

Abstract
Background and Purpose
The histaminergic tuberomamillary nucleus (TMN) of the posterior hypothalamus controls the cognitive aspects of vigilance which is reduced by common sedatives and anxiolytics. The receptors targeted by these drugs in histaminergic neurons are unknown. TMN neurons express nine different subunits of the GABAA receptor (GABAAR) with three α- (α1, α2 and α5) and two γ- (γ1, γ 2) subunits, which confer different pharmacologies of the benzodiazepine-binding site.
Experimental Approach
We investigated the actions of zolpidem, midazolam, diazepam, chlordiazepoxide, flumazenil (Ro15-1788) and methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate (DMCM) in TMN neurons using mouse genetics, electrophysiological and molecular biological methods.
Key Results
We find the sensitivity of GABAAR to zolpidem, midazolam and DMCM significantly reduced in TMN neurons from γ2F77I mice, but modulatory activities of diazepam, chlordiazepoxide and flumazenil not affected. Potencies and efficacies of these compounds are in line with the dominance of α2- and α1-subunit containing receptors associated with γ2- or γ1-subunits. Functional expression of the γ1-subunit is supported by siRNA-based knock-down experiments in γ2F77I mice.
Conclusions and Implications
GABAAR of TMN neurons respond to a variety of common sedatives with a high affinity binding site (γ2F77I) involved. The γ1-subunit likely contributes to the action of common sedatives in TMN neurons. This study is relevant for understanding the role of neuronal histamine and benzodiazepines in disorders of sleep and metabolism.
Linked Articles
This article is part of a themed issue on Histamine Pharmacology Update. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2013.170.issue-1
Keywords: GABA, whole-cell patch-clamp, microelectrode array, histamine, zolpidem, single-cell RT-PCR
Introduction
Most frequently used drugs for the treatment of insomnia and anxiety are acting at the benzodiazepine-site of GABAA receptors (GABAAR). These receptors are pentameric assemblies of subunits that form a central ion channel (in mammals: α1–6, β1–3, γ1–3, δ, π, ρ1–3, ε, θ). The GABA-binding pocket is formed at the α/β subunit interface, whereas the modulatory benzodiazepine binding site is located at the α/γ interface (Sigel, 2002) in the subunits arrangement γ-β-α-β-α (Baumann et al., 2002; Farrant and Nusser, 2005). Nineteen known GABAAR subunits co-assemble with a restricted number of preferred combinations. For example, the prevailing GABAAR type in the hypothalamus, striatum and amygdala is composed of α2, β3 and γ2-subunits (Wisden et al., 1992). This molecular structure provides the basis for selective pharmacological modulation of inhibition within and between diverse neuronal networks of the brain. The preference for one of the α-subunit types by some benzodiazepine-site modulators can mark different behavioural actions: sedation and hypnosis [α1 preferring (Anaclet et al., 2012)] or anxiolysis [α2 preferring (Rudolph and Mohler, 2006)]. Benzodiazepine-site ligands at the GABAAR have a long clinical history for the treatment of insomnia and support a role of GABA in sleep (Wafford and Ebert, 2008). GABA is an evolutionary old and conserved ‘sleep’ transmitter. Numerous studies in many species describe sleep-active GABAergic neurons inhibiting wake-active neurons during sleep (Saper et al., 2005; Agosto et al., 2008; Parisky et al., 2008; Zimmerman et al., 2008). A small number of neurons maintain an active state in the fruit fly (Parisky et al., 2008), several thousands of aminergic and peptidergic neurons orchestrate waking in vertebrates (Wafford and Ebert, 2008). The histaminergic neurons in the tuberomamillary nucleus (TMN) of the posterior hypothalamus are the only aminergic neurons exhibiting a strict wake-on firing pattern being entirely silent during sleep. Histamine plays an important role for the cognitive aspects of vigilance and supports exploratory activity in a novel environment (Parmentier et al., 2002; Anaclet et al., 2009; Zecharia et al., 2012), last but not least synaptic plasticity and learning are influenced by histamine (Haas and Panula, 2003; Haas et al., 2008). TMN neurons receive a dense innervation from GABAergic sleep-active neurons in the preoptic area (Sherin et al., 1998) and express at least nine different GABAAR subunits: α1, α2, α5, β1, β2, β3, γ1, γ2 and ε (Sergeeva et al., 2002; 2005; 2010; Yanovsky et al., 2012b, a). The GABAAR γ2-subunit is enriched at synaptic sites (Essrich et al., 1998; Farrant and Nusser, 2005). Deletion of this subunit in selected types of neurons can lead to behavioural abnormalities as a consequence of the impaired synaptic inhibition (Wulff et al., 2007; 2009). Zecharia et al. (2012) generated a novel genetic mouse model with a selective deletion, only in histaminergic neurons, of the γ2-subunit and report a marked reduction of synaptic GABAergic currents in TMN neurons from the knock-out mice compared with littermate controls, but a normal sleep-wake cycle. The authors conclude that the GABAAR on TMN neurons plays no role for sleep. GABA-currents remaining after γ2-subunit deletion were not analysed in TMN neurons; however, their presence is expected (Gunther et al., 1995; Lorez et al., 2000). Information is needed (i) whether γ2-containing GABAAR play a dominant role for the pharmacology of all TMN neurons and (ii) how the presence of the three α subunits (α1, α2, α5) affects benzodiazepine-site pharmacology here. Our study aims at providing answers to these questions. Mutation at the γ2-subunit F77I (γ2F77I) eliminates high affinity (IC50∼0.2 μM) negative allosteric modulation by methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate (DMCM) and the modulation of α1- and α2-containing GABAAR by zolpidem (N,N-dimethyl-2-(6-methyl-2-p-tolylimidazo[1,2-a]pyridin-3-yl)acetamide; Buhr et al., 1997; Wingrove et al., 1997). Zolpidem is inactive at α5-containing GABAAR (Puia et al., 1991; Ramerstorfer et al., 2010). The benzodiazepine-site pharmacology was investigated now in TMN neurons obtained from wild type (WT) and mutant γ2F77I mice generated by W. Wisden (Cope et al., 2004).
Methods
Experimental animals and slice preparation procedures
Experiments were conducted according to the Animal Protection Law of the Federal Republic of Germany (Tierschutzgesetz BGBI.I, S.1206, revision 2006), European Communities Council directive regarding care and use of animals for experimental procedures (86/609/EEC) and the local guidelines (LANUV FB Tierschutz, Bezirksregierung, Duesseldorf, Germany). All efforts were made to minimize the number of animals and their suffering. Mice were maintained on a 12 h light-12 h dark cycle (light from 0700 h) with food and water available ad libitum. Slices were prepared between 0900 and 1200 h from 1 to 6 month old male mice carrying a point mutation on GABAAR γ2 subunit (γ2F77I) further referred as KI (knock-in) mice and their WT littermates (offspring of heterozygote breeding pairs). Genotyping was performed as previously described (Cope et al., 2004). Coronal brain slices containing the posterior hypothalamus were prepared as described previously (Yanovsky et al., 2012a, b). During preparation, NaCl was replaced by 207 mM sucrose in the ice-cold modified artificial cerebrospinal fluid saturated with carbogen (pH 7.4). Slices containing the TMN region were selected and incubated for 1 h at room temperature in a solution containing (in mM): NaCl 125, KCl 3.7, CaCl2 1.0, MgCl2 1.0, NaH2PO4 1.3, NaHCO3 23, d-glucose 10, bubbled with carbogen (pH 7.4). The same solution was used for the patch-clamp recordings from TMN neurons in slices. For the preparation of the acutely isolated neurons, the area including TMN was dissected and incubated with papain in crude form (0.3–0.5 mg mL−1) for 10–30 min at 37°C. Dissociation of cells and whole-cell patch-clamp recordings (as in Sergeeva et al., 2010; Yanovsky et al., 2012a, b) were done in a solution with the following composition (in mM): NaCl 150, KCl 3.7, CaCl2 2.0, MgCl2 2.0, HEPES 10, glucose 10 (pH 7.4). Sterile electrodes were filled with the following solution (in mM): 140 KCl, 2 MgCl2, 0.5 CaCl2, 5 EGTA, 10 HEPES/KOH (pH 7.2). A fast perfusion technique was used for application of ligands and modulators (Vorobjev et al., 1996). Currents were recorded and analysed with TIDA for Windows software (HEKA, Lambrecht, Germany). The cells were voltage-clamped by an EPC-9 amplifier. The holding potential was −50 mV. Only neurons with no leak current and series resistance lower than 15MΩ were selected for recording. TMN neurons responded to GABA with EC50s varying between 3.5 and 100 μM in a gabazine-sensitive way. GABA concentrations of 1–5 μM (below the EC30) were chosen for analysis of the modulatory potency and efficacy of benzodiazepine-site ligands. The maximal GABA response (to 500 μM) was determined at the end of each experiment and the relative control response amplitude calculated (ECx). Exact ECX values are given where appropriate; a group comparison was done if these values did not differ significantly. For the construction of concentration-response curves for the GABAAR-modulator the control GABA response (ECx) was subtracted from the potentiated responses. All responses were normalized to the maximal potentiation over control, referred to as ‘relative potentiation’. Data are given as the mean ± SEM. Statistical analysis was done with the non-parametrical Mann–Whitney U-test and Fisher's exact probability test. Significance level was set at P < 0.05.
Cell attached recording from TMN neurons in slices was done as previously described (Yanovsky et al., 2012c). Neurons with large somata (major axes >15 μm) and at the typical location (around third ventricle: TMN medial; or at the ventral brain surface: TMN ventral) were approached and the cell-attached configuration (holding potential 0mV) was obtained. Spontaneous action potential currents were recorded (Perkins, 2006). At the end of experiments, the histamine 3 receptor agonist R-α-methylhistamine (RAMH) was applied. Only cells responding to RAMH with significant reduction of firing frequency were considered for the analysis.
Single-cell reverse transcription (RT)-PCR was performed after whole-cell patch-clamp recordings from acutely isolated TMN neurons to identify them post hoc through the expression of histidine decarboxylase (HDC), the histamine-producing enzyme, primers and protocols are published in Sergeeva et al., (2002) and Parmentier et al. (2009). Transcripts encoding for the GABAAR subunits were amplified with primers published in Sergeeva et al. (2010). For the RT-PCR analysis of γ1- and γ2-subunit expression the following primers were used: γDg Se: 5′-TAT GT(GAT) AAC AGC ATT GG(TA) CC(TA) GT- 3′ taken together either with γ1 Ase: 5′-ATC GAA GAG TAT AGA GAA CCC TTC C-3′ (PCR product of 262 b.p. size) or with γ2 Ase: 5′-AAC ATC ATT CCA AAT TCT CAG CAT-3′ (size of amplimer 234 b.p.). A heat dissociation protocol (PE Biosystems 5700 Software; Applied Biosystems Inc., Darmstadt, Germany) was performed at the end of each PCR amplification. Standard curves were obtained with the sequential dilution of one cDNA sample. From these curves, the linear regression coefficient (r = −0.98) and efficiency (∼2.0) were calculated for the amplification of cDNAs encoding the γ1- or γ2-subunit. As previous studies have shown big changes in β–actin as well as other house-keeping genes expression between embryonic, newborn and adult brain (Mauric et al., 2013), we compared γ1–subunit expression with the γ2–subunit (ΔCt = Ct γ1–Ct γ2), as both PCRs showed the same efficiency and the γ2–subunit displayed constant and widespread expression through development (Laurie et al., 1992). The same amount of template (100ng) was taken in all reactions and relative level of γ1 mRNA was estimated by the 2–ΔCt method.
Primary dissociated cultures, electrophysiological recordings and siRNA-based knock-down technique
Primary dissociated cultures of the posterior hypothalamus were prepared from newborn mice according to the protocols previously described (Sergeeva et al., 2005). Dissociated cells were plated at a density of 1 to 2 × 105·cm–2 onto polyethylenimine-coated microelectrode arrays (MEAs) in a volume of 100 μL (Multi Channel Systems, Reutlingen, Germany) or on coverslips (for patch-clamp recordings) and cultured in an incubator with 5% CO2, 95% air and 98% relative humidity, at 37 ± 0.5°C. On the second day serum-free neurobasal medium containing supplement B27 (2%) was added to the final volume of 1 mL. Extracellular potentials were recorded on MEAs with a square grid of 60 planar Ti/TiN-microelectrodes (30 μm diameter, 200 μm spacing) at 37°C. Signals from all 60 electrodes were simultaneously sampled at 25 kHz, visualized and stored using the standard software MCRack provided by Multi Channel Systems. Spike detection was performed offline using the software SpAnNer (RESULT Medizinische Analyseverfahren, Tönisvorst, Germany). At the beginning of experiments, the basal medium was replaced by a magnesium-free HEPES-based recording solution (see above) and measurements were started after a 20 min adaptation phase. Every measurement comprised three recordings – control, test substance and washout (second control) – each 2 min long and separated by an intermediate period of 30 s. Whole-cell voltage clamp recordings were performed from non-identified hypothalamic neurons using an application system adapted for adherent cells (Sergeeva et al., 2005). In knock-down experiments, the culture medium was changed on day 10–12 either to transfection medium alone or to transfection medium with four siRNAs (100 μM, Accell SMART pool, Thermo Scientific, Cat# E-059012-00) directed towards the target sequences on the mouse GABAAR γ1-subunit. A non-targeting siRNA pool or transfection medium alone were used as negative controls. Recordings of neuronal activity were done from day 6 to 28 after plating. For each treatment, cultures of about the same age (days in vitro, Div) were selected (averaged Div is provided as mean ± SEM), the difference in Div between compared groups was not significant (unpaired t-test).
Drugs and chemicals
GABA, gabazine (SR95531), diazepam (7-chloro-1,3-dihydro-1-methyl-5-phenyl-1,4-benzodiazepin-2(3H)-one), midazolam (8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine), chlordiazepoxide (7-chloro-2-methylamino-5-phenyl-3H-1,4-benzodiazepine-4-oxide) and ZnCl2 were obtained from Sigma/RBI (Deisenhofen, Germany); DMCM, flumazenil (Ro15-1788; Ethyl 8-fluoro-5-methyl-6-oxo-5,6-dihydro-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate), RAMH, CGS 20625 and zolpidem from Biotrend (Koeln, Germany).
Results
Benzodiazepine-site pharmacology demands presence of a γ-subunit in the GABAAR (Sigel, 2002; Farrant and Nusser, 2005). GABAAR lacking γ-subunits are blocked by ZnCl2 10 μM, whereas γ-subunit-containing receptors are weakly or not affected by this concentration (Draguhn et al., 1990; Herb et al., 1992). In contrast to the rat TMN neurons which are all zinc resistant (Sergeeva et al., 2002), we found that, in about 30% of the mouse TMN neurons identified by the expression of HDC, GABA-responses are about halved by ZnCl2 10 μM (Kletke et al., 2013). In the present study, pyrazolopyridine CGS 20625, a two to six times more potent positive modulator of α1β2 than of α1β2γ receptors (Khom et al., 2006), confirmed γ-subunit-deficiency of zinc-sensitive TMN neurons [CGS 20625's EC50 = 1.1 ± 0.1 μM (n = 5, 28% of total cell number], which is significantly different (P = 0.0055) from the zinc-resistant cells (EC50 = 3.4 ± 0.4 μM, n = 13, Supporting Information Figure S1). Histaminergic neurons with the zinc-sensitive GABAAR were excluded from further analysis.
TMN neurons isolated from WT or γ2 F77I KI mice showed similar sensitivity to GABA (EC50 and nHill calculated in 16 neurons: 14.6 ± 1.1 μM and 1.7 ± 0.2 vs. 13 ± 0.7 μM and 1.5 ± 0.1, respectively). In all TMN neurons from WT mice, zolpidem potentiated GABA-responses with the half-maximal concentration (EC50) 0.08 ± 0.01 μM (nHill 0.6 ± 0.05) and maximal modulation 275 ± 40% of control (Figure 1). This indicates that α5-containing GABAAR, which are not modulated by zolpidem, are not dominant in TMN neurons. In mutant γ2 F77I mice, a significant potentiation of GABA-evoked responses by zolpidem (1 μM) was only seen in 40% of TMN neurons. In KI neurons responding to zolpidem, the maximal potentiation was significantly smaller compared to WT mice (171 ± 10% of control, P = 0.0165, Figure 1). At 1 μM, zolpidem enhanced GABA-evoked responses to 129 ± 7% of control in KI neurons which was significantly different from the WT neurons (242 ± 39%, P < 0.005). The potency of zolpidem was approximately 10 times lower (P = 0.037) in KI mice compared to the WT littermates (EC50 = 0.86 ± 0.2 μM, nHill = 0.8 ± 0.12). Thus, zolpidem modulation of GABA-currents in TMN neurons from γ2 F77I mice was significantly different from the WT neurons, supporting a functional presence of the γ2-subunit. These experiments indicate expression of another γ-containing receptor population besides the γ2-GABAAR in 40% of TMN neurons.
Figure 1.
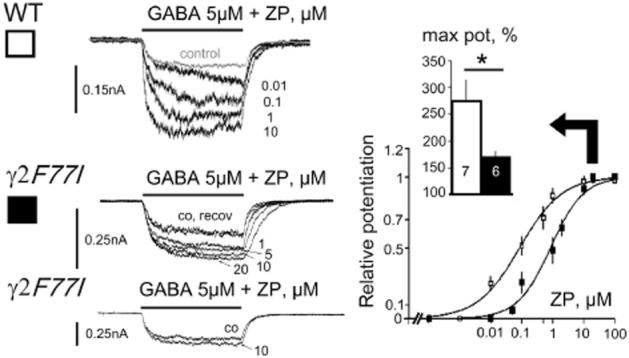
Modulation of GABA-evoked currents by zolpidem in tuberomamillary nucleus neurons from wild type (WT) and mutant γ2 F77I mice. Representative responses to GABA in control and in the presence of zolpidem (ZP) in WT and mutant mice (left) and averaged concentration-response diagrams obtained from neurons with significant ZP modulation (right). Maximal potentiation of control response taken as 100% is shown in insert. Neurons with a modulation smaller than 15% (left, bottom) were excluded from the knock-in group. Size of analysed neuronal groups is shown in columns. GABA taken at EC20.
Diazepam potentiated GABA-responses with an EC50 = 0.1 ± 0.01 μM (n = 6, nHill: 1.7 ± 0.2) in WT and with 0.07 ± 0.003 μM (n = 5, nHill: 1.4 ± 0.2) in γ2 F77I mice (no difference between genotypes). The potency of water soluble diazepam analogue, chlordiazepoxide, also did not show difference: GABA-responses were potentiated with an EC50 = 0.74 ± 0.15 μM (nHill: 0.98 ± 0.2) in WT and with 0.89 ± 0.14 μM (nHill: 0.8 ± 0.1) in γ2 F77I mice (Supporting Information Figure S2B).
The modulatory potency of midazolam in TMN neurons differed significantly (P = 0.018) between WT (EC50 = 0.16 ± 0.04 μM, nHill = 0.8 ± 0.1, n = 6) and KI neurons (EC50 = 1.0 ± 0.2 μM, nHill = 0.9 ± 0.2, n = 5), whereas maximal potentiation (357 ± 76 vs. 286 ± 62% of control, respectively) did not differ between the two genotypes (Figure 2). The rightward shift in midazolam potency in TMN neurons from γ2 F77I- compared to WT-mice can be explained by the presence of γ1-containing receptors (Khom et al., 2006), but contribution of γ3 (Herb et al., 1992) or mutant γ2 F77I GABAAR (Ogris et al., 2004) cannot be excluded.
Figure 2.
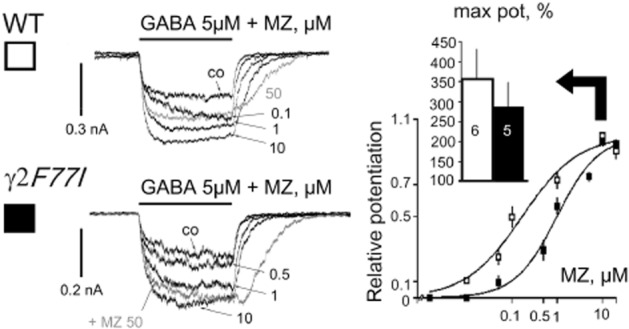
Modulation of GABA-evoked currents by midazolam (MZ) in mouse tuberomamillary nucleus neurons. Examples of representative recordings (left) and averaged concentration-response diagrams for the relative potentiation of GABA-responses by MZ (right). Number of investigated histaminergic neurons is shown in columns. GABA taken at EC16.
Flumazenil (Ro15-1788) potentiates some types of GABAAR including those composed of α2,β3,γ2- (Ramerstorfer et al., 2010), α1,β2,γ1- (Khom et al., 2006) or α1,β1- (Malherbe et al., 1990) subunits. Interestingly, potentiation at α2β3γ2 receptors disappears after mutation γ2F77I (Ramerstorfer et al., 2010). Flumazenil at concentrations 10 and 100 μM potentiated GABA-evoked currents in TMN neurons to the same extent in WT and KI mice (Figure 3), indicating that low affinity modulation is independent of the γ2F77-site in native neurons.
Figure 3.
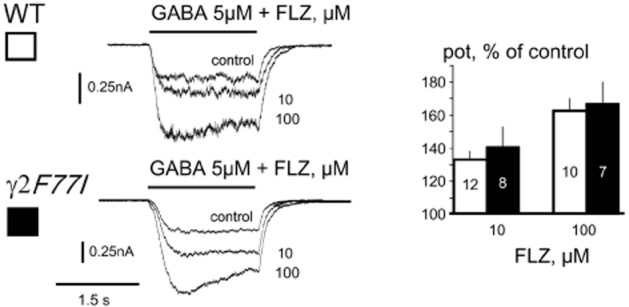
Flumazenil (FLZ; Ro15-1788) modulates GABA-evoked currents to the same extent in tuberomamillary nucleus neurons from wild type (WT) and knock-in (KI) mice. Representative current traces show recordings from one WT and one KI neuron, which responded to GABA and GABA+ FLZ at indicated concentrations. Averages of potentiated current amplitude relative to the control (% of control) are shown at the right. Number of investigated histaminergic neurons is shown in columns. GABA taken at EC15.
In WT mice, DMCM (from 0.05 to 1 μM) progressively inhibited GABA-responses recorded from histaminergic neurons. In the majority of cells, a further increase in DMCM concentration (up to 100 μM) resulted in an apparent reduction of the inhibition of the GABA-current, caused by the superimposition of a potentiation (Figure 4A). The inhibitory half-maximal concentration [IC50 = 0.2 ± 0.01 μM, Hill coefficient 1.1 ± 0.06 (n = 5)] was obtained when DMCM concentrations below 2 μM were considered for construction of the concentration – response curve. At DMCM concentrations higher than 2 μM, GABA-response modulation was significantly different from the modulation by 1 μM (Figure 4A). DMCM potentiated GABA – evoked responses in γ2 F77I mice with an EC50 = 2.1 ± 0.5 μM (Hill coefficient 1.0 ± 0.2, n = 5) (Figure 4B) with maximal potentiation achieved in the majority of the cells at 10 μM. In some KI neurons, modulation of GABA-responses by DMCM was negligible and these neurons were excluded from the concentration-response diagram shown in Figure 4B.
Figure 4.
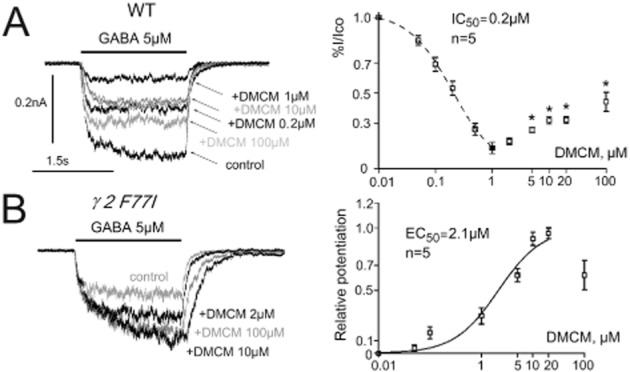
Methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate (DMCM) modulation of GABA-evoked currents in tuberomamillary nucleus neurons. (A) Recordings of DMCM – mediated biphasic modulation of GABA-currents in a wild type neuron (left) and averages from five neurons representing the concentration-dependent action of DMCM on GABA-currents (right). At concentrations upto 1 μM DMCM progressively inhibits GABA-currents, whereas at higher concentrations the reduction of the GABA-current is partially reversed, indicating the involvement of two modulatory sites with high (negative modulation) and low (positive modulation) affinity. Effects of high DMCM concentrations were compared with the modulation by 1 μM DMCM. Significance is indicated by stars (*P < 0.05). (B) Potentiation of GABA-evoked currents by DMCM in histaminergic neuron from a knock-in (KI) mouse. At the right: averaged dose-response curve for the positive DMCM modulation of GABA-responses in KI mice obtained from seven neurons and fitted with logistic equation.
Pharmacological analysis of benzodiazepine-site ligands at GABAAR expressed by mouse histaminergic neurons indicated presence of a further γ-subunit in addition to γ2. In order to test the possibility that the zolpidem and DMCM induced potentiation of GABA-evoked responses involves γ1-containing receptors, a correlation analysis between the expression pattern of GABAARs (scRT-PCR) and modulation of GABA-responses by these two compounds was done in KI neurons. Only cells expressing one or two γ-subunits were considered. DMCM potentiated GABA-evoked responses in neurons expressing only the γ2 subunit by 23 ± 6% (n = 10) and in neurons expressing γ1 and γ2 subunits by 67 ± 22% (n = 13) (P = 0.009, Mann–Whitney U-test). Complete single-cell RT-PCR analysis of GABAAR expression was successfully performed in 16 WT and 34 KI TMN neurons. Histaminergic cells expressed α1 (25 and 32% of neurons from WT and KI group, respectively), α2 (100 and 94%), α5 (25 and 18%), β1 (31 and 38%), β2 (13 and 15%), β3 (88 and 88%), γ1 (38 and 47%) and γ2 (81 and 79%) subunits (no significant difference in occurrence of any subunit between WT and KI neurons, Fisher's exact probability test). The γ3 subunit was present in positive control (TMN whole) but not detected in individual neurons. Sequencing of selected PCR products confirmed their identity to the known mouse sequences of GABAAR subunits. We found no difference (P = 0.28) between relative levels of γ1-subunit transcripts in TMN of WT (0.81 ± 0.1, n = 6) and KI (0.99 ± 0.1, n = 7) mice. The γ1-subunit transcripts were only slightly (∼20%) less abundant than γ2-transcripts in the TMN region of adult mouse.
Next, we performed knock-down experiments using siRNA technology to test the γ1-subunit function in hypothalamic neurons. We screened GABAAR modulators (at 10 μM) for effects on firing properties in MEA recordings. Zolpidem and flumazenil did not affect firing rate significantly in hypothalamic cultures derived from WT or KI mice (Figure 5A). Midazolam suppressed spontaneous firing in both WT and γ2 F77I groups. Identified TMN neurons recorded in brain slices from adult WT and KI mice showed similar maximal responses to midazolam (10 μM): the firing was inhibited to 58 ± 7% of control in WT (five neurons) and to 70 ± 11% of control in γ2 F77I mice (six neurons). Recovery after midazolam withdrawal was delayed in WT compared to KI mice (Supporting Information Figure S3), indicating loss of the high affinity midazolam binding site in KI neurons, which is dependent on the γ2-subunit. Due to the low potency modulation by midazolam of γ3- and γ2 F77I-containing GABAAR (see above), this compound was considered unsuitable for the knock-down experiments.
Figure 5.
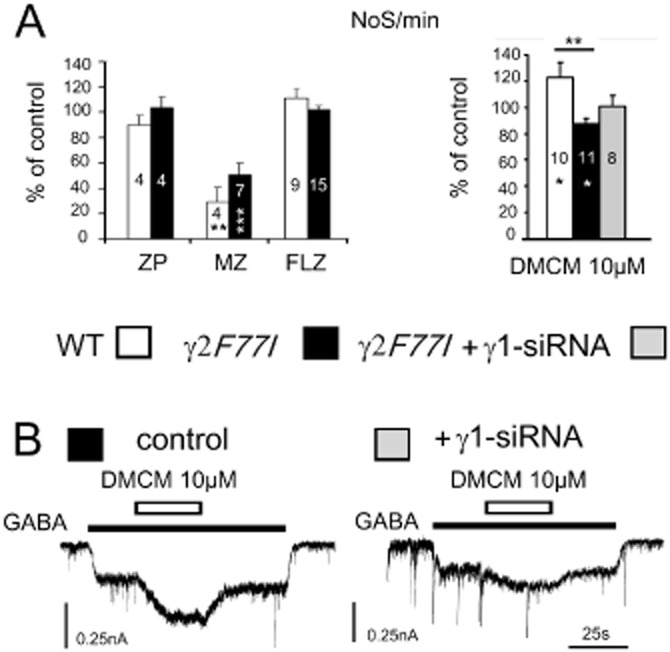
Microelectrode array (MEA) recordings from posterior hypothalamic cultures reveal functional role of γ1-subunit in methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate (DMCM) action. (A) Firing frequency of hypothalamic neurons (number of spikes recorded from the whole MEA in 1 min: NoS min−1) is not significantly affected in the presence of zolpidem (ZP) [Div 11.3 ± 1.4 for the wild type (WT) and 12.5 ± 1 for the knock-in (KI) cultures] and flumazenil (FLZ) (Div 8.7 ± 0.5 for the WT and 9.9 ± 0.5 for the KI), whereas midazolam (MZ) suppressed neuronal activity (Div 8.8 ± 1.1 for the WT and 10.7 ± 0.8 for the KI). Right: DMCM increases firing frequency in WT (Div 13 ± 2) and suppresses it in KI neurons (Div 13.1 ± 1). Treatment with γ1-siRNA abolishes inhibitory activity of DMCM (Div 15.5 ± 1). Significant difference between groups is indicated with stars on top of columns, significant difference from baseline activity within columns. Number of investigated cultures is given within columns. All modulators were used at 10 μM. (B) Whole-cell voltage clamp (-50mV) recordings from two cultured neurons grown in parallel with those on the MEAs shown in A. DMCM modulation is significantly reduced in a neuron treated with γ1-siRNA. Note: preservation of synaptic GABAergic currents in neuron treated with γ1-siRNA (right).
In cultured hypothalamic neurons, the action of DMCM (10 μM, Figure 5A) on spontaneous firing frequency differed significantly between WT and γ2 F77I mice in accordance with GABAAR block versus positive modulation, respectively. Importantly, this different action on neuronal firing reflected the behavioural action of DMCM: convulsions in WT versus sedation in γ2 F77I mice (Leppa et al., 2011). The action of DMCM was further investigated in γ1-knock-down experiments.
Posterior hypothalamic primary dissociated cultures containing TMN neurons were treated for 2–3 days with γ1-siRNA, non-targeting siRNA or incubated in transfection (Accell) medium. Data obtained with two different controls (non-targeting siRNA or vehicle) were pooled, as they were not different. Relative levels of γ1-subunit transcripts dropped from 3 ± 0.8 (n = 4) in control to 0.29 ± 0.05 (n = 4, P = 0.03) in cultures treated with γ1-siRNA. Suppression of neuronal firing by DMCM 10 μM in KI cultures was abolished after γ1-siRNA treatment (Figure 5). Whole-cell patch-clamp recordings showed a significant (P < 0.05) reduction of DMCM-induced positive modulation of GABA-evoked currents after γ1-siRNA treatment in neurons from KI mice (Figure 5B): under control conditions DMCM potentiated GABA-evoked currents (EC9 ± 1) to 157 ± 7% of control (n = 25), whereas, after γ1-siRNA treatment, potentiation of the same amplitude control currents (EC9 ± 1) amounted to 137 ± 4% of control (n = 26). Zolpidem (10 μM) modulation of the control GABA currents declined after γ1-siRNA treatment from 168 ± 13% of control (n = 5) to 108 ± 3% of control (n = 9, P < 0.005). Spontaneous synaptic GABAergic currents were preserved after γ1-siRNA treatment (Figure 5B). Thus GABAAR-benzodiazepine-site modulators investigated in the present study have a differential impact on GABAergic control of neuronal activity: midazolam and DMCM affect firing frequency of hypothalamic neurons whereas zolpidem and flumazenil are inactive. Location and function of different GABAAR types on TMN neurons might be responsible for this difference.
Discussion and conclusions
We present a variety of clinically important benzodiazepine-site ligands increasing GABAA receptor-mediated currents on native hypothalamic TMN neurons, whose activity is thus decreased and expected to lead to a decline in cognitive performance and vigilance. A mutation within the high-affinity benzodiazepine binding site γ2 F77I changed the sensitivity of TMN neurons to zolpidem, midazolam and DMCM, but not to diazepam, chlordiazepoxide and flumazenil. If among the three known γ-subunits only the γ2-subunit would be functionally present in TMN neurons, the mutation γ2 F77I should have abolished the allosteric positive modulation of GABA-evoked currents by zolpidem and reduced the efficacy of diazepam. However, an additional site for the high potency zolpidem modulation was detected in KI (γ2 F77I) neurons, whose occurrence coincided with the expression of the γ1-subunit and with a strong potentiation of GABA-currents by DMCM. Knock-down of the γ1-subunit in hypothalamic neurons reduced the modulatory activity of DMCM and zolpidem in γ2 F77I mutant TMN neurons. Thus multiple types of GABAAR are affected by benzodiazepine-site ligands in TMN neurons.
Our results support a dominant role of α2- or α1-, but not α5- subunits for the benzodiazepine-site pharmacology in mouse TMN neurons. Cerebellar Purkinje neurons expressing the GABAAR α1-subunit display a typical zolpidem potency of about 30nM, whereas striatal neurons expressing the α2-subunit are characterized by a zolpidem potency of about 200nM (Itier et al., 1996). Zolpidem modulation of GABA-responses in TMN neurons occurs at 80nM, indicating the presence of mixed α1, α2-containing receptor populations. In mutant γ2 F77I mice, zolpidem enhanced GABA-currents only in 40% of investigated neurons, with lower potency and efficacy than in WT neurons. Mutated γ2-containing GABAARs might respond to zolpidem at micromolar concentrations through the anaesthetics-binding site, like it was previously shown for the diazepam modulation of αβ-receptors (Walters et al., 2000). Alternatively, receptors containing the γ1-subunit (Puia et al., 1991; Wafford et al., 1993) or γ3-subunit (Herb et al., 1992) could mediate the response to zolpidem. No potentiation of GABAAR by zolpidem (1 or 10 μM) is observed in oocytes transfected with α1/2, β2/3, γ2F77I-subunits (Buhr et al., 1997; Ramerstorfer et al., 2010; Kletke et al., 2013), indicating that the ‘anaesthetics-binding site’ present on β2 and β3 subunits is not involved. The modulatory potency and efficacy of diazepam and chlordiazepoxide was indifferent between WT and KI neurons in our study, whereas at recombinant α1β2γ2F77I receptors the efficacy of diazepam is reduced (Ramerstorfer et al., 2010). Presence of an additional diazepam-responsive receptor population could explain this difference. Receptors composed of α1, α2 or α5, β1 and γ1-subunits are positively modulated by diazepam (Puia et al., 1991) although to a smaller extent than the corresponding γ2-containing receptors. We did not detect γ3-subunit expression in individual TMN neurons of the mouse in this and in our previous studies (Sergeeva et al., 2010; Kletke et al., 2013), whereas the whole TMN region used as positive control contained γ3-subunit transcripts. This subunit confers zinc-resistance to the recombinant GABAA receptors and benzodiazepine-site ligands modulate them, although with very low potencies and efficacies (Herb et al., 1992). Therefore a role of this subunit for the benzodiazepine-pharmacology in TMN neurons cannot be excluded.
The structure of the GABAAR benzodiazepine-binding site is complex, but well characterized. Mutational analysis has identified the amino acid residues H101, Y161, T206 and Y209 on the α1-subunit and F77 and M130 on the γ2-subunit as putative parts forming the benzodiazepine-binding pocket (Buhr et al., 1996; Sigel, 2002). Mutation at the position F77 of the γ2-subunit separates diazepam effects from zolpidem/DMCM effects: the former being unchanged or reduced in efficacy, the latter abolished (Buhr et al., 1996; 1997; Wingrove et al., 1997; Ramerstorfer et al., 2010). The γ1-subunit carries different amino acid residues (I79 and L132) at the positions analogous to F77 and M130 of the γ2-subunit and this may be the reason for the poor modulation by benzodiazepine-site agonists. The α-type subunits influence this modulation and it was reported that association of the γ1-subunit with the α1- and β-subunits results in a greater zolpidem modulation of GABA-evoked currents compared to the α2-containing receptors (Puia et al., 1991). Recombinant receptors composed of α2, β1 and γ1 subunits are not modulated by zolpidem [see Supporting Information Figure S3 in Kletke et al., (2013)]. The α1-subunit thus plays an important role for the zolpidem modulation of GABA-responses in mutant mice. Further studies with α1-preferring benzodiazepine-site modulators (Anaclet et al., 2012) and with mutant α1H101R mice (Crestani et al., 2002) are warranted to determine the role of this subunit in TMN neurons.
We used DMCM for the pharmacological identification of γ1-subunit expression in TMN neurons (Puia et al., 1991; Sergeeva et al., 2002). Interpretation of the results is, however, complicated by several DMCM binding sites on GABAAR (Puia et al., 1991; Stevenson et al., 1995). The mutation γ2F77I abolishes the negative modulation (inverse agonism) through the high-affinity binding site, unmasking a low-affinity positive allosteric modulation, which is believed to be mediated either through the γ1- and β1-containing receptors (Puia et al., 1991) or through the β2/3-containing receptors (Stevenson et al., 1995). In the recombinant receptors containing the β-subunit with a mutation in the anaesthetics-binding site (e.g. β2 N265M), the loreclezole-like action of DMCM is abolished (Stevenson et al., 1995). In Xenopus Oocytes, GABAAR composed of α2-, β3- and γ2F77I- subunits DMCM at 10 and 100 μM potentiates GABA-responses to 136 and 540% of control, respectively (n = 5) (O. Kletke and O.A. Sergeeva, unpublished), whereas analogous β1-containing receptors are insensitive to DMCM 10 μM. Thus, the prevailing expression of the β1-subunit over the β3-subunit in histaminergic neurons (Sergeeva et al., 2010; Yanovsky et al., 2012a) devoid of the γ1-subunit can explain the lack of positive modulation of GABA-currents by DMCM or zolpidem. Experiments with transient γ1-subunit knock-down performed in hypothalamic cultures revealed that positive modulation by DMCM may be dependent on both sites: α/γ1 or β3N265, as it was not abolished after γ1-siRNA treatment. Further experiments with the mutant β3N265M or double mutant β3N265M/γ2F77I mice may shed light on the relative participation of each site to DMCM modulation.
Khom et al., (2006) reported that the pyrazolopyridine CGS 20625 is the most efficient positive modulator (when compared to the variety of benzodiazepine-site agonists) of the γ1-containing recombinant α1β2γ1-GABAAR, which enhances GABA responses to more than 600% over control. They did not study GABAAR containing the β1 subunit, which dominate the pharmacological properties of TMN neurons (Sergeeva et al., 2010; Yanovsky et al., 2012a). Another pyrazolopyridine, tracazolate, modulates β1-containing GABAAR poorly, in contrast to the receptors composed of α1β3-subunits (Thompson et al., 2002; Kletke et al., 2013). In zinc-resistant TMN neurons, the modulatory potency of CGS 20625 changed from 2.5 μM in WT to 4.4 μM in γ2 F77I mice in line with the two times lower potency of this modulator at γ1- than at γ2-containing GABAAR (Khom et al., 2006). Modulatory efficacy of CGS 20625 in histaminergic neurons was three times lower than in recombinant α1β2γ1 receptors (Khom et al., 2006), which could be due to the different properties of CGS 20625 at β1- versus at β3-containing GABAAR.
Another benzodiazepine-site ligand reported to cause pronounced positive modulation of γ1-containing GABAAR is flumazenil (Ro15-1788) (Khom et al., 2006). Flumazenil potentiates recombinant α2β3γ2 receptors (but not if α1- or α5- are present) and the mutation γ2 F77I abolishes this potentiation (Ramerstorfer et al., 2010). However, α1β1 receptors can be modulated by flumazenil (Malherbe et al., 1990). Flumazenil has a beneficial action in patients suffering from hepatic encephalopathy (HE) (Laccetti et al., 2000; Dursun et al., 2003) with mechanisms poorly understood. Our study revealed that low potency flumazenil modulation does not depend on the benzodiazepine-site formed by the γ2 – subunit. Symptoms of subclinical and advanced HE are worsened by midazolam used for the anaesthesia (Assy et al., 1999; Haq et al., 2012). Potentiating extrasynaptic GABAAR in hippocampus by midazolam suppresses neuronal firing. In contrast, zolpidem, mainly enhancing phasic (synaptic) inhibition, does not affect neuronal firing rate (Farrant and Nusser, 2005). Our data obtained on hypothalamic neurons are in line with these findings in the hippocampus. We also show that midazolam-suppression of the firing rate of adult histaminergic neurons is not abolished but shortened in γ2 F77I mice compared to the WT mice revealing the lack of a high affinity modulatory site. Thus mutant γ2 F77I mice could represent a good model for studies on HE neuropathology under midazolam. Hyperammonemia, the major pathogenic factor of HE, triggers brain taurine release, which is neuroprotective through GABAA and glycine receptors. Our finding that the mutation γ2 F77I impairs GABAAR gating by taurine (Kletke et al., 2013) will complicate interpretation of data obtained from this model. The changed benzodiazepine-site pharmacology in histaminergic neurons from γ2 F77I mice highlights a yet unexplored structural complexity of GABAAR, whose modification under pathological conditions is far from being understood (Sergeeva, 2013). Mice with mutated or deleted γ-subunits should provide a clue to the cellular mechanisms underlying changed GABAergic transmission in several neuropsychiatric diseases including HE.
TMN neurons express different GABAAR types, which respond to the clinically important benzodiazepine-site agonists. Different synaptic pathways impinging on these neurons may be involved in regulation of different forms of behaviour. We have shown previously that GABAergic axons from sleep-active neurons of the preoptic area form synapses on TMN neurons mainly carrying β1-containing GABAAR, which are not very sensitive to propofol, whereas receptors highly sensitive to propofol contain the β3-subunit and are unlikely to play a role in this pathway (Yanovsky et al., 2012a). Further studies are warranted to delineate the receptor types involved in different forms of behaviour. The recently generated HDC-cre mice (Zecharia et al., 2012; Yanovsky et al., 2012c) allow selective genetic manipulations of GABAAR in TMN neurons. Deletion of the γ2-subunit only in TMN neurons impairs the habituation to a novel environment, but does not affect sleep-wake patterns with GABAergic synaptic currents strongly compromised (Zecharia et al., 2012). The role of remaining extrasynaptic receptors waits to be analysed. The importance of GABAAR on TMN neurons controlling wakefulness may become more obvious during pathological states accompanied by reduced vigilance and increased GABAergic tone. High histamine levels during hyperammonemia (Yanovsky et al., 2012c) and loss of coordination among GABAergic inputs likely contribute to the slowed frequencies of oscillatory activity of the brain and cognitive deficits seen in hepatic encephalopathy (Sergeeva, 2013). In conclusion, benzodiazepine-site pharmacology depends not solely on γ2-containing GABAAR and its understanding is a prerequisite for rational therapeutic interventions in disorders of sleep and metabolism.
Acknowledgments
Supported by Deutsche Forschungsgemeinschaft SE 1767, SFB 575/3 and 8, Forschungskommission HHU Düsseldorf and a Heisenberg fellowship to O. A. S. We are grateful to Dr. W. Wisden for the donation of γ2F77I mice.
Glossary
- Chlordiazepoxide
7-chloro-2-methylamino-5-phenyl-3H-1,4-benzodiazepine-4-oxide
- Diazepam
7-chloro-1,3-dihydro-1-methyl-5-phenyl-1,4-benzodiazepin-2(3H)-one
- Div
days in vitro
- DMCM
methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate
- Flumazenil
Ethyl 8-fluoro-5-methyl-6-oxo-5,6-dihydro-4H-benzo[f]imidazo[1,5-a][1,4]diazepine-3-carboxylate
- GABAAR
GABA receptor type A
- HE
hepatic encephalopathy
- KI
knock-in
- MEA
microelectrode array
- Midazolam
8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
- RAMH
R-α-methylhistamine
- TMN
tuberomamillary nucleus
- WT
wild type
- Zolpidem
N,N-dimethyl-2-(6-methyl-2-p-tolylimidazo[1,2-a]pyridin-3-yl)acetamide
Conflict of interest
All authors state they have no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Modulation of GABA-evoked currents by CGS 20625 in mouse tuberomamillary nucleus neurons. (A) Representative recordings [upper panel shows recordings from zinc-sensitive wild type (WT) neuron and lower panel from zinc-resistant knock-in (KI) neuron]. (B) Averaged concentration-response diagrams for the relative potentiation of GABA-responses by CGS 20625 in pooled KI and WT zinc-sensitive (EC50 = 1.1 ± 0.1 μM, n = 5) versus zinc-resistant neurons (EC50 = 3.4 ± 0.4 μM, n = 13). Red and blue curves without symbols are fitted data obtained in zinc resistant WT (EC50 = 2.5 ± 0.4 μM, n = 6) and KI neurons(EC50 = 4.4 ± 0.5 μM, n = 7), respectively. Note that maximal potentiation did not differ between different neuronal groups. GABA taken at EC13–16.
Figure S2 Modulation of GABA-evoked currents by Diazepam (DZ) and chlordiazepoxide (CDZ) in mouse histaminergic neurons. (A) Examples of representative recordings (left) and averaged concentration-response diagrams for the relative potentiation of GABA-responses by diazepam (right). Total number of investigated neurons is shown in columns. GABA response amplitude (ECx) normalized to maximal control amplitude in these experiments amounted to 16 ± 4% and 12 ± 2% in wild type (WT) and knock-in (KI) neurons, respectively. (B) Examples of representative recordings (left) and averaged concentration-response diagrams for the relative potentiation of GABA-responses by CDZ (right). Total number of investigated neurons is shown in columns. The GABA ECx in these experiments amounted to 19 ± 5% and 14 ± 2% in WT and KI neurons, respectively.
Figure S3 Midazolam 10μM inhibits firing of tuberomamillary nucleus neurons identified by the R-α-methylhistamine (RAMH) in mouse brain slices. Averaged time course diagrams show firing frequency normalized to the 7 min control period (cell-attached voltage-clamp mode) in the presence of midazolam (upper plot) or the histamine 3 receptor agonist RAMH 2μM (lower plot) in wild type and knock-in (KI) (γ2F77I) mice. Note: no difference in response to RAMH but faster recovery to control after midazolam withdrawal in KI mice.
References
- Agosto J, Choi JC, Parisky KM, Stilwell G, Rosbash M, Griffith LC. Modulation of GABAA receptor desensitization uncouples sleep onset and maintenance in Drosophila. Nat Neurosci. 2008;11:354–359. doi: 10.1038/nn2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anaclet C, Parmentier R, Ouk K, Guidon G, Buda C, Sastre JP, et al. Orexin/hypocretin and histamine: distinct roles in the control of wakefulness demonstrated using knock-out mouse models. J Neurosci. 2009;29:14423–14438. doi: 10.1523/JNEUROSCI.2604-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anaclet C, Zhang M, Zhao C, Buda C, Seugnet L, Lin JS. Effects of GF-015535-00, a novel alpha1 GABA A receptor ligand, on the sleep-wake cycle in mice, with reference to zolpidem. Sleep. 2012;35:103–111. doi: 10.5665/sleep.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assy N, Rosser BG, Grahame GR, Minuk GY. Risk of sedation for upper GI endoscopy exacerbating subclinical hepatic encephalopathy in patients with cirrhosis. Gastrointest Endosc. 1999;49:690–694. doi: 10.1016/s0016-5107(99)70283-x. [DOI] [PubMed] [Google Scholar]
- Baumann SW, Baur R, Sigel E. Forced subunit assembly in alpha1beta2gamma2 GABAA receptors. Insight into the absolute arrangement. J Biol Chem. 2002;277:46020–46025. doi: 10.1074/jbc.M207663200. [DOI] [PubMed] [Google Scholar]
- Buhr A, Baur R, Malherbe P, Sigel E. Point mutations of the alpha 1 beta 2 gamma 2 gamma-aminobutyric acid(A) receptor affecting modulation of the channel by ligands of the benzodiazepine binding site. Mol Pharmacol. 1996;49:1080–1084. [PubMed] [Google Scholar]
- Buhr A, Baur R, Sigel E. Subtle changes in residue 77 of the gamma subunit of alpha1beta2gamma2 GABAA receptors drastically alter the affinity for ligands of the benzodiazepine binding site. J Biol Chem. 1997;272:11799–11804. doi: 10.1074/jbc.272.18.11799. [DOI] [PubMed] [Google Scholar]
- Cope DW, Wulff P, Oberto A, Aller MI, Capogna M, Ferraguti F, et al. Abolition of zolpidem sensitivity in mice with a point mutation in the GABAA receptor gamma2 subunit. Neuropharmacology. 2004;47:17–34. doi: 10.1016/j.neuropharm.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Crestani F, Assandri R, Tauber M, Martin JR, Rudolph U. Contribution of the alpha1-GABA(A) receptor subtype to the pharmacological actions of benzodiazepine site inverse agonists. Neuropharmacology. 2002;43:679–684. doi: 10.1016/s0028-3908(02)00159-4. [DOI] [PubMed] [Google Scholar]
- Draguhn A, Verdorn TA, Ewert M, Seeburg PH, Sakmann B. Functional and molecular distinction between recombinant rat GABAA receptor subtypes by Zn2+ Neuron. 1990;5:781–788. doi: 10.1016/0896-6273(90)90337-f. [DOI] [PubMed] [Google Scholar]
- Dursun M, Caliskan M, Canoruc F, Aluclu U, Canoruc N, Tuzcu A, et al. The efficacy of flumazenil in subclinical to mild hepatic encephalopathic ambulatory patients. A prospective, randomised, double-blind, placebo-controlled study. Swiss Med Wkly. 2003;133:118–123. doi: 10.4414/smw.2003.10107. [DOI] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Gunther U, Benson J, Benke D, Fritschy JM, Reyes G, Knoflach F, et al. Benzodiazepine-insensitive mice generated by targeted disruption of the gamma 2 subunit gene of gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 1995;92:7749–7753. doi: 10.1073/pnas.92.17.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas H, Panula P. The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev Neurosci. 2003;4:121–130. doi: 10.1038/nrn1034. [DOI] [PubMed] [Google Scholar]
- Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88:1183–1241. doi: 10.1152/physrev.00043.2007. [DOI] [PubMed] [Google Scholar]
- Haq MM, Faisal N, Khalil A, Haqqi SA, Shaikh H, Arain N. Midazolam for sedation during diagnostic or therapeutic upper gastrointestinal endoscopy in cirrhotic patients. Eur J Gastroenterol Hepatol. 2012;24:1214–1218. doi: 10.1097/MEG.0b013e328356ae49. [DOI] [PubMed] [Google Scholar]
- Herb A, Wisden W, Luddens H, Puia G, Vicini S, Seeburg PH. The third gamma subunit of the gamma-aminobutyric acid type A receptor family. Proc Natl Acad Sci U S A. 1992;89:1433–1437. doi: 10.1073/pnas.89.4.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itier V, Depoortere H, Scatton B, Avenet P. Zolpidem functionally discriminates subtypes of native GABAA receptors in acutely dissociated rat striatal and cerebellar neurons. Neuropharmacology. 1996;35:137–145. doi: 10.1016/0028-3908(95)00158-1. [DOI] [PubMed] [Google Scholar]
- Khom S, Baburin I, Timin EN, Hohaus A, Sieghart W, Hering S. Pharmacological properties of GABAA receptors containing gamma1 subunits. Mol Pharmacol. 2006;69:640–649. doi: 10.1124/mol.105.017236. [DOI] [PubMed] [Google Scholar]
- Kletke O, Gisselmann G, May A, Hatt H, Sergeeva OA. Partial agonism of taurine at gamma-containing native and recombinant GABAA receptors. PLoS ONE. 2013;8:e61733. doi: 10.1371/journal.pone.0061733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laccetti M, Manes G, Uomo G, Lioniello M, Rabitti PG, Balzano A. Flumazenil in the treatment of acute hepatic encephalopathy in cirrhotic patients: a double blind randomized placebo controlled study. Dig Liver Dis. 2000;32:335–338. doi: 10.1016/s1590-8658(00)80027-4. [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Wisden W, Seeburg PH. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci. 1992;12:4151–4172. doi: 10.1523/JNEUROSCI.12-11-04151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppa E, Linden AM, Rabe H, Vekovischeva OY, Wulff P, Luddens H, et al. Actions of two GABAA receptor benzodiazepine-site ligands that are mediated via non-gamma2-dependent modulation. Eur J Pharmacol. 2011;666:111–121. doi: 10.1016/j.ejphar.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Lorez M, Benke D, Luscher B, Mohler H, Benson JA. Single-channel properties of neuronal GABAA receptors from mice lacking the 2 subunit. J Physiol. 2000;527(Pt 1):11–31. doi: 10.1111/j.1469-7793.2000.t01-1-00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malherbe P, Draguhn A, Multhaup G, Beyreuther K, Mohler H. GABAA-receptor expressed from rat brain alpha- and beta-subunit cDNAs displays potentiation by benzodiazepine receptor ligands. Brain Res Mol Brain Res. 1990;8:199–208. doi: 10.1016/0169-328x(90)90017-8. [DOI] [PubMed] [Google Scholar]
- Mauric V, Molders A, Harmel N, Heimrich B, Sergeeva OA, Klocker N. Ontogeny repeats the phylogenetic recruitment of the cargo exporter cornichon into AMPA receptor signaling complexes. Mol Cell Neurosci. 2013;56C:10–17. doi: 10.1016/j.mcn.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Ogris W, Poltl A, Hauer B, Ernst M, Oberto A, Wulff P, et al. Affinity of various benzodiazepine site ligands in mice with a point mutation in the GABA(A) receptor gamma2 subunit. Biochem Pharmacol. 2004;68:1621–1629. doi: 10.1016/j.bcp.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Parisky KM, Agosto J, Pulver SR, Shang Y, Kuklin E, Hodge JJ, et al. PDF cells are a GABA-responsive wake-promoting component of the Drosophila sleep circuit. Neuron. 2008;60:672–682. doi: 10.1016/j.neuron.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmentier R, Ohtsu H, Djebbara-Hannas Z, Valatx JL, Watanabe T, Lin JS. Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control. J Neurosci. 2002;22:7695–7711. doi: 10.1523/JNEUROSCI.22-17-07695.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmentier R, Kolbaev S, Klyuch BP, Vandael D, Lin JS, Selbach O, et al. Excitation of histaminergic tuberomamillary neurons by thyrotropin-releasing hormone. J Neurosci. 2009;29:4471–4483. doi: 10.1523/JNEUROSCI.2976-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins KL. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods. 2006;154:1–18. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puia G, Vicini S, Seeburg PH, Costa E. Influence of recombinant gamma-aminobutyric acid-A receptor subunit composition on the action of allosteric modulators of gamma-aminobutyric acid-gated Cl- currents. Mol Pharmacol. 1991;39:691–696. [PubMed] [Google Scholar]
- Ramerstorfer J, Furtmuller R, Vogel E, Huck S, Sieghart W. The point mutation gamma 2F77I changes the potency and efficacy of benzodiazepine site ligands in different GABAA receptor subtypes. Eur J Pharmacol. 2010;636:18–27. doi: 10.1016/j.ejphar.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Mohler H. GABA-based therapeutic approaches: GABAA receptor subtype functions. Curr Opin Pharmacol. 2006;6:18–23. doi: 10.1016/j.coph.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA. GABAergic transmission in hepatic encephalopathy. Arch Biochem Biophys. 2013 doi: 10.1016/j.abb.2013.04.005. doi: 10.1016/j.abb.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA, Eriksson KS, Sharonova IN, Vorobjev VS, Haas HL. GABA(A) receptor heterogeneity in histaminergic neurons. Eur J Neurosci. 2002;16:1472–1482. doi: 10.1046/j.1460-9568.2002.02221.x. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA, Andreeva N, Garret M, Scherer A, Haas HL. Pharmacological properties of GABAA receptors in rat hypothalamic neurons expressing the epsilon-subunit. J Neurosci. 2005;25:88–95. doi: 10.1523/JNEUROSCI.3209-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeeva OA, Kletke O, Kragler A, Poppek A, Fleischer W, Schubring SR, et al. Fragrant dioxane derivatives identify beta1-subunit-containing GABAA receptors. J Biol Chem. 2010;285:23985–23993. doi: 10.1074/jbc.M110.103309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998;18:4705–4721. doi: 10.1523/JNEUROSCI.18-12-04705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel E. Mapping of the benzodiazepine recognition site on GABA(A) receptors. Curr Top Med Chem. 2002;2:833–839. doi: 10.2174/1568026023393444. [DOI] [PubMed] [Google Scholar]
- Stevenson A, Wingrove PB, Whiting PJ, Wafford KA. beta-Carboline gamma-aminobutyric acidA receptor inverse agonists modulate gamma-aminobutyric acid via the loreclezole binding site as well as the benzodiazepine site. Mol Pharmacol. 1995;48:965–969. [PubMed] [Google Scholar]
- Thompson SA, Wingrove PB, Connelly L, Whiting PJ, Wafford KA. Tracazolate reveals a novel type of allosteric interaction with recombinant gamma-aminobutyric acid(A) receptors. Mol Pharmacol. 2002;61:861–869. doi: 10.1124/mol.61.4.861. [DOI] [PubMed] [Google Scholar]
- Vorobjev VS, Sharonova IN, Haas HL. A simple perfusion system for patch-clamp studies. J Neurosci Methods. 1996;68:303–307. doi: 10.1016/0165-0270(96)00097-0. [DOI] [PubMed] [Google Scholar]
- Wafford KA, Ebert B. Emerging anti-insomnia drugs: tackling sleeplessness and the quality of wake time. Nat Rev Drug Discov. 2008;7:530–540. doi: 10.1038/nrd2464. [DOI] [PubMed] [Google Scholar]
- Wafford KA, Bain CJ, Whiting PJ, Kemp JA. Functional comparison of the role of gamma subunits in recombinant human gamma-aminobutyric acidA/benzodiazepine receptors. Mol Pharmacol. 1993;44:437–442. [PubMed] [Google Scholar]
- Walters RJ, Hadley SH, Morris KD, Amin J. Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat Neurosci. 2000;3:1274–1281. doi: 10.1038/81800. [DOI] [PubMed] [Google Scholar]
- Wingrove PB, Thompson SA, Wafford KA, Whiting PJ. Key amino acids in the gamma subunit of the gamma-aminobutyric acidA receptor that determine ligand binding and modulation at the benzodiazepine site. Mol Pharmacol. 1997;52:874–881. doi: 10.1124/mol.52.5.874. [DOI] [PubMed] [Google Scholar]
- Wisden W, Laurie DJ, Monyer H, Seeburg PH. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J Neurosci. 1992;12:1040–1062. doi: 10.1523/JNEUROSCI.12-03-01040.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff P, Goetz T, Leppa E, Linden AM, Renzi M, Swinny JD, et al. From synapse to behavior: rapid modulation of defined neuronal types with engineered GABAA receptors. Nat Neurosci. 2007;10:923–929. doi: 10.1038/nn1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff P, Ponomarenko AA, Bartos M, Korotkova TM, Fuchs EC, Bahner F, et al. Hippocampal theta rhythm and its coupling with gamma oscillations require fast inhibition onto parvalbumin-positive interneurons. Proc Natl Acad Sci U S A. 2009;106:3561–3566. doi: 10.1073/pnas.0813176106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanovsky Y, Schubring S, Fleischer W, Gisselmann G, Zhu XR, Lubbert H, et al. GABAA receptors involved in sleep and anaesthesia: beta1- versus beta3-containing assemblies. Pflugers Arch. 2012a;463:187–199. doi: 10.1007/s00424-011-0988-4. [DOI] [PubMed] [Google Scholar]
- Yanovsky Y, Schubring SR, Yao Q, Zhao Y, Li S, May A, et al. Waking action of ursodeoxycholic acid (UDCA) involves histamine and GABAA receptor block. PLoS ONE. 2012b;7:e42512. doi: 10.1371/journal.pone.0042512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanovsky Y, Zigman JM, Kernder A, Bein A, Sakata I, Osborne-Lawrence S, et al. Proton- and ammonium-sensing by histaminergic neurons controlling wakefulness. Front Syst Neurosci. 2012c;6:23. doi: 10.3389/fnsys.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecharia AY, Yu X, Gotz T, Ye Z, Carr DR, Wulff P, et al. GABAergic inhibition of histaminergic neurons regulates active waking but not the sleep-wake switch or propofol-induced loss of consciousness. J Neurosci. 2012;19:13062–13075. doi: 10.1523/JNEUROSCI.2931-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman JE, Naidoo N, Raizen DM, Pack AI. Conservation of sleep: insights from non-mammalian model systems. Trends Neurosci. 2008;31:371–376. doi: 10.1016/j.tins.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.