antagonists, inverse agonist and (partial/full/biased) agonist bound], whereas for the other aminergic GPCRs only one crystal structure bound to an inverse agonist or antagonist has been solved (Jacobson and Costanzi, 2012; Katritch et al., 2012). An overview of all aminergic GPCR crystal structures can be found in Supporting Information Table S2.
The conserved fold of GPCRs has enabled the construction of protein homology models including histamine receptor models (Wieland et al., 1999; Kelley et al., 2001; Jongejan et al., 2005; 2008; Schlegel et al., 2007; Jojart et al., 2008; Kiss et al., 2008b; Igel et al., 2009; Strasser et al., 2009; Lim et al., 2010; Werner et al., 2010; Istyastono et al., 2011b; Schultes et al., 2012; Sirci et al., 2012; Seifert et al., 2013) to predict GPCR-ligand interactions (de Graaf and Rognan, 2009; Kooistra et al., 2013). GPCR homology models have furthermore been successfully used to discover new ligands by structure-based virtual screening (de Graaf and Rognan, 2009; Kooistra et al., 2013), as demonstrated for H3R (Schlegel et al., 2007; Sirci et al., 2012) and H4R (Kiss et al., 2008a; Istyastono, 2012). Most aminergic GPCR models so far have been constructed based on the bovine rhodopsin (Palczewski et al., 2000) or β2AR crystal structure (Figure 2E) (Cherezov et al., 2007), but the recently solved GPCR crystal structures now in principle offer more (closely related) templates to construct higher resolution homology models (Istyastono et al., 2011b; Schultes et al., 2012). For example, H4R models based on the H1R crystal structure could better explain the fact that 2-aminopyrimidines (33 and 34) can accommodate larger substituents than indolecarboxamides (32 and 31) (Figure 4), than H4R homology models based on the β2AR crystal structure template (Schultes et al., 2012). However, β2AR- and H1R-based H4R homology models performed comparable in retrospective virtual screening studies (Istyastono, 2012), and in recent prospective virtual screening runs against the D3R crystal structure and a D3R homology model similarly high numbers of novel ligands were discovered (Carlsson et al., 2011). The recent GPCR crystal structures have nevertheless opened up new opportunities for structure-based virtual screenings studies to identify (more fragment-like) ligands with higher hit rates, as illustrated for the aminergic H1R (de Graaf et al., 2011), β2AR (Kolb et al., 2009), and D3R (Carlsson et al., 2011) receptors. The H1R crystal structure was for example used in combination with a customized virtual screening approach for the identification of novel fragment-like compounds (including compound 13, see Figure 4B) (de Graaf et al., 2011).
Figure 2.
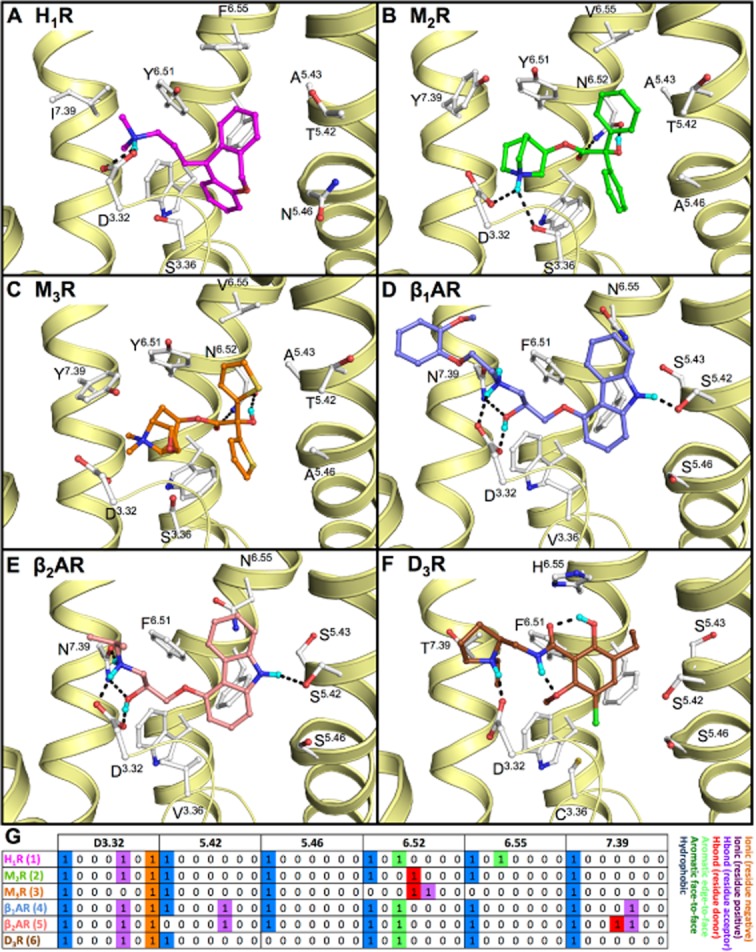
Binding mode of (A) doxepin (1, magenta carbon atoms) in human H1R (PDB code 3RZE (Shimamura et al., 2011)), (B) (R)-3-quinuclidinylbenzilate (2, green carbon atoms) in human M2R (PDB code 3UON (Haga et al., 2012)), (C) tiotropium (3, orange carbon atoms) in rat M3R (PDB code 4DAJ (Kruse et al., 2012)), (D) (S)-carvedilol (4, blue carbon atoms) in turkey β1AR (PDB code 4AMJ (Warne et al., 2012)), (E) (S)-carazolol (5, red carbon atoms) in human β2AR (PDB code 2RH1 (Cherezov et al., 2007)) and (F) (S)-eticlopride [(6, brown carbon atoms in D3R (PDB code 3PBL (Chien et al., 2010)]. The yellow ribbons represent parts of the backbone of transmembrane (TM) helices 2, 3, 5, 6 and 7. Selected binding site residues are depicted as ball-and-sticks with light grey carbon atoms. Oxygen, nitrogen, sulphur, hydrogen and chlorine atoms are coloured red, blue, yellow, cyan and green, respectively. Hydrogen bonds are depicted by black dashes. Polar hydrogen atoms of the ligand are shown, but are omitted for the pocket residues. The labels for W6.48 are omitted for all structures as well as F6.52 for H1R, β1AR, β2AR and D3R for clarity purposes. (G) Molecular interaction fingerprint (IFP) (Marcou and Rognan, 2007) bitstrings describing the binding poses of 1–6 (A–F), encoding different interaction types (negatively charged, positively charged, H-bond acceptor, H-bond donor, aromatic face-to-edge, aromatic-face-to-face and hydrophobic) for each residue in the binding site. For reasons of clarity, only the bit strings of residues D3.32, 5.42, 5.46, 6.52, 6.55, and 7.39 are shown. All binding modes are presented in a similar fashion throughout the manuscript. 2D structures of the molecules are presented in Figure 3A.
Figure 4.
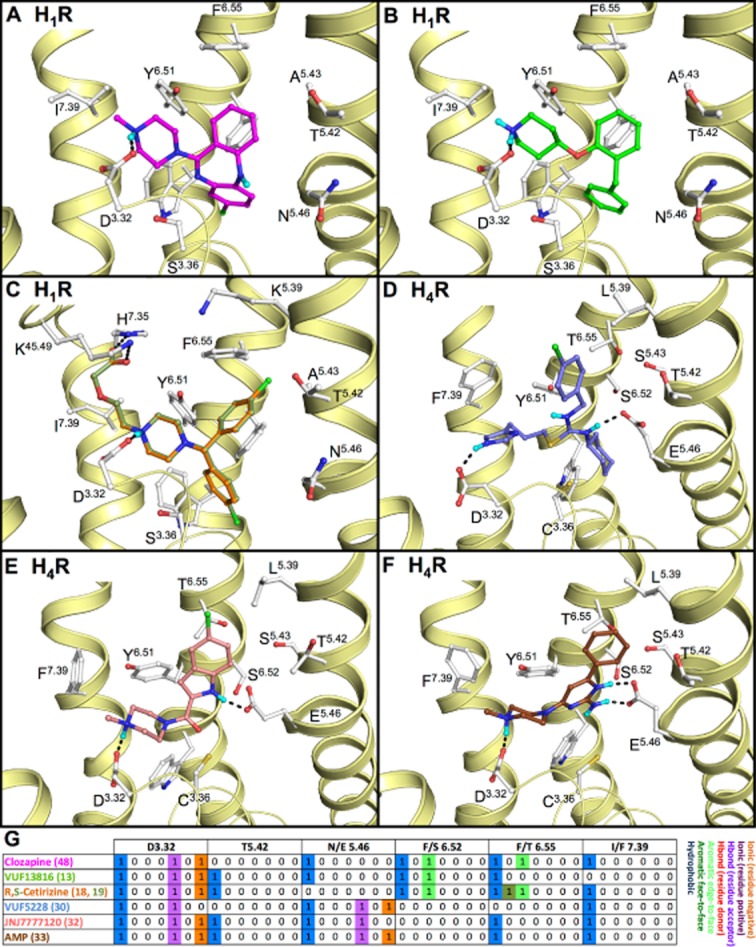
Proposed binding modes, based on SAR and SDM studies, of (A) clozapine (48) (magenta carbon atoms) in human H1R, (B) SBVS hit VUF13816 (13) (de Graaf et al., 2011) (green carbon atoms) in human H1R, (C) R-cetirizine (18) (green carbon atoms) and S-cetirizine (19) (orange carbon atoms) in human H1R, (D) VUF5228 (30) (blue carbon atoms) in a human H4R homology model (Istyastono et al., 2011b), (E) JNJ7777120 (32) (red carbon atoms) and (F) aminopyrimidine 33 (brown carbon atoms) (Schultes et al., 2012). Rendering and colour-coding are the same as in Figure 2. H4R homology models were build using the H1R (E, F) and β2AR (D) crystal structures as templates. (G) Molecular interaction fingerprint (IFP) bit strings describing the binding poses of 48, 13, 18, 19, 30, 32, 33 (A–F), encoding different interaction types with D3.32, 5.42, 5.46, 6.52, 6.55 and 7.39 (colour-coding as described in Figure 2). 2D structures of the molecules are presented in Figure 3B.
Structural chemogenomics analyses of (hist)aminergic ligand binding sites
Over several decades mutagenesis studies have been extensively performed on GPCR targets in order to identify residues that are important for ligand binding (Shi and Javitch, 2002; Surgand et al., 2006; Vroling et al., 2011). Arguably, mutagenesis studies were needed to compensate for the lack of useful GPCR crystal structures. With the emerging GPCR crystal structures, mutation data should be revisited and evaluated in the new context of structural biology. Binding affinities from mutation studies of the TM binding pocket (Figure 1C) of histamine receptors (i.e. H1R, H2R, H3R and H4R) and crystallized aminergic GPCRs (i.e. D3R, M2R, M3R, β1AR and β2AR) are reported in Table 1 and Supporting Information Table S1. It must be noted that mutations can not only have an effect on ligand binding, but can also have an effect on the structure of the binding site/protein. Supporting Information Table S1 contains 1420 reported single point mutations for 128 individual amino acid positions in the biogenic amine GPCRs (Supporting Information Figure S3). Most of the data (47%) correspond to residues located in the major pocket (between TM3, TM4, TM5 and TM6), while only 23 and 7% of the data are associated with residues located in the minor pocket (between TM1, TM2, TM3 and TM7) and in the second extracellular loop (EL2) respectively. The conserved residue D3.32, the main interaction anchor of aminergic GPCRs, has been studied in 104 mutation studies. TM5 and TM6 have also been frequently studied (306 and 325 data points, respectively), in particular positions 5.39 (53), 5.42 (92), 5.46 (62), 6.34 (115), 6.52 (51) and 6.55 (41). Based on ligand binding pockets in the aminergic GPCR crystal structures and the analysis of the available aminergic receptor mutation data (Table 1, Supporting Information Table S1) and SAR, four important ligand interaction hot spots in aminergic GPCRs will be discussed that systematically cover the different regions of the GPCR ligand binding site: (i) the conserved ionic interaction anchor D3.32; (ii) the aromatic cluster in TM6; (iii) functional selectivity via TM5; (iv) allosteric contacts with the minor pocket and extracellular loops. We illustrate how systematic mining of aminergic GPCR-ligand interaction space can give insights into how conserved and selective aminergic GPCR interaction hot spots in different regions of the receptor binding site can accommodate different chemical scaffolds observed in aminergic GPCR ligands and histamine receptor ligands in particular.
The conserved ionic interaction anchor D3.32
The negatively charged and conserved aspartate residue in TM3 (D3.32) of aminergic GPCRs is generally proposed as a key anchor for the basic moieties of aminergic ligands (Shi and Javitch, 2002; Surgand et al., 2006). The aminergic GPCR crystal structures (Figure 2) show subtle differences in the binding sites and ligand binding modes around this conserved D3.32 residue. Structural chemogenomics analyses can help to explain the ligand and receptor dependent effects of mutation of D3.32 and rationalize structure-affinity relationships of basic amine groups in aminergic GPCR ligands.
Mutation of D3.32 in aminergic receptors, including histamine receptors (Gantz et al., 1992; Ohta et al., 1994; Nonaka et al., 1998; Shin et al., 2002; Bruysters et al., 2004; Jongejan et al., 2008), often leads to a significant decrease in ligand binding affinity (Table 1, Supporting Information Table S1). This is in line with the currently available crystal structures of aminergic GPCRs (H1R, M2R, β1AR, β2AR, D3R) in which co-crystallized ligands (Figure 2) make an ionic interaction as well as a hydrogen bond with this residue. The quaternary amine moiety of tiotropium (3) in M3R cannot form a hydrogen bond (Figure 2C), but is nevertheless within ionic interaction distance (4.3 Å) from the carboxylate group of D1483.32. The experimentally supported ionic/hydrogen bond interaction with D3.32 has been used to (i) guide the in silico prediction of ligand binding modes in (hist)aminergic receptor crystal structures and homology models [e.g. Figures 4 and 5 (Istyastono et al., 2011b; Schultes et al., 2012)]; and (ii) to select docking poses in structure-based virtual screening studies to identify new (hist)aminergic receptor ligands (e.g. H1R ligand VUF13816, 13, Figure 4B) (de Graaf et al., 2011). Although the orientation of D3.32 changes only slightly between crystal structures, it should be noted that the residues around the D3.32 vary between aminergic GPCRs, creating distinct microenvironments in this area of the binding site. Such microenvironment can still be a subtle but important determinant of ligand selectivity. For example, the large aromatic Y7.39 residue in M2R and M3R covers a deep hydrophobic binding pocket that fits the quinuclidine and N-methylscopine amine moieties in 2 (Figure 2B) and 3 (Figure 2C) respectively. This ligand binding mode is further accommodated by an alternative rotamer conformation of W6.48 (that is not observed in the other aminergic GPCR crystal structures) that creates more space for these relatively bulky and rigid ring systems (Figure 2). Quinuclidine is indeed identified as a common substructure in muscarinic receptor specific ligands (van der Horst et al., 2009). The ethanolamine group in (S)-carvedilol (4, Figure 2D) and (S)-carazolol (5, Figure 2E) is a common substructure in (selective) beta-adrenergic receptor ligands (van der Horst et al., 2009) that can form a tight hydrogen bond network with D3.32 and the medium sized polar N7.39 residue in β1AR and β2AR. N7.39 is a beta-adrenergic receptor-specific and stereoselective (Rosenbaum et al., 2007; Katritch et al., 2009; Seifert and Dove, 2009) recognition site for ethanolamines (e.g. the affinity of R-isoproterenol, 7, is 40 fold higher than S-isoproterenol, 50, see Table 1) and therefore an attractive feature to include in protein-based pharmacophore screening studies (Sanders et al., 2011; 2012). Interestingly, while the D3R mimicking N3227.39T mutation in β2AR diminishes ligand binding (Suryanarayana and Kobilka, 1993), the beta-adrenergic receptor mimicking F3697.39N mutation in the alpha-2-adrenergic receptor promotes stronger binding for aryloxyalkylamine ligands such as S-alprenolol (58), pindolol (57) and R-propranolol (55) (Suryanarayana et al., 1991). All these data indicate the important role of this residue in aminergic subtype selectivity (Ericksen et al., 2012). Most H1R ligands contain linear alkylamines [like doxepin (1), Figure 2A] or cyclic amines such as piperazines and piperidines (Simons and Simons, 2011). Most high affinity H3R ligands (Celanire et al., 2005), as well as compounds that have so far been tested in clinical trials contain cyclic amines including pyrrolidines and (homo)piperidines (Kuhne et al., 2011). Many H4R receptor ligands contain a N-methylpiperazine moiety, but also other basic cyclic amines are an alternative to N-methylpiperazines including azetidines, aminopyrrolidines and piperazines (Engelhardt et al., 2009; Smits et al., 2009; Istyastono et al., 2011a). While H1R SAR studies show a preference for tertiary amines over secondary and primary amines (Shah et al., 2009), N-methylation can be used as a subtle chemical switch to modulate H3R affinity (Smits et al., 2012) and H3R/H4R selectivity (Lim et al., 2005; Govoni et al., 2006). Changing piperazine to a N-methylpiperazine (Smits et al., 2012) increases H3R affinity while maintaining similar affinity for H4R, but changing immepip (23) into methimepip (24) (Lim et al., 2005), or changing imbutamine (25) to N, N-dimethylimbutamine (26) (Govoni et al., 2006) increases H3R selectivity over H4R.
Figure 5.
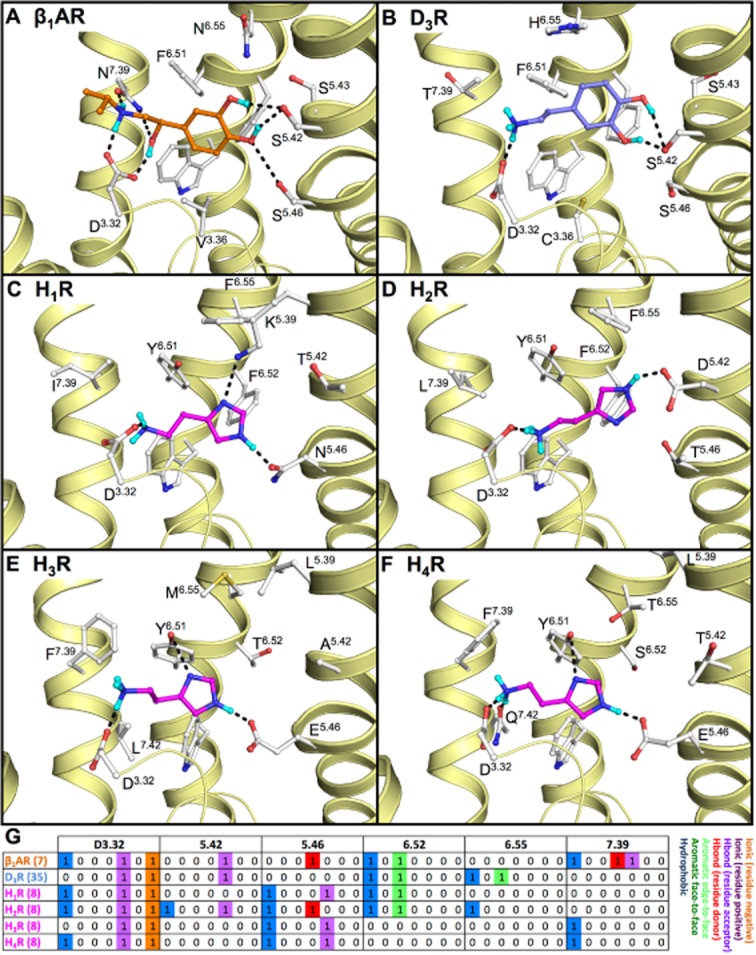
Binding mode of (A) isoproterenol (7, orange carbon atoms) in the turkey β1AR (PDB code 2Y03 (Warne et al., 2012)). Proposed binding modes of endogenous agonists, based on SDM studies, of (B) dopamine (35, blue carbon atoms) in the human D3R and the proposed binding modes of histamine (8, magenta carbon atoms) in the different human histamine receptors; (C) H1R, (D) H2R, (E) H3R and (F) H4R. Minor changes in the H1R crystal structure (i.e. rotation of K1915.39 and N1985.46) allow for accommodation of histamine. The H2R, H3R and H4R models are based on the H1R crystal structure. Rendering and colour-coding are the same as in Figure 2. (G) Molecular interaction fingerprint (IFP) bit strings describing the binding poses of 7–8 and 35 (A–F), encoding different interaction types with D3.32, 5.42, 5.46, 6.52, 6.55 and 7.39 (colour-coding as described in Figure 2). Two-dimensional structures of the molecules are presented in Figure 3A and B.
This preference for specific amine moieties might be explained by differences between the histamine receptor binding sites in amino acid composition in the region close to D3.32, including positions 7.39 (I in H1R, F in H3R and H4R) and 7.42 (G in H1R, L in H3R, and Q in H4R). In silico guided mutagenesis studies have indeed identified Q/L7.42 as one of the molecular determinants of H3R over H4R selectivity (Istyastono et al., 2011b). Residues at position 7.39 however, have so far not yet been subjected to mutagenesis studies in histamine receptors (Table 1 and Supporting Information Table S1).
It should finally be noted that negatively charged residues like D3.32 also play an important role in binding the basic amine moieties of small molecule ligands in other GPCR subfamilies (Surgand et al., 2006), as demonstrated by the crystal structures of delta (Granier et al., 2012), kappa (Wu et al., 2012), and mu (Manglik et al., 2012)-opioid receptors (i.e. D3.32) and the chemokine receptor CXCR4 (Wu et al., 2010) (i.e. D972.63 and E2887.39). As a result, several positively ionized privileged structures, like orthoalkoxy-N-phenylpiperazines (Rognan, 2007), are known to bind both aminergic and opioid receptors, most probably via ionic interactions with the shared D3.32 residue. This is emphasized once more by ligand overlap between aminergic and opioid receptors, e.g. H1R and the μ/κ/δ-opioid receptors share multiple ligands (32%, 29% and 25% of 57, 42, 32 tested ligands respectively). Fluphenazine (62) and Chlorpromazine (63) for example have affinity for these three opioid receptors and they were also found to have affinity for 22 out of 26 and 19 out of 24 tested aminergic receptors respectively. A similar pattern was obtained by (Poulain et al., 2001) who screened a panel of 47 compounds against multiple aminergic as well as opioid receptors and showed that several ligands, including ligand 64, bind to the NOP receptor (nociceptin), μ/κ/δ-opioid receptors, H2R, β1AR, M2R, D2R and D3R amongst others.
From all these combined data and chemogenomic analyses, it can be concluded that D3.32 is a key interaction partner in (hist)aminergic receptors, but that subtle differences in the binding site around this residue can in principle be used to design ligands with specific selectivity profiles, as demonstrated by position F/N7.39 in alpha and beta-adrenergic receptors (vide supra).
The aromatic cluster in TM6
Aminergic GPCRs contain a conserved cluster of aromatic residues in TM6: W6.48, F/Y6.51 and F6.52 (Figures 1C and 2) that play an important role in the binding of agonists and antagonists to aminergic receptors (Table 1, Table Supporting Information and Figure S5), including histamine receptors (Wieland et al., 1999; Shin et al., 2002; Bruysters et al., 2004). The new GPCR crystal structures, however, show that the binding modes of aromatic ring systems of aminergic GPCR ligands in this conserved aromatic region is ligand dependent and chemogenomics analyses suggest that small differences in this region can determine receptor selectivity.
Mutation studies of W6.48, F/Y6.51 and F6.52 have been used to guide the construction of three-dimensional aminergic receptor-ligand models, including histamine receptor models (Wieland et al., 1999; Jongejan et al., 2005; 2008; Schlegel et al., 2007; Kiss et al., 2008b; Strasser et al., 2009; Lim et al., 2010; Istyastono et al., 2011b; Schultes et al., 2012; Sirci et al., 2012), that propose essential apolar interactions between this aromatic cluster and the different aromatic ring systems in the ligands (e.g. Figures 5). These binding mode hypotheses are confirmed by all currently available crystal structures of aminergic GPCRs in which ligands indeed make aromatic stacking and/or hydrophobic interactions with this aromatic cluster (Figure 2). In addition to the conserved D3.32 ionic link, this conserved aromatic and hydrophobic pocket contributes to the low selectivity of many aminergic ligands (Figures 6C and 7), including the atypical antipsychotic drug clozapine (48, Figure 4A) which has considerable affinity for many aminergic receptors (Selent et al., 2008), including H1R, H4R, D3R, M2R, M3R and to a less extent for β2AR (Bolden et al., 1992; Schotte et al., 1996; Lim et al., 2005). Nevertheless, several subtle and local differences in the pharmacophoric properties and shape of this hydrophobic pocket in aminergic receptors can be used to explain and obtain receptor selectivity for specific (hetero)aromatic ligand scaffolds. For example, most high affinity H1R ligands contain (tricyclic) hydrophobic aromatic systems that are proposed to form multiple complementary stacking interactions with W4286.48, F4326.52 and F4356.55, as demonstrated for doxepin (1) in the H1R crystal structure (Shimamura et al., 2011) and predicted for levocetirizine (18) (Figure 4C). Mutation of these aromatic residues in TM6 of H1R diminish histamine (8) and [3H]mepyramine (14) binding (Table 1, Supporting Information Table S1) (Wieland et al., 1999; Bruysters et al., 2004). Several high affinity H4R ligands on the other hand contain heteroaromatic systems including indoles (e.g. JNJ7777120 (32), Figure 4E, Table 1) and aminopyrimidines (e.g. compound 33, Figure 4F, Table 1) that are complementary to the larger and more polar binding pocket between S6.52 and T6.55 and form hydrogen bonds and/or ionic interactions with the H3R/H4R specific E5.46 residue, as demonstrated by in silico guided mutagenesis studies (Jongejan et al., 2008; Schultes et al., 2012). Doxepin (1) adopts a butterfly-shape in the H1R, and interacts extensively with W4286.48 in the lower binding pocket (Figure 2A) (Shimamura et al., 2011). Similarly, clozapine (48), the SBVS hit VUF13816 (13) (de Graaf et al., 2011) and R- (18) and S-cetirizine (19) are proposed to all adopt this butterfly conformation in the H1R binding site (Figure 4A–C). While most aminergic receptors contain a phenylalanine at position 6.52 (74%), M2R and M3R do not contain a phenylalanine at position 6.52 as the other receptors, but a polar (and less bulky) asparagine residue. This difference in local amino acid composition results in a clear change in ligand binding mode: (R)-3-quinuclidindyl-benzilate (2) and tiotropium (3) adopt an inverse butterfly-shape in M2R and M3R crystal structures respectively (Figure 2B,C) (Haga et al., 2012; Kruse et al., 2012), compared to doxepin (1) in the H1R co-crystal structure (Figure 2A) (Shimamura et al., 2011), and probably also other tricyclic aromatic ligands in aminergic receptors that contain F6.52 (Supporting Information Figure S2). Mutation studies demonstrate the essential role of this asparagine residue in binding (-)scopolamine (46), the N4046.52D mutant has a 28 000-fold lower affinity than wild-type M3R) (Bluml et al., 1994). Mutation studies suggest that the aromatic rings of the endogenous agonists histamine (8) and dopamine (35) interact with F4356.55 (H1R, Figure 5C) (Wieland et al., 1999; Bruysters et al., 2004) and H6.55 (D3R, Figure 5A) (Lundstrom et al., 1998) respectively.
Figure 6.

(A) The percentage (%) of the pairwise sequence identity between the ligand binding pockets (i.e. the selected 55 residues) of the histamine receptors as well as the aminergic receptors with a crystal structure available. The percentage (%) of the pairwise sequence identity for the TM helices is described in Supporting Information Table S3. (B) The average ligand similarity as calculated by EDprints (Kooistra et al., 2010) by comparing ligands from the ChEMBL database for each of the histamine receptors as well as the aminergic receptors with a crystal structure available (the scores are based on the average of the highest similarity scores). (C) The ligand overlap by comparing 9903 ligands from the ChEMBL database with annotated affinity for one or more of the discussed aminergic receptors (expressed as a percentage of the total number of ligands with experimentally determined binding affinity, i.e. if the experimentally determined radioligand displacement Ki or IC50 value is 10 μM or lower, for one or both receptors). (D) The structural distance between the ligand binding pockets as calculated by SiteAlign (Schalon et al., 2008) (i.e. the selected 54 residues using distance-3) for the crystallized aminergic GPCRs. The gradient from blue to white to red indicates a low to high similarity of sequences (A), similarity of ligands (B), ligand overlap (C) and similarity of structures (D) respectively. One has to be warned for the values with a grey colour (B, C) as this indicates a low number of ligands available for this analysis (n < 45) therefore they might be misleading.
Figure 7.

Examples of ligand overlap within histamine receptor subtypes (H1R/H3R/H4R) as observed in the ChEMBL database (Gaulton et al., 2012) and the VU-MedChem fragment library (de Graaf et al., 2013). Compounds with annotated affinities (Ki/IC50) and a confidence factor of 8 or higher for H1R, H3R or H4R were retrieved from the ChEMBL database and considered as ligands for a receptor if the affinity for that receptor was ≤10 μM. The median affinity was used when multiple values were reported. Two-dimensional structures of some subtype selective as well as subtype unselective histamine ligands are presented.
The rotameric state of W6.48, a conserved residue in aminergic receptors and most other GPCRs [e.g. A2AR (Jaakola et al., 2008) and CB1R (Singh et al., 2002)], has been postulated to be associated with the activation state of GPCRs via a ‘rotamer toggle switch’ (Schwartz et al., 2006; Holst et al., 2010), like proposed for H1R (Jongejan et al., 2005; Sansuk et al., 2011), H4R (Jongejan et al., 2008) and β2AR (Shi et al., 2002). The W6.48 side chain shows relatively small conformational changes; however, when comparing the currently available agonist and antagonist bound GPCR crystal structures. Interestingly, W6.48 adopts a different conformation (oriented more perpendicular to TM6) in the inactive antagonist bound M2R and M3R crystal structures compared to all other crystal structures (Figure 2B,C) (Katritch et al., 2012). The χ2 angle of W6.48 rotates 60 degrees (compared to W6.48 in H1R) while the χ1 remains similar, which is compatible with the bulky quinuclidine and N-methylscopine amine groups and the ‘inverted’ butterfly shape of the benzene/thiophene rings of the M2R/M3R ligands 2 and 3 as discussed above (Figure 2B,C). Interestingly, the chemically related clozapine (48) acts as an inverse agonist on H1R, but is a full agonist of H4R (Lim et al., 2005; Bakker et al., 2007), suggesting slightly different binding modes and receptor activation mechanisms for these histamine receptors. Taken together, this data clearly shows the importance of the aromatic cluster in (hist)aminergic receptors.
Selective binding to TM5
The sequence diversity of the binding site residues in TM5 is relatively high among aminergic receptors (Supporting Information Figure S1). Structural and chemogenomics analyses of aminergic GPCRs indicate that interactions with these residues are important determinants of ligand function and receptor selectivity. In particular, positions 5.39 (A/G/K/L/V), 5.42/5.43 (A/D/G/S/T) and 5.46 (A/E/N/S/T) have been associated with functional and receptor selectivity in aminergic receptors (Figures 1C and 2) (Shi and Javitch, 2002), as demonstrated for histamine receptors (Gantz et al., 1992; Leurs et al., 1994; Ohta et al., 1994; Moguilevsky et al., 1995; Shin et al., 2002; Uveges et al., 2002; Bruysters et al., 2004; Jongejan et al., 2008). Natural aminergic receptor agonists (e.g. histamine 8 (Figure 5C–F), dopamine 35 (Figure 5B), adrenaline 54) are proposed to interact with polar residues at positions 5.42 and/or 5.46, as shown in the β1AR (biased) agonist bound crystal structures (Figures 2D and 5A) (Warne et al., 2011; 2012) as well as the agonist (BI-167107, 60) bound β2AR crystal structure (Rasmussen et al., 2011a; 2011b), and supported by mutagenesis studies (e.g. compound 8, 35, 42 and 53 in Table 1 and Supporting Information Table S1) (Gantz et al., 1992; Wess et al., 1993; Ohta et al., 1994; Moguilevsky et al., 1995; Sartania and Strange, 1999; Liapakis et al., 2000; Gillard et al., 2002; Shin et al., 2002; Uveges et al., 2002; Bakker et al., 2004; Bruysters et al., 2004; Jongejan et al., 2008). Interestingly, mutagenesis studies suggest that similar agonists adopt aminergic receptor specific binding modes. While the catechol hydroxyl groups of beta-adrenergic receptor agonist isoproterenol (7) form hydrogen bonds with both S2285.42 and S2325.46 in the β1AR crystal structure (Figure 5A) (Warne et al., 2012), mutagenesis studies indicate that the catechol moiety of dopamine (35) interacts with S1925.42 but not with S1965.46 in D3R (Table 1) (Sartania and Strange, 1999). Mutation studies also suggest that histamine (8) has similar binding modes in H1R, H3R, and H4R by donating a hydrogen bond to N5.46 (H1R) (Leurs et al., 1994; Ohta et al., 1994; Moguilevsky et al., 1995; Bruysters et al., 2004) or E5.46 (H3R, H4R) (Shin et al., 2002; Uveges et al., 2002; Jongejan et al., 2008) with its Nτ imidazole nitrogen atom (Figures 3 and 5), but adopts a different binding orientation in H2R in which Nτ and Nπ form hydrogen bonds with D1865.42 and T1905.46 respectively (Figures 3 and 5) (Gantz et al., 1992). In H3R and H4R, the negatively charged E5.46 can furthermore form a stronger ionic/hydrogen bond with Nτ than N1985.46 in H1R, explaining the higher affinity of histamine for H3R and H4R compared to H1R. Although there is a negatively charged aspartate at position 5.42 in the H2R, it has been hypothesized that an increase in distance to the Nτ nitrogen atom might lead to the reduced affinity for this receptor compared to the H3R and H4R (Shin et al., 2002). The symmetric distributions of complementary pharmacophore features in H2R, H3R and H4R binding sites (i.e. D3.32 in TM3 and D985.42/E2065.46/E1825.46 in TM5) and histamine receptor ligands that contain two basic groups, makes binding mode prediction challenging (Lorenzi et al., 2005; Schlegel et al., 2007; Jongejan et al., 2008; Kiss et al., 2008b; Ishikawa et al., 2010; Istyastono et al., 2011b; Schultes et al., 2012). The binding modes of several H4R ligands, including isothioureas (e.g. 29, 30), indolecarboxamides (e.g. 31, 32) and aminopyrimidines (e.g. 33, 34), in H4R have been investigated by combining complementary in silico and in vitro approaches (Istyastono et al., 2011b; Schultes et al., 2012). Extensive SAR, mutagenesis, docking and MD simulation studies indicated that indolecarboxamides and aminopyrimidines form an H-bond with D943.32 via their piperazine amine moiety, while making H-bond interactions with E1825.46 with their indole and aminopyrimidine groups respectively (Figure 4D–F) (Jongejan et al., 2008; Schultes et al., 2012). A comparable ligand-steered, experimentally supported protein-modelling approach combining 3D-quantitative structure-activity relationship (QSAR), MD simulations, SAR and mutagenesis studies indicated that clobenpropit (29) can bind H4R in two distinct binding modes (forming H-bonds with D943.32 and E1825.46 with their imidazole and isothiourea groups). The addition of a cyclohexyl group to the clobenpropit isothiourea moiety, however, allows VUF5228 (30) to adopt only one specific binding mode in the H4R binding pocket, in which its imidazole interacts with D943.32 and its isothiourea group interacts with E1825.46 (Figure 4D) (Istyastono et al., 2011b). Indolecarboxamides (31 and 32) and aminopyrimidines (33 and 34) show predominantly one binding mode in which the methylpiperazine moiety forms a hydrogen bond with D943.32 (Figure 4E,F) (Schultes et al., 2012). Mutation studies based on a guinea pig H1R homology model identified K2005.39 as an important interaction site for the zwitterionic ligands acrivastine (17) and (to lesser extent) S-cetirizine (19) (Wieland et al., 1999). This H1R-specific anion-binding subpocket, consisting of the residues K17945.49, K1915.39 and H4507.35, was indeed confirmed by the H1R crystal structure in which this pocket is occupied by a phosphate ion (Shimamura et al., 2011). L1755.39 is an important interaction point for the chlorine substituents of indolecarboxamide 32 and aminopyrimidines 34 in H4R that forms a subpocket between the extracellular region of TM5 and EL2 which determines subtle differences in SAR between these two ligand classes (Schultes et al., 2012). The same L1755.39 residue furthermore is an important molecular determinant of H4R species selectivity that explains differences in binding affinities of JNJ7777120 (32) and clozapine (48) for human (L1755.39) and monkey (V1755.39) H4R orthologs (Lim et al., 2010).
Figure 3.
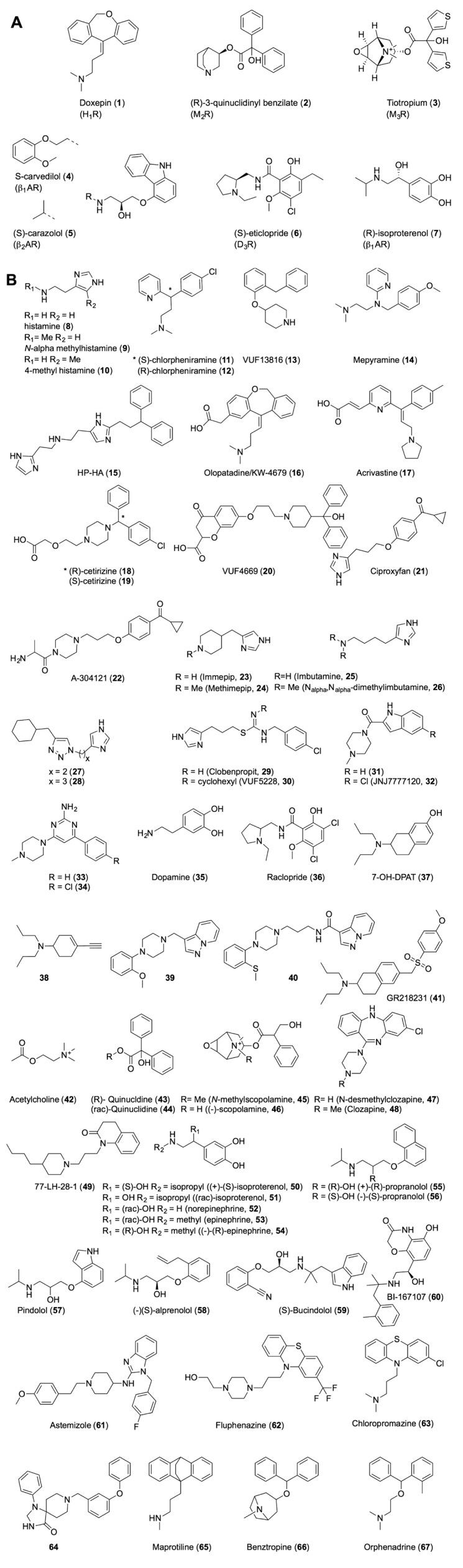
2D-structures of (A) co-crystallized ligands in the crystal structures (Figure 2) and (B) ligands shown in Table 1 or mentioned in the text. Most of the ligands in panel B are ordered according to their primary aminergic GPCR target: histamine receptors H1R (8, 11–20), H2R (8), H3R/H4R (8–10, 20–34), dopamine receptor D3R (35–41), muscarinic receptors M2R/M3R (42–49), beta-adrenergic receptors β1AR/β2AR (50–60), but it should be noted that many of the ligands bind to multiple aminergic GPCR subfamilies (e.g. 48, 61, 62, 63).
Stereoselective binding is observed for R- (18) and S-cetirizine (19) and R- (11) and S-chlorpheniramine (12), where both the R enantiomers have a higher affinity for H1R. Interestingly, the T1945.42A mutant increases binding of S-cetirizine (19), but not of R-cetirizine (18) (Table 1) (Leurs et al., 1994; Gillard et al., 2002). This stereoisomer specific mutational effect can be explained by docking studies in the H1R crystal structure which indicate that only S-cetirizine (19) is sterically hindered by T1945.42 (Figure 4C).
Although polar residues in TM5 of aminergic receptor are proposed to specifically interact with agonists, mutation studies and crystal structures indicate that also some antagonists can form hydrogen bonds interactions with some of these residues. For example, co-crystallized antagonists (S)-carvedilol (4) and S-carazolol (5) make polar interactions with TM5 (S5.42) in β1AR and β2AR respectively, but not with S5.46 (Cherezov et al., 2007; Warne et al., 2012). It is therefore the combination of interactions that make small molecules behave as either (inverse) agonists or antagonists (de Graaf and Rognan, 2008).
Allosteric interactions with the minor pocket and extracellular loops
Many GPCRs have allosteric sites and ligands binding there can alter orthosteric ligand affinity and/or efficacy, thereby offering additional opportunities for specific modulation of GPCR signalling (May et al., 2007). Currently all ligands in aminergic GPCR co-crystal structures bind primarily in the major pocket between TM3-6, like doxepin (1) in H1R (Figure 2A). Some ligands also extend towards the minor pocket between TM1-3 and TM7 (Figure 1B,C), including (S)-carvedilol (4) (Figure 2D), (S)-bucindolol (59), and BI-167 107 (60) in beta-adrenergic receptors (Rasmussen et al., 2011a,b; Warne et al., 2012). The methoxyphenoxy moiety of (S)-carvedilol (4) for example is stacked between the aromatic rings W1343.28 and W3647.40 and makes hydrophobic contacts with G1152.61, L1182.64 and V1192.65 in the minor pocket of β1AR (Figure 2D). In silico modelling studies suggest that also other large aminergic receptor ligands can extend towards the minor pocket, as for example proposed for (R)- (18) and S-cetirizine (19) (Figure 4C). Mutation studies indicate that residues in this region can determine ligand affinity and selectivity for aminergic GPCRs (Table 1), and can for example explain species selectivity of HP-HA (15) and VUF4669 (20) for guinea pig (S842.61) versus human (N842.61) H1R (Bruysters et al., 2005). Mutation studies in the dopamine D2 and D4 receptors showed that D2/D4 receptor selectivity is not determined by specific single amino acids but rather by a cluster of divergent aromatic residues in TM2, TM3 and TM7 (Simpson et al., 1999). The ELs of GPCRs covering the TM binding site (in particular EL2) show a high diversity in residue composition as well as length (Supporting Information Figure S1) (Peeters et al., 2012). The currently available GPCR crystal structures indeed confirm the high structural diversity in EL2 regions (Peeters et al., 2012; Wheatley et al., 2012). Mutagenesis studies have first of all shown that the conserved disulphide bridge between C3.25 in TM3 and C45.50 in EL2 is essential to maintain a proper fold of this loop over the GPCR ligand binding site (Shi and Javitch, 2002; de Graaf et al., 2008; Peeters et al., 2011). The crystal structure of β2AR for example explains that only mutation of C19145.50 but also mutation of C19045.49 causes a significant decrease in ligand binding affinity (Fraser, 1989; Dohlman et al., 1990) as the latter residue forms another disulphide bridge with C18445.43 that stabilizes a short helix in EL2 (Cherezov et al., 2007). Mutation studies have furthermore identified several residues (especially in the region around C45.50) that are of great importance for ligand binding to aminergic GPCRs (including M1R, M2R, M3R, α1AAR, β2AR, D2R, 5-HT1D and H4R, Table 1 and Supporting Information Table S1) ((Lim et al., 2008; de Graaf et al., 2008) and references therein). For example, systematic mutation of 10 residues of the EL2 of D2R indicated several residues that were involved in ligand binding (Shi and Javitch, 2004), and this was later confirmed by the crystal structure of the highly homologous D3R (Chien et al., 2010). Residues of EL2 in M2R (Huang et al., 2005) and M3R (Krejci and Tucek, 2001) are also involved in ligand binding. Furthermore, based on the recently solved H1R crystal structure it can be hypothesized that the positively charged and H1R-specific lysine residue at position 45.49 might be involved in binding of second-generation antihistamines such as R- (18) and S-cetirizine (19) (vide supra) (Figure 4C).
Despite the apparent role of the minor pocket and extracellular loops in aminergic receptor ligand recognition (de Graaf et al., 2008; Shi and Javitch, 2002), it should be noted that the IT1t bound CXCR4 crystal structure (Wu et al., 2010) is currently the only GPCR crystal structure that shows a ligand bound solely in the minor pocket. Furthermore, there are several co-crystal structures solved of (the N-terminal) extracellular domains of (especially class B and class C) GPCRs bound to polypeptide ligands, but so far no GPCR crystal structure has been solved in which a small molecule is bound to only the extracellular loop region. Mutation studies indicate that the minor pockets of certain GPCR subfamilies [including chemokine receptors (Scholten et al., 2012) and prostanoid receptors (Kedzie et al., 1998)] can indeed be specifically targeted by small molecules that do not bind the major pocket. The minor binding site and/or extracellular loop regions is therefore an interesting site to allosterically modulate aminergic receptors. Only few small molecule allosteric modulators of aminergic receptors have been reported (e.g. for muscarinic receptors (Gregory et al., 2010; Mohr et al., 2010), dopamine receptors (Hoare et al., 2000), alpha-adrenergic receptors (Leppik et al., 1998), and beta-adrenergic receptors (Steinfeld et al., 2011), and serotonin receptors (Im et al., 2003; Mohr et al., 2010). M2R is so far the only aminergic GPCR for which the extracellular allosteric binding site has been systematically mapped (located between TM2 (Y802.61), EL2 (E17245.46, D17345.47, E17545.49, Y17745.51) and TM7/EL3 (N4197.32, T4237.36) (Gregory et al., 2010). This has facilitated the design of dualsteric ligands (Mohr et al., 2010); compounds that bind simultaneously to both the orthosteric and allosteric binding pockets. Dualsteric ligands for, for example, H1R (Bruysters et al., 2005), H2R (Birnkammer et al., 2012) and H1R/H3R (Procopiou et al., 2011) have been described, but to the best of our knowledge true allosteric modulators of histamine receptors (that do not bind the orthosteric site) have so far not been identified. The emerging GPCR crystal structures now offer new opportunities to further explore allosteric binding sites in aminergic receptors, and to try to target these pockets with structure-based ligand discovery and design approaches. In particular fragment-based ligand design approaches, including growing, merging and linking of fragments can be an attractive strategy to target different binding sites (Smits et al., 2008; de Kloe et al., 2009). It should be noted however that (orthosteric and allosteric) ligand binding sites in aminergic GPCRs can be rather flexible, as suggested by long-term (Dror et al., 2011; Kruse et al., 2012) and random acceleration molecular dynamic simulation (Wang and Duan, 2009) studies of β2AR, M2R and M3R crystal structures that identified several (transient) ligand access and exit channels. Such computational simulations can be used to map the binding pathway in order to steer experimental studies (e.g. mutagenesis studies, biophysical measurements) to ultimately derive structure kinetic relationships (Miller et al., 2012) and rationally achieve receptor subtype selectivity, as has for example been suggested for the adrenergic and muscarinic receptors (Dror et al., 2011; Kruse et al., 2012). Interestingly, the H1R antagonist levocetirizine (18) (Figure 4C) shows a decrease in binding affinity and a reduced dissociation half-life at the H1R K1915.39A mutant (Gillard et al., 2002), while recently an analogue of VUF14480 (Supporting Information Figure S7) was designed that covalently binds to C3.36 (but not S3.36) in H4R (Nijmeijer et al., 2013). These H1R and H4R ligands and mutants are valuable tools to study structure kinetic relationships in histamine receptors.
Links between aminergic GPCR binding site, ligand similarity and receptor-ligand selectivity
The previous sections have provided an extensive analysis of receptor-ligand interaction hotspots and binding modes in aminergic GPCR binding sites within the context of receptor mutagenesis studies and ligand structure-affinity and selectivity relationships. This final section brings together systematic bioinformatics and chemoinformatics analyses of aminergic receptor binding site similarity ( Figure 6A,D), ligand similarity (Figure 6B) and experimental receptor-ligand selectivity data (Figures 6C and 7). This integrative analysis gives complementary insights into the possibilities and challenges in the discovery and design of ligands with specific selectivity profiles within and between aminergic GPCR subfamilies. Our analysis highlights the current gaps in experimentally determined GPCR-ligand selectivity profiles and illustrates how complete all-against-all ligand-protein affinity matrices can give new insights into the atomic details of (selective) protein-ligand interactions. The affinity profiles across aminergic GPCRs (Figures 3B and 7) and ligand similarity analyses (Figure 6B) allow the identification of affinity cliffs (two chemically similar fragments of which one shows affinity and the other shows no (or much lower) affinity for a specific protein target) and selectivity cliffs (two chemically similar fragments of which one has affinity for a set of protein targets and the other shows no affinity for at least one of these targets) that can be used for (in silico guided) hit optimization. Furthermore, our ligand similarity analysis reflects to what extent the binding pockets of different aminergic GPCRs can accommodate/have been probed with different ligand scaffolds, and offer opportunities and challenges to identify new ligand chemotypes for specific receptors. This combined chemogenomics analysis provides important information for the development of histamine receptor ligands, and can offer new opportunities to develop, for example, dual H1R-H3R or H1R-H4R ligands with synergistic anti-inflammatory properties (Thurmond et al., 2008; Kuhne et al., 2011; Leurs et al., 2011).
Ligand binding site similarity
Within the sequence identity matrix of crystallized aminergic GPCRs (H1R, β1AR, β2AR, M2R, M3R, D3R) and the other three (H2R, H3R, H4R) histamine receptors (Figure 6A) three clusters can be identified: a histamine receptor cluster, a muscarinic receptor cluster and a cluster including β1AR, β2AR and D3R.
It should be noted that the histamine receptors are not as closely related (especially H2R and H4R) as the muscarinic or the beta-adrenergic receptors. The binding sites of H3R and H4R are relatively similar and explain the difficulty to develop H3R/H4R selective ligands (Celanire et al., 2005; Istyastono et al., 2011a). Figure 6A shows however that the overall sequence similarities between H1R and M2R/M3R and between H2R and β1AR/β2AR are significantly higher than the binding site similarities between H1R and H3R/H4R and between H2R and H3R/H4R. These data are in line with previous phylogenetic analyses of human GPCRs (Vassilatis et al., 2003; Surgand et al., 2006; Gloriam et al., 2009). The H1R crystal structure furthermore has a relatively high structural similarity (Schalon et al., 2008) to the crystal structures of other aminergic receptors (Figure 6D). Although the overall sequence similarity between D3R and β1AR and β2AR is relatively high, the subtle differences of the pharmacophoric properties of T3697.39 (in D3R) versus N7.39 (in β1AR and ADBR2) is apparently crucial for dopamine/beta-adrenergic receptor binding selectivity (vide supra), as reflected by the slightly lower structural similarity (Figure 6D) and the low ligand overlap (Figures 6C and 7, vide infra). The high but expected sequence similarity in sequence and structure for both M2R-M3R and β1AR-β2AR strongly highlight their close kinship and once more underlines the difficulties in developing receptor subtype selective ligands (Baker, 2010; Mohr et al., 2012).
Ligand similarity and ligand selectivity profiles
Similarity analyses of the sequences and structures of the receptors (Figure 6A,D) often correlate with chemical similarity and receptor selectivity of ligand sets (Figures 6B,C and 7). H1R for example shares the highest sequence identity with M2R and H2R (41%), which is correlated with a high ligand similarity and ligand overlap with both targets (Supporting Information Figure S4). Multiple ligands, in particular tricyclic amines (i.e. doxepin, 1, clozapine, 48 and maprotiline, 65) but also other ligands like benztropine (66) and orphenadrine (67), have been found to bind the H1R, H2R as well as both muscarinic receptors (Supporting Information Figure S6). Within the histamine subfamily, the highest sequence identity, ligand similarity and ligand overlap (Figure 6A–C) are however observed between H3R and H4R. Imidazole containing compounds are considered to be non-selective H3R and H4R binders as illustrated by the ligand structures in Figure 3B, and the ligand overlap analyses presented in Figure 7. For example, 90 and 80% of the imidazole containing compounds binding at least H3R or H4R in the ChEMBL database and VU-MedChem fragment sets, respectively, bind both receptors. It should be noted however, that the relative affinities of specific imidazole containing ligands for H3R and H4R can be very different. While histamine has equal affinity for H3R and H4R, methylsubstitution of the amine group (Nα-methylhistamine, 9) gives a 100-fold selectivity for H3R, while methyl substitution of the four position of histamine (4-methyl-histamine, 10) gives more than 100-fold selectivity for H4R (Istyastono et al., 2011a). Other interesting examples of strong changes in H3R/H4R selectivity by subtle changes in chemical structure are immepip (23) (35-fold selectivity for H3R) versus the methyl-substituted analogue methimepip (24) (3500-fold selectivity for H3R) (Istyastono et al., 2011a) and ligands 27 (10-fold selectivity for H3R) versus 28 (15-fold selectivity for H4R) that differ by only one carbon atom in the linker between their imidazole and triazole moieties (Wijtmans et al., 2011). Another example of a steep H3R/H4R selectivity cliff is the addition of a cyclohexyl group to clobenpropit (29), that is VUF5228 (30), which results in more than 60-fold selectivity for H3R compared to clobenpropit (29) (Istyastono et al., 2011b). Interestingly, the isoquinoline fragment 69 only binds H1R, while several chemically related quinazoline and aminopyrimidine-containing fragment-like compounds have been reported to show submicromolar affinities for H3R and H4R (Smits et al., 2008; 2012; de Graaf et al., 2013; Schultes et al., 2012), illustrating how small changes in heteroaromatic ring systems can affect binding of specific histamine receptors. Stereoisomers 71 and 72 represent interesting examples of a stereoisomer specific histamine receptor selectivity switch (Figure 7) (Ishikawa et al., 2010). While 71 has a 4-fold selectivity for H1R over H3R (and 6-fold selectivity over H4R), 72 has a 20-fold selectivity for H3R over H1R (and 93-fold selectivity over H4R).
Several H4R/H3R selectivity hotspots have been identified by combining (3D) QSAR, protein homology modelling, molecular dynamics simulations and site-directed mutagenesis studies (i.e. N/Y4.57, T/M6.55, Q/L7.42) that give insights into the molecular determinants of H4R/H3R ligand selectivity (Istyastono et al., 2011b). Another interesting example is the histamine receptor selectivity cliff between the pyrrolidine fragment 68, which binds all three histamine receptors, and the related piperidine fragment 23, which only has affinity for H3R and H4R (Figure 7). Finally, it should be noted that there are also examples where chemical similarity and selectivity analyses of ligand sets show more pronounced differences between receptors than suggested by the receptor sequence and structure similarity analyses (Figure 6A,D). This is exemplified for the beta-adrenergic ligand similarity and overlap that share many ligands that contain the beta-adrenergic privileged scaffold ethanolamine (432 out of 532) that in 14 out of the 15 cases (with the exception of carvedilol) do not bind other aminergic GPCRs that share sequence similarity, like D3R (Figure 6B,D). The discovery of high affinity ligands for adrenergic receptors that do not contain this scaffold is therefore challenging, but might be facilitated by experimental screening of diverse sets of fragment-like libraries (de Graaf et al., 2013) or structure-based virtual screening studies (Kolb et al., 2009).
Data (in)completeness in chemogenomic analyses
The ligand overlap analyses presented in Figures 6 and 7 provide insight in the molecular determinants of receptors binding and selectivity, but also illustrate that data incompleteness is the Achilles' heel of chemogenomic analyses and predictions (Mestres et al., 2008). While there is a wealth of ligand affinity data available for individual receptors and receptor selectivity data for specific receptor pairs are available (e.g. H3R and H4R), there are many (large) gaps in the all-against-all aminergic GPCR ligand affinity matrix that could give new insights into the molecular determinants of ligand selectivity. For example, Figure 7 shows the ligand overlap of 280 ligands that were tested on H3R and H4R, while for only 56 of these 280 ligands also the H1R affinity is known (Figure 7D). This is even more clearly illustrated by Supporting Information Table S3 describing the number of compounds tested on different receptor combinations. Supporting Information Table S3 shows that there are almost no H3R and H4R ligands for which binding data for M2R, M3R, β1AR, β2AR or D3R is available (coloured grey in Figure 6). Complete all receptor against all ligand selectivity profiles are valuable data sets to guide chemogenomic analyses and enable the identification of protein-ligand interactions (Besnard et al., 2012; de Graaf et al., 2013; Stumpfe and Bajorath, 2012). However, up to now, most chemogenomic studies have focused on the analysis of large, lead-like molecules from high-throughput screening data or (often incomplete) published ligand affinity data (Lipinski et al., 2001; Macarron, 2006). Fragment-like molecules can probe their respective chemical space more efficiently (Murray et al., 2012), which allows for the construction of more accurate maps of protein-ligand binding sites as well as the construction of complete selectivity profiles (de Graaf et al., 2013). Using our in-house VU-MedChem library complete screenings against multiple proteins (e.g. GPCRs and an ion channel) have been performed giving unique insights in ligand overlap but also selectivity, which is already observed on a fragment level (de Graaf et al., 2013). This complete fragment library, containing 1010 compounds, has been screened against H1R, H3R and H4R (Figure 7) and include 120 fragment hits that bind individual or a combination of histamine receptors (vs. 56 ligands present in ChEMBL database for which H1R, H3R, and H4R is reported). These fragment screening data provide additional surprising examples of subtle selectivity switches, as illustrated by the H3R selective fragment 70 (Figure 7), which is a substructure of JNJ777120 (32) which has more than 300-fold selectivity for H4R over H3R (Lim et al., 2005). Systematic determination of ligand affinities against multiple protein targets (Besnard et al., 2012; de Graaf et al., 2013) furthermore can yield more surprising cases of ligand overlap between aminergic GPCRs and other GPCR subfamilies or even other protein families, like ligand-gated ion channels, kinases or phosphodiesterases (Keiser et al., 2009; Lin et al., 2012; Sanders et al., 2012; de Graaf et al., 2013). These cases offer useful information for the consideration of potential side effects associated with anti-targets (Vaz and Klabunde 2008) and/or offer new opportunities for designing ligands with polypharmacological effects (Besnard et al., 2012; Morphy and Rankovic, 2005).
Conclusion
In summary, we have performed chemogenomic studies to identify links between (hist)aminergic receptor ligands and their binding sites and binding modes. This GPCR receptor subfamily shares a conserved structural fold around the transmembrane binding pocket and a large amount of experimental ligand affinity data is available to derive protein and ligand SAR. The increasing number of aminergic GPCR crystal structures now for the first time allowed us to combine ligand affinity data, receptor mutagenesis studies, and amino acid sequence analyses to high-resolution structural analyses of GPCR-ligand interactions. This combined approach enabled us to identify the molecular and structural determinants of ligand affinity and selectivity in several regions of the crystallized aminergic GPCRs as well as all histamine receptors. Further investigations highlighted interesting correlations and differences between ligand similarity/overlap between different receptors and ligand binding site sequence/structure similarity. Apparent discrepancies could be explained by detailed analysis of crystallized or predicted protein-ligand binding modes and local differences in essential pharmacophore features in the ligand binding sites of different receptors. Currently, chemogenomic analyses are however limited by the incompleteness of experimental (all ligand against all protein) affinity data sets. Fragment-based approaches, however, can be used to overcome this problem as they probe their respective chemical space more efficiently, which allows systematic experimental screening of the same small chemical library against multiple protein targets. Chemogenomic analysis of these bioaffinity profiles can be used to identify structure-selectivity relationships that improve our understanding of ligand binding to (hist)aminergic receptors and hence can be used in future GPCR ligand design.
Acknowledgments
This research was financially supported by The Netherlands Organization for Scientific Research (NWO VENI Grant 700.59.408 to C.de.G.).
Conflicts of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Table S1 Binding affinities from site directed mutagenesis studies on histamine receptors and crystallized aminergic GPCRs (see separate excel file). The two-dimensional structures of all the compounds are depicted in Figures 3 and Supporting Information Figure S7. The single-letter amino acid codes indicated in the grey rows are the human wild-type residues. aSDM study on guinea pig H1R. bOnly functional SDM data available for histamine (8) in H2R. cSDM study on rat M3R.
Table S2 Aminergic GPCR crystal structures available in the PDB.
Table S3 The number of ligands for which the activity for both receptors is known (as annotated in the ChEMBL).
Table S4 The percentage (%) of the pairwise sequence identity between the TM domains of the histamine receptors as well as the aminergic receptors with a crystal structure available. The gradient from blue to white to red indicates a low to high sequence similarity.
Figure S1 Full sequence alignment of the histamine and all crystallized aminergic GPCR receptors.
Figure S2 Sequence alignment of the selected pocket residues for all aminergic GPCR receptors.
Figure S3 Snakeplot (http://www.ssfa-7tmr.de/ssfe/snakes/snakes_designer.php) showing the helices of H1R. All B&W numbers are given within the circles as well as the amino acids in H1R, all B&W positions within the TM helices that were mutated during single-point SDM studies for the aminergic GPCRs from Supporting Information Table S1 are shown in orange. The most conserved residues (i.e. the numbers as indicated by TM.50 in the Ballesteros-Weinstein numbering scheme) are circled in green.
Figure S4 Venn diagram visualizing the overlap between the ligands of H1R and all crystallized GPCR receptors according to ligand binding data obtained from the ChEMBL.
Figure S5 (A) front and (B) bottom view displaying the changes in the orientation of the TM helices of β2AR upon activation. The structure of the inactive (PDB-code 2RH1) and active and (PDB-code 3P0G) β2AR structure are shown in red and light blue respectively.
Figure S6 Venn diagram visualizing the overlap between the ligands of H1R, H2R, M2R and M3R.
Figure S7 2D structures for most of the ligands for which SDM data is annotated in Table S1. The structures omitted here are depicted in Figure 3.
References
- Alberts GL, Pregenzer JF, Im WB. Contributions of cysteine 114 of the human D3 dopamine receptor to ligand binding and sensitivity to external oxidizing agents. Br J Pharmacol. 1998;125:705–710. doi: 10.1038/sj.bjp.0702136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosio C, Molinari P, Cotecchia S, Costa T. Catechol-binding serines of beta(2)-adrenergic receptors control the equilibrium between active and inactive receptor states. Mol Pharmacol. 2000;57:198–210. [PubMed] [Google Scholar]
- Attwood TK, Findlay JB. Fingerprinting G-protein-coupled receptors. Protein Eng. 1994;7:195–203. doi: 10.1093/protein/7.2.195. [DOI] [PubMed] [Google Scholar]
- Baker JG. The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. Br J Pharmacol. 2010;160:1048–1061. doi: 10.1111/j.1476-5381.2010.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker RA, Weiner DM, ter Laak T, Beuming T, Zuiderveld OP, Edelbroek M, et al. 8R-lisuride is a potent stereospecific histamine H1-receptor partial agonist. Mol Pharmacol. 2004;65:538–549. doi: 10.1124/mol.65.3.538. [DOI] [PubMed] [Google Scholar]
- Bakker RA, Nicholas MW, Smith TT, Burstein ES, Hacksell U, Timmerman H, et al. In vitro pharmacology of clinically used central nervous system-active drugs as inverse H(1) receptor agonists. J Pharmacol Exp Ther. 2007;322:172–179. doi: 10.1124/jpet.106.118869. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, et al. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnkammer T, Spickenreither A, Brunskole I, Lopuch M, Kagermeier N, Bernhardt G, et al. The bivalent ligand approach leads to highly potent and selective acylguanidine-type histamine H(2) receptor agonists. J Med Chem. 2012;55:1147–1160. doi: 10.1021/jm201128q. [DOI] [PubMed] [Google Scholar]
- Bluml K, Mutschler E, Wess J. Functional role in ligand binding and receptor activation of an asparagine residue present in the sixth transmembrane domain of all muscarinic acetylcholine receptors. J Biol Chem. 1994;269:18870–18876. [PubMed] [Google Scholar]
- Bolden C, Cusack B, Richelson E. Antagonism by antimuscarinic and neuroleptic compounds at the five cloned human muscarinic cholinergic receptors expressed in Chinese hamster ovary cells. J Pharmacol Exp Ther. 1992;260:576–580. [PubMed] [Google Scholar]
- Bondensgaard K, Ankersen M, Thogersen H, Hansen BS, Wulff BS, Bywater RP. Recognition of privileged structures by G-protein coupled receptors. J Med Chem. 2004;47:888–899. doi: 10.1021/jm0309452. [DOI] [PubMed] [Google Scholar]
- Brianso F, Carrascosa MC, Oprea TI, Mestres J. Cross-pharmacology analysis of G protein-coupled receptors. Curr Top Med Chem. 2011;11:1956–1963. doi: 10.2174/156802611796391285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruysters M, Pertz HH, Teunissen A, Bakker RA, Gillard M, Chatelain P, et al. Mutational analysis of the histamine H1-receptor binding pocket of histaprodifens. Eur J Pharmacol. 2004;487:55–63. doi: 10.1016/j.ejphar.2004.01.028. [DOI] [PubMed] [Google Scholar]
- Bruysters M, Jongejan A, Gillard M, van de Manakker F, Bakker RA, Chatelain P, et al. Pharmacological differences between human and guinea pig histamine H1 receptors: Asn84 (2.61) as key residue within an additional binding pocket in the H1 receptor. Mol Pharmacol. 2005;67:1045–1052. doi: 10.1124/mol.104.008847. [DOI] [PubMed] [Google Scholar]
- Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, et al. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat Chem Biol. 2011;7:769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celanire S, Wijtmans M, Talaga P, Leurs R, de Esch IJP. Keynote review: histamine H3 receptor antagonists reach out for the clinic. Drug Discov Today. 2005;10:1613–1627. doi: 10.1016/S1359-6446(05)03625-1. [DOI] [PubMed] [Google Scholar]
- Chen X, Lin Y, Gilson MK. The binding database: overview and user's guide. Biopolymers. 2001;61:127–141. doi: 10.1002/1097-0282(2002)61:2<127::AID-BIP10076>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman HG, Caron MG, DeBlasi A, Frielle T, Lefkowitz RJ. Role of extracellular disulfide-bonded cysteines in the ligand binding function of the beta 2-adrenergic receptor. Biochemistry. 1990;29:2335–2342. doi: 10.1021/bi00461a018. [DOI] [PubMed] [Google Scholar]
- Dorfler M, Tschammer N, Hamperl K, Hubner H, Gmeiner P. Novel D3 selective dopaminergics incorporating enyne units as nonaromatic catechol bioisosteres: synthesis, bioactivity, and mutagenesis studies. J Med Chem. 2008;51:6829–6838. doi: 10.1021/jm800895v. [DOI] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, et al. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci U S A. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich K, Gotz A, Bollinger S, Tschammer N, Bettinetti L, Harterich S, et al. Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific recesptor binding. J Med Chem. 2009;52:4923–4935. doi: 10.1021/jm900690y. [DOI] [PubMed] [Google Scholar]
- Engelhardt H, Smits RA, Leurs R, Haaksma E, de Esch IJP. A new generation of anti-histamines: histamine H4 receptor antagonists on their way to the clinic. Curr Opin Drug Discov Devel. 2009;12:628–643. [PubMed] [Google Scholar]
- Ericksen SS, Cummings DF, Teer ME, Amdani S, Schetz JA. Ring substituents on substituted benzamide ligands indirectly mediate interactions with position 7.39 of transmembrane helix 7 of the D4 dopamine receptor. J Pharmacol Exp Ther. 2012;342:472–485. doi: 10.1124/jpet.112.193979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM. Site-directed mutagenesis of beta-adrenergic receptors. Identification of conserved cysteine residues that independently affect ligand binding and receptor activation. J Biol Chem. 1989;264:9266–9270. [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Gantz I, DelValle J, Wang LD, Tashiro T, Munzert G, Guo YJ, et al. Molecular basis for the interaction of histamine with the histamine H2 receptor. J Biol Chem. 1992;267:20840–20843. [PubMed] [Google Scholar]
- Garland SL, Gloriam DE. A ligand's view of target similarity: chemogenomic binding site-directed techniques for drug discovery. Curr Top Med Chem. 2011a;11:1872–1881. doi: 10.2174/156802611796391276. [DOI] [PubMed] [Google Scholar]
- Garland SL, Gloriam DE. Methods for the successful application of chemogenomics to GPCR drug design. Curr Top Med Chem. 2011b;11:1870–1871. doi: 10.2174/156802611796391294. [DOI] [PubMed] [Google Scholar]
- Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40(Database issue):D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillard M, Van Der Perren C, Moguilevsky N, Massingham R, Chatelain P. Binding characteristics of cetirizine and levocetirizine to human H(1) histamine receptors: contribution of Lys(191) and Thr(194) Mol Pharmacol. 2002;61:391–399. doi: 10.1124/mol.61.2.391. [DOI] [PubMed] [Google Scholar]
- Gloriam DE, Foord SM, Blaney FE, Garland SL. Definition of the G protein-coupled receptor transmembrane bundle binding pocket and calculation of receptor similarities for drug design. J Med Chem. 2009;52:4429–4442. doi: 10.1021/jm900319e. [DOI] [PubMed] [Google Scholar]
- Gloriam DE, Wellendorph P, Johansen LD, Thomsen AR, Phonekeo K, Pedersen DS, et al. Chemogenomic discovery of allosteric antagonists at the GPRC6A receptor. Chem Biol. 2011;18:1489–1498. doi: 10.1016/j.chembiol.2011.09.012. [DOI] [PubMed] [Google Scholar]
- Govoni M, Lim HD, El-Atmioui D, Menge WM, Timmerman H, Bakker RA, et al. A chemical switch for the modulation of the functional activity of higher homologues of histamine on the human histamine H3 receptor: effect of various substitutions at the primary amino function. J Med Chem. 2006;49:2549–2557. doi: 10.1021/jm0504353. [DOI] [PubMed] [Google Scholar]
- de Graaf C, Rognan D. Selective structure-based virtual screening for full and partial agonists of the beta2 adrenergic receptor. J Med Chem. 2008;51:4978–4985. doi: 10.1021/jm800710x. [DOI] [PubMed] [Google Scholar]
- de Graaf C, Rognan D. Customizing G Protein-coupled receptor models for structure-based virtual screening. Curr Pharm Des. 2009;15:4026–4048. doi: 10.2174/138161209789824786. [DOI] [PubMed] [Google Scholar]
- de Graaf C, Foata N, Engkvist O, Rognan D. Molecular modeling of the second extracellular loop of G-protein coupled receptors and its implication on structure-based virtual screening. Proteins. 2008;71:599–620. doi: 10.1002/prot.21724. [DOI] [PubMed] [Google Scholar]
- de Graaf C, Kooistra AJ, Vischer HF, Katritch V, Kuijer M, Shiroishi M, et al. Crystal structure-based virtual screening for fragment-like ligands of the human histamine H(1) receptor. J Med Chem. 2011;54:8195–8206. doi: 10.1021/jm2011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf C, Vischer HF, de Kloe GE, Kooistra AJ, Nijmeijer S, Kuijer M, et al. Small and colourful stones make beautiful mosaics: fragment-Based Chemogenomics. Drug Discov Today. 2013;18:323–330. doi: 10.1016/j.drudis.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, et al. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Hall NE, Tobin AB, Sexton PM, Christopoulos A. Identification of orthosteric and allosteric site mutations in M2 muscarinic acetylcholine receptors that contribute to ligand-selective signaling bias. J Biol Chem. 2010;285:7459–7474. doi: 10.1074/jbc.M109.094011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitz F, Holzwarth JA, Gies JP, Pruss RM, Trumpp-Kallmeyer S, Hibert MF, et al. Site-directed mutagenesis of the putative human muscarinic M2 receptor binding site. Eur J Pharmacol. 1999;380:183–195. doi: 10.1016/s0014-2999(99)00439-2. [DOI] [PubMed] [Google Scholar]
- Hoare SR, Coldwell MC, Armstrong D, Strange PG. Regulation of human D(1), d(2(long)), d(2(short)), D(3) and D(4) dopamine receptors by amiloride and amiloride analogues. Br J Pharmacol. 2000;130:1045–1059. doi: 10.1038/sj.bjp.0703370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst B, Nygaard R, Valentin-Hansen L, Bach A, Engelstoft MS, Petersen PS, et al. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J Biol Chem. 2010;285:3973–3985. doi: 10.1074/jbc.M109.064725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst E, Okuno Y, Bender A, IJzerman AP. Substructure mining of GPCR ligands reveals activity-class specific functional groups in an unbiased manner. J Chem Inf Model. 2009;49:348–360. doi: 10.1021/ci8003896. [DOI] [PubMed] [Google Scholar]
- Huang XP, Prilla S, Mohr K, Ellis J. Critical amino acid residues of the common allosteric site on the M2 muscarinic acetylcholine receptor: more similarities than differences between the structurally divergent agents gallamine and bis(ammonio)alkane-type hexamethylene-bis-[dimethyl-(3-phthalimidopropyl)ammonium]dibromide. Mol Pharmacol. 2005;68:769–778. doi: 10.1124/mol.105.014043. [DOI] [PubMed] [Google Scholar]
- Hulme EC, Curtis CA, Page KM, Jones PG. The role of charge interactions in muscarinic agonist binding, and receptor–response coupling. Life Sci. 1995;56:891–898. doi: 10.1016/0024-3205(95)00025-2. [DOI] [PubMed] [Google Scholar]
- Igel P, Geyer R, Strasser A, Dove S, Seifert R, Buschauer A. Synthesis and structure-activity relationships of cyanoguanidine-type and structurally related histamine H4 receptor agonists. J Med Chem. 2009;52:6297–6313. doi: 10.1021/jm900526h. [DOI] [PubMed] [Google Scholar]
- Im WB, Chio CL, Alberts GL, Dinh DM. Positive allosteric modulator of the human 5-HT2C receptor. Mol Pharmacol. 2003;64:78–84. doi: 10.1124/mol.64.1.78. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Watanabe T, Kudo T, Yokoyama F, Yamauchi M, Kato K, et al. Investigation of the histamine H3 receptor binding site. Design and synthesis of hybrid agonists with a lipophilic side chain. J Med Chem. 2010;53:6445–6456. doi: 10.1021/jm100643t. [DOI] [PubMed] [Google Scholar]
- Istyastono EP. 2012. Structure-based virtual fragment screening for ligands of the G protein-coupled histamine H4 receptor. PhD Thesis. Division of medicinal chemistry – VU University Amsterdam.
- Istyastono EP, de Graaf C, de Esch IJP, Leurs R. Molecular determinants of selective agonist and antagonist binding to the histamine H(4) receptor. Curr Top Med Chem. 2011a;11:661–679. doi: 10.2174/1568026611109060661. [DOI] [PubMed] [Google Scholar]
- Istyastono EP, Nijmeijer S, Lim HD, van de Stolpe A, Roumen L, Kooistra AJ, et al. Molecular determinants of ligand binding modes in the histamine H(4) receptor: linking ligand-based three-dimensional quantitative structure-activity relationship (3D-QSAR) models to in silico guided receptor mutagenesis studies. J Med Chem. 2011b;54:8136–8147. doi: 10.1021/jm201042n. [DOI] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Costanzi S. New insights for drug design from the X-ray crystallographic structures of G-protein-coupled receptors. Mol Pharmacol. 2012;82:361–371. doi: 10.1124/mol.112.079335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. The 7 TM G-protein-coupled receptor target family. ChemMedChem. 2006;1:761–782. doi: 10.1002/cmdc.200600134. [DOI] [PubMed] [Google Scholar]
- Jacoby E, editor. Chemogenomics: Methods and Applications. New York: Humana Press; 2009. [Google Scholar]
- Johansson H, Bøgeløv Jørgensen T, Gloriam DE, Bräuner-Osborne H, Sejer Pedersen D. 3-Substituted 2-phenyl-indoles: privileged structures for medicinal chemistry. RSC Adv. 2013;3:945–960. [Google Scholar]
- Jojart B, Kiss R, Viskolcz B, Keseru GM. Activation mechanism of the human histamine H4 receptor – an explicit membrane molecular dynamics simulation study. J Chem Inf Model. 2008;48:1199–1210. doi: 10.1021/ci700450w. [DOI] [PubMed] [Google Scholar]
- Jongejan A, Bruysters M, Ballesteros JA, Haaksma E, Bakker RA, Pardo L, et al. Linking agonist binding to histamine H1 receptor activation. Nat Chem Biol. 2005;1:98–103. doi: 10.1038/nchembio714. [DOI] [PubMed] [Google Scholar]
- Jongejan A, Lim HD, Smits RA, de Esch IJP, Haaksma E, Leurs R. Delineation of agonist binding to the human histamine H4 receptor using mutational analysis, homology modeling, and ab initio calculations. J Chem Inf Model. 2008;48:1455–1463. doi: 10.1021/ci700474a. [DOI] [PubMed] [Google Scholar]
- Katritch V, Abagyan R. GPCR agonist binding revealed by modeling and crystallography. Trends Pharmacol Sci. 2011;32:637–643. doi: 10.1016/j.tips.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Reynolds KA, Cherezov V, Hanson MA, Roth CB, Yeager M, et al. Analysis of full and partial agonists binding to beta2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J Mol Recognit. 2009;22:307–318. doi: 10.1002/jmr.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Structure-Function of the G Protein-Coupled Receptor Superfamily. Annu Rev Pharmacol Toxicol. 2012;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzie KM, Donello JE, Krauss HA, Regan JW, Gil DW. A single amino-acid substitution in the EP2 prostaglandin receptor confers responsiveness to prostacyclin analogs. Mol Pharmacol. 1998;54:584–590. doi: 10.1124/mol.54.3.584. [DOI] [PubMed] [Google Scholar]
- Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25:197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, et al. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley MT, Burckstummer T, Wenzel-Seifert K, Dove S, Buschauer A, Seifert R. Distinct interaction of human and guinea pig histamine H2-receptor with guanidine-type agonists. Mol Pharmacol. 2001;60:1210–1225. doi: 10.1124/mol.60.6.1210. [DOI] [PubMed] [Google Scholar]
- Kiss R, Keseru GM. Histamine H4 receptor ligands and their potential therapeutic applications: an update. Expert Opinion On Therapeutic Patents. 2012;22:205–221. doi: 10.1517/13543776.2012.665447. [DOI] [PubMed] [Google Scholar]
- Kiss R, Kiss B, Konczol A, Szalai F, Jelinek I, Laszlo V, et al. Discovery of novel human histamine H4 receptor ligands by large-scale structure-based virtual screening. J Med Chem. 2008a;51:3145–3153. doi: 10.1021/jm7014777. [DOI] [PubMed] [Google Scholar]
- Kiss R, Noszal B, Racz A, Falus A, Eros D, Keseru GM. Binding mode analysis and enrichment studies on homology models of the human histamine H4 receptor. Eur J Med Chem. 2008b;43:1059–1070. doi: 10.1016/j.ejmech.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Klabunde T. Chemogenomic approaches to drug discovery: similar receptors bind similar ligands. Br J Pharmacol. 2007;152:5–7. doi: 10.1038/sj.bjp.0707308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klabunde T, Giegerich C, Evers A. Sequence-derived three-dimensional pharmacophore models for G-protein-coupled receptors and their application in virtual screening. J Med Chem. 2009;52:2923–2932. doi: 10.1021/jm9001346. [DOI] [PubMed] [Google Scholar]
- de Kloe GE, Bailey D, Leurs R, de Esch IJP. Transforming fragments into candidates: small becomes big in medicinal chemistry. Drug Discov Today. 2009;14:630–646. doi: 10.1016/j.drudis.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, et al. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011;39(Database issue):D1035–D1041. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakowski LF., Jr GCRDb: a G-protein-coupled receptor database. Receptors Channels. 1994;2:1–7. [PubMed] [Google Scholar]
- Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK. Structure-based discovery of beta2-adrenergic receptor ligands. Proc Natl Acad Sci U S A. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra AJ, Binsl TW, van Beek JH, de Graaf C, Heringa J. Electron density fingerprints (EDprints): virtual screening using assembled information of electron density. J Chem Inf Model. 2010;50:1772–1780. doi: 10.1021/ci1002608. [DOI] [PubMed] [Google Scholar]
- Kooistra AJ, Roumen L, Leurs R, de Esch IJP, de Graaf C. From heptahelical bundle to hits from the Haystack: structure-based virtual screening for GPCR ligands. Methods Enzymol. 2013;522:279–336. doi: 10.1016/B978-0-12-407865-9.00015-7. [DOI] [PubMed] [Google Scholar]
- Krejci A, Tucek S. Changes of cooperativity between N-methylscopolamine and allosteric modulators alcuronium and gallamine induced by mutations of external loops of muscarinic M-3 receptors. Mol Pharmacol. 2001;60:761–767. [PubMed] [Google Scholar]
- Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhne S, Wijtmans M, Lim HD, Leurs R, de Esch IJP. Several down, a few to go: histamine H3 receptor ligands making the final push towards the market? Expert Opin Investig Drugs. 2011;20:1629–1648. doi: 10.1517/13543784.2011.625010. [DOI] [PubMed] [Google Scholar]
- Leppik RA, Lazareno S, Mynett A, Birdsall NJ. Characterization of the allosteric interactions between antagonists and amiloride analogues at the human alpha2A-adrenergic receptor. Mol Pharmacol. 1998;53:916–925. [PubMed] [Google Scholar]
- Leurs R, Smit MJ, Meeder R, Ter Laak AM, Timmerman H. Lysine200 located in the fifth transmembrane domain of the histamine H1 receptor interacts with histamine but not with all H1 agonists. Biochem Biophys Res Commun. 1995;214:110–117. doi: 10.1006/bbrc.1995.2263. [DOI] [PubMed] [Google Scholar]
- Leurs R, Smit MJ, Tensen CP, Ter Laak AM, Timmerman H. Site-directed mutagenesis of the histamine H1-receptor reveals a selective interaction of asparagine207 with subclasses of H1-receptor agonists. Biochem Biophys Res Commun. 1994;201:295–301. doi: 10.1006/bbrc.1994.1701. [DOI] [PubMed] [Google Scholar]
- Leurs R, Vischer HF, Wijtmans M, de Esch IJP. En route to new blockbuster anti-histamines: surveying the offspring of the expanding histamine receptor family. Trends Pharmacol Sci. 2011;32:250–257. doi: 10.1016/j.tips.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Liapakis G, Ballesteros JA, Papachristou S, Chan WC, Chen X, Javitch JA. The forgotten serine. A critical role for Ser-2035.42 in ligand binding to and activation of the beta 2-adrenergic receptor. J Biol Chem. 2000;275:37779–37788. doi: 10.1074/jbc.M002092200. [DOI] [PubMed] [Google Scholar]
- Lim HD, van Rijn RM, Ling P, Bakker RA, Thurmond RL, Leurs R. Evaluation of histamine H1-, H2-, and H3-receptor ligands at the human histamine H4 receptor: identification of 4-methylhistamine as the first potent and selective H4 receptor agonist. J Pharmacol Exp Ther. 2005;314:1310–1321. doi: 10.1124/jpet.105.087965. [DOI] [PubMed] [Google Scholar]
- Lim HD, Jongejan A, Bakker RA, Haaksma E, de Esch IJP, Leurs R. Phenylalanine 169 in the second extracellular loop of the human histamine H4 receptor is responsible for the difference in agonist binding between human and mouse H4 receptors. J Pharmacol Exp Ther. 2008;327:88–96. doi: 10.1124/jpet.108.140343. [DOI] [PubMed] [Google Scholar]
- Lim HD, de Graaf C, Jiang W, Sadek P, McGovern PM, Istyastono EP, et al. Molecular determinants of ligand binding to H4R species variants. Mol Pharmacol. 2010;77:734–743. doi: 10.1124/mol.109.063040. [DOI] [PubMed] [Google Scholar]
- Lin X, Huang XP, Chen G, Whaley R, Peng S, Wang Y, et al. Life beyond kinases: structure-based discovery of sorafenib as nanomolar antagonist of 5-HT receptors. J Med Chem. 2012;55:5749–5759. doi: 10.1021/jm300338m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Lorenzi S, Mor M, Bordi F, Rivara S, Rivara M, Morini G, et al. Validation of a histamine H3 receptor model through structure-activity relationships for classical H3 antagonists. Bioorg Med Chem. 2005;13:5647–5657. doi: 10.1016/j.bmc.2005.05.072. [DOI] [PubMed] [Google Scholar]
- Lundstrom K, Turpin MP, Large C, Robertson G, Thomas P, Lewell XQ. Mapping of dopamine D3 receptor binding site by pharmacological characterization of mutants expressed in CHO cells with the Semliki Forest virus system. J Recept Signal Transduct Res. 1998;18:133–150. doi: 10.3109/10799899809047741. [DOI] [PubMed] [Google Scholar]
- Macarron R. Critical review of the role of HTS in drug discovery. Drug Discov Today. 2006;11:277–279. doi: 10.1016/j.drudis.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, et al. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcou G, Rognan D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J Chem Inf Model. 2007;47:195–207. doi: 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- Mestres J, Gregori-Puigjane E, Valverde S, Sole RV. Data completeness – the Achilles heel of drug-target networks. Nat Biotechnol. 2008;26:983–984. doi: 10.1038/nbt0908-983. [DOI] [PubMed] [Google Scholar]
- Miller DC, Lunn G, Jones P, Sabnis Y, Davies NL, Driscoll P. Investigation of the effect of molecular properties on the binding kinetics of a ligand to its biological target. Medchemcomm. 2012;3:449–452. [Google Scholar]
- Moguilevsky N, Varsalona F, Guillaume JP, Noyer M, Gillard M, Daliers J, et al. Pharmacological and functional characterisation of the wild-type and site-directed mutants of the human H1 histamine receptor stably expressed in CHO cells. J Recept Signal Transduct Res. 1995;15:91–102. doi: 10.3109/10799899509045210. [DOI] [PubMed] [Google Scholar]
- Mohr K, Trankle C, Kostenis E, Barocelli E, De Amici M, Holzgrabe U. Rational design of dualsteric GPCR ligands: quests and promise. Br J Pharmacol. 2010;159:997–1008. doi: 10.1111/j.1476-5381.2009.00601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr K, Schmitz J, Schrage R, Trankle C, Holzgrabe U. Molecular Alliance-From Orthosteric and Allosteric Ligands to Dualsteric/Bitopic Agonists at G Protein Coupled Receptors. Angew Chem Int Ed Engl. 2012;52:508–516. doi: 10.1002/anie.201205315. [DOI] [PubMed] [Google Scholar]
- Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- Murray CW, Verdonk ML, Rees DC. Experiences in fragment-based drug discovery. Trends Pharmacol Sci. 2012;33:224–232. doi: 10.1016/j.tips.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Nijmeijer S, Engelhardt H, Schultes S, van de Stolpe AC, Lusink V, de Graaf C, et al. Design and pharmacologicalcharacterization of VUF14480, a covalent partial agonist thatinteracts with cysteine 98(3.36) of the human histamine H(4) receptor. Br J Pharmacol. 2013 doi: 10.1111/bph.12113. doi: 10.1111/bph.12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka H, Otaki S, Ohshima E, Kono M, Kase H, Ohta K, et al. Unique binding pocket for KW-4679 in the histamine H1 receptor. Eur J Pharmacol. 1998;345:111–117. doi: 10.1016/s0014-2999(97)01620-8. [DOI] [PubMed] [Google Scholar]
- Ohta K, Hayashi H, Mizuguchi H, Kagamiyama H, Fujimoto K, Fukui H. Site-directed mutagenesis of the histamine H1 receptor: roles of aspartic acid107, asparagine198 and threonine194. Biochem Biophys Res Commun. 1994;203:1096–1101. doi: 10.1006/bbrc.1994.2295. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Paolini GV, Shapland RH, van Hoorn WP, Mason JS, Hopkins AL. Global mapping of pharmacological space. Nat Biotechnol. 2006;24:805–815. doi: 10.1038/nbt1228. [DOI] [PubMed] [Google Scholar]
- Parsons ME, Ganellin CR. Histamine and its receptors. Br J Pharmacol. 2006;147(Suppl. 1):S127–S135. doi: 10.1038/sj.bjp.0706440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters MC, van Westen GJ, Li Q, IJzerman AP. Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol Sci. 2011;32:35–42. doi: 10.1016/j.tips.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Peeters MC, Wisse LE, Dinaj A, Vroling B, Vriend G, IJzerman AP. The role of the second and third extracellular loops of the adenosine A1 receptor in activation and allosteric modulation. Biochem Pharmacol. 2012;84:76–87. doi: 10.1016/j.bcp.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Poulain R, Horvath D, Bonnet B, Eckhoff C, Chapelain B, Bodinier MC, et al. From hit to lead. Analyzing structure-profile relationships. J Med Chem. 2001;44:3391–3401. doi: 10.1021/jm010878g. [DOI] [PubMed] [Google Scholar]
- Procopiou PA, Browning C, Buckley JM, Clark KL, Fechner L, Gore PM, et al. The discovery of phthalazinone-based human H1 and H3 single-ligand antagonists suitable for intranasal administration for the treatment of allergic rhinitis. J Med Chem. 2011;54:2183–2195. doi: 10.1021/jm1013874. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011a;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011b;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognan D. Chemogenomic approaches to rational drug design. Br J Pharmacol. 2007;152:38–52. doi: 10.1038/sj.bjp.0707307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Sanders MP, Verhoeven S, de Graaf C, Roumen L, Vroling B, Nabuurs SB, et al. Snooker: a structure-based pharmacophore generation tool applied to class A GPCRs. J Chem Inf Model. 2011;51:2277–2292. doi: 10.1021/ci200088d. [DOI] [PubMed] [Google Scholar]
- Sanders MP, Roumen L, van der Horst E, Lane JR, Vischer HF, van Offenbeek J, et al. A prospective cross-screening study on G-protein-coupled receptors: lessons learned in virtual compound library design. J Med Chem. 2012;55:5311–5325. doi: 10.1021/jm300280e. [DOI] [PubMed] [Google Scholar]
- Sansuk K, Deupi X, Torrecillas IR, Jongejan A, Nijmeijer S, Bakker RA, et al. A structural insight into the reorientation of transmembrane domains 3 and 5 during family A G protein-coupled receptor activation. Mol Pharmacol. 2011;79:262–269. doi: 10.1124/mol.110.066068. [DOI] [PubMed] [Google Scholar]
- Sartania N, Strange PG. Role of conserved serine residues in the interaction of agonists with D3 dopamine receptors. J Neurochem. 1999;72:2621–2624. doi: 10.1046/j.1471-4159.1999.0722621.x. [DOI] [PubMed] [Google Scholar]
- Schalon C, Surgand JS, Kellenberger E, Rognan D. A simple and fuzzy method to align and compare druggable ligand-binding sites. Proteins. 2008;71:1755–1778. doi: 10.1002/prot.21858. [DOI] [PubMed] [Google Scholar]
- Schlegel B, Laggner C, Meier R, Langer T, Schnell D, Seifert R, et al. Generation of a homology model of the human histamine H(3) receptor for ligand docking and pharmacophore-based screening. J Comput Aided Mol Des. 2007;21:437–453. doi: 10.1007/s10822-007-9127-x. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Li B, Bloodworth L, Erlenbach I, Zeng FY, Wess J. Random mutagenesis of the M3 muscarinic acetylcholine receptor expressed in yeast. Identification of point mutations that “silence” a constitutively active mutant M3 receptor and greatly impair receptor/G protein coupling. J Biol Chem. 2003;278:30248–30260. doi: 10.1074/jbc.M304991200. [DOI] [PubMed] [Google Scholar]
- Scholten DJ, Canals M, Maussang D, Roumen L, Smit MJ, Wijtmans M, et al. Pharmacological modulation of chemokine receptor function. Br J Pharmacol. 2012;165:1617–1643. doi: 10.1111/j.1476-5381.2011.01551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, et al. Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (Berl) 1996;124:57–73. doi: 10.1007/BF02245606. [DOI] [PubMed] [Google Scholar]
- Schultes S, Nijmeijer S, Engelhardt H, Kooistra AJ, Vischer HF, de Esch IJP, et al. Mapping histamine H4 receptor-ligand binding modes. Medchemcomm. 2012;4:193–204. [Google Scholar]
- Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE. Molecular mechanism of 7TM receptor activation – a global toggle switch model. Annu Rev Pharmacol Toxicol. 2006;46:481–519. doi: 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- Seifert R, Dove S. Functional selectivity of GPCR ligand stereoisomers: new pharmacological opportunities. Mol Pharmacol. 2009;75:13–18. doi: 10.1124/mol.108.052944. [DOI] [PubMed] [Google Scholar]
- Seifert R, Strasser A, Schneider EH, Neumann D, Dove S, Buschauer A. Molecular and cellular analysis of human histamine receptor subtypes. Trends Pharmacol Sci. 2013;34:33–58. doi: 10.1016/j.tips.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selent J, Lopez L, Sanz F, Pastor M. Multi-receptor binding profile of clozapine and olanzapine: a structural study based on the new beta2 adrenergic receptor template. ChemMedChem. 2008;3:1194–1198. doi: 10.1002/cmdc.200800074. [DOI] [PubMed] [Google Scholar]
- Shah JR, Mosier PD, Roth BL, Kellogg GE, Westkaemper RB. Synthesis, structure-affinity relationships, and modeling of AMDA analogs at 5-HT2A and H1 receptors: structural factors contributing to selectivity. Bioorg Med Chem. 2009;17:6496–6504. doi: 10.1016/j.bmc.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Javitch JA. The binding site of aminergic G protein-coupled receptors: the transmembrane segments and second extracellular loop. Annu Rev Pharmacol Toxicol. 2002;42:437–467. doi: 10.1146/annurev.pharmtox.42.091101.144224. [DOI] [PubMed] [Google Scholar]
- Shi L, Javitch JA. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc Natl Acad Sci U S A. 2004;101:440–445. doi: 10.1073/pnas.2237265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, et al. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin N, Coates E, Murgolo NJ, Morse KL, Bayne M, Strader CD, et al. Molecular modeling and site-specific mutagenesis of the histamine-binding site of the histamine H4 receptor. Mol Pharmacol. 2002;62:38–47. doi: 10.1124/mol.62.1.38. [DOI] [PubMed] [Google Scholar]
- Simons FE, Simons KJ. Histamine and H1-antihistamines: celebrating a century of progress. J Allergy Clin Immunol. 2011;128:1139–1150. doi: 10.1016/j.jaci.2011.09.005. e1134. [DOI] [PubMed] [Google Scholar]
- Simpson MM, Ballesteros JA, Chiappa V, Chen J, Suehiro M, Hartman DS, et al. Dopamine D4/D2 receptor selectivity is determined by A divergent aromatic microdomain contained within the second, third, and seventh membrane-spanning segments. Mol Pharmacol. 1999;56:1116–1126. doi: 10.1124/mol.56.6.1116. [DOI] [PubMed] [Google Scholar]
- Singh R, Hurst DP, Barnett-Norris J, Lynch DL, Reggio PH, Guarnieri F. Activation of the cannabinoid CB1 receptor may involve a W6 48/F3 36 rotamer toggle switch. J Pept Res. 2002;60:357–370. doi: 10.1034/j.1399-3011.2002.21065.x. [DOI] [PubMed] [Google Scholar]
- Sirci F, Istyastono EP, Vischer HF, Kooistra AJ, Nijmeijer S, Kuijer M, et al. Virtual fragment screening: discovery of histamine H(3) receptor ligands using ligand-based and protein-based molecular fingerprints. J Chem Inf Model. 2012;52:3308–3324. doi: 10.1021/ci3004094. [DOI] [PubMed] [Google Scholar]
- Smits RA, Lim HD, Hanzer A, Zuiderveld OP, Guaita E, Adami M, et al. Fragment based design of new H4 receptor-ligands with anti-inflammatory properties in vivo. J Med Chem. 2008;51:2457–2467. doi: 10.1021/jm7014217. [DOI] [PubMed] [Google Scholar]
- Smits RA, Leurs R, de Esch IJP. Major advances in the development of histamine H4 receptor ligands. Drug Discov Today. 2009;14:745–753. doi: 10.1016/j.drudis.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Smits RA, Lim HD, van der Meer T, Kuhne S, Bessembinder K, Zuiderveld OP, et al. Ligand based design of novel histamine H(4) receptor antagonists; fragment optimization and analysis of binding kinetics. Bioorg Med Chem Lett. 2012;22:461–467. doi: 10.1016/j.bmcl.2011.10.104. [DOI] [PubMed] [Google Scholar]
- Steinfeld T, Hughes AD, Klein U, Smith JA, Mammen M. THRX-198321 is a bifunctional muscarinic receptor antagonist and beta2-adrenoceptor agonist (MABA) that binds in a bimodal and multivalent manner. Mol Pharmacol. 2011;79:389–399. doi: 10.1124/mol.110.069120. [DOI] [PubMed] [Google Scholar]
- Strasser A, Wittmann HJ, Kunze M, Elz S, Seifert R. Molecular basis for the selective interaction of synthetic agonists with the human histamine H1-receptor compared with the guinea pig H1-receptor. Mol Pharmacol. 2009;75:454–465. doi: 10.1124/mol.108.053009. [DOI] [PubMed] [Google Scholar]
- Stumpfe D, Bajorath J. Exploring activity cliffs in medicinal chemistry. J Med Chem. 2012;55:2932–2942. doi: 10.1021/jm201706b. [DOI] [PubMed] [Google Scholar]
- Surgand JS, Rodrigo J, Kellenberger E, Rognan D. A chemogenomic analysis of the transmembrane binding cavity of human G-protein-coupled receptors. Proteins. 2006;62:509–538. doi: 10.1002/prot.20768. [DOI] [PubMed] [Google Scholar]
- Suryanarayana S, Kobilka BK. Amino acid substitutions at position 312 in the seventh hydrophobic segment of the beta 2-adrenergic receptor modify ligand-binding specificity. Mol Pharmacol. 1993;44:111–114. [PubMed] [Google Scholar]
- Suryanarayana S, Daunt DA, Von Zastrow M, Kobilka BK. A point mutation in the seventh hydrophobic domain of the alpha 2 adrenergic receptor increases its affinity for a family of beta receptor antagonists. J Biol Chem. 1991;266:15488–15492. [PubMed] [Google Scholar]
- Thurmond RL, Gelfand EW, Dunford PJ. The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nat Rev Drug Discov. 2008;7:41–53. doi: 10.1038/nrd2465. [DOI] [PubMed] [Google Scholar]
- Uveges AJ, Kowal D, Zhang Y, Spangler TB, Dunlop J, Semus S, et al. The role of transmembrane helix 5 in agonist binding to the human H3 receptor. J Pharmacol Exp Ther. 2002;301:451–458. doi: 10.1124/jpet.301.2.451. [DOI] [PubMed] [Google Scholar]
- Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE, Mortrud MT, et al. The G protein-coupled receptor repertoires of human and mouse. Proc Natl Acad Sci U S A. 2003;100:4903–4908. doi: 10.1073/pnas.0230374100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz RJ, Klabunde T, editors. Antitargets: Prediction and Prevention of Drug Side Effects. Weinheim, Germany: Wiley-VCH; 2008. [Google Scholar]
- Vroling B, Sanders M, Baakman C, Borrmann A, Verhoeven S, Klomp J, et al. GPCRDB: information system for G protein-coupled receptors. Nucleic Acids Res. 2011;39:D309–D319. doi: 10.1093/nar/gkq1009. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Duan Y. Ligand entry and exit pathways in the beta2-adrenergic receptor. J Mol Biol. 2009;392:1102–1115. doi: 10.1016/j.jmb.2009.07.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, et al. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Edwards PC, Leslie AG, Tate CG. Crystal structures of a stabilized beta1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure. 2012;20:841–849. doi: 10.1016/j.str.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weill N. Chemogenomic approaches for the exploration of GPCR space. Curr Top Med Chem. 2011;11:1944–1955. doi: 10.2174/156802611796391212. [DOI] [PubMed] [Google Scholar]
- Weill N, Rognan D. Development and validation of a novel protein-ligand fingerprint to mine chemogenomic space: application to G protein-coupled receptors and their ligands. J Chem Inf Model. 2009;49:1049–1062. doi: 10.1021/ci800447g. [DOI] [PubMed] [Google Scholar]
- Werner T, Sander K, Tanrikulu Y, Kottke T, Proschak E, Stark H, et al. In silico characterization of ligand binding modes in the human histamine H4 receptor and their impact on receptor activation. Chembiochem. 2010;11:1850–1855. doi: 10.1002/cbic.201000180. [DOI] [PubMed] [Google Scholar]
- Wess J, Gdula D, Brann MR. Site-directed mutagenesis of the m3 muscarinic receptor: identification of a series of threonine and tyrosine residues involved in agonist but not antagonist binding. EMBO J. 1991;10:3729–3734. doi: 10.1002/j.1460-2075.1991.tb04941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess J, Nanavati S, Vogel Z, Maggio R. Functional role of proline and tryptophan residues highly conserved among G protein-coupled receptors studied by mutational analysis of the m3 muscarinic receptor. EMBO J. 1993;12:331–338. doi: 10.1002/j.1460-2075.1993.tb05661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley M, Wootten D, Conner MT, Simms J, Kendrick R, Logan RT, et al. Lifting the lid on GPCRs: the role of extracellular loops. Br J Pharmacol. 2012;165:1688–1703. doi: 10.1111/j.1476-5381.2011.01629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland K, Zuurmond HM, Krasel C, IJzerman AP, Lohse MJ. Involvement of Asn-293 in stereospecific agonist recognition and in activation of the beta 2-adrenergic receptor. Proc Natl Acad Sci U S A. 1996;93:9276–9281. doi: 10.1073/pnas.93.17.9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland K, Laak AM, Smit MJ, Kuhne R, Timmerman H, Leurs R. Mutational analysis of the antagonist-binding site of the histamine H(1) receptor. J Biol Chem. 1999;274:29994–30000. doi: 10.1074/jbc.274.42.29994. [DOI] [PubMed] [Google Scholar]
- Wijtmans M, de Graaf C, de Kloe G, Istyastono EP, Smit J, Lim H, et al. Triazole ligands reveal distinct molecular features that induce histamine H4 receptor affinity and subtly govern H4/H3 subtype selectivity. J Med Chem. 2011;54:1693–1703. doi: 10.1021/jm1013488. [DOI] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao BB, Hutchins CW, Carr TL, Cassar S, Masters JN, Bennani YL, et al. Molecular modeling and pharmacological analysis of species-related histamine H(3) receptor heterogeneity. Neuropharmacology. 2003;44:773–786. doi: 10.1016/s0028-3908(03)00056-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.