

Abstract
Background and Purpose
An alanine to valine exchange at amino acid position 280 (A280V) in the third intracellular loop of the human histamine H3 receptor was first identified in a patient suffering from Shy–Drager syndrome and later reported as a risk factor for migraine. Here, we have compared the pharmacological and signalling properties of wild-type (hH3RWT) and A280V mutant (hH3RA280V) receptors stably expressed in CHO-K1 cells.
Experimental Approach
The hH3RA280V cDNA was created by overlapping extension PCR amplification. Receptor expression and affinity were assessed by radioligand (N-α-[methyl-3H]-histamine) binding to cell membranes, and receptor function by the inhibition of forskolin-induced cAMP accumulation and stimulation of ERK1/2 phosphorylation in intact cells, as well as stimulation of [35S]-GTPγS binding to cell membranes.
Key Results
Both receptors were expressed at similar levels with no significant differences in their affinities for H3 receptor ligands. Upon activation the hH3RWT was significantly more efficacious to inhibit forskolin-induced cAMP accumulation and to stimulate [35S]-GTPγS binding, with no difference in pEC50 estimates. The hH3RWT was also more efficacious to stimulate ERK1/2 phosphorylation, but this difference was not significant. The inverse agonist ciproxifan was more efficacious at hH3RWT to reduce [35S]-GTPγS binding but, for both receptors, failed to enhance forskolin-induced cAMP accumulation.
Conclusions and Implications
The A280V mutation reduces the signalling efficacy of the human H3 receptor. This effect may be relevant to the pathophysiology of disorders associated with the mutation.
Linked Articles
This article is part of a themed issue on Histamine Pharmacology Update. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2013.170.issue-1
Keywords: histamine, H3 receptor, migraine, orthostatic hypotension, neuromodulation, CHO-K1 cells
Introduction
In the mammalian brain histamine regulates several functions by acting at four different GPCRs, H1–H4 (Haas et al., 2008; receptor nomenclature follows Alexander et al., 2011). Histamine H3 receptors are primarily expressed on nerve terminals, where they regulate the synthesis and release of histamine as well as the release of other neuroactive substances namely ACh, dopamine, noradrenaline, 5-HT, glutamate, GABA and substance P (Feuerstein, 2008). There is also evidence for post-synaptic H3 receptors in the striatum, cerebral cortex and hippocampus (Pillot et al., 2002).
Histamine H3 receptors couple to Gαi/o proteins and thus trigger several signalling pathways that include inhibition of adenylyl cyclase, inhibition of voltage-operated Ca2+ channels, activation of phospholipase A2 (PLA2), modulation of the MAPK pathway and activation of the Akt/glycogen synthase kinase-3β axis (Bongers et al., 2007)
The originally cloned human H3 (hH3) receptors cDNA encoded a 445 amino acid protein (Lovenberg et al., 1999) and in 2002, Wiedemann et al. reported a variation in the genomic sequence of the H3 receptors of a patient suffering from Shy–Drager syndrome, currently classified as multiple system atrophy with orthostatic hypotension (Jecmenica-Lukic et al., 2012). The identified variation in the hH3 receptors was a cytosine to thymine transition at nucleotide position 839 with respect to the start codon, which results in an alanine to valine exchange at amino acid position 280 in the third intracellular loop (Wiedemann et al., 2002; see Supporting Information Figure S1). Further, a recent report by Millán-Guerrero et al. (2011) indicated that in a Mexican population (147 cases and 186 controls) the A280V mutation was associated with increased risk of migraine.
In this work we therefore set out to compare the pharmacological and signalling properties of wild-type (hH3RWT) and A280V mutant (hH3RA280V) receptors by stably expressing them in CHO-K1 cells. A preliminary account of this work has been presented in abstract form to the European Histamine Research Society (Flores-Clemente et al., 2012).
Methods
Generation of the A280V mutant hH3 receptor
The hH3RA280V cDNA was created by overlapping extension PCR amplification essentially as described by Horton et al. (1993). Amplifications were carried out in 25 μL incubations containing 0.8 mM dNTPs, 0.4 μM each primer, 0.5 U Pfu DNA polymerase (Fermentas, Glen Burnie, MD, USA) and either 100 ng of hH3RWT for initial amplifications or gel purified fragments from the amplifications as templates. The mutated DNA fragment was reintroduced into hH3RWT backbone after digestion of unique restriction sites BstEII (position 405 relative to start codon) and NotI (vector polylinker). The sequence of the amplified fragment was verified both by restriction analysis and automated sequencing performed at FESI-UNAM., Mexico.
Cell culture and transfection
CHO-K1 cells were cultured in DMEM Nutrient F-12 (DMEM-F12) mix supplemented with 10% FBS and 1% antibiotic/antimycotic solution (Gibco Life Technologies, Grand Island, NY, USA). For stable transfection, CHO-K1 cells (70–80% confluence) were transfected with cDNA constructs using Lipofectamine 2000 (Invitrogen Life Technologies, Grand Island, NY, USA), selected in 600 μg·mL−1 geneticin (G-418), tested for receptor expression by radioligand binding and maintained in the presence of 400 μg·mL−1 geneticin.
N-α-(methyl-3H)-histamine [(3H)-NMHA] binding assays
Cells grown in plastic Petri dishes (100 mm diameter) were scrapped and homogenized in ice-cold hypotonic buffer (10 mM Tris-HCl, 1 mM EGTA, pH 7.4) and lysates centrifuged (42,000× g, 20 min at 4°C). Pellets were re-suspended in incubation buffer (50 mM Tris-HCl, 5 mM MgCl2, pH 7.4) and binding determinations (20 μg protein aliquots) were carried out and analysed as described in detail elsewhere (Osorio-Espinoza et al., 2011). Protein contents were determined by the bicinchoninic acid assay (BCA; Pierce, Rockford, IL, USA), using BSA as standard.
Measurement of cAMP accumulation in intact cells
Cells, grown in 24-well plates, were incubated (37°C) in 250 μL Krebs–Ringer–HEPES (KRH) buffer (composition (mM): NaCl 113, NaHCO3 25, KCl 3, MgCl2 1, KH2PO4 1, CaCl2 1.8, d-glucose 11, HEPES 20; pH 7.4 with NaOH) containing 1 mM IBMX. After 15 min, forskolin was added in a 10 μL volume and incubations continued for a further 15 min. Final forskolin concentrations were 10 or 3 μM for the test of agonists or inverse agonists, respectively, and where required H3 receptor agonists were added 5 min before forskolin.
Incubations were terminated with 25 μL ice-cold 1 M HCl. After neutralization with 25 μL 1 M NaOH and 100 μL 1 M Tris-HCl (pH 7.4), endogenous cAMP was determined by a competition assay for which 50 μL samples were incubated in 125 μL of incubation buffer (composition (mM): 50 Tris-HCl, 100 NaCl, 5 EDTA, 5 mg·mL−1 BSA, pH 7.0 at 4°C) containing the PKA regulatory subunit (0.5 UI per sample) and [3H]cAMP (10 nM). After 2.5 h at 4°C, incubations were terminated by filtration over GF/B filters pre-soaked in 0.3% polyethylenimine followed by three subsequent washes with 1 mL ice-cold deionized water. Retained radioactivity was determined by liquid scintillation and the amount of endogenous cAMP present in each sample was calculated by using a standard cAMP curve (10−12–10−5 M).
[35S]-GTPγS binding assay
Assays were carried out essentially as described by Gardner et al. (1996). Briefly, cells were scraped and lysed in ice-cold hypotonic buffer (20 mM HEPES, 2 mM EDTA, pH 7.4) containing a protease inhibitor cocktail (1:100 v : v; Sigma Aldrich, Mexico City, Mexico), followed by centrifugation at 42,000× g (20 min, 4°C). The pellet (membranes) was re-suspended in 10 mL HEPES buffer (20 mM HEPES, 100 mM NaCl, pH 7.4) containing adenosine deaminase (10 UI·mL−1) and incubated for 30 min at 30°C. Ice-cold HEPES buffer (20 mL) was added and the membrane suspension was centrifuged as described earlier. The resulting pellet was re-suspended and incubated (20 μg protein aliquots) in the absence and presence of drugs under test in 500 μL HEPES buffer containing 10 mM MgCl2, 0.1 mM dithiothreitol, 10 μM GDP, 1 mg·mL−1 BSA, 0.2 mM EDTA, 10 mg·mL−1 saponin and 50 pM [35S]-GTPγS. Non-specific binding was determined in the presence of 10 μM unlabelled GTPγS. After 30 min at 30°C, the reaction was terminated by rapid filtration through GF/B filters with four washes of 1 mL ice-cold HEPES buffer. Radioactivity bound to the filters was quantified by liquid scintillation counting.
Detection of H3 receptor-induced phosphorylation of ERK1/2 (p44/p42-ERK) by Western blotting
Cells, incubated overnight in DMEM-F12 medium containing 0.1% FBS, were washed with KRH buffer and incubated at 37°C with the H3 receptor agonist (R)-α-methylhistamine (RAMH). After rinsing twice with pre-warmed KRH solution and once with ice-cold solution, cells were scraped into lysis buffer [20 mM Tris-HCl, pH 8.0, 0.5 mM EDTA, 0.1% Triton X-100, 10% deoxycholate, 10% SDS, 1% glycerol, 150 mM NaCl and 1% (v : v) protease inhibitor cocktail]. The samples were cleared by centrifugation and samples (30 μg protein) were resolved by SDS-PAGE (10% gel) and then transferred onto a PVDF membrane.
Blots were blocked (1 h at 22°C) with Tris-buffered saline containing Tween-20 (0.1% v : v; T-BST) and 5% non-fat dry milk, before incubation overnight at 4°C with primary antibodies (rabbit anti phospho-ERK1/2 or anti total-ERK1/2, Cell Signaling, Danvers, MA, USA; 1:1000 dilution in T-BST containing 1.5% BSA). After rinsing with T-BST, membranes were incubated for 1 h at 22°C with a secondary antibody (goat anti-rabbit IgG coupled to HRP; 1:5000 in T-BST/1.5% BSA). Membranes were rinsed with T-BST, developed with chemiluminescence (Enhanced ChemiLuminescent Substrate; Biorad, Hercules, CA, USA) and quantified by densitometry analysis with the Kodak Image Station 4000R (Eastman Kodak, Rochester, NY, USA) and ImageJ software (National Institutes of Health, USA). Target bands were expressed quantitatively by normalization to the intensity of the non-phosphorylated ERK1/2 signal on the same lane. Protein loading was also controlled by actin immunodetection.
Data analysis
Results are presented as the means ± s.e.m. of the indicated number of experiments. Statistical comparisons were performed with Student's unpaired t-test or one-way ANOVA followed by Dunnett's test, as appropriate (Prism 5.0 software; GraphPad Software, San Diego, CA, USA). Differences were considered significant at P < 0.05.
Materials
The following drugs and reagents were purchased from Sigma Aldrich: A-331440 (4′-[3-[(3(R)-dimethylamino-1-pyrrolidinyl]propoxy]-[1,1-biphenyl]-4′-carbonitrile dihydrochloride), adenosine deaminase (from bovine spleen), cAMP, ciproxifan hydrochloride, clobenpropit dihydrobromide, DTT, GDP, IBMX, geneticin, GTPγS, histamine dihydrochloride, PKA regulatory subunit (from bovine heart), (R)-α-methylhistamine dihydrochloride and saponin. N-α-[methyl-3H]-histamine (85.4 Ci·mmol−1), [35S]GTPγS (1250 Ci·mmol−1) and [3H]-cAMP ([2,8-3H]-adenosine 3′,5′-cyclic phosphate, 34 Ci·mmol−1) were from Perkin Elmer (Boston, MA, USA).
Results
Binding characteristics of hH3 receptors stably expressed in CHO-K1 cells
Specific [3H]-NMHA binding to cell membranes, yielded maximum binding (Bmax) 203 ± 34 and 176 ± 26 fmol·mg·protein−1 (mean ± SEM; four experiments, Figure 1A) for hH3RWT and hH3RA280V, respectively, with no statistical difference (P = 0.25, Student's t-test). Estimates for the equilibrium dissociation constant (Kd) were 0.86 ± 0.23 and 0.83 ± 0.24 nM respectively (P = 0.46).
Figure 1.
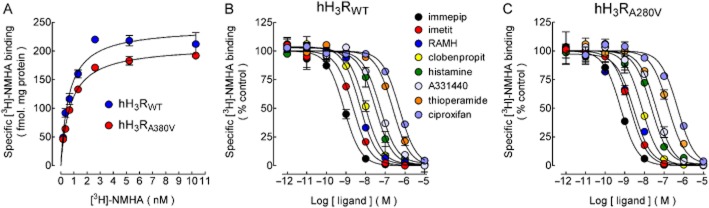
Binding of N-α-[methyl-3H]histamine ([3H]-NMHA) to membranes from CHO-K1-hH3RWT or CHO-K1-hH3RA280V cells. (A) Saturation binding. Cell membranes were incubated with the indicated concentrations of [3H]-NMHA and specific receptor binding was determined by subtracting the binding in the presence of 10 μM histamine from total binding. Points are means ± SEM from triplicate determinations from a single experiment, which was repeated a further three times. The line drawn is the best fit to a hyperbola. Best-fit values for the equilibrium dissociation constant (Kd) and maximum binding (Bmax) are given in the text. (B,C) Inhibition by H3 receptor ligands. Membranes from CHO-K1-hH3RWT (B) or CHO-K1-hH3RA280V cells (C) were incubated with 1.5 nM [3H]-NMHA and the indicated drug concentrations. Values are expressed as percentage of control specific binding and are averages ± ranges of duplicates from a single experiment, repeated a further two to four times with similar results. The line drawn is the best fit to a logistic equation for a one-site model. pKi values calculated from the best-fit IC50 estimates are compared in Table 1.
For both receptors, a set of agonists (histamine, imetit, immepip and RAMH) and antagonists/inverse agonists (A-331440, ciproxifan, clobenpropit and thioperamide) inhibited [3H]-NMHA binding in a concentration-dependent manner (Figure 1B,C). Estimates for the inhibition constants (-log Ki, pKi; Cheng and Prusoff, 1973) are shown in Table 1. There was no significant difference for any of the ligands tested between the native (wild type) and mutant receptors. Further, the affinities of both receptors were in good accord with values previously reported for the expression of cloned hH3 receptors in CHO-K1 cells (Ligneau et al., 2000; Cogé et al., 2001; Table 1).
Table 1.
Affinities of hH3RWT or hH3RA280V for selective ligands
| pKi values | ||||
|---|---|---|---|---|
| This study | Ligneau et al., 2000 | Cogé et al., 2001 | ||
| hH3RWT | hH3RA280V | |||
| Agonists | ||||
| Histamine | 8.30 ± 0.10 | 8.38 ± 0.12 (P = 0.623, n = 4) | 7.89 | 8.24 ± 0.01 |
| Immepip | 9.51 ± 0.03 | 9.58 ± 0.01 (P = 0.091, n = 3) | ND | 9.60 ± 0.15 |
| Imetit | 9.23 ± 0.19 | 9.29 ± 0.05 (P = 0.775, n = 3) | ND | 9.37 ± 0.22 |
| RAMH | 8.92 ± 0.11 | 9.05 ± 0.10 (P = 0.431, n = 3) | 8.56 | 8.73 ± 0.09 |
| Antagonists/inverse agonists | ||||
| A-331440 | 7.75 ± 0.04 | 7.77 ± 0.08 (P = 0.834, n = 3) | ND | ND |
| Ciproxifan | 7.17 ± 0.04 | 7.21 ± 0.10 (P = 0.729, n = 3) | 7.33 | 7.26 ± 0.02 |
| Clobenpropit | 8.79 ± 0.08 | 8.64 ± 0.12 (P = 0.329, n = 5) | 8.62 | 9.27 ± 0.05 |
| Thioperamide | 7.34 ± 0.13 | 7.37 ± 0.05 (P = 0.840, n = 3) | 7.22 | 7.70 ± 0.07 |
Values are pKi estimates (-Log10 of the inhibition constant, Ki) and correspond to means ± SEM from the indicated number of determinations. Figures in parentheses are P values (Student's t-test) for the comparison between hH3RWT and hH3RA280V cells. Determinations reported in Ligneau et al. (2000) and Cogé et al. (2001) were performed with the hH3 receptor stably expressed in CHO-K1 cells. Values from Ligneau et al. (2000) were transformed to pKi estimates from the Ki values provided. ND, not determined.
Agonist-induced inhibition of forskolin-induced cAMP accumulation
Forskolin-stimulated cAMP accumulation in a similar manner for CHO-K1-hH3RWT and CHO-K1-hH3RA280V cells (Supporting Information Figure S2), and the effect of 10 μM of forskolin was significantly reduced by the H3 receptor agonist RAMH (Figure 2A). However, maximum inhibition was significantly lower in cells expressing the mutant receptor (Table 2). The estimates for half-maximal effective concentrations (pEC50) showed a tendency to a lower potency for CHO-K1-hH3RA280V cells, but the difference was not statistically significant, as shown in Table 2.
Figure 2.
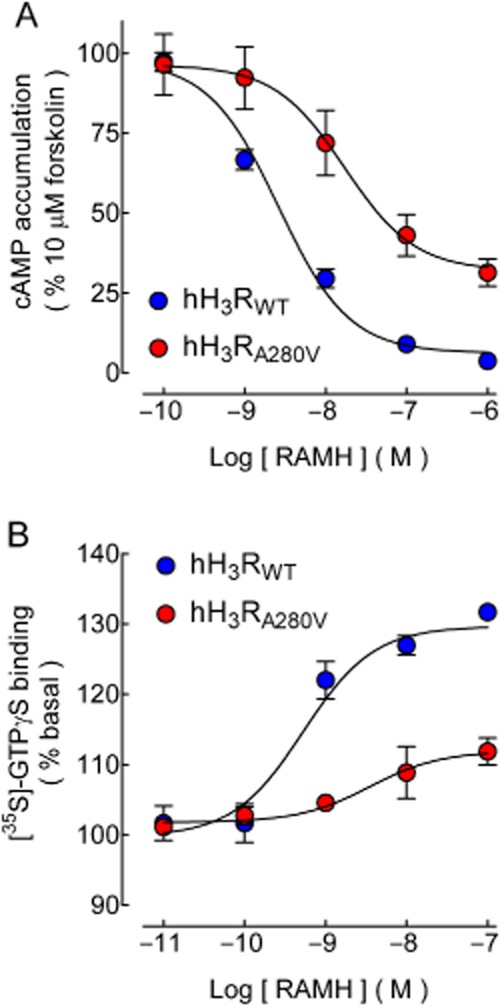
Effect of H3 receptor activation on forskolin-induced cAMP accumulation and [35S]-GTPγS binding. (A) Forskolin-induced cAMP accumulation. CHO-K1-hH3RWT or CHO-K1-hH3RA280V cells were pre-incubated (15 min) with 1 mM IBMX and then exposed for 15 min to forskolin (10 μM). Where required the H3 receptor agonist RAMH was added 5 min before forskolin. Values are expressed as percentage of the response to forskolin, after basal subtraction, and correspond to the means ± SEM from four replicates from a representative experiment, repeated a further four times with similar results. The line drawn is the best fit to a logistic equation. Estimates for maximum effect (Emax) and pEC50 are compared in Table 2. Raw data from a representative experiment are presented in Supporting Information Figure S2. (B). [35S]-GTPγS binding. Membranes from CHO-K1-hH3RWT or CHO-K1-hH3RA280V cells were incubated (30 min) with 50 pM [35S]-GTPγS in the presence and absence of the indicated concentrations of RAMH. Values are expressed as percentage of basal [35S]-GTPγS binding after subtraction of non-specific binding and correspond to the means ± SEM from three replicates from a representative experiment, repeated a further seven times with similar results. The line drawn is the best fit to a logistic equation. Estimates for maximum stimulation and pEC50 are compared in Table 2. Basal [35S]-GTPγS binding was 4993 ± 226 and 5179 ± 43 dpm for membranes from CHO-K1-hH3RWT or CHO-K1-hH3RA280V cells, respectively. Raw data from a representative experiment are presented in Supporting Information Figure S3.
Table 2.
Effect of receptor activation on forskolin-induced cAMP accumulation and [35S]-GTPγS binding
| hH3RWT | hH3RA280V | P | |
|---|---|---|---|
| cAMP accumulation | |||
| Emax (%) | −93.9 ± 2.6 | −57.2 ± 8.4 | 0.003 |
| pEC50 | 8.22 ± 0.35 | 7.75 ± 0.05 | 0.224 |
| [35S]-GTPγS binding | |||
| Emax (% basal) | 129.2 ± 2.6 | 112.6 ± 2.7 | <0.001 |
| pEC50 | 8.50 ± 0.14 | 9.00 ± 0.32 | 0.174 |
| ERK1/2 phosphorylation | |||
| Emax (% basal) | 302 ± 63 | 217 ± 31 | 0.155 |
| pEC50 | 7.94 ± 0.17 | 7.49 ± 0.19 | 0.128 |
Values are means ± SEM from five (cAMP accumulation), eight ([35S]-GTPγS binding) or four (ERK1/2 phosphorylation) experiments. Statistical comparisons were carried out with Student's t-test.
Agonist-induced [35S]-GTPγS binding
The ability of GPCRs to activate G-proteins is also studied by determining [35S]-GTPγS binding to cell membranes. Maximum stimulation evoked by the agonist RAMH was significantly higher in membranes from CHO-K1-hH3RWT cells (Figure 2B and Table 2, and Supporting Information Figure S3A), with no significant difference in the pEC50 estimates.
Agonist-induced ERK1/2 phosphorylation
Activation of transfected H3 receptors leads to ERK1/2 phosphorylation (Bongers et al., 2007), and in both CHO-K1-hH3RWT and CHOK1-hH3RA280V cells, incubation (1–15 min) with the agonist RAMH resulted in increased ERK1/2 phosphorylation over basal levels that reached a peak between 2.5 and 5 min to decay steadily afterwards (Supporting Information Figure S4A). At 5 min incubations, the effect was concentration-dependent (Supporting Information Figure S4B), with a tendency for higher efficacy in CHOK1-hH3RWT cells, although the effect was not significant (Table 2).
Effect of inverse agonists on [35S]-GTPγS binding and forskolin-induced cAMP accumulation
Native (wild type) and cloned H3 receptors may exhibit spontaneous or constitutive activity (Arrang et al., 2007), and for hH3 receptors expressed in CHO cells such an activity is apparent in cells expressing 250–400 fmol mg−1 protein of the hH3 receptor (Rouleau et al., 2002). Because the reduced efficacy of hH3RA280V observed in our experiments may rely on higher intrinsic activity, the effect of antagonists/inverse agonists was tested in functional assays.
For both native and mutant receptors, the H3 receptor antagonist/inverse agonist ciproxifan (Leurs et al., 2005) reduced [35S]-GTPγS binding to cell membranes. The effect was modest (Figures 3A and Supporting Information Figure S3B), but significantly higher for membranes from CHO-K1-hH3RWT cells (−12.03 ± 0.29 vs. −5.85 ± 0.60% of basal for CHOK1-hH3RA280V membranes; P < 0.001, n = 4, Student's t-test), without a significant effect in agonist potency (pEC50 values 9.06 ± 0.45 and 9.09 ± 0.73, respectively; P = 0.964). The errors associated to pEC50 values are large, most likely due to the small window for the ciproxifan effect which hampers the estimate accuracy.
Figure 3.
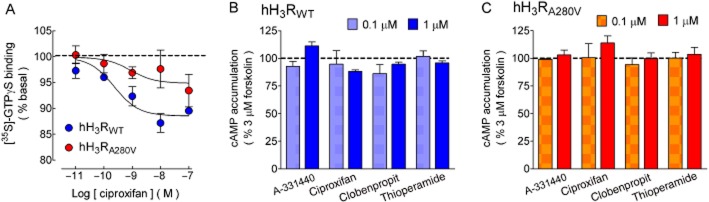
Effect of the H3 receptor inverse agonists on [35S]-GTPγS binding and forskolin-induced cAMP accumulation. (A) [35S]-GTPγS binding. Membranes from CHO-K1-hH3RWT or CHO-K1-hH3RA280V cells were incubated (30 min) with 50 pM [35S]-GTPγS in the presence and absence of the indicated concentrations of ciproxifan. Values are expressed as percentage of basal [35S]-GTPγS binding after subtraction of non-specific binding and correspond to the means ± SEM from three replicates from a representative experiment, repeated a further three times with similar results. The line drawn is the best fit to a logistic equation. Best-fit values for maximum effect (Emax) and pEC50 are given in the text. Basal [35S]-GTPγS binding was 3527 ± 54 and 4487 ± 77 dpm for membranes from CHO-K1-hH3RWT or CHO-K1-hH3RA280V cells respectively. Raw data from a representative experiment are presented in Supporting Information Figure S3. (B,C) Forskolin-induced cAMP accumulation. CHO-K1-hH3RWT or CHO-K1-hH3RA280V cells were pre-incubated (15 min) with 1 mM IBMX and then exposed for 15 min to forskolin (3 μM). Where required, H3 receptor inverse agonists (0.1 and 1 μM) were added 5 min before forskolin. Values are expressed as percentage of the response to forskolin, after basal subtraction, and correspond to the means ± SEM from three or four experiments for 0.1 or 1 μM of the inverse agonists, respectively. None of the values in the presence of H3 receptor inverse agonists was significantly different from control values (P > 0.05, one-way ANOVA and post hoc Dunnett's test).
Forskolin-stimulated cAMP accumulation increased in a concentration-dependent manner (Supporting Information Figure S2), but the slope was significantly higher between 1 and 10 μM than between 10 and 30 μM (2.05 ± 0.21 and 0.53 ± 0.13, respectively; P < 0.001, Student's t-test). For testing receptor constitutive activity, the forskolin concentration was therefore set at 3 μM in order to reliably assess the possible enhancing action of inverse agonists.
In contrast to [35S]-GTPγS binding to cell membranes, for both CHO-K1-hH3RWT and CHOK1-hH3RA280V cells ciproxifan had no significant effect on forskolin-induced cAMP accumulation over a wide range of concentrations (10−11–10−6 M, three experiments; data not shown). Other H3 receptor inverse agonists (clobenpropit, thioperamide and A-331440) also failed to modify cAMP accumulation at concentrations of 0.1 and 1 μM, as shown in Figure 3C (panels B and C).
Discussion and conclusions
The main finding of this work is that the A280V mutation on the hH3 receptor modifies its signalling efficacy without affecting the binding characteristics.
Effect of the A280V mutation on H3 receptor binding and functional characteristics
Binding determinations indicated that the A280V mutation did not affect hH3 receptor expression or its affinity for a series of selective ligands. The latter result was not unexpected because the mutation is not located on any of the receptor regions primarily involved in ligand binding, namely trans-membrane domains 3, 5 and 6 (Uveges et al., 2002; Ishikawa et al., 2010; Kim et al., 2011). In contrast, the mutant receptor was less efficacious to inhibit forskolin-stimulated cAMP accumulation and to stimulate [35S]-GTPγS binding, indicating that the mutation did affect the signalling properties of the receptor.
There was also a trend for lower efficacy for the mutant receptor for the stimulation of ERK1/2 phosphorylation, but the effect did not reach statistical significance. Receptor-induced ERK1/2 phosphorylation can be due either to Gβγ subunits or β-arrestins bound to activated receptors acting as a signalling scaffold (Gutkind, 2000; DeWire et al., 2007), but the fast response induced by hH3RWT and hH3RA280V indicates that the effect is more likely to rely on βγ complexes than on β-arrestin recruitment because the latter is a delayed response with a peak at 30 min incubations (Rosethorne and Charlton, 2011). The lack of difference in the efficacy of hH3RWT and hH3RA280V for the stimulation of ERK1/2 phosphorylation may reflect the distinct sensitivity of the pathways to the signalling molecules activated upon receptor stimulation.
Native and cloned H3 receptors show constitutive activity (Arrang et al., 2007), and in CHO cells spontaneous activity of the hH3 receptor, evaluated by ciproxifan-induced inhibition of basal [35S]-GTPγS binding is apparent from a density of 250 fmol receptor per mg protein (Rouleau et al., 2002). Our [35S]-GTPγS binding data indicate that both receptors do have constitutive activity when expressed in CHO-K1 cells, although to a modest extent. However, H3 receptor inverse agonists failed to enhance forskolin-induced cAMP accumulation. This discrepancy may rely on the activated G proteins detected by [35S]-GTPγS binding not yielding the concentration required for a significant inhibitory effect upon adenylyl cyclases stimulated by forskolin.
There is no direct evidence from this study as to the molecular mechanism responsible for the effect of the A280V mutation on H3 receptor function, although the change in agonist efficacy but not in potency suggests that the mutation is hampering the receptor ability to activate G-proteins. The A280V mutation is located in the receptor third intracellular loop (Figure S1) which plays a key role in determining receptor/G-protein coupling (Wess, 1998). Activated receptors catalyse GDP release from G-protein heterotrimers and this is the rate-limiting step in G-protein activation and, consequently, the activation of downstream signalling proteins (Oldham and Hamm, 2007). Because both alanine and valine are neutral amino acids, the A280V mutation confers no change in charge, but in size (71.1 and 99.1 daltons, and 0.067 and 0.105 nm3, respectively; Zamyatnin, 1972), and one possible explanation for the reduced efficacy of the mutant receptor is a steric reduction in the access of the C-terminus of the Gαi/o protein to sites in the cytoplasmic face of the receptor trans-membrane helices required for G-protein stimulation (Oldham and Hamm, 2007).
Possible implications of the A280V mutation for the pathophysiology of the Shy–Drager syndrome and migraine
This study originated from reports that the A280V mutation may be involved in the pathophysiology of two neurological disorders, the Shy–Drager syndrome (Wiedemann et al., 2002) and migraine (Millán-Guerrero et al., 2011).
The Shy–Drager syndrome is now recognized as a form of multiple system atrophy, a sporadic and progressive neurological disease characterized by autonomic dysfunction, Parkinsonism and ataxia in any combination (Jecmenica-Lukic et al., 2012). In the patient in whom the A280V mutation was first identified, a reduction in both noradrenaline plasma level and urinary excretion of the amine and its metabolites was found, and a link between the H3 receptor mutation and altered noradrenaline release was suggested (Wiedemann et al., 2002). Because H3 receptor activation reduces depolarization-evoked noradrenaline release from sympathetic nerve terminals (Silver et al., 2002), the decrease in noradrenaline plasma levels cannot be directly explained by the reduced efficacy of the mutant receptor reported herein, which would instead lead to increased noradrenaline release from peripheral neurones, in analogy with the enhanced release from sympathetic nerve endings observed in mice lacking H3 receptors (Koyama et al., 2003). Further, orthostatic hypotension has been mainly related to dysfunction of the catecholaminergic rostral ventrolateral medulla nuclei (Colosimo, 2011), and one possibility to explain the alterations in arterial pressure is that the reduced signalling efficacy of mutant H3 auto-receptors enhances histamine release resulting in augmented activation of H3 hetero-receptors located on central noradrenergic/adrenergic neurones (Pillot et al., 2002) and thus in inhibition of neuronal firing. Alternatively, reduced receptor efficacy might lead to diminished H3 receptor-mediated inhibition of neurotransmitter release and increased auto-receptor activation in cathecolaminergic neurones resulting in decreased neuronal activity. In turn, reduced activity of the rostral ventrolateral medulla nuclei would result in decreased sympathetic vasomotor tone (Guyenet, 2006).
Migraine is a neurovascular disorder involving activation of the trigemino-vascular system with the primary dysfunction located in brainstem centres regulating vascular tone and pain sensation (Cutrer, 2010). Its frequency is significantly higher in patients with allergic (histamine-driven) disease (Ku et al., 2006; Aamodt et al., 2007) and plasma histamine levels are significantly elevated in migraine patients (Heatley et al., 1982). Recently, Millán-Guerrero et al. (2011) reported that the A280V mutation in the hH3 receptor was significantly associated with increased risk of migraine with V allele frequencies of 6.46% and 2.68% for cases and controls, respectively (odds ratio 2.67), and frequencies of the V/V and V/A genotypes 12.92% and 3.22% in migraine patients and controls (odds ratio 4.45). This group also reported that S.C. injections of low doses of histamine or the H3 receptor agonist NAMH (twice a week over 12 weeks) reduced pain intensity, frequency and length of attacks as well as painkiller use in migraine patients, relating these actions to the activation of H3 receptors controlling the release of histamine and other neurotransmitters (Millán-Guerrero et al., 2006; 2009).
Alterations in the release of glutamate and neuroactive peptides have also been linked to migraine. Higher levels of glutamate have been found in plasma and in saliva samples in migraine patients when compared with healthy controls (Tajti et al., 2011), and trigeminal nociceptive fibres release the vasoactive neuropeptides, substance P, CGRP and neurokinin A (Cutrer, 2010). Pre-synaptic H3 receptors reduce substance P and neurokinin release from sensory nerve endings as well as glutamate release in several areas of the rat brain (see Feuerstein, 2008 and Osorio-Espinoza et al., 2010). Thus, the reduced efficacy of the mutant hH3 receptor reported herein may be also related to a diminished inhibitory action of endogenous histamine on the release of glutamate and neuroactive peptides, leading to increased susceptibility to migraine attacks.
In conclusion, we have here provided evidence that a single point mutation in the third intracellular loop of the human histamine H3 receptor modifies its functional properties. This effect may have relevance for the pathophysiology of disorders associated with the mutation, namely migraine and multiple system atrophy with orthostatic hypotension, but further research on this issue is clearly required.
Acknowledgments
Supported by Cinvestav, Conacyt (grants 128205 to J.-A.A.-M and 131778 to J.-M.A.) and PAPIIT-UNAM (grant IN214509 to J.-M.A.). We thank Raul Gonzalez-Pantoja for excellent technical assistance.
Glossary
- NHMA
N-α-methyl histamine
- RAMH
(R)-α-methyl histamine
Conflicts of interest
The authors disclose no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Location of the A280V mutation in the human H3R445. A cytosine to thymine transition at nucleotide position 839 with respect to the start codon results in an alanine to valine exchange at amino acid position 280 in the third intracellular loop (red circle).
Figure S2 Characteristics of forskolin-induced cAMP accumulation and hH3 receptor-mediated inhibition. In all determinations cells were pre-incubated (15 min) with 1 mM IBMX and then exposed for 15 min to forskolin. (A,B) Concentration-response curve in CHO-K1 cells. (A) Representative experiment. Values are means ± SEM from four replicates. (B) Analysis of the concentration–response curve from four experiments. Values are expressed as pmol cAMP per sample after subtraction of basal accumulation, and correspond to the means ± SEM. Lines are data best-fit by linear regression analysis. (C,D) Representative determinations of H3 receptor-mediated inhibition of forskolin-induced cAMP accumulation in CHO-K1-hH3RWT (C) or CHO-K1-hH3RA280V (D) cells. Cells were incubated for 15 min with forskolin (10 μM) in the absence and presence of the H3 receptor agonist RAMH, added 5 min before forskolin. Values are means ± SEM from three to four replicates from representative determinations. The experiments were repeated a further 4 times with similar results.
Figure S3 Representative determinations for [35S]-GTPγS binding. Membranes from CHO-K1-hH3RWT or CHO-K1-hH3RA280V cells were incubated (30 min) with 50 pM [35S]-GTPγS in the presence and absence of the indicated drug concentrations. (A) Stimulation by the agonist RAMH. (B) Inhibition by the inverse agonist ciproxifan. For both panels values are means ± SEM from four replicates from a representative experiment after subtraction of non-specific binding. The line drawn is the best fit to a logistic equation. The experiment was repeated a further four times with similar results.
Figure S4 Effect of H3 receptor activation on ERK1/2 phosphorylation. ERK1/2 phosphorylation was analysed by Western blot. (A) Time course. CHO-K1-hH3RWT or CHO-K1-hH3RA280V cells were incubated for 1–15 min with the agonist RAMH (100 nM). A1) Representative blot. Similar results were obtained in two independent determinations for each cell line. P-ERK1/2, phosphorylated ERK1/2. A2) Densitometric analysis. Western blot target bands were quantified by densitometry analysis and normalized according to the intensity of the non-phosphorylated ERK1/2 signal on the same lane. Values are expressed as percentage of basal phosphorylation and correspond to the means from two determinations. (B) Concentration-response curves. Cells were incubated for 5 min with the indicated concentrations of the agonist RAMH. B1) Representative blot. Similar results were obtained in three other independent experiments. B2) Analysis from four determinations. Values (means ± SEM) are expressed as percentage of basal phosphorylation. The lines drawn are the best fit to a logistic equation. Estimates for maximum stimulation and EC50 are given in the text.
References
- Aamodt AH, Stovner LJ, Langhammer A, Hagen K, Zwart JA. Is headache related to asthma, hay fever, and chronic bronchitis? The Head-HUNT study. Headache. 2007;47:204–212. doi: 10.1111/j.1526-4610.2006.00597.x. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrang JM, Morisset S, Gbahou F. Constitutive activity of the histamine H3 receptor. Trends Pharmacol Sci. 2007;28:350–357. doi: 10.1016/j.tips.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Bongers G, Bakker RA, Leurs R. Molecular aspects of the histamine H3 receptor. Biochem Pharmacol. 2007;73:1195–1204. doi: 10.1016/j.bcp.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cogé F, Guénin SP, Audinot V, Renouard-Try A, Beauverger P, Macia C, et al. Genomic organization and characterization of splice variants of the human histamine H3 receptor. Biochem J. 2001;355:279–288. doi: 10.1042/0264-6021:3550279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo C. Nonmotor presentations of multiple system atrophy. Nat Rev Neurol. 2011;7:295–298. doi: 10.1038/nrneurol.2011.5. [DOI] [PubMed] [Google Scholar]
- Cutrer FM. Pathophysiology of migraine. Semin Neurol. 2010;30:120–130. doi: 10.1055/s-0030-1249222. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. β-Arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Feuerstein TJ. Presynaptic receptors for dopamine, histamine, and serotonin. Handb Exp Pharmacol. 2008;184:289–338. doi: 10.1007/978-3-540-74805-2_10. [DOI] [PubMed] [Google Scholar]
- Flores-Clemente C, Osorio-Espinoza A, Escamilla-Sánchez J, Arias JM, Arias-Montaño JA. Comparison of the pharmacological and signaling properties of wild-type and A280V mutant human histamine H3 receptors expressed in CHO-K1 cells. Inflamm Res. 2012;61(Suppl. 2):S77. [Google Scholar]
- Gardner B, Hall DA, Strange PG. Pharmacological analysis of dopamine stimulation of [35S]-GTPγS binding via human D2short and D2long dopamine receptors expressed in recombinant cells. Br J Pharmacol. 1996;118:1544–1550. doi: 10.1111/j.1476-5381.1996.tb15572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutkind JS. Regulation of mitogen-activated protein kinase signaling networks by G protein-coupled receptors. Sci STKE. 2000;2000:re1. doi: 10.1126/stke.2000.40.re1. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88:1183–1241. doi: 10.1152/physrev.00043.2007. [DOI] [PubMed] [Google Scholar]
- Heatley RV, Denburg JA, Bayer N, Bienenstock J. Increased plasma histamine levels in migraine patients. Clin Allergy. 1982;12:145–149. doi: 10.1111/j.1365-2222.1982.tb01633.x. [DOI] [PubMed] [Google Scholar]
- Horton RM, Ho SN, Pullen JK, Hunt HD, Cai Z, Pease LR. Gene splicing by overlap extension. Methods Enzymol. 1993;217:270–279. doi: 10.1016/0076-6879(93)17067-f. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Watanabe T, Kudo T, Yokoyama F, Yamauchi M, Kato K, et al. Investigation of the histamine H3 receptor binding site. Design and synthesis of hybrid agonists with a lipophilic side chain. J Med Chem. 2010;53:6445–6456. doi: 10.1021/jm100643t. [DOI] [PubMed] [Google Scholar]
- Jecmenica-Lukic M, Poewe W, Tolosa E, Wenning GK. Premotor signs and symptoms of multiple system atrophy. Lancet Neurol. 2012;11:361–368. doi: 10.1016/S1474-4422(12)70022-4. [DOI] [PubMed] [Google Scholar]
- Kim SK, Fristrup P, Abrol R, Goddard WA., 3rd Structure-based prediction of subtype selectivity of histamine H3 receptor selective antagonists in clinical trials. J Chem Inf Model. 2011;51:3262–3274. doi: 10.1021/ci200435b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama M, Seyedi N, Fung-Leung WP, Lovenberg TW, Levi R. Norepinephrine release from the ischemic heart is greatly enhanced in mice lacking histamine H3 receptors. Mol Pharmacol. 2003;63:378–382. doi: 10.1124/mol.63.2.378. [DOI] [PubMed] [Google Scholar]
- Ku M, Silverman B, Prifti N, Ying W, Persaud Y, Schneider A. Prevalence of migraine headaches in patients with allergic rhinitis. Ann Allergy Asthma Immunol. 2006;97:226–230. doi: 10.1016/S1081-1206(10)60018-X. [DOI] [PubMed] [Google Scholar]
- Leurs R, Bakker RA, Timmerman H, de Esch IJ. The histamine H3 receptor: from gene cloning to H3 receptor drugs. Nat Rev Drug Discov. 2005;4:107–120. doi: 10.1038/nrd1631. [DOI] [PubMed] [Google Scholar]
- Ligneau X, Morisset S, Tardivel-Lacombe J, Gbahou F, Ganellin CR, Stark H, et al. Distinct pharmacology of rat and human histamine H3 receptors: role of two amino acids in the third transmembrane domain. Br J Pharmacol. 2000;131:1247–1250. doi: 10.1038/sj.bjp.0703712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, et al. Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol. 1999;55:1101–1107. [PubMed] [Google Scholar]
- Millán-Guerrero RO, Isais-Millán R, Benjamín TH, Tene CE. Nα-methyl histamine safety and efficacy in migraine prophylaxis: phase III study. Can J Neurol Sci. 2006;33:195–199. doi: 10.1017/s0317167100004960. [DOI] [PubMed] [Google Scholar]
- Millán-Guerrero RO, Isais-Millán S, Barreto-Vizcaíno S, Rivera-Castaño L, Rios-Madariaga C. Subcutaneous histamine versus botulinum toxin type A in migraine prophylaxis: a randomized, double-blind study. Eur J Neurol. 2009;16:88–94. doi: 10.1111/j.1468-1331.2008.02352.x. [DOI] [PubMed] [Google Scholar]
- Millán-Guerrero RO, Baltazar-Rodríguez LM, Cárdenas-Rojas MI, Ramírez-Flores M, Isais-Millán S, Delgado-Enciso I, et al. A280V polymorphism in the histamine H3 receptor as a risk factor for migraine. Arch Med Res. 2011;42:44–47. doi: 10.1016/j.arcmed.2011.01.009. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. How do receptors activate G proteins? Adv Protein Chem. 2007;74:67–93. doi: 10.1016/S0065-3233(07)74002-0. [DOI] [PubMed] [Google Scholar]
- Osorio-Espinoza A, Ramos-Jimenez J, Arias-Montaño JA. Presynaptic control by histamine H3 receptors of neurotransmitter release. In: Shahid M, Khardori N, Kahan RA, Tripathi T, editors. Biomedical Aspects of Histamine: Current Perspectives. London: Springer; 2010. pp. 339–370. [Google Scholar]
- Osorio-Espinoza A, Alatorre A, Ramos-Jimenez J, Garduño-Torres B, García-Ramírez M, Querejeta E, et al. Pre-synaptic histamine H3 receptors modulate glutamatergic transmission in rat globus pallidus. Neuroscience. 2011;176:20–31. doi: 10.1016/j.neuroscience.2010.12.051. [DOI] [PubMed] [Google Scholar]
- Pillot C, Heron A, Cochois V, Tardivel-Lacombe J, Ligneau X, Schwartz JC, et al. A detailed mapping of the histamine H3 receptor and its gene transcripts in rat brain. Neuroscience. 2002;114:173–193. doi: 10.1016/s0306-4522(02)00135-5. [DOI] [PubMed] [Google Scholar]
- Rosethorne EM, Charlton SJ. Agonist-biased signaling at the histamine H4 receptor: JNJ7777120 recruits β-arrestin without activating G proteins. Mol Pharmacol. 2011;79:749–757. doi: 10.1124/mol.110.068395. [DOI] [PubMed] [Google Scholar]
- Rouleau A, Ligneau X, Tardivel-Lacombe J, Morisset S, Gbahou F, Schwartz JC, et al. Histamine H3-receptor-mediated [35S]GTPγ[S] binding: evidence for constitutive activity of the recombinant and native rat and human H3 receptors. Br J Pharmacol. 2002;135:383–392. doi: 10.1038/sj.bjp.0704490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RB, Poonwasi KS, Seyedi N, Wilson SJ, Lovenberg TW, Levi R. Decreased intracellular calcium mediates the histamine H3-receptor-induced attenuation of norepinephrine exocytosis from cardiac sympathetic nerve endings. Proc Natl Acad Sci USA. 2002;99:501–506. doi: 10.1073/pnas.012506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajti J, Párdutz A, Vámos E, Tuka B, Kuris A, Bohár Z, et al. Migraine is a neuronal disease. J Neural Transm. 2011;118:511–524. doi: 10.1007/s00702-010-0515-3. [DOI] [PubMed] [Google Scholar]
- Uveges AJ, Kowal D, Zhang Y, Spangler TB, Dunlop J, Semus S, et al. The role of transmembrane helix 5 in agonist binding to the human H3 receptor. J Pharmacol Exp Ther. 2002;301:451–458. doi: 10.1124/jpet.301.2.451. [DOI] [PubMed] [Google Scholar]
- Wess J. Molecular basis of receptor/G-protein-coupling selectivity. Pharmacol Ther. 1998;80:231–264. doi: 10.1016/s0163-7258(98)00030-8. [DOI] [PubMed] [Google Scholar]
- Wiedemann P, Bönisch H, Oerters F, Brüss M. Structure of the human histamine H3 receptor gene (HRH3) and identification of naturally occurring variations. J Neural Transm. 2002;109:443–453. doi: 10.1007/s007020200036. [DOI] [PubMed] [Google Scholar]
- Zamyatnin AA. Protein volume in solution. Prog Biophys Mol Biol. 1972;24:107–123. doi: 10.1016/0079-6107(72)90005-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.