

Abstract
Pain and inflammation are major therapeutic areas for drug discovery. Current drugs for these pathologies have limited efficacy, however, and often cause a number of unwanted side effects. In the present study, we identify the non-steroid anti-inflammatory drug, carprofen, as a multi-target-directed ligand that simultaneously inhibits cyclooxygenase-1 (COX-1), COX-2 and fatty acid amide hydrolase (FAAH). Additionally, we synthesized and tested several racemic derivatives of carprofen, sharing this multi-target activity. This may result in improved analgesic efficacy and reduced side effects (Naidu, et al (2009) J Pharmacol Exp Ther 329, 48-56; Fowler, C.J. et al. (2012) J Enzym Inhib Med Chem Jan 6; Sasso, et al (2012) Pharmacol Res 65, 553). The new compounds are among the most potent multi-target FAAH/COXs inhibitors reported so far in the literature, and thus may represent promising starting points for the discovery of new analgesic and anti-inflammatory drugs.
Introduction
Pain and inflammation remain areas of substantial unmet patient need.1-7 Current drugs used to treat these conditions have, however, moderate efficacy and can produce a variety of untoward side effects, such as gastrointestinal bleeding and ulceration, renal dysfunction, nausea and vomiting. Therefore, the search for novel and more effective analgesics able to overcome these limitations is the subject of intense efforts in both academia and industry.
Non-steroidal anti-inflammatory drugs (NSAIDs) are commonly used to treat acute and chronic pain. NSAIDs produce their beneficial action by inhibiting the two isoforms of the cyclooxygenase (COX) enzyme, COX-1 and COX-2.8, 9 These enzymes convert arachidonic acid into prostaglandins and thromboxane, which are important physiological and pathological effectors. Different tissues express varying levels of COX-1 and COX-2. COX-1 is a constitutive enzyme found in most mammalian cells. COX-2, on the other hand, is an inducible enzyme whose expression can be strongly stimulated by pro-inflammatory stimuli in macrophages and other cells.10 There are several well-known classes of NSAIDs, which are either non-selective for COX-1 and COX-2 or selective for COX-2.11 Both classes exert, however, a number of potentially serious side effects.12 In the gastrointestinal tract, COX-1 inhibition blocks the synthesis of tissue-protecting prostaglandins such as PGE2, facilitating the development of peptic ulceration and dyspepsia. Selective COX-2 inhibitors have raised major concerns because of increased cardiovascular risk. A notable example is rofecoxib, which was withdrawn from the market in 2004 because of such – still debated – concerns.13, 14
Fatty acid amide hydrolase (FAAH) has been proposed as a promising target for the discovery of new drugs to treat pain, inflammation and other pathologies.15-19 FAAH is an intracellular serine hydrolase responsible for the deactivating hydrolysis of a family of naturally occurring fatty-acid ethanolamides, such as its main substrate anandamide, which acts as an endogenous cannabinoid agonist.20-22 Interestingly, it has been suggested that drugs currently marketed as analgesics may derive some of their efficacy from inhibition of FAAH, which further highlights the potential of this target for drug discovery.23, 24 Several classes of FAAH inhibitors have been discovered during the last decade – including α-ketoheterocycles, carbamate-, piperidine- and piperazine urea based molecules – some of which are undergoing pre-clinical and clinical studies. 25-31
Several in vivo studies suggest that the simultaneous inhibition of COX and FAAH activities produces super-additive pharmacological effects and lowered toxicity in animal models. Naidu et al. showed that the FAAH inhibitor URB59732 and the non-selective COX inhibitor diclofenac act synergistically to reduce visceral pain in mice.33 Similar results were obtained by Sasso et al. using the peripherally restricted FAAH inhibitor URB597 and the NSAID indomethacin.34 Importantly, both studies showed that FAAH blockade lowers the ulcerogenic activity of COX inhibitors.11 These findings suggest that multi-target-directed-ligands35 able to inhibit simultaneously FAAH and COX activities might offer certain advantages over traditional single-target drugs and/or drug combinations. These include: (i) improved efficacy, due to the synergistic interaction between FAAH and COX blockade, (ii) improved safety, due to the lowering of COX-mediated side effects produced by FAAH inhibition, and (iii) reduced uncertainty in clinical development with respect to drug cocktails or multicomponent drugs, due to the avoided risk of drug-drug interactions.35-38 It is worth remembering that some very successful drugs act via multiple target mechanisms (e.g. quetiapine, imatinib);
Here, we report on the discovery of new multi-target inhibitors that show improved potency compared to previously reported mixed FAAH/COXs compounds.23, 24, 39 We used docking calculations to identify putative FAAH/COXs inhibitors starting from known COX-targeting drugs. In vitro pharmacological tests identified carprofen (Figure 1) as a multi-target FAAH/COXs hit. Based on this finding, we designed several carprofen derivatives that showed significant multi-target inhibitory activity, highlighting the potential of the carprofen scaffold as a source for new effective and safe analgesics.
Figure 1.
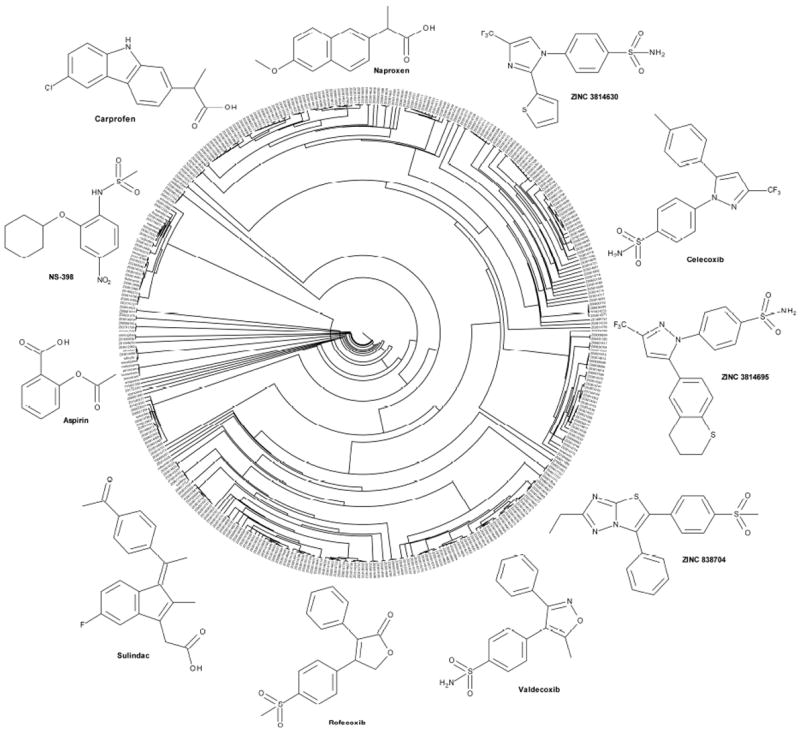
Circular tree based on pairwise Tanimoto distances between Daylight fingerprints of 382 diverse known COXs inhibitors. To help in the interpretation, only selected molecules, belonging to different clusters, are depicted in proximity of their positions in the tree to highlight the structural diversity of the set. Carprofen is shown in the upper left corner.
Results
Identification of carprofen as a multi-target FAAH/COX inhibitor
We selected 382 COXs inhibitors retrieved from DrugBank40 and DUD41 and docked them into the structure of FAAH (see the Experimental Section). Several clinically approved drugs were found among the top-ranking molecules. The entire assembled set was clustered according to pairwise Tanimoto distances, using a description based on the Daylight fingerprints (Figure 1). A clustering threshold of 0.4 resulted in 84 clusters, which highlighted the structural diversity within the set. The top 100 scored molecules were visually inspected. Among them, indomethacin was ranked no. 4, flurbiprofen no. 10 and celecoxib no. 16 (the putative binding mode of these three COX inhibitors at the active site of FAAH is reported in Figure S2). Based on their commercial availability, we purchased 25 compounds for testing (see Figure 2). These molecules were also clustered according to their topological distance and physico-chemical similarity, as highlighted in Figure 2. According to the reported tree, a clustering threshold of 0.3 originated 19 clusters, underlining the absence of trivially similar compounds. This unanticipated chemotype richness enabled us to probe FAAH binding thoroughly, and maximize the chance to find a multi-target FAAH/COX inhibitor among the 25 selected compounds. The in vitro results identified carprofen (1) as the best multi-target FAAH/COXs inhibitor in the set of compounds tested. Carprofen inhibited FAAH with a median effective concentration (IC50) = 79 μM, COX-1 with an IC50 = 22 μM and COX-2 with an IC50 = 4 μM. As the profile against the three targets turned out to be rather balanced, compound 1 emerged as a promising starting point for multi-target FAAH/COXs lead discovery.
Figure 2.
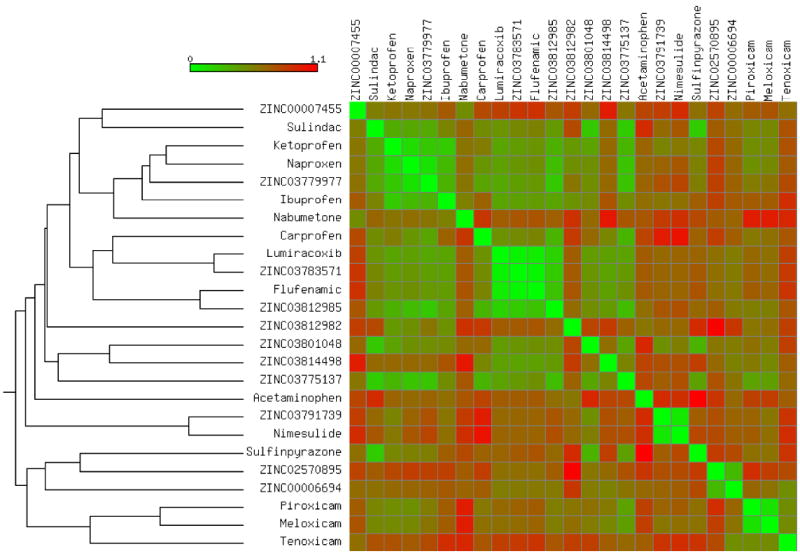
Tree based on the pairwise Tanimoto-fingerprint distances between the 25 COX inhibitors tested in the present study. The heat map highlights the distances calculated in the first 5 principal components space (% variance explained > 90%) originating from 10 physico-chemical descriptors (i.e. net charge, MW, LogP, LogS, HBD, HBA, PSA, no. of atoms, no. of rings and no. of rotatable bonds).
Chemistry
Several possible chemical variations of 1 were considered (Scheme 1). The preparation of the corresponding des-chlorinated derivative (2) was performed by hydrogenation of 1, using a flow hydrogenator apparatus (Scheme 2).
Scheme 1.
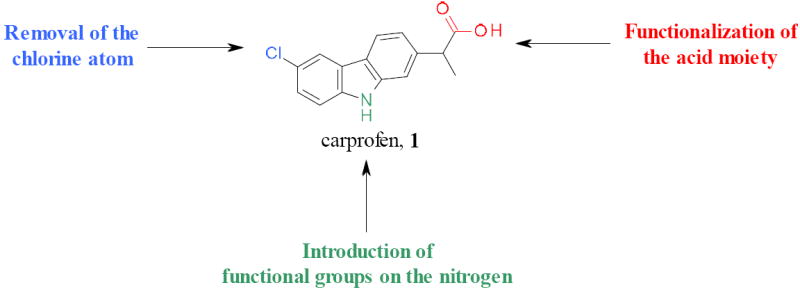
Planned chemical variations of carprofen, 1
Scheme 2.
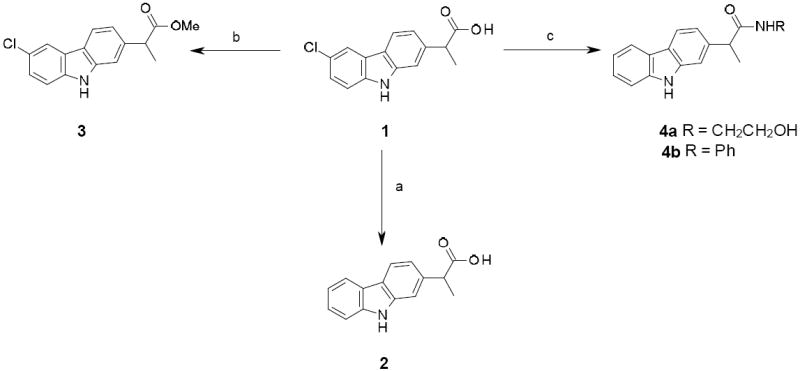
Synthesis of des-chlorinated compound 2, ester 3 and amides 4a
a Reagents and conditions: (a) H-Cube, H2, 10 % Pd/C, EtOH, EtOAc, 60 °C, 40 %; (b) MeOH, H2SO4, rt, 12 h, quant; (c) CDI, pyridine, 60 °C, 12 h, 85-86 %.
Syntheses of the ester and amide derivatives of 1 were achieved using standard reaction conditions (Scheme 2, for representative examples). The esterification of 1 in acidic methanol gave the corresponding methyl ester 3 in quantitative yield. The amides 4a and 4b were efficiently prepared by the reaction of 1 in pyridine in the presence of CDI, respectively with ethanolamine and aniline.
The functionalization of the nitrogen atom of 1 was achieved using ester 3 as common intermediate (Scheme 3). The preparation of different N-alkyl derivatives was performed using the appropriate alkyl halides in the presence of Cs2CO3 in MeCN to provide compounds 5 in moderate to good yields. Saponification of the ester with LiOH, followed by acidic treatment, gave the corresponding carboxylic acids 6 in good yields.
Scheme 3.
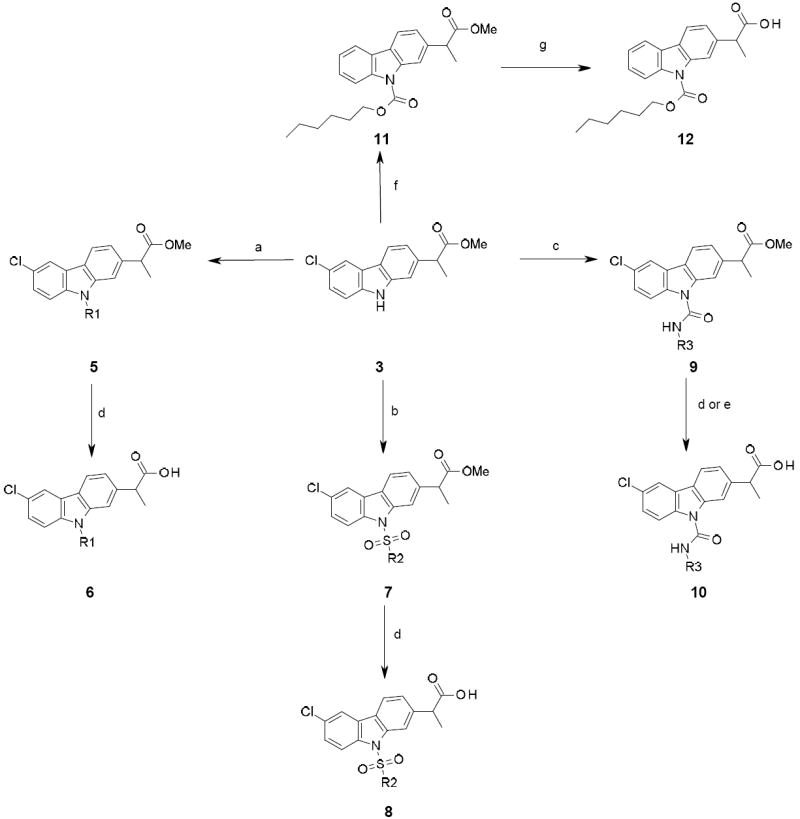
Synthesis of compounds 6, 8, 10 and 12a
aReagents and conditions: (a) R1-X, Cs2CO3, MeCN, reflux, 12 h, 32-99 %; (b) R2-SO2Cl, Et3N, DMAP, THF, reflux, 5 h or 100 °C, 3 h, MW, 35-78 %; (c) R3-NCO, Et3N, DMAP, THF, 100 °C, MW, 10 h, 51-81 %; (d) LiOH, MeOH, THF, H2O, 12 h, 21-85%; (e) 6M HCl, THF, rt, 5 days, 80 %; (f) hexyl chloroformate, Et3N, DMAP, THF, 100 °C, 3 h, MW, 85 %; (g) 6M HCl, THF, rt, 3 days, 55 %.
Preparation of the sulfonamide derivatives 7 was achieved by reaction of 3 with the appropriate sulfonyl chlorides in THF, in the presence of DMAP and Et3N, under thermal heating or microwave irradiation. In a similar manner, as described before, saponification of the ester group produced the corresponding acid 8 in good yields.
Reaction of compound 3 with the appropriate isocyanates in THF, in the presence of DMAP and Et3N, under microwave conditions, afforded the urea derivatives 9. Compounds 9 were then hydrolyzed under basic conditions to give the corresponding acid derivatives 10 in moderate to good yields. In the case of 9d, bearing a para-chlorophenyl moiety, the methyl ester hydrolysis was performed in acidic media (due to the observed cleavage of the urea bond of 9d under basic conditions) to provide 10d in a good 80 % yield.
The carbamate derivative 12 was prepared under standard conditions (Scheme 3). Compound 3 was reacted in THF with hexyl chloroformate, in presence of Et3N and DMAP, to give intermediate 11, which was further hydrolyzed under acidic condition to provide 12.
The preparation of the acyl derivatives was performed using a different protecting group on the carboxylic functionality (Scheme 4). In our initial experiments, it was indeed observed that N-acetylated derivatives were not stable under basic and acidic conditions, typically used for the hydrolysis of the methyl ester. The use of tert-butyl ester was also exploited without any synthetic success.42 We therefore decided to protect 1 as an O-benzyl ester, using standard reaction conditions (BnBr, K2CO3, DMF).43 The acylation reactions of the benzyl ester 13 proceeded well in MeCN in the presence of DMAP and Et3N, with various acyl chlorides, to afford esters 14.
Scheme 4.
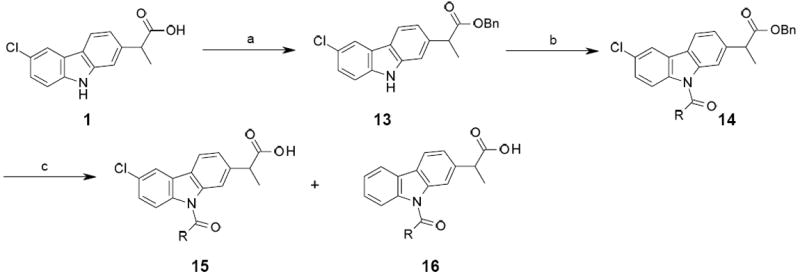
Synthesis of compounds 15a
aReagents and conditions: (a) BnBr, K2CO3, DMF, rt, 3 h, 84 %; (b) Acyl chloride, DMAP, Et3N, MeCN, rt, 2 h, 76-97 % ; (c) H-Cube, H2, 1 or 5 % Pd/C, rt, THF, 15-42 %.
The last debenzylation step to obtain the corresponding acids 15 was performed using the H-Cube apparatus, under optimized conditions in order to reduce the formation of the undesired des-chloro derivatives 16. In this context, the influence of the solvent (EtOH:EtOAc (1:1) and THF), the temperature of the reaction (from room temperature to 60 °C), the catalyst loading (1%, 5% and 10% Pd/C cartridge), and the hydrogenation conditions were evaluated. After several trials, two different conditions appeared to be satisfactory and were applied for the preparation of our targeted compounds. The first method relied on passing a solution of 14 in THF through a 5 % Pd/C cartridge, at room temperature and 1 bar H2 pressure (H-Cube “full H2 mode”). In this case, complete conversion of the benzyl ester 14 was observed and the expected 15 was formed in approximately 3:1 ratio with respect to 16. The second method was performed using a 1 % Pd/C cartridge at room temperature and 1 bar H2 pressure, in a closed-loop system with the H-Cube (where the out-coming solution was directly re-injected in the in-coming solution). By carefully monitoring the advancement of the reaction by UPLC/MS analysis, it was possible, after 4-5 h of hydrogenation, to reach a stage where the conversion of 14 was satisfactory (generally around 70 %) and where the formation of 16 was maintained low compared to 15; the reaction was stopped when compounds 15:16 ratio reached approximately 9:1. 15a-j were prepared using this procedure with the exception of 15k. In this case, we observed that the final hydrogenation step did not proceed, probably because of the presence of the thiazole ring, which poisoned the catalyst. We therefore decided to use the methyl ester 3 as starting material and to carry the hydrolysis in acidic media, which led to the formation of 15k in a low yield.
Structure-Activity Relationship (SAR) exploration
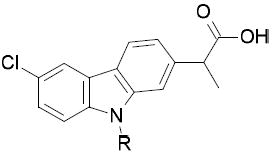
The inhibitory activities against FAAH, COX-1 and COX-2 of the first set of derivatives of 1 are summarized in Table 1. The IC50 values were not determined for compounds showing less than 50% inhibition at concentrations of 100 μM for FAAH and COXs.
Table 1.
FAAH-1, COX-1 and COX-2 activities of carprofen derivativesa
| ||||||
|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | IC50 (μM)
± SDb
|
||
| FAAH | COX-1 | COX-2 | ||||
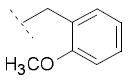
| 1 | Cl | OH | H | 78.6±19.7 | 22.3±6.6 | 3.9±1.0 |
| 2 | H | OH | H | 4.6±0.3 | / | / |
| 3 | Cl | OCH3 | H | 22.1±2.4 | / | / |
| 4a | Cl | HOCH2CH2NH | H | / | / | / |
| 4b | Cl | C6H5NH | H | 2.9±0.2 | / | / |
| 6a | Cl | OH | CH3 | 15.5±2.5 | / | / |
| 6b | Cl | OH |
|
23.3±2.5 | / | / |
| 6c | Cl | OH |
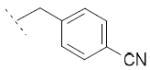
|
33.4±15.7 | / | / |
| 6d | Cl | OH |
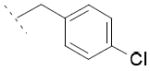
|
10.2±0.9 | / | / |
| 6e | Cl | OH |
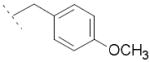
|
8.4±2.4 | / | / |
| 6f | Cl | OH |
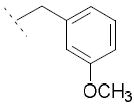
|
32.5±7.1 | / | / |
| 6g | Cl | OH |
|
/ | / | / |
| 8a | Cl | OH |
|
50.3±2.3 | / | / |
| 8b | Cl | OH |
|
4.8±4.5 | / | / |
| 10a | Cl | OH |
|
3.2±1.7 | / | / |
| 10b | Cl | OH |
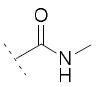
|
/ | / | / |
| 10c | Cl | OH |
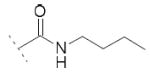
|
/ | / | / |
| 10d | Cl | OH |
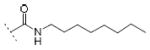
|
26.8±8.1 | 15.4±8.8 | / |
| 10e | Cl | OH |
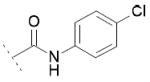
|
102.0±6.1 | 41.3±6.9 | / |
| 10f | Cl | OH |
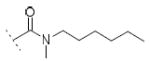
|
/ | / | / |
| 10g | Cl | OH |
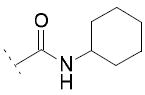
|
/ | / | / |
| 12 | Cl | OH |
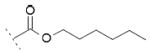
|
31.6±0.8 | 17.2±1.9 | / |
| 15a | Cl | OH |
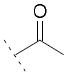
|
/ | / | / |
| 15b | Cl | OH |
|
25.4±1.9 | / | / |
| 15c | Cl | OH |
|
22.0±4.2 | 74.3±28.0 | 72.3±28.0 |
Values are means of ≥3 experiments performed in duplicate;
/ : IC50 >100μM
Compound 1 inhibited FAAH, COX-1 and COX-2 with a rather balanced profile (Table 1). Although an improved potency against FAAH was observed with 2 (IC50 = 5 μM), the removal of the chlorine atom was detrimental for the inhibition of both COX-1 and COX-2. Also, the pivotal role of the carboxylic function for COXs inhibition was confirmed by the lack of activity of the ester derivative 3 and the amides, 4a and 4b. On the other hand, we observed a 26-fold increase in FAAH potency for 4b (IC50 = 3 μM) compared to 1. Based on these results, we decided to continue the exploration exclusively on derivatives bearing both the chlorine atom and the carboxylic function.
We investigated the functionalization of the nitrogen atom of the carbazole core. The N-alkylated compounds 6a-g and the N-arylsulfonylated derivatives 8a-b were devoid of COX inhibitory activity. On the other hand, the compounds showed a slightly better FAAH inhibitory potency compared to 1. The introduction of a methyl group (6a) led to an IC50 = 15 μM against FAAH, while a benzyl group on the carbazole nitrogen gave a 3-fold improvement in FAAH inhibition compared to 1 (6b, IC50 = 23 μM). We also evaluated the influence of the substitution on the phenyl ring. Also compound 6c, which bears a CN group in the para position, showed a slight decrease in FAAH potency (IC50 = 33 μM). Conversely, the p-Cl derivative 6d and the p-OCH3 derivative 6e showed greater potency against FAAH, with IC50 values of 10 μM and 8 μM, respectively. Moreover, we observed that the para-substitution was preferential: the p-OCH3 derivative 6e was more potent against FAAH (IC50 = 8 μM) than the m-OCH3 derivative 6f (IC50 = 32 μM) and the o-OCH3 derivative 6g (which showed no activity at 30 μM). Within the series of sulfonamides, 8a and 8b were active against FAAH, displaying IC50 values of 50 μM and 5 μM, respectively. It is worth mentioning that N-alkylated and N-arylsulfonylated carprofen derivatives were previously investigated for their ability to inhibit γ-secretase activity.43
A number of urea derivatives were also investigated. Here too, it was observed that inhibitory activity was generally lost on both COX isoforms, while FAAH inhibitory activity was highly dependent on the type of substituent on the urea moiety. Indeed, the best FAAH inhibitor of this series was 10a (IC50 = 3 μM), bearing a linear C6 chain. Shorter linear chains, from C1 to C4, showed no inhibition on FAAH, as exemplified by 10b and 10c. Longer chains brought to less potent FAAH inhibitors, but interestingly led to an increase in COX-1 inhibition. For example 10d showed an IC50 = 27 μM against FAAH and an IC50 = 15 μM against COX-1. Compound 10e (bearing a 4-chlorobenzyl moiety), showed weak potency against FAAH (IC50 = 102 μM), and an IC50 value of 41 μM against COX-1. N, N-disubstituted derivatives or ureas bearing cyclic moiety, such as 10f and 10g, were also prepared but they all did not show any improved activities against the targets. Compound 12 showed a 10-fold weaker potency against FAAH (IC50 = 32 μM) compared to its carbamate analog 10a (IC50 = 3 μM), while it showed an IC50 = 17 μM on COX-1.
We then analyzed a set of acyl derivatives. The acetyl derivative 15a did not show any inhibitory potency. The compound 15b was active only against FAAH (IC50 = 25 μM). Compound 15c, which bears a p-Cl benzoyl group, showed promising inhibitory potency on FAAH (IC50 = 22 μM) and COX (COX-1 IC50 = 74 μM and COX-2 IC50 = 72 μM).
In light of these promising results, we came to the conclusion that a carbonyl function linked to the nitrogen atom of the carbazole ring of 1 could be important to obtain a multi-target inhibition. Therefore, we decided to further expand the SAR around 15c, obtaining a second series of molecules, whose activities are summarized in Table 2.
Table 2.
SAR exploration around 15ca
| ||||
|---|---|---|---|---|
| Compd | R | IC50 (μM)
± SDb
|
||
| FAAH | COX-1 | COX-2 | ||
| 15c |
|
22.0±4.2 | 74.3±28.0 | 72.3±28.0 |
| 15d |
|
30.9±10.6 | / | / |
| 15e |
|
10.6±2.6 | / | / |
| 15f |
|
20.1±2.6 | / | / |
| 15g |
|
59.6±15.5 | / | / |
| 15h |
|
/ | / | / |
| 15i |
|
84.8±10.6 | 30.0±12.1 | 27.8±9.7 |
| 15j |
|
5.6±2.9 | 12.8±8.9 | / |
| 15k |
|
/ | / | / |
Values are means of ≥3 experiments performed in duplicate;
/ : IC50 >100μM
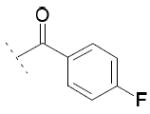
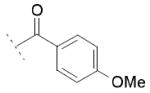
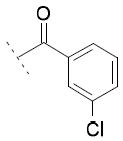
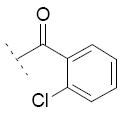
Focusing on the benzoyl core of 15c, we prepared the p-F-benzoyl and p-OCH3-benzoyl analogs 15d and 15e, which showed IC50 values on FAAH of 31 μM and 11 μM, respectively. However, the compounds did not inhibit COX-1 or COX-2 activity. We then analyzed the influence of the position of the chlorine atom on the phenyl ring. Interestingly, as we reported before in the N-alkyls series, the para substitution was the most tolerated. The m-Cl derivative 15f (FAAH IC50 = 20 μM), and the o-Cl derivative 15g (FAAH IC50 = 60 μM) retained a similar inhibitory potency for FAAH compared to 15c, while inhibitory activity against COXs was lost. A total loss of activity for the three enzymes was observed when we tried to combine the most tolerated para- and m-Cl substitutions, as shown with 15h.
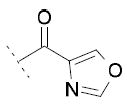
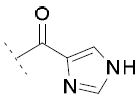
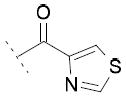
The inhibitory activity on the three enzymes was also highly dependent on the nature of the heteroaromatic ring. The 4-oxazole substitution of 15i showed to be well tolerated, with FAAH IC50 = 85 μM, COX-1 IC50 = 30 μM and COX-2 IC50 = 28 μM. Compound 15j, bearing a 4-imidazole ring, showed FAAH IC50 = 6 μM and COX-1 IC50 = 13 μM, but no relevant inhibition of COX-2. The introduction of a 4-thiazole ring in 15k was detrimental for the inhibition of FAAH and COXs enzymes.
We finally analyzed the potency of the single enantiomers of the most active compounds of the present series, namely 1, 15c and 15i. The racemic mixtures were submitted to enantiomeric separation by chiral HPLC. The results obtained are summarized in Table 3. For 1, the S-(+) enantiomer was the only active against the three targets. For 15c and 15i, the S-(+) enantiomer was the only active against COXs, while the inhibitory activity against FAAH was obtained with the R-(-) enantiomer, for both compounds. These findings are consistent with the known ability of the R-enantiomers of flurbiprofen, indomethacin and celecoxib to inhibit FAAH.44, 45 In addition, the inactivity of R-(-) enantiomers of 1, 15c and 15i against COXs was expected, in agreement with previous results showing a preferential COXs inhibition by S-(+) enantiomers.46
Table 3.
FAAH, COX-1 and COX-2 activities of single enantiomers of compounds 1, 15c and 15i a
| Compd | IC50 (μM)
± SDb
|
||
|---|---|---|---|
| FAAH | COX-1 | COX-2 | |
| (±)-1 | 78.6±19.7 | 22.3±6.6 | 3.9±1.0 |
| S-(+)-1 | 64.2±3.6 | 5.6±0.1 | 5.3±3.0 |
| R-(-)-1 | / | / | / |
| (±)-15c | 22.0±4.2 | 74.3±28.0 | 72.3±28.0 |
| S-(+)-15c | / | 45.0±0.3 | 46.5±4.3 |
| R-(-)-15c | 14.9±1.6 | / | / |
| (±)-15i | 84.8±10.6 | 30.0±12.1 | 27.8±9.7 |
| S-(+)-15i | / | 4.1±2.8 | 2.5±1.4 |
| R-(-)-15i | 53.2±22.6 | / | / |
Values are means of ≥3 experiments performed in duplicate;
/ : IC50 >100μM
Discussion
The present results identify carprofen (compound 1) as a multi-target FAAH/COX inhibitor. SAR studies on the scaffold of the molecule show that different functionalizations at the nitrogen atom of the carbazole ring of this molecule yield additional active compounds, such as 15c and 15i, which inhibit FAAH/COX activities in the low μM range (see Tables 1 and 2). The ability of NSAIDs to inhibit anandamide hydrolysis has been previously described by Fowler and coworkers in several informative studies.39, 47, 48 These investigators described new molecules that simultaneously target FAAH and COX activities. In particular, the compound ibu-am5 (N-(3-methylpyridin-2-yl)-2-(4-isobutylphenyl)-propionamide) was shown to be a promising multi-target FAAH/COX inhibitor.23, 24, 39, 49 This compound displayed an IC50 value for FAAH of ~0.5 μM (in EtOH), and IC50 values for COX-1 of ~60 μM and ~240 μM in DMSO and ethanol, respectively. These data for COXs inhibition were generated using an oxygen electrode assay for peroxidase (POX) activity.24 Under these experimental conditions, ibu-am5 was inactive against COX-2. However, if anandamide was used as COX-2 substrate in the assay, instead of arachidonic acid, ibu-am5 showed a COX-2 IC50 ~ 20 μM, suggesting a substrate-dependency in COX-2 inhibition.24 Under our present experimental conditions, ibu-am5 inhibited FAAH with an IC50 of 66 μM. COX inhibition was measured with an enzyme immunoassay using a prostaglandin-specific antibody (see Methods), which yielded a IC50 for COX-1 of 170 μM. The potency toward COX-2 was very low, as ibu-am5 produced only 30% inhibition at 30 μM. We also tested ibuprofen as an additional reference compound, which showed IC50 of 5 μM and 36 μM against COX-1 and COX-2, respectively, while it was very weak against FAAH, in reasonably good agreement with previous findings.47, 50
Therefore, carprofen and its racemic derivatives described in the present study are among the most potent multi-target FAAH/COX inhibitors reported so far. These molecules also show a well-balanced multi-target profile, implying that they may provide a starting point for the discovery of new FAAH/COXs inhibitors to treat pain, inflammation and potentially, as recently discussed, cancer.51, 52 In addition, these compounds may provide a pharmacological tool to characterize the synergistic effect produced by the simultaneous inhibition of FAAH and COXs in different pathological conditions.34
Despite being evolutionary unrelated, FAAH and COXs share several structural similarities, which might help in the rationalization of the multi-target activity of carprofen and its derivatives. Such common features of the catalytic pocket of FAAH and COXs could also be anticipated in light of the similarity of their endogenous substrates. The main product of FAAH activity on anandamide is arachidonic acid, which is a substrate for COXs. In addition, it has been shown that anandamide can be metabolized by COX-2.49 Because of the presence of such common structural features, useful to discuss also the basis of biological promiscuity,53 we mapped FAAH and COX-2 with two probes able to pinpoint hydrophilic and hydrophobic spots of the proteins (Figure 3). The acyl chain binding channel is the most hydrophobic portion of FAAH while, in proximity of the oxyanion hole, the hydrophilic features become preponderant due to both the presence of hydroxyl groups and the proximity of the cytosolic channel. We found a similar pattern in COX-2. In the case of this enzyme, the core of the binding site is mainly hydrophobic. In fact, this is the volume that hosts the lipophilic core of virtually all known COX inhibitors. Approaching the exit of the site, the hydrophilic character increases. The polar tails of several well-known COX inhibitors lie in this area, establishing H-bond interactions with Arg120 and Tyr355. The most important difference can be observed for the hydrophobic volume, which is greater in the case of FAAH (Figure 3).
Figure 3.
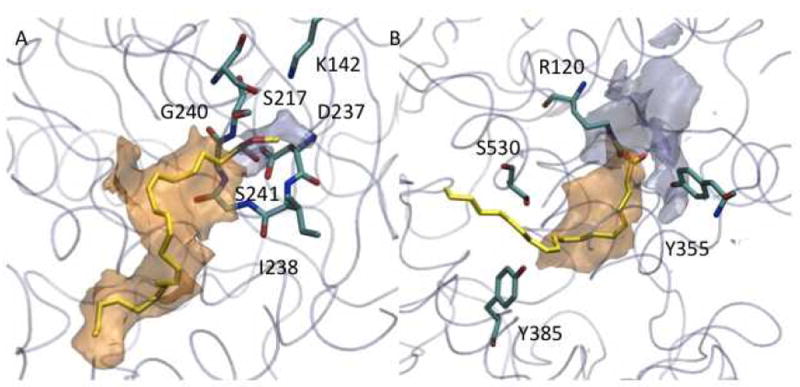
Hydrophilic (light blue) and hydrophobic (orange) isocontour surfaces of FAAH-1 (A) and COX-2 (B). For the sake of clarity, relevant residues are highlighted as stick models with C atoms colored in cyan. The protein is shown as transparent cyan tube. Substrates are methyl arachidonyl fluorophosphonate in FAAH and arachidonic acid in COX-2.
On the basis of our docking results, we are quite confident on the binding mode of 1 in COXs, since it is consistent with the experimentally observed binding mode of several arylpropionic acid derivatives in complex with either of the two COX isoforms.9 On the other hand, in FAAH, 1 could be accommodated into the acyl chain binding channel and establish H-bond interactions with the oxyanion hole through its carboxylic group. However, we cannot rule out alternative binding modes, in different regions of the large binding cavities of FAAH’s catalytic site (see Figure S3). Notably, 1 is a small molecule bearing fragment-like physicochemical features.54
Finally, it is worth mentioning that the multi-target FAAH/COXs activity described in this study could in principle explain why 1 is reported to have reduced ulcerogenic side effects in humans, compared to other NSAIDs.55 The ability of this compound to block FAAH activity might attenuate the ulcerogenic effects of COX inhibition, as shown in mice for the combination URB937 plus indomethacin.34
Conclusions
We reported on the identification of multi-target inhibitors that block simultaneously FAAH, COX-1 and COX-2 activities. The concomitant inhibition of these enzymes has recently been shown to produce, in vivo, improved analgesic response and diminished side effects in animal models of pain. Despite their still moderate activity, the present series of compounds comprise the most active multi-target FAAH/COXs inhibitors reported in the literature so far. They could serve, therefore, both as starting points for future drug discovery efforts, and as tools to further characterize the synergistic effects obtained by the simultaneous inhibition of FAAH and COX activities.
Experimental section
Cox inhibitors dataset
A large collection of known COXs inhibitors was retrieved, in a ready-to-dock format, from DUD41 and from DrugBank.40 The grand total of unique molecules collected, after eliminating double occurrences, was 382. Some of these are currently marketed drugs mostly in use as anti-inflammatory agents, while others have been reported to inhibit at least one of the two COX isoforms, with diverse potencies. For years, COXs inhibitors have been classified according to their binding preferences and mechanism of action. A clear-cut distinction between classes can be sometimes misleading and formally incorrect. In general, there can be selective, non-selective and isoenzyme-preferring binders that can act through i) an irreversible mechanism (e.g. aspirin), ii) a reversible substrate competitive binding (e.g. piroxicam), iii) a slow, time-dependent non covalent action (e.g. indomethacin), and iv) a slow, time-dependent COX-2 irreversible type of inhibition (e.g. rofecoxib/Vioxx®, celecoxib/Celebrex®). Very recently, Marnett and colleagues showed that the (R) enantiomers of several well known COX inhibitors such as naproxen, flurbiprofen and ibuprofen, considered to be COX-2 inactive, are, in fact, ‘substrate-selective-inhibitors’.56
The dataset reported here comprises all the classes of inhibitors mentioned above, among which we can find classic NSAIDs, such as fenac- (e.g. sulindac) and profen-derivatives (e.g. ketoprofen), oxicames (e.g. tenoxicam) as well as some classes of COX-2 selective binders, such as di(hetero)aryl (thio)ethers (e.g. NS-398, nimesulide), carbocycles and heterocycles with vicinal aryl substitutions (e.g. valdecoxib, celecoxib and the recently retired from the market xib), just to mention a few. However, the collection presented here is meant to be a set of readily accessible molecules known to inhibit at least one of the two COX isoforms, and to be used as starting points toward the search for dual FAAH/COX inhibitors, rather than being an exhaustive collection of COXs inhibitors.
The entire assembled set was clustered according to pairwise Tanimoto distances, using a description based on the Daylight fingerprints (Figure 1). A clustering threshold of 0.4 resulted in 84 clusters, which highlighted the structural diversity within the set. The chemotype richness found among the selected COX inhibitors is convenient when looking for binders of an evolutionary unrelated target (i.e., FAAH), such as in this case. To better characterize the molecular diversity in the dataset, we looked at specific physico-chemical features of each compound. The distribution observed for nine common physico-chemical descriptors further describes the diversity found over the entire dataset. Normal distributions can be observed for most of the selected descriptors. An exception was the total charge descriptor. In fact, apart from a few negatively charged molecules, the vast majority of entries were neutral. Also, most compounds showed a number of HB donors of either 0 or 2 (see Figure S1).
Molecular descriptors and fingerprint similarity
Molecular properties for each fragment were calculated by means of ICM3.7 (Molsoft LLC, San Diego, CA). Molecular patterns were calculated and hashed into bitmaps according to the Daylight algorithm for fingerprint generation (Daylight Chemical Information Systems Inc., Laguna Niguel, CA) as available in ICM3.7 (Molsoft LLC, San Diego, CA). Similarity between molecules was calculated as the difference between 1 and the Tanimoto coefficient, Tc, where Tc = c/(a + b - c), where c counts the common bits on in molecule 1 and molecule 2, a counts the bits on in molecule 1, and b counts the bits on in molecule 2. Tc spans from 0 to 1, with 1 indicating that two molecules share the same fingerprint.
Docking and binding site mapping
Chain A of the X-ray structure of rFAAH, covalently bound to a molecule of methyl arachidonyl fluorophosphonate, as available at the protein databank (PDBid 1mt5)57 and chain A of the X-ray structure of COX-2, in complex with flurbiprofen (PDBid 3pgh)58 were used for the docking study. Missing sidechains were added through SCWRL 4.0.59 Then, water and co-crystallized ligands were removed, and the catalytic serine (S241) was treated as negatively charged. The so-obtained structures were energy minimized with NAMD 2.660 program until a 0.3 Å RMSD convergence criterion on heavy atoms was reached. The Amber force field parm99SB61 was employed throughout. The final protein models and the selected known COXs inhibitors were then converted through the Python scripts available in the ADT suite of programs62 into a suitable format for AutoDock Vina v.1.0.63
AutoDock Vina v.1.063 was used to dock 382 known COXs inhibitors at the FAAH binding site. The search volume, centered on the phosphate atom of the co-crystallized inhibitor, spanned 24 Å in the three directions. The algorithm search exhaustiveness was set to 4 and the maximum number of output poses was set to 10. Finally, the top 100 ranked molecules (approx. 25% of the distribution), were visually inspected to eliminate hits that did not directly bind in proximity of the oxyanion hole. We checked the commercial availability of each of the top 100 scored molecules and we eventually purchased 25 compounds for experimental testing (see Figure 2). Some molecules such as acetaminophen and aspirin that exceeded the rank threshold (rank no. 350 and 326, respectively) were considered because they were readily available. Despite their low scores, we also included molecules belonging to an underrepresented chemical class, the oxicams (i.e. piroxicam, meloxicam end tenoxicam). When a top ranked molecule was not available we purchased alternative molecules that were close in the Tanimoto-fingerprint space, regardless of their estimated binding energies. For example, diclofenac and etodolac were purchased in place of ZINC03814788 (rank no. 61) and ketorolac (rank no.53), respectively.
General methods for synthesis
Solvents and reagents were obtained from commercial suppliers and were used without further purification. For simplicity, solvents and reagents were indicated as follows: acetyl chloride (AcCl), acetonitrile (MeCN), ammonium chloride (NH4Cl), benzyl bromide (BnBr), carbonyldiimidazole (CDI), cesium carbonate (Cs2CO3), cyclohexane (Cy), chloroform (CHCl3), dichloromethane (DCM), dimethylsulfoxide (DMSO), diethyl ether (Et2O), N,N-diisopropylethylamine (DIPEA), 4-(dimethylamino)-pyridine (DMAP), di-tert-butyl dicarbonate (Boc2O), ethanol (EtOH), ethyl acetate (EtOAc), hydrochloric acid (HCl), sulfuric acid (H2SO4), iodomethane (MeI), N,N-dimethylformamide (DMF), lithium hydroxide (LiOH), magnesium sulfate (MgSO4), methanol (MeOH), sodium bicarbonate (NaHCO3), tetrabutylammonium iodide (TBAI), tetrahydrofuran (THF), triethylamine (Et3N), trifluoroacetic acid (TFA).
Automated column chromatography purifications were done using a Teledyne ISCO apparatus (CombiFlash® Rf) with pre-packed silica gel columns of different sizes (from 4 g until 120 g). Mixtures of increasing polarity of cyclohexane and ethyl acetate or dichloromethane and methanol were used as eluents. Preparative TLC were performed using Macherey-Nagel pre-coated 0.05 mm TLC plates (SIL G-50 UV254). Hydrogenation reactions were performed using H-Cube® continuous hydrogenation equipment (SS-reaction line version), employing disposable catalyst cartridges (CatCart®) preloaded with the required heterogeneous catalyst. Microwave heating was performed using Explorer®-48 positions instrument (CEM). NMR experiments were run on a Bruker Avance III 400 system (400.13 MHz for 1H, and 100.62 MHz for 13C), equipped with a BBI probe and Z-gradients. Spectra were acquired at 300 K, using deuterated dimethylsulfoxide (DMSO-d6) or deuterated chloroform (CDCl3) as solvents. Chemical shifts for 1H and 13C spectra were recorded in parts per million using the residual non-deuterated solvent as the internal standard (for DMSO-d6: 2.50 ppm, 1H; 39.52 ppm, 13C; for CDCl3: 7.26 ppm, 1H and 77.16 ppm, 13C). Data are reported as follows: chemical shift (ppm), multiplicity (indicated as: bs, broad signal; s, singlet; d, doublet; t, triplet; q, quartet; p, quintet, sx, sextet; m, multiplet and combinations thereof), coupling constants (J) in Hertz (Hz) and integrated intensity. UPLC/MS analyses were run on a Waters ACQUITY UPLC/MS system consisting of a SQD (Single Quadropole Detector) Mass Spectrometer equipped with an Electrospray Ionization interface and a Photodiode Array Detector. PDA range was 210-400 nm. Analyses were performed on an ACQUITY UPLC BEH C18 column (50×2.1 mmID, particle size 1.7 μm) with a VanGuard BEH C18 pre-column (5×2.1 mmID, particle size 1.7 μm). Mobile phase was either 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN-H2O (95:5) at pH 5 (B) or 5 mM NH4OAc + 0.25 % AcOH in H2O (A) and 5 mM NH4OAc + 0.25 % AcOH in MeOH (B) for compounds 5. Electrospray ionization in positive and negative mode was applied. Purifications by preparative HPLC/MS were run on a Waters Autopurification system consisting of a 3100 Single Quadropole Mass Spectrometer equipped with an Electrospray Ionization interface and a 2998 Photodiode Array Detector. HPLC system included a 2747 Sample Manager, 2545 Binary Gradient Module, System Fluidic Organizer and 515 HPLC Pump. PDA range was 210-400 nm. Purifications were performed on a XBridgeTM Prep C18 OBD column (100×19 mmID, particle size 5 μm) with a XBridgeTM Prep C18 (10×19 mmID, particle size 5 μm) Guard Cartridge. Mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN-H2O (95:5) at pH 5 (B). Electrospray ionization in positive and negative mode was used. Analyses by chiral HPLC were run on a Waters Alliance HPLC instrument consisting of an e2695 Separation Module and a 2998 Photodiode Array Detector. PDA range was 210-400 nm. Analyses were performed isocratic on a Daicel ChiralPak AD column (250×4.6 mmID, particle size 10 μm). Mobile phase was 0.1 % TFA Heptane/2-Propanol (75:25). Separations by preparative chiral HPLC were run on a Waters Alliance HPLC instrument consisting of a 1525 Binary HPLC Pump, Waters Fraction Collector III and a 2998 Photodiode Array Detector. UV detection was at 240 nm. Purifications were performed isocratic on a Daicel ChiralPak AD column (250×10mmID, particle size 10 μm). Mobile phase was 0.1 % TFA Heptane/2-Propanol (75:25). Optical rotations were measured on a Rudolf Research Analytical Autopol II Automatic polarimeter using a sodium lamp (589 nm) as the light source; concentrations expressed in g/100 mL using EtOAc or MeOH as a solvent and a 1 dm cell. All final compounds displayed ≥ 95% purity as determined by NMR and UPLC/MS analysis.
2-(9H-carbazol-2-yl)propanoic acid 2
A solution of carprofen 1 (106 mg, 0.39 mmol) in a 1:1 mixture of EtOH/EtOAc (20 ml) was hydrogenated with the ThalesNano H-Cube™ using a 10% Pd/C catalyst, at 1 mL/min flow rate, room temperature and H2 atmosphere (1 atm) (full H2 mode). After three consecutive runs, the solvent was removed in vacuo and the crude oil was purified by preparative HPLC to yield the corresponding des-chlorinated compound 2 (37 mg, 40 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.45 (d, J = 7.1 Hz, 3H), 3.83 (q, J = 7.1 Hz, 1H), 7.09 (dd, J = 8.1, 1.3 Hz, 1H), 7.14 (m, 1H), 7.38 (m, 2H), 7.47 (d, J = 8.1 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 7.8 Hz, 1H), 11.18 (s, 1H), 12.25 (s, 1H). MS (ES) C15H13NO2 requires 239, found 240 [M+H]+.
Methyl 2-(6-chloro-9H-carbazol-2-yl)propanoate 3
To a suspension of carprofen 1 (1.055 g, 3.85 mmol) in MeOH (40 mL) was added H2SO4 (0.1 mL). The mixture became homogeneous and was stirred at room temperature overnight. After evaporation of MeOH, the residue was taken up in EtOAc and washed with a saturated aqueous NaHCO3 solution (3 × 20 mL) and brine (10 mL). After separation, the organic phase was dried over MgSO4 and concentrated in vacuo to give 3 (1.15 g, quant.) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 1.59 (d, J = 7.2 Hz, 3H), 3.68 (s, 3H), 3.88 (q, J = 7.2 Hz, 1H), 7.18 (dd, J = 8.1, 1.4 Hz, 1H), 7.37 (m, 3H), 7.95 (d, J = 8.1 Hz, 1H), 7.99 (m, 1H), 8.02 (s, 1H). MS (ES) C16H14 ClNO2 requires 287, found 286 [M-H]-.
2-(6-chloro-9H-carbazol-2-yl)-N-(2-hydroxyethyl)propanamide 4a
To a solution of carprofen 1 (112 mg, 0.41 mmol, 1 eq) in pyridine (2 mL) was added CDI (133 mg, 0.82 mmol, 2 eq) portionwise. The mixture was stirred for 2 h at room temperature, before addition of ethanolamine (50 μL, 0.82 mmol, 2 eq) and then the reaction was heated at 55°C overnight. Pyridine was then evaporated, the residue taken up in EtOAc and washed with a saturated aqueous NH4Cl solution (5mL). After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (DCM/MeOH) to give amide 4a (110 mg, 85 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.41 (d, J = 7.0 Hz, 3H), 3.03-3.20 (m, 2H), 3.38 (dt, J = 9.4, 5.8 Hz, 2H), 3.78 (q, J = 7.0 Hz, 1H), 4.64 (t, J = 5.3 Hz, 1H), 7.15 (dd, J = 8.2, 1.3 Hz, 1H), 7.34-7.37 (m, 1H), 7.45 (s, 1H), 7.47-7.49 (m, 1H), 7.96 (t, J = 5.5 Hz, 1H), 8.05 (d, J = 8.1 Hz, 1H), 8.16 (d, J = 2.0 Hz, 1H), 11.33 (s, 1H). MS (ES) C17H17ClN2O2 requires 316, found 317 [M+H]+.
2-(6-chloro-9H-carbazol-2-yl)-N-phenylpropanamide 4b
To a solution of carprofen 1 (126 mg, 0.46 mmol, 1 eq) in pyridine (2 mL) was added CDI (149 mg, 0.92 mmol, 2 eq) portionwise. The mixture was stirred for 2 h at room temperature, before addition of aniline (84 μL, 0.92 mmol, 2 eq) and then the reaction was heated at 55 °C overnight. Pyridine was then evaporated, the residue taken up in EtOAc and washed with a saturated aqueous NH4Cl solution (5mL). After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (DCM/MeOH) to give amide 4b (138 mg, 86 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.51 (d, J = 7.0 Hz, 3H), 4.00 (q, J = 6.9 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 7.23 (dd, J = 8.2, 1.3 Hz, 1H), 7.26-7.30 (m, 2H), 7.35-7.37 (m, 1H), 7.47-7.49 (m, 1H), 7.52 (s, 1H), 7.61 (d, J = 8.5 Hz, 2H), 8.10 (d, J = 8.1 Hz, 1H), 8.17 (d, J = 2.0 Hz, 1H), 10.07 (s, 1H), 11.36 (s, 1H). MS (ES) C21H17ClN2O requires 348, found 349 [M+H]+.
General procedure A
To a solution of 3 in MeCN (0.05 M solution) were successively added the alkyl halide (3 to 5 eq) and Cs2CO3 (5 eq). The mixture was heated under reflux overnight. After cooling down to room temperature, the mixture was filtered. EtOAc and H2O were added to the filtrate. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to provide the alkylated product 5.
Methyl 2-(6-chloro-9-methyl-9H-carbazol-2-yl)propanoate 5a
Following general procedure A, alkylation of 3 (113 mg, 0.39 mmol) in the presence of MeI (0.12 mL, 1.96 mmol) and Cs2CO3 (756 mg, 1.96 mol) afforded the methylated product 5a (118 mg, quant.) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.62 (d, J = 7.1 Hz, 3H), 3.69 (s, 3H), 3.83 (s, 3H), 3.93 (q, J = 7.1 Hz, 1H), 7.18 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.8 Hz, 1H), 7.33 (s, 1H), 7.30-7.42 (m, 1H), 7.97 (d, J = 8.1 Hz, 1H), 8.01 (d, J = 2.0 Hz, 1H). MS (ES) C17H16ClNO2 requires 301, found 302 [M+H]+.
Methyl 2-(9-benzyl-6-chloro-9H-carbazol-2-yl)propanoate 5b
Following general procedure A, alkylation of 3 (103 mg, 0.39 mmol) in the presence of BnBr (0.13 mmol, 1,08 mmol) and Cs2CO3 (692 mg, 1.79 mmol) afforded the benzylated product 5b (55 mg, 40 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.55 (d, J = 7.3 Hz, 3H), 3.64 (s, 3H), 3.87 (q, J = 7.1 Hz, 1H), 5.49 (s, 2H), 7.11 (dd, J = 7.3, 2.1 Hz, 2H), 7.19-7.29 (m, 6H), 7.33-7.35 (m, 1H), 8.01 (d, J = 8.1 Hz, 1H), 8.04 (d, J = 2.1 Hz, 1H). MS (ES) C23H20ClNO2 requires 377, found 378 [M+H]+.
Methyl 2-(6-chloro-9-(4-cyanobenzyl)-9H-carbazol-2-yl)propanoate 5c
Following general procedure A, alkylation of 3 (112 mg, 0.39 mmol) in the presence of 4-cyanobenzyl bromide (380 mg, 1.94 mmol) and Cs2CO3 (785 mg, 1.94 mmol) afforded the 4-cyanobenzylated product 5c (149 mg, 95 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.58 (d, J = 7.1 Hz, 3H), 3.66 (s, 3H), 3.89 (q, J = 7.1 Hz, 1H), 5.55 (s, 2H), 7.17 (d, J = 8.7 Hz, 1H), 7.20 (d, J = 8.5 Hz, 2H), 7.24-7.28 (m, 2H), 7.38 (dd, J = 8.6, 2.1 Hz, 1H), 7.58-7.60 (m, 2H), 8.04 (dd, J = 7.9, 0.8 Hz, 1H), 8.07 (d, J = 2.0 Hz, 1H). MS (ES) C24H19ClN2O2 requires 402, found 401 [M-H]-.
Methyl 2-(6-chloro-9-(4-methoxybenzyl)-9H-carbazol-2-yl)propanoate 5e
Following general procedure A, alkylation of 3 (122 mg, 0.42 mmol) in the presence of 4-methoxybenzyl bromide (0.31 mL, 2.11 mmol) and Cs2CO3 (816 mg, 2.11 mmol) afforded the 4-methoxybenzylated product 5e (99 mg, 55 %) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 1.57 (d, J = 7.2 Hz, 3H), 3.65 (s, 3H), 3.75 (s, 3H), 3.88 (q, J = 7.2 Hz, 1H), 5.42 (s, 2H), 6.79-6.81 (m, 2H), 7.06 (d, J = 8.7 Hz, 2H), 7.20 (dd, J = 8.1, 1.4 Hz, 1H), 7.25-7.27 (m, 1H), 7.29-7.31 (m, 1H), 7.33-7.35 (m, 1H), 8.00 (d, J = 8.1 Hz, 1H), 8.03 (d, J = 2.0 Hz, 1H). MS (ES) C24H22ClNO3 requires 407, found 408 [M+H]+.
General procedure B
To a solution of the methyl ester in a 1:1:1 MeOH:THF:H2O (0.03 M solution) was added LiOH (4 eq). The mixture was stirred at room temperature overnight, then the organic solvents were evaporated. The aqueous solution was then acidified to pH 1 with 6M HCl. The precipitate formed was either 1) filtered, washed with H2O and dried under vacuum or 2) redissolved in EtOAc, dried over MgSO4 and concentrated.
2-(6-chloro-9-methyl-9H-carbazol-2-yl)propanoic acid 6a
Following general procedure B, hydrolysis of ester 5a (86 mg, 0.28 mmol) in the presence of LiOH (27 mg, 1.13 mmol) furnished acid 6a (69 mg, 85 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.48 (d, J = 7.1 Hz, 3H), 3.86 (q, J = 7.1 Hz), 3.87 (s, 3H), 7.16 (dd, J = 8.2, 1.5 Hz, 1H), 7.45 (dd, J = 8.7, 2.1 Hz, 1H), 7.50 (d, J = 1.5 Hz, 1H), 7.61 (d, J = 8.7 Hz, 1H), 8.12 (d, J = 8.0 Hz, 1H), 8.21 (d, J = 2.1 Hz, 1H). MS (ES) C16H14ClNO2 requires 287, found 242 [M-H-CO2]-.
2-(9-benzyl-6-chloro-9H-carbazol-2-yl)propanoic acid 6b
Following general procedure B, hydrolysis of ester 5b (40 mg, 0.10 mmol) in the presence of LiOH (10 mg, 0.42 mmol) furnished acid 6b (27 mg, 73 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.44 (d, J = 7.1 Hz, 3H), 3.83 (q, J = 7.1 Hz, 1H), 5.60–5.74 (m, 2H), 7.13–7.20 (m, 3H), 7.19–7.33 (m, 3H), 7.42 (dd, J = 8.7, 2.1 Hz, 1H), 7.58 (s, 1H), 7.62 (d, J = 8.7 Hz, 1H), 8.17 (d, J = 8.1 Hz, 1H), 8.25 (d, J = 2.0 Hz, 1H), 12.31 (s, 1H). MS (ES) C22H18ClNO2 requires 287, found 363 [M-H-CO2]-, 362 [M-H]-.
2-(6-chloro-9-(4-cyanobenzyl)-9H-carbazol-2-yl)propanoic acid 6c
Following general procedure B, hydrolysis of ester 5c (104 mg, 0.26 mmol) in the presence of LiOH (25 mg, 1.04 mmol), followed by purification by preparative TLC (DCM/MeOH: 95/5), furnished acid 6c (52 mg, 67 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.43 (d, J = 7.1 Hz, 3H), 3.82 (q, J = 7.0 Hz, 1H), 5.80 (s, 2H), 7.19 (dd, J = 8.2, 1.4 Hz, 1H), 7.26 (d, J = 8.1 Hz, 2H), 7.43 (dd, J = 8.8, 2.1 Hz, 1H), 7.55 (d, J = 1.4 Hz, 1H), 7.60 (d, J = 8.7 Hz, 1H), 7.75-7.77 (m, 2H), 8.18 (d, J = 8.1 Hz, 1H), 8.28 (d, J = 2.1 Hz, 1H), 12.28 (s, 1H). MS (ES) C23H17ClN2 O2 requires 388, found 343 [M-H-CO2]-.
2-(6-chloro-9-(4-chlorobenzyl)-9H-carbazol-2-yl)propanoic acid 6d
To a solution of 3 (144 mg, 0.5 mmol, 1 eq) in MeCN (5 mL) were successively added 4-chlorobenzyl bromide (308 mg, 1.50 mmol, 3 eq), Cs2CO3 (814 mg, 5 mmol, 5 eq) and TBAI (92 mg, 0.25 mmol, 0.5 eq). The mixture was stirred at room temperature overnight. The suspension was then passed through a silica plug and washed with EtOAc. After concentration in vacuo, the residue was dissolved in a 1:1:1 MeOH:THF:H2O solution (3 mL). LiOH (24 mg, 1 mmol, 2 eq) was added and the mixture was stirred at room temperature overnight. The solution was then acidified with 2M HCl and extracted with EtOAc. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was crystallised in Et2O to obtain acid 6d (64 mg, 32 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.44 (d, J = 7.1 Hz, 3H), 3.83 (q, J = 7.1 Hz, 1H), 5.67 (s, 2H), 7.15 (d, J = 8.4 Hz, 2H), 7.18 (dd, J = 8.2, 1.3 Hz, 1H), 7.34 (d, J = 8.4 Hz, 2H), 7.42 (dd, J = 8.7, 2.1 Hz, 1H), 7.56 (d, J = 1.3 Hz, 1H), 7.61 (d, J = 8.7 Hz, 1H), 8.16 (d, J = 8.1 Hz, 1H), 8.25 (d, J = 2.0 Hz, 1H), 12.27 (s, 1H). MS (ES) C22H17Cl2NO2 requires 397, found 352 [M-H-CO2]-.
2-(6-chloro-9-(4-methoxybenzyl)-9H-carbazol-2-yl)propanoic acid 6e
Following general procedure B, hydrolysis of ester 5e (55 mg, 0.13 mmol) in the presence of LiOH (12 mg, 0.52 mmol), followed by purification by preparative HPLC, furnished acid 6e (18 mg, 35 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.45 (d, J = 7.1 Hz, 3H), 3.68 (s, 3H), 3.84 (q, J = 7.1 Hz, 1H), 5.54-5.62 (m, 2H), 6.82-6.84 (m, 2H), 7.12-7.18 (m, 3H), 7.42 (dd, J = 8.7, 2.1 Hz, 1H), 7.61-7.65 (m, 2H), 8.15 (d, J = 8.1 Hz, 1H), 8.24 (d, J = 2.1 Hz, 1H), 12.29 (s, 1H). MS (ES) C23H20ClNO3 requires 393, found 348 [M-H-CO2]-.
2-(6-chloro-9-(3-methoxybenzyl)-9H-carbazol-2-yl)propanoic acid 6f
To a solution of 3 (144 mg, 0.5 mmol, 1 eq) in MeCN (5 mL) were successively added 3-methoxybenzyl bromide (0.21, 1.50 mmol, 3 eq), Cs2CO3 (814 mg, 5 mmol, 5 eq) and TBAI (92 mg, 0.25 mmol, 0.5 eq). The mixture was stirred at room temperature overnight. The suspension was then passed through a silica plug and washed with EtOAc. After concentration in vacuo, the residue was dissolved in a 1:1:1 MeOH:THF:H2O solution (3 mL). LiOH (24 mg, 1 mmol, 2 eq) was added and the mixture was stirred at room temperature overnight. The solution was then acidified with 2M HCl and extracted with EtOAc. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by preparative HPLC to obtain acid 6f (42 mg, 21 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.44 (d, J = 7.1 Hz, 3H), 3.66 (s, 2H), 3.84 (q, J = 7.0 Hz, 1H), 5.62 (m, 3H), 6.66 (d, J = 7.6 Hz, 1H), 6.79 (m, 2H), 7.16 (m, 2H), 7.41 (dd, J = 8.7, 1.9 Hz, 1H), 7.59 (s, 1H), 7.62 (d, J = 8.8 Hz, 1H), 8.16 (d, J = 8.1 Hz, 1H), 8.24 (d, J = 1.8 Hz, 1H), 12.28 (s, 1H). MS (ES) C23H20ClNO3 requires 393, found 348 [M-H-CO2]-.
2-(6-chloro-9-(2-methoxybenzyl)-9H-carbazol-2-yl)propanoic acid 6g
To a solution of 3 (144 mg, 0.5 mmol, 1 eq) in MeCN (5 mL) were successively added 2-methoxybenzyl bromide (235 mg, 1.50 mmol, 3 eq), Cs2CO3 (814 mg, 5 mmol, 5 eq) and TBAI (92 mg, 0.25 mmol, 0.5 eq). The mixture was stirred at room temperature overnight. The suspension was then passed through a silica plug and washed with EtOAc. After concentration in vacuo, the residue was dissolved in a 1:1:1 MeOH:THF:H2O solution (3 mL). LiOH (24 mg, 1 mmol, 2 eq) was added and the mixture was stirred at room temperature overnight. The solution was then acidified with 2M HCl and extracted with EtOAc. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by preparative HPLC to obtain acid 6g (46 mg, 23 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.42 (d, J = 7.1 Hz, 3H), 3.83 (q, J = 7.2 Hz, 1H), 3.86 (s, 3H), 5.55 (s, 2H), 6.65 (dd, J = 7.6, 1.7 Hz, 1H), 6.75 (td, J = 7.4, 1.0 Hz, 1H), 7.05 (dd, J = 8.3, 0.9 Hz, 1H), 7.16 (dd, J = 8.2, 1.4 Hz, 1H), 7.23 (ddd, J = 8.2, 7.4, 1.8 Hz, 1H), 7.40 (dd, J = 8.7, 2.1 Hz, 1H), 7.51 (d, J = 1.3 Hz, 1H), 7.56 (d, J = 8.7 Hz, 1H), 8.15 (d, J = 8.1 Hz, 1H), 8.24 (d, J = 2.0 Hz, 1H), 12.28 (s, 1H). MS (ES) C23H20ClNO3 requires 393, found 348 [M-H-CO2]-.
Methyl 2-(6-chloro-9-(hexylsulfonyl)-9H-carbazol-2-yl)propanoate 7a
To a solution of 3 (105 mg, 0.36 mmol, 1 eq) in THF (5 mL) were successively added 1-hexanesulfonyl chloride (0.17 mL, 1.09 mmol, 3 eq), DMAP (133 mg, 1.09 mmol, 3 eq) and Et3N (0.15 mmol, 1.09 mmol, 3 eq). The mixture was heated under reflux overnight, then filtered and concentrated. The residue was purified by column chromatography (Cy/EtOAc) to give sulfonamide 7a (55 mg, 35 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 0.81 (t, J = 7.0 Hz, 3H), 1.11-1.30 (m, 6H), 1.58-1.68 (m, 5H), 3.19-3.23 (m, 2H), 3.71 (s, 3H), 3.94 (q, J = 7.2 Hz, 1H), 7.41 (dd, J = 8.1, 1.3 Hz, 1H), 7.45 (dd, J = 8.9, 2.1 Hz, 1H), 7.93 (d, J = 8.1 Hz, 1H), 7.96 (d, J = 2.0 Hz, 1H), 8.08-8.11 (m, 2H). MS (ES) C22H26ClNO4S requires 435, found 436 [M+H+], 453 [M+NH4+].
Methyl 2-(6-chloro-9-((4-chlorophenyl)sulfonyl)-9H-carbazol-2-yl)propanoate 7b
To a solution of 3 (110 mg, 0.38 mmol, 1 eq) in THF (5 mL) were successively added 4-chlorobenzene sulfonyl chloride (241 mg, 1.14 mmol, 3 eq), DMAP (140 mg, 1.14 mml, 3 eq) and Et3N (0.16 mL, 1.14 mmol, 3 eq). The mixture was heated in the microwave at 100 °C for 3 h, then filtrated through a pad of celite, washed with EtOAc and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to furnish sulfonamide 7b (137 mg, 78 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.65 (d, J = 7.2 Hz, 3H), 3.74 (s, 3H), 3.96 (q, J = 7.2 Hz, 1H), 7.32 (d, J = 8.7 Hz, 2H), 7.36 (dd, J = 8.1, 1.4 Hz, 1H), 7.47 (dd, J = 8.9, 2.1 Hz, 1H), 7.73 (d, J = 8.7 Hz, 2H), 7.82 (d, J = 8.1 Hz, 1H), 7.86 (d, J = 2.1 Hz, 1H), 8.24-8.27 (m, 2H). MS (ES) C22H17Cl2NO4S requires 461, found 460, 462 [M-H]-.
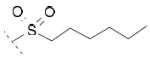
2-(6-chloro-9-(hexylsulfonyl)-9H-carbazol-2-yl)propanoic acid 8a
Following general procedure B, hydrolysis of ester 7a (55 mg, 0.13 mmol) in the presence of LiOH (12 mg, 0.50 mmol), followed by purification by column chromatography (Cy/EtOAc) and trituration with Et2O, furnished acid 8a (15 mg, 28 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 0.71 (t, J = 6.9 Hz, 3H), 0.99-1.08 (m, 4H), 1.15 (p, J = 7.3 Hz, 2H), 1.41-1.48 (m, 5H), 3.56 (t, J = 7.5 Hz, 2H), 3.91 (q, J = 7.0 Hz, 1H), 7.42 (d, J = 8.1 Hz, 1H), 7.58 (dd, J = 8.9, 2.2 Hz, 1H), 8.00 (s, 1H), 8.03 (d, J = 8.9 Hz, 1H), 8.24 (d, J = 8.1 Hz, 1H), 8.37 (d, J = 2.1 Hz, 1H), 12.44 (s, 1H). MS (ES) C21H24ClNO4S requires 421, found 376 [M-H-CO2]-, 420 [M-H]-.
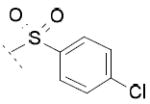
2-(6-chloro-9-((4-chlorophenyl)sulfonyl)-9H-carbazol-2-yl)propanoic acid 8b
Following general procedure B, hydrolysis of ester 7b (133 mg, 0.29 mmol) in the presence of LiOH (27 mg, 1.15 mmol), followed by trituration with Et2O, furnished acid 8b (51 mg, 40 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.49 (d, J = 7.0 Hz, 3H), 3.97 (q, J = 6.8 Hz, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.57-7.63 (m, 3H), 7.82 (d, J = 8.6 Hz, 2H), 8.14-8.17 (m, 2H), 8.25 (d, J = 8.9 Hz, 1H), 8.30 (s, 1H), 12.44 (s, 1H). MS (ES) C21H15Cl2NO4S requires 447, found 402, 404 [M-H-CO2]-, 446, 448 [M-H]-.
Methyl 2-(6-chloro-9-(hexylcarbamoyl)-9H-carbazol-2-yl)propanoate 9a
To a solution of 3 (100 mg, 0.35 mmol, 1 eq) in THF (5 mL) were successively added hexyl isocyanate (0.10 mL, 0.70 mmol, 2 eq), DMAP (85 mg, 0.70 mmol, 2 eq) and Et3N (0.15 mL, 1.05 mmol, 3 eq). The mixture was heated under reflux for 60 h and then concentrated. The residue was taken up in EtOAc and a saturated aqueous NH4Cl solution (5mL) was added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to furnish the urea 9a (106 mg, 70 %) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 0.92 (t, J = 6.6 Hz, 3H), 1.38 (dt, J = 7.6, 3.7 Hz, 4H), 1.46-1.50 (m, 2H), 1.60 (d, J = 7.1 Hz, 3H), 1.74 (p, J = 7.3 Hz, 2H), 3.56 (q, J = 6.7 Hz, 2H), 3.68 (s, 3H), 3.90 (q, J = 7.2 Hz, 1H), 5.67 (t, J = 5.6 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.40 (dd, J = 8.8, 2.0 Hz, 1H), 7.90-7.94 (m, 4H). MS (ES) C23H27ClN2O3 requires 414, found 415 [M+H]+, 433 [M+NH4]+.
Methyl 2-[6-chloro-9-(methylcarbamoyl)carbazol-2-yl]propanoate 9b
To a solution of Boc2O (0.392 mg, 1.79 mmol, 3 eq) in MeCN (3 mL) were successively added DMAP (219 mg, 1.79 mmol, 3 eq) and methylamine (2M solution in THF, 0.89 mL, 1.79 mmol, 3 eq). The mixture was stirred for 30 min at room temperature and then added to a solution of 3 (170 mg, 0.60 mmol, 1 eq) in MeCN (2 mL). The mixture was heated in the microwave at 100 °C for 3 h. EtOAc (10mL) and a saturated aqueous NH4Cl solution (5mL) were then added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to furnish the urea 9b (139 mg, 68%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.48 (d, J = 7.1 Hz, 3H), 2.94 (d, J = 4.4 Hz, 3H), 3.60 (s, 3H), 3.99 (q, J = 7.1 Hz, 1H), 7.26 (dd, J = 1.3, 8.1 Hz, 1H), 7.50 (dd, J = 2.2, 8.8 Hz, 1H), 7.85 (s, 1H), 7.89 (d, J = 8.8 Hz, 1H), 8.18 (d, J = 8.1 Hz, 1H), 8.25 (q, J = 4.1 Hz, 1H), 8.28 (d, J = 2.1 Hz, 1H). MS (ES) C18H17ClN2O3 requires 344, found 345 [M+H]+.
Methyl 2-(9-(butylcarbamoyl)-6-chloro-9H-carbazol-2-yl)propanoate 9c
To a solution of 3 (105 mg, 0.36 mmol, 1 eq) in THF (5 mL) were successively added butyl isocyanate (0.12 mL, 1.09 mmol, 3 eq), DMAP (133 mg, 1.09 mml, 3 eq) and Et3N (0.15 mL, 1.09 mmol, 3 eq). The mixture was heated in the microwave at 100 °C for 10 h. EtOAc (10mL) and a saturated aqueous NH4Cl solution (5mL) were then added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to furnish the urea 9c (97 mg, 80 %) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 1.03 (t, J = 7.3 Hz, 3H), 1.51 (m, 2H), 1.60 (d, J = 7.2 Hz, 3H), 1.73 (m, 2H), 3.58 (td, J = 7.1, 5.5 Hz, 2H), 3.69 (s, 3H), 3.91 (q, J = 7.3 Hz, 1H), 5.66 (t, J = 5.7 Hz, 1H), 7.29 (m, 1H), 7.41 (dd, J = 8.9, 2.0 Hz, 1H), 7.93 (m, 4H). MS (ES) C21H23ClN2O3 requires 386, found 387 [M+H]+, 404 [M+NH4]+.
Methyl 2-(6-chloro-9-(octylcarbamoyl)-9H-carbazol-2-yl)propanoate 9d
To a solution of 3 (106 mg, 0.37 mmol, 1 eq) in THF (5 mL) were successively added octyl isocyanate (0.19 mL, 1.10 mmol, 3 eq), DMAP (135 mg, 1.10 mml, 3 eq) and Et3N (0.15 mL, 1.10 mmol, 3 eq). The mixture was heated in the microwave at 100 °C for 10 h. EtOAc (10 mL) and a saturated aqueous NH4Cl solution (5mL) were then added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to furnish the urea 9d (170 mg, 70 %) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 0.86-0.91 (m, 3H), 1.26-1.47 (m, 10H), 1.60 (d, J = 7.2 Hz, 3H), 1.70-1.77 (m, 2H), 3.56 (td, J = 7.2, 5.5 Hz, 2H), 3.68 (s, 3H), 3.90 (q, J = 7.2 Hz, 1H), 5.68 (t, J = 5.4 Hz, 1H), 7.29 (dd, J = 8.0, 1.5 Hz, 1H), 7.40 (dd, J = 8.9, 2.1 Hz, 1H), 7.90-7.93 (m, 4H). MS (ES) C25H31ClN2O3 requires 442, found 443 [M+H]+, 460 [M+NH4]+.
Methyl 2-(6-chloro-9-((4-chlorophenyl)carbamoyl)-9H-carbazol-2-yl)propanoate 9e
To a solution of 3 (116 mg, 0.40 mmol, 1 eq) in THF (5 mL) were successively added 4-chlorophenyl isocyanate (0.16 mL, 1.21 mmol, 3 eq), DMAP (148 mg, 1.21 mml, 3 eq) and Et3N (0.17 mL, 1.21 mmol, 3 eq). The mixture was heated in the microwave at 100 °C for 10 h. EtOAc (10 mL) and a saturated aqueous NH4Cl solution (5 mL) were then added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to furnish the urea 9e (143 mg, 81 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.60 (d, J = 7.2 Hz, 3H), 3.69 (s, 3H), 3.91 (q, J = 7.2 Hz, 1H), 7.33 (dd, J = 8.0, 1.3 Hz, 1H), 7.40-7.44 (m, 3H), 7.54-7.59 (m, 3H), 7.92-7.98 (m, 4H). MS (ES) C23H18Cl2N2O3 requires 440, found 441, 443 [M+H]+, 458, 460 [M+NH4]+.
Methyl 2-[6-chloro-9-[hexyl(methyl)carbamoyl]carbazol-2-yl]propanoate 9f
To a solution of 3 (124 mg, 0.43 mmol, 1 eq) in THF (5 mL) were successively added N-hexyl-N-methyl-carbamoyl chloride (460 mg, 2.58 mmol, 6 eq, freshly prepared from N-methylhexylamine (1.0 eq) and triphosgene (0.33 eq) in presence of pyridine (1.0 eq) in dry DCM), DMAP (158 mg, 1.29 mmol, 3 eq) and Et3N (0.18 mL, 1.27 mmol, 3 eq). The mixture was heated in the microwave at 100 °C for 3 h. EtOAc (10mL) and a saturated aqueous NH4Cl solution (5mL) were then added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to furnish the urea 9f (88 mg, 48%) as a colourless oil. 1H NMR (400 MHz, DMSO-d6) δ 0.75 (q, J = 6.7 Hz, 2H), 1.04 - 1.23 (m, 6H), 1.47 (dd, J = 1.3, 7.1 Hz, 3H), 1.59 (p, J = 7.2 Hz, 2H), 3.00 (d, J = 5.2 Hz, 3H), 3.35 - 3.48 (m, 2H), 3.60 (d, J = 1.4 Hz, 3H), 4.01 (q, J = 7.1 Hz, 1H), 5.75 (s, 1H), 7.26 (ddd, J = 1.5, 2.6, 8.2 Hz, 1H), 7.39 - 7.44 (m, 1H), 7.47 - 7.54 (m, 2H), 8.19 (d, J = 8.1 Hz, 1H), 8.30 (dd, J = 0.9, 1.8 Hz, 1H). MS (ES) C24H29ClN2O3 requires 428, found 429 [M+H]+.
Methyl 2-[6-chloro-9-(cyclohexylcarbamoyl)carbazol-2-yl]propanoate 9g
To a solution of 3 (103 mg, 0.36 mmol, 1 eq) in THF (5 mL) were successively added cyclohexyl isocyanate (0.10 mL, 0.70 mmol, 2 eq), DMAP (85 mg, 0.70 mmol, 2 eq) and Et3N (0.15 mL, 1.05 mmol, 3 eq). The mixture was heated under reflux for 60 h and then concentrated. EtOAc (10mL) and a saturated aqueous NH4Cl solution (5mL) were then added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to furnish the urea 9g (120 mg, 80%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.18-1.57 (m, 5H), 1.60 (d, J = 7.2 Hz, 3H), 1.70 (dt, J = 3.7, 12.8 Hz, 1H), 1.82 (dt, J = 3.7, 13.0 Hz, 2H), 2.18 (d, J = 11.6 Hz, 2H), 3.69 (s, 3H), 3.91 (q, J = 7.2 Hz, 1H), 4.01 (tdt, J = 3.9, 7.8, 11.6 Hz, 1H), 5.54 (d, J = 7.5 Hz, 1H), 7.29 (dd, J = 1.4, 8.1 Hz, 1H), 7.41 (dd, J = 2.2, 8.8 Hz, 1H), 7.92 (d, J = 4.9 Hz, 1H), 7.93-7.96 (m, 3H). MS (ES) C23H25ClN2O3 requires 412, found 413 [M+H]+.
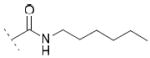
2-(6-chloro-9-(hexylcarbamoyl)-9H-carbazol-2-yl)propanoic acid 10a
Following general procedure B, hydrolysis of ester 9a (74 mg, 0.25 mmol) in the presence of LiOH (24 mg, 1 mmol), followed by trituration with EtOAc at 40 °C, furnished acid 10a (28 mg, 39 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 0.90 (t, J = 6.9 Hz, 3H), 1.33-1.36 (m, 4H), 1.37 (d, J = 7.2 Hz, 3H), 1.40-1.45 (m, 2H), 1.65 (p, J = 7.2 Hz, 2H), 3.34-3.40 (m, 2H), 3.37 (q, J = 7.2 Hz, 1H), 7.29 (d, J = 8.1 Hz, 1H), 7.45 (dd, J = 8.9, 2.2 Hz, 1H), 7.80 (s, 1H), 7.84 (d, J = 8.8 Hz, 1H), 8.05 (d, J = 8.1 Hz, 1H), 8.21 (d, J = 2.2 Hz, 1H), 8.34 (t, J = 5.5 Hz, 1H). MS (ES) C22H25ClN2O3 requires 400, found 401 [M+H]+, 418 [M+NH4]+.
2-[6-chloro-9-(methylcarbamoyl)carbazol-2-yl]propanoic acid 10b
To a solution of ester 9b (60 mg, 0.17 mmol) in THF (6 mL) was added a solution of 6M HCl (4 mL). The solution was stirred for 3 days at room temperature. EtOAc and H2O were then added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by trituration with Et2O/pentane to furnish acid 10f (43 mg, 75 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.45 (d, J = 7.1 Hz, 3H), 2.94 (d, J = 4.4 Hz, 3H), 3.86 (q, J = 7.0 Hz, 1H), 7.28 (d, J = 8.1 Hz, 1H), 7.49 (dd, J = 2.1, 8.8 Hz, 1H), 7.86 (s, 1H), 7.89 (d, J = 8.8 Hz, 1H), 8.17 (d, J = 8.1 Hz, 1H), 8.25 (q, J = 4.0 Hz, 2H), 8.27 (d, J = 2.1 Hz, 1H). MS (ES) C17H15ClN2O3 requires 330, found 331 [M+H]+.
2-(9-(butylcarbamoyl)-6-chloro-9H-carbazol-2-yl)propanoic acid 10c
Following general procedure B, hydrolysis of ester 9b (80 mg, 0.20 mmol) in the presence of LiOH (24 mg, 1 mmol), followed by purification by preparative HPLC, furnished acid 10c (45 mg, 60 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 0.98 (t, J = 7.3 Hz, 3H), 1.47 (m, 5H), 1.66 (p, J = 7.2 Hz, 2H), 3.39 (dd, J = 13.3, 6.4 Hz, 2H), 3.86 (q, J = 7.0 Hz, 1H), 7.29 (d, J = 8.1 Hz, 1H), 7.51 (dd, J = 8.8, 2.0 Hz, 1H), 7.85-7.87 (m, 2H), 8.18 (d, J = 8.1 Hz, 1H), 8.28 (d, J = 1.9 Hz, 1H), 8.39 (t, J = 5.4 Hz, 1H), 12.34 (s, 1H). MS (ES) C20H21ClN2O3 requires 372, found 371 [M-H]-.
2-(6-chloro-9-(octylcarbamoyl)-9H-carbazol-2-yl)propanoic acid 10d
Following general procedure B, hydrolysis of ester 9c (120 mg, 0.27 mmol) in the presence of LiOH (26 mg, 1.08 mmol), followed by trituration with Et2O, furnished acid 10d (62 mg, 53 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 0.87 (t, J = 6.7 Hz, 3H), 1.28-1.44 (m, 10H), 1.46 (d, J = 7.1 Hz, 3H), 1.66 (p, J = 7.0 Hz, 2H), 3.34-3.40 (m, 2H), 3.86 (q, J = 7.0 Hz, 1H), 7.29 (dd, J = 8.1, 1.7 Hz, 1H), 7.50 (dd, J = 8.8, 2.2 Hz, 1H), 7.84-7.87 (m, 2H), 8.18 (d, J = 8.0 Hz, 1H), 8.28 (d, J = 2.1 Hz, 1H), 8.40 (t, J = 5.5 Hz, 1H), 12.31 (s, 1H). MS (ES) C24H29ClN2O3 requires 428, found 429 [M+H]+, 447 [M+NH4]+.
2-(6-chloro-9-((4-chlorophenyl)carbamoyl)-9H-carbazol-2-yl)propanoic acid 10e
To a solution of ester 9d (140 mg, 0.32 mmol) in THF (5 mL) was added a solution of 6M HCl (5 mL). The solution was stirred for 5 days at room temperature. EtOAc (10 mL) and H2O (5 mL) were then added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was triturated with Et2O/pentane to furnish acid 10e (105 mg, 80 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.46 (d, J = 7.1 Hz, 3H), 3.90 (q, J = 7.0 Hz, 1H), 7.35 (dd, J = 8.1, 1.2 Hz, 1H), 7.50 (d, J = 8.9 Hz, 2H), 7.55 (m, 1H), 7.73 (d, J = 8.9 Hz, 2H), 7.88 (s, 1H), 7.92 (d, J = 8.8 Hz, 1H), 8.24 (d, J = 8.1 Hz, 1H), 8.34 (s, 1H), 10.74 (s, 1H). MS (ES) C22H16Cl2N2O3 requires 426, found 427, 429 [M+H]+, 444, 446 [M+NH4]+.
2-[6-chloro-9-[hexyl(methyl)carbamoyl]carbazol-2-yl]propanoic acid 10f
Following general procedure B, hydrolysis of ester 9f (60 mg, 0.14 mmol) in the presence of LiOH (8 mg, 0.35 mmol), followed by trituration with DCM/MeOH, furnished acid 10f (45 mg, 77 %) as a colourless oil. 1H NMR (400 MHz, DMSO-d6) δ 0.71 - 0.79 (m, 2H), 1.16 (m, 6H), 1.44 (dd, J = 2.8, 7.1 Hz, 3H), 1.60 (t, J = 7.1 Hz, 2H), 3.00 (d, J = 1.7 Hz, 3H), 3.41 (h, J = 7.2 Hz, 2H), 3.87 (q, J = 7.1 Hz, 1H), 5.75 (s, 1H), 7.28 (dt, J = 1.6, 8.2 Hz, 1H), 7.43 (d, J = 1.3 Hz, 1H), 7.46 - 7.53 (m, 2H), 8.18 (d, J = 8.1 Hz, 1H), 8.29 (dd, J = 0.9, 1.8 Hz, 1H), 12.36 (s, 1H). MS (ES) C23H27ClN2O3 requires 414, found 415 [M+H]+.
2-[6-chloro-9-(cyclohexylcarbamoyl)carbazol-2-yl]propanoic acid 10g
Following general procedure B, hydrolysis of ester 9g (60 mg, 0.14 mmol) in the presence of LiOH (8 mg, 0.35 mmol), followed by trituration with Et2O, furnished acid 10g (51 mg, 89 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.17-1.31 (m, 2H), 1.43 (dd, J = 9.1, 16.1 Hz, 6H), 1.63 (d, J = 12.8 Hz, 1H), 1.72 - 1.84 (m, 2H), 2.02 (d, J = 9.6 Hz, 2H), 3.68-3.81 (m, 1H), 3.85 (q, J = 7.1 Hz, 1H), 7.27 (dd, J = 1.3, 8.1 Hz, 1H), 7.49 (dd, J = 2.2, 8.8 Hz, 1H), 7.79-7.87 (m, 2H), 8.16 (d, J = 8.1 Hz, 1H), 8.27 (d, J = 2.1 Hz, 1H), 8.35 (d, J = 7.5 Hz, 1H), 12.34 (s, 1H). MS (ES) C22H23ClN2O3 requires 398, found 399 [M+H]+.
Hexyl 6-chloro-2-(1-methoxy-1-oxopropan-2-yl)-9H-carbazole-9-carboxylate 11
To a solution of 3 (140 mg, 0.49 mmol, 1 eq) in THF (5 mL) were successively added hexyl chloroformate (freshly prepared, 215 mg, 1.47 mmol, 3 eq), DMAP (179 mg, 1.47 mml, 3 eq) and Et3N (0.20 mL, 1.47 mmol, 3 eq). The mixture was heated in the microwave at 100 °C for 3 h. The reaction mixture was then filtrated and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to furnish the carbamate 11 (173 mg, 85 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 0.89 (t, J = 6.9 Hz, 3H), 1.33-1.37 (m, 4H), 1.48-1.53 (m, 5H), 1.84-1.91 (m, 2H), 3.61 (s, 3H), 4.02 (q, J = 7.0 Hz, 1H), 4.52 (t, J = 6.5 Hz, 2H), 7.37 (dd, J = 8.0, 1.5 Hz, 1H), 7.56 (dd, J = 8.9, 2.2 Hz, 1H), 8.20-8.25 (m, 3H), 8.31 (d, J = 2.1 Hz, 1H). MS (ES) C23H26ClNO4 requires 415, found 433 [M+NH4]+.
2-(6-chloro-9-((hexyloxy)carbonyl)-9H-carbazol-2-yl)propanoic acid 12
To a solution of ester 11 (87 mg, 0.21 mmol) in THF (2.5 mL) was added a solution of 6M HCl (2.5 mL). The solution was stirred for 5 days at room temperature. EtOAc (10 mL) and H2O (5 mL) were then added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to furnish acid 111 (46 mg, 55 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 0.89 (t, J = 6.9 Hz, 3H), 1.31-139 (m, 4H), 1.46 (d, J = 7.1 Hz, 3H), 1.49-1.54 (m, 2H), 1.84-1.91 (m, 2H), 3.88 (q, J = 7.1 Hz, 1H), 4.52 (t, J = 6.5 Hz, 2H), 7.38 (dd, J = 8.1, 1.5 Hz, 1H), 7.55 (dd, J = 8.9, 2.2 Hz, 1H), 8.19 (d, J = 8.1 Hz, 1H), 8.22-8.24 (m, 2H), 8.30 (d, J = 2.2 Hz, 1H), 12.38 (s, 1H). MS (ES) C22H24ClNO4 requires 401, found 400 [M-H]-, 356 [M-H-CO2]-.
Benzyl 2-(6-chloro-9H-carbazol-2-yl)propanoate 13.43
To a solution of 1 (773 mg, 2.82 mmol, 1 eq) in DMF (10 mL) was added K2CO3 (1.17 g, 8.47 mmol, 3 eq). The mixture was stirred at room temperature for 30 min, then BnBr (0.37 mL, 3.10 mmol, 1.1 eq) was added. After 3 h at room temperature, EtOAc was added (20mL) and the mixture was washed with a saturated aqueous NH4Cl solution (5 mL) and with H2O (5 mL). The organic phase was then dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to yield the corresponding benzyl ester 13 (860 mg, 84%). 1H NMR (400 MHz, CDCl3) δ 1.61 (d, J = 7.2 Hz, 3H), 3.94 (q, J = 7.1 Hz, 1H), 5.13 (q, J = 12.5 Hz, 2H), 7.19 (dd, J = 8.1, 1.3 Hz, 1H), 7.23-7.25 (m, 2H), 7.28-7.36 (m, 6H), 7.94 (d, J = 8.1 Hz, 1H), 7.99 (d, J = 1.8 Hz, 1H), 8.03 (s, 1H). MS (ES) C22H18ClNO2 requires 363, found 362 [M-H]-.
General procedure C
To a solution of 13 (or 3 in the case of 14j) in MeCN (5 mL) were successively added the acyl chloride (3 eq, either obtained from commercial source or freshly prepared from the corresponding acid), DMAP (3 eq) and Et3N (3 eq). The mixture was stirred at room temperature for 2 h, and then a saturated aqueous NH4Cl solution and H2O were added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc).
Benzyl 2-(9-acetyl-6-chloro-9H-carbazol-2-yl)propanoate 14a
Following general procedure C, acylation of 13 (185 mg, 0.51 mmol) in the presence of AcCl (0.11 mL, 1.52 mmol), DMAP (186 mg, 1.52 mmol) and Et3N (0.21 mL, 1.52 mmol) furnished the acetyl derivative 14a (200 mg, 97 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.64 (d, J = 7.2 Hz, 3H), 2.82 (s, 3H), 3.98 (q, J = 7.2 Hz, 1H), 5.12-5.19 (m, 2H), 7.25-7.32 (m, 5H), 7.37 (dd, J = 8.0, 1.3 Hz, 1H), 7.44 (dd, J = 8.9, 2.2 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.94 (d, J = 2.1 Hz, 1H), 8.08 (s, 1H), 8.24 (d, J = 8.9 Hz, 1H). MS (ES) C24H20ClNO3 requires 405, found 406 [M+H+], 423 [M+NH4+].
Benzyl 2-(9-benzoyl-6-chloro-9H-carbazol-2-yl)propanoate 14b
Following general procedure C, acylation of 13 (105 mg, 0.29 mmol) in the presence of benzoyl chloride (0.10 mL, 0.87 mmol), DMAP (106 mg, 0.87 mmol) and Et3N (0.12 mL, 0.87 mmol) furnished the benzoyl derivative 14f (123 mg, 91 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.47 (d, J = 7.2 Hz, 3H), 3.81 (q, J = 7.1 Hz, 1H), 5.06-5.14 (m, 2H), 7.23-7.26 (m, 2H), 7.29-7.34 (m, 5H), 7.38 (s, 1H), 7.50-7.54 (m, 3H), 7.65-7.70 (m, 3H), 7.91 (d, J = 8.0 Hz, 1H), 7.96 (d, J = 2.1 Hz, 1H). MS (ES) C29H22ClNO3 requires 467, found 468 [M+H+], 485 [M+NH4+].
Benzyl 2-(6-chloro-9-(4-chlorobenzoyl)-9H-carbazol-2-yl)propanoate 14c
Following general procedure C, acylation of 13 (98 mg, 0.27 mmol) in the presence of 4-chlorobenzoyl chloride (0.10 mL, 0.80 mmol), DMAP (98 mg, 0.80 mmol) and Et3N (0.11 mL, 0.80 mmol) furnished the 4-chlorobenzoyl derivative 14c (119 mg, 88 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.47 (d, J = 7.2 Hz, 3H), 3.81 (q, J = 7.2 Hz, 1H), 5.06-5.13 (m, 2H), 7.21-7.24 (m, 2H), 7.28-7.33 (m, 5H), 7.40 (s, 1H), 7.47 (dd, J = 8.6, 5.6 Hz, 3H), 7.62 (d, J = 8.5 Hz, 2H), 7.89 (d, J = 8.1 Hz, 1H), 7.94 (d, J = 2.1 Hz, 1H). MS (ES) C29H21Cl2NO3 requires 501, found 502 [M+H+], 519 [M+NH4+].
Benzyl 2-(6-chloro-9-(4-fluorobenzoyl)-9H-carbazol-2-yl)propanoate 14d
Following general procedure C, acylation of 13 (98 mg, 0.27 mmol) in the presence of 4-fluorobenzoyl chloride (0.10 mL, 0.80 mmol), DMAP (98 mg, 0.80 mmol) and Et3N (0.11 mL, 0.80 mmol) furnished the 4-fluorobenzoyl derivative 14d (104 mg, 88 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.49 (d, J = 7.2 Hz, 3H), 3.83 (q, J = 7.1 Hz, 1H), 5.07-5.14 (m, 2H), 7.18 (t, J = 8.6 Hz, 2H), 7.23-7.25 (m, 2H), 7.31-7.35 (m, 5H), 7.40 (s, 1H), 7.52 (d, J = 8.9 Hz, 1H), 7.72 (dd, J = 8.8, 5.3 Hz, 1H), 7.92 (d, J = 8.1 Hz, 2H), 7.97 (d, J = 1.9 Hz, 1H). MS (ES) C29H21ClFNO3 requires 485, found 486 [M+H+].
Benzyl 2-(6-chloro-9-(4-methoxybenzoyl)-9H-carbazol-2-yl)propanoate 14e
Following general procedure C, acylation of 13 (137 mg, 0.37 mmol) in the presence of 4-methoxybenzoyl chloride (0.15 mL, 1.12 mmol), DMAP (138 mg, 1.12 mmol) and Et3N (0.16 mL, 1.12 mmol) furnished the 4-methoxybenzoyl derivative 14e (156 mg, 85 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.49 (d, J = 7.2 Hz, 3H), 3.83 (q, J = 7.1 Hz, 1H), 3.90 (s, 3H), 5.05-5.13 (m, 2H), 6.96 (d, J = 8.8 Hz, 2H), 7.21-7.23 (m, 2H), 7.26-7.31 (m, 5H), 7.46-7.49 (m, 2H), 7.67 (d, J = 4.9 Hz, 2H), 7.90 (d, J = 8.1 Hz, 1H), 7.94 (d, J = 2.0 Hz, 1H). MS (ES) C30H24ClNO4 requires 497, found 498 [M+H]+.
Benzyl 2-(6-chloro-9-(3-chlorobenzoyl)-9H-carbazol-2-yl)propanoate 14f
Following general procedure C, acylation of 13 (112 mg, 0.32 mmol) in the presence of 3-chlorobenzoyl chloride (0.12 mL, 0.96 mmol), DMAP (117 mg, 0.96 mmol) and Et3N (0.13 mL, 0.96 mmol) furnished the 3-chlorobenzoyl derivative 14f (124 mg, 77 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.46 (d, J = 7.2 Hz, 3H), 3.80 (q, J = 7.1 Hz, 1H), 5.04-5.13 (m, 2H), 7.21-7.23 (m, 2H), 7.28-7.34 (m, 6H), 7.42 (m, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.61-7.63 (m, 1H), 7.67 (t, J = 1.7 Hz, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.94 (d, J = 2.1 Hz, 1H). MS (ES) C29H21Cl2NO3 requires 501, found 502 [M+H+], 519 [M+NH4+].
Benzyl 2-(6-chloro-9-(2-chlorobenzoyl)-9H-carbazol-2-yl)propanoate 14g
Following general procedure C, acylation of 13 (105 mg, 0.29 mmol) in the presence of 2-chlorobenzoyl chloride (0.11 mL, 0.86 mmol), DMAP (105 mg, 0.86 mmol) and Et3N (0.12 mL, 0.86 mmol) furnished the 2-chlorobenzoyl derivative 14g (129 mg, 88 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.42 (d, J = 7.0 Hz, 3H), 3.77 (q, J = 7.1 Hz, 1H), 5.04-5.12 (m, 2H), 7.22-7.33 (m, 8H), 7.43-7.57 (m, 5H), 7.86 (d, J = 8.1 Hz, 1H), 7.92 (d, J = 2.1 Hz, 1H). MS (ES) C29H21Cl2NO3 requires 501, found 502 [M+H+], 519 [M+NH4+].
Benzyl 2-(6-chloro-9-(3,4-dichlorobenzoyl)-9H-carbazol-2-yl)propanoate 14h
Following general procedure C, acylation of 13 (120 mg, 0.33 mmol) with 3,4-dichlorobenzoyl chloride (freshly prepared from 3,4-dichlorobenzoic acid, 1 mmol), DMAP (122 mg, 1 mmol) and Et3N (0.14 mL, 1 mmol) furnished the 3,4-dichlorobenzoyl derivative 14h (157 mg, 89 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.48 (d, J = 7.2 Hz, 3H), 3.83 (q, J = 7.1 Hz, 1H), 5.05-5.14 (m, 2H), 7.22-7.25 (m, 2H), 7.29-7.34 (m, 5H), 7.40 (s, 1H), 7.46-7.50 (m, 2H), 7.54-7.56 (m, 1H), 7.79 (d, J = 1.9 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 2.0 Hz, 1H). MS (ES) C29H20Cl3NO3 requires 535, found 536, 538 [M+H+], 553, 555 [M+NH4+].
Benzyl 2-(6-chloro-9-(oxazole-4-carbonyl)-9H-carbazol-2-yl)propanoate 14i
Following general procedure C, acylation of 13 (144 mg, 0.39 mmol) with 4-oxazole carbonyl chloride (freshly prepared from 4-oxazole carboxylic acid, 1.20 mmol), DMAP (145 mg, 1.20 mmol) and Et3N (0.16 mL, 1.20 mmol) furnished the 4-oxazole carbonyl derivative 14i (135 mg, 76 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.53 (d, J = 7.2 Hz, 3H), 3.86 (q, J = 7.1 Hz, 1H), 5.06-5.14 (m, 2H), 7.23-7.25 (m, 2H), 7.28-7.35 (m, 5H), 7.58 (d, J = 0.9 Hz, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.93 (m, 2H), 8.33 (s, 1H). MS (ES) C26H19ClN2O4 requires 458, found 459 [M+H+], 476 [M+NH4+].
Tert-butyl 4-(2-(1-(benzyloxy)-1-oxopropan-2-yl)-6-chloro-9H-carbazole-9-carbonyl)-1H-imidazole-1-carboxylate 14j
To a suspension of imidazole-4-carboxylic acid (143 mg, 1.28 mmol, 1 eq) in DMF (1.5 mL) were successively added Et3N (0.35 mL, 2.56 mmol, 2 eq) and a solution of Boc2O (307 mg, 1.08 mmol, 1.1 eq) in DMF (1.5 mL). After 4 h at room temperature, the solution was concentrated. The residue was taken up in EtOAc and washed with H2O. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was suspended in DCM (5 mL) and oxalyl chloride (0.26 mL, 3.07 mmol, 2.4 eq) followed by DMF (0.05 mL) were added. The solution was stirred for 2 h at room temperature and then concentrated in vacuo to provide the crude 1-Boc imidazole-4-carbonyl chloride as yellow oil. To a solution of 13 (120 mg, 0.33 mmol, 1 eq) in MeCN (3 mL) were successively added the crude acyl chloride described previously (1.28 mmol, 3.9 eq) in MeCN (2 mL), DMAP (156 mg, 1.28 mmol, 3.9 eq) and Et3N (0.18 mL, 1.28 mmol, 3.9 eq). The mixture was stirred at room temperature for 2 h, then a saturated aqueous NH4Cl solution and H2O were added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (Cy/EtOAc) to give the N-Boc carbonylimidazole compound 14j (46 mg, 25%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.56 (d, J = 7.2 Hz, 3H), 1.70 (s, 9H), 3.89 (q, J = 7.1 Hz, 1H), 5.06-5.17 (m, 2H), 7.24-7.34 (m, 7H), 7.65 (d, J = 8.8 Hz, 1H), 7.68 (s, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.94 (d, J = 1.9 Hz, 1H), 8.13 (d, J = 1.2 Hz, 1H), 8.17 (d, J = 1.2 Hz, 1H). MS (ES) C31H28ClN3O5 requires 557, found 558 [M+H]+.
Methyl 2-(6-chloro-9-(thiazole-4-carbonyl)-9H-carbazol-2-yl)propanoate 14k
Following general procedure C, acylation of 3 (117 mg, 0.41 mmol) with thiazole-4-carbonyl chloride (freshly prepared from thiazole-4-carboxylic acid, 1.22 mmol), DMAP (149 mg, 1.22 mmol) and Et3N (0.17 mL, 1.22 mmol) furnished the 4-thiazole carbonyl derivative 14k (115 mg, 71 %) as a white solid. 1H NMR (400 MHz, CDCl3) δ 1.49 (d, J = 7.2 Hz, 3H), 3.67 (s, 3H), 3.80 (q, J = 7.1 Hz, 1H), 7.27 – 7.35 (m, 3H), 7.48 (d, J = 8.8 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.93 (d, J = 2.1 Hz, 1H), 8.27 (d, J = 2.0 Hz, 1H), 8.94 (d, J = 2.0 Hz, 1H). MS (ES) C20H15ClN2O3S requires 398, found 399 [M+H]+.
2-(9-acetyl-6-chloro-9H-carbazol-2-yl)propanoic acid 15a
A solution of benzyl ester 14a (100 mg, 0.25 mmol) in THF (30 mL) was hydrogenated with the ThalesNano H-Cube™ using a 1% Pd/C catalyst, at 1 mL/min flow rate, room temperature and H2 atmosphere (1 atm) (full H2 mode), in a closed system for 5 h. The solvent was then removed in vacuo and the product was crystallized from DCM/pentane to give acid 15a (15 mg, 19 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.46 (d, J = 7.1 Hz, 3H), 2.88 (s, 3H), 3.91 (q, J = 7.1 Hz, 1H), 7.37-7.39 (m, 1H), 7.53 (dd, J = 8.9, 2.2 Hz, 1H), 8.20-8.25 (m, 3H), 8.31 (d, J = 2.2 Hz, 1H), 12.39 (s, 1H). MS (ES) C17H14ClNO3 requires 315, found 314 [M-H]-, 270 [M-H-CO2]-.
General procedure D
A solution of benzyl ester 14 in THF (5 mM solution) was hydrogenated with the ThalesNano H-Cube™ using a 1 % or 5 % Pd/C catalyst, at 1 mL/min flow rate, room temperature and H2 atmosphere (1 atm) (full H2 mode). The solvent was then removed in vacuo and the crude oil was purified by preparative HPLC to yield the corresponding acid 15.
2-(9-benzoyl-6-chloro-9H-carbazol-2-yl)propanoic acid 15b
Following general procedure D (5 % Pd/C catalyst), ester 14b (120 mg, 0.25 mmol) was hydrogenated to yield acid 15b (29 mg, 31 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.29 (d, J = 7.1 Hz, 3H), 3.72 (q, J = 7.1 Hz, 1H), 7.34-7.37 (m, 3H), 7.39-7.42 (m, 1H), 7.60-7.64 (m, 2H), 7.72-7.78 (m, 3H), 8.22 (d, J = 8.4 Hz, 1H), 8.34 (d, J = 2.0 Hz, 1H), 12.31 (s, 1H). MS (ES) C22H16ClNO3 requires 377, found 378 [M+H+], 395 [M+NH4+].
2-(6-chloro-9-(4-chlorobenzoyl)-9H-carbazol-2-yl)propanoic acid 15c
Following general procedure D (5 % Pd/C catalyst), ester 14c (119 mg, 0.23 mmol) was hydrogenated to yield acid 15c (40 mg, 42 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.32 (d, J = 7.1 Hz, 3H), 3.76 (q, J = 7.0 Hz, 1H), 7.33–7.40 (m, 2H), 7.39–7.47 (m, 2H), 7.69 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.5 Hz, 2H), 8.22 (d, J = 8.0 Hz, 1H), 8.34 (d, J = 2.1 Hz, 1H), 12.35 (s, 1H). MS (ES) C22H15ClNO3 requires 411, found 366 [M-H-CO2]-.
2-(6-chloro-9-(4-fluorobenzoyl)-9H-carbazol-2-yl)propanoic acid 15d
Following general procedure D (5 % Pd/C catalyst), ester 14d (104 mg, 0.21 mmol) was hydrogenated to yield acid 15d (35 mg, 42 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.32 (d, J = 7.1 Hz, 3H), 3.75 (q, J = 7.0 Hz, 1H), 7.35-7.39 (m, 3H), 7.42-7.47 (m, 3H), 7.83 (dd, J = 8.6, 5.4 Hz, 2H), 8.22 (d, J = 8.6 Hz, 1H), 8.34 (d, J = 2.0 Hz, 1H), 12.33 (s, 1H). MS (ES) C22H15ClFNO3 requires 395, found 396 [M+H+], 413 [M+NH4+].
2-(6-chloro-9-(4-methoxybenzoyl)-9H-carbazol-2-yl)propanoic acid 15e
Following general procedure D (1 % Pd/C catalyst), ester 14e (0.156 g, 0.31 mmol) was hydrogenated to yield acid 15e (28 mg, 22 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.33 (d, J = 7.1 Hz, 3H), 3.76 (q, J = 7.0 Hz, 1H), 3.90 (s, 3H), 7.13 (d, J = 8.8 Hz, 2H), 7.33 (t, J = 8.9 Hz, 2H), 7.39-7.42 (m, 1H), 7.47 (s, 1H), 7.71 (d, J = 8.8 Hz, 2H), 8.21 (d, J = 8.0 Hz, 1H), 8.33 (d, J = 2.1 Hz, 1H), 12.32 (s, 1H). MS (ES) C23H18ClNO4 requires 407, found 408 [M+H]+.
2-(6-chloro-9-(3-chlorobenzoyl)-9H-carbazol-2-yl)propanoic acid 15f
Following general procedure D (1 % Pd/C catalyst), ester 14f (197 mg, 0.39 mmol) was hydrogenated to yield acid 15f (25 mg, 15 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.30 (d, J = 7.1 Hz, 3H), 3.74 (q, J = 7.1 Hz, 1H), 7.33-7.37 (m, 2H), 7.40-7.45 (m, 2H), 7.62-7.66 (m, 1H), 7.68-7.70 (m, 1H), 7.81-7.85 (m, 2H), 8.21 (d, J = 8.0 Hz, 1H), 8.34 (s, 1H), 12.33 (s, 1H). MS (ES) C22H15Cl2NO3requires 411, found 412 [M+H+], 434 [M+Na+].
2-(6-chloro-9-(2-chlorobenzoyl)-9H-carbazol-2-yl)propanoic acid 15g
Following general procedure D (1 % Pd/C catalyst), ester 14g (125 mg, 0.25 mmol) was hydrogenated to yield acid 15g (22 mg, 21 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.26 (d, J = 7.1 Hz, 3H), 3.71 (q, J = 6.9 Hz, 1H), 7.23-7.25 (m, 1H), 7.38 (d, J = 7.7 Hz, 1H), 7.44 (d, J = 6.6 Hz, 2H), 7.63-7.67 (m, 1H), 7.71-7.77 (m, 2H), 7.84 (d, J = 7.2 Hz, 1H), 8.21 (d, J = 8.0 Hz, 1H), 8.35 (s, 1H), 12.34 (s, 1H). MS (ES) C22H15Cl2NO3 requires 411, found 412 [M+H+], 429 [M+NH4]+.
2-(6-chloro-9-(3,4-dichlorobenzoyl)-9H-carbazol-2-yl)propanoic acid 15h
Following general procedure D (5 % Pd/C catalyst), ester 14h (157 mg, 0.29 mmol) was hydrogenated to yield acid 15h (35 mg, 28 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.32 (d, J = 7.1 Hz, 3H), 3.78 (q, J = 7.0 Hz, 1H), 7.37 - 7.44 (m, 4H), 7.74 (dd, J = 8.3, 2.0 Hz, 1H), 7.89 (d, J = 8.3 Hz, 1H), 8.08 (d, J = 1.9 Hz, 1H), 8.22 (d, J = 8.0 Hz, 1H), 8.34 (d, J = 1.7 Hz, 1H), 12.35 (s, 1H). MS (ES) C22H14Cl3NO3 requires 445, found 400, 402 [M-H-CO2]-.
2-(6-chloro-9-(oxazole-4-carbonyl)-9H-carbazol-2-yl)propanoic acid 15i
Following general procedure D (1 % Pd/C catalyst), ester 14i (130 mg, 0.28 mmol) was hydrogenated to yield acid 15i (21 mg, 20 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.38 (d, J = 7.1 Hz, 3H), 3.82 (q, J = 7.0 Hz, 1H), 7.37 (dd, J = 8.1, 1.1 Hz, 1H), 7.44 (dd, J = 8.9, 2.2 Hz, 1H), 7.58 - 7.63 (m, 2H), 8.21 (d, J = 8.0 Hz, 1H), 8.33 (d, J = 2.1 Hz, 1H), 8.69 (s, 1H), 9.06 (s, 1H), 12.38 (s, 1H). MS (ES) C19H13ClN2O4 requires 368, found 323 [M-H-CO2]-.
2-(6-chloro-9-(1H-imidazole-4-carbonyl)-9H-carbazol-2-yl)propanoic acid hydrochloride 15j
To a solution of 14j (84 mg, 0.15 mmol) in acetone (2 mL) 2M HCl was added (2 mL). The mixture was stirred at room temperature overnight. After evaporation of the solvents, the residue was then dissolved in THF (40 mL) and the solution was hydrogenated using the ThalesNano H-Cube™ using a 5 % Pd/C catalyst, at 1 mL/min flow rate, room temperature and H2 atmosphere (1 atm) (full H2 mode). The solvent was then removed in vacuo and the crude oil was purified by preparative HPLC. After evaporation of the solvents, the residue was dissolved in a 1/1 mixture of MeCN/H2O and 0.5 mL of concentrated HCl. The solution was lyophilized overnight to afford the acid 15j (11 mg, 18 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.39 (d, J = 7.1 Hz, 3H), 3.81 (q, J = 7.0 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.42 (dd, J = 8.9, 2.0 Hz, 1H), 7.55 (d, J = 8.9 Hz, 1H), 7.60 (s, 1H), 8.16 (s, 1H), 8.20 (d, J = 8.1 Hz, 1H), 8.27 (s, 1H), 8.30 (d, J = 1.9 Hz, 1H). MS (ES) C19H14ClN3O3 requires 367, found 322 [M-H-CO2]-, 366 [M-H]-.
2-(6-chloro-9-(thiazole-4-carbonyl)-9H-carbazol-2-yl)propanoic acid 15k
To a solution of ester 14k (115 mg, 0.29 mmol) in THF (5 mL) was added a solution of 6M HCl (5 mL). The solution was stirred for 5 days at room temperature. EtOAc and H2O were then added. After separation, the organic phase was dried over MgSO4 and concentrated in vacuo. The residue was purified by preparative HPLC to furnish acid 15k (18 mg, 16 %) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.34 (d, J = 7.1 Hz, 3H), 3.76 (q, J = 7.1 Hz, 1H), 7.20 (s, 1H), 7.34 - 7.41 (m, 2H), 7.45 (dd, J = 8.9, 2.2 Hz, 1H), 8.21 (d, J = 8.0 Hz, 1H), 8.34 (s, 1H), 8.74 (s, 1H), 9.29 (s, 1H), 12.35 (s, 1H). MS (ES) C19H13ClN2O3S requires 384, found 339 [M-H-CO2]-.
Separation of enantiomers
Compounds 1, 15c and 15i were subjected to enantiomeric separation by chiral HPLC. For carprofen 1, the absolute configuration of each enantiomer was assigned by measuring the optical rotation and comparing the values to the data described in the literature.42 The absolute configuration of each enantiomer of compounds 15c and 15i was assigned by chemical correlation. To this purpose, each enantiomer of 15c and 15i was separately hydrolysed with 6M HCl in THF for 3 days at 40 °C to give (S)- and (R)-1. The absolute configuration of each enantiomer was assigned by comparison of the chiral HPLC chromatogram with that of samples of (+)-(S)- and (-)-(R)-1. The results were consistent with the formation of a single enantiomer of 1 in each experiment.
(2S)-2-(6-chloro-9H-carbazol-2-yl)propanoic acid (+)-1
[α]D = + 58.32 (c = 0.1, MeOH), ee > 99.5 % (detector UV 240 nm); 1H NMR (400 MHz, DMSO-d6) δ 1.45 (d, J = 7.1 Hz, 3H), 3.84 (q, J = 7.0 Hz, 1H), 7.12 (dd, J = 8.1, 1.3 Hz, 1H), 7.37 (dd, J = 8.6, 2.1 Hz, 1H), 7.41 (s, 1H), 7.49 (d, J = 8.6 Hz, 1H), 8.09 (d, J = 8.1 Hz, 1H), 8.18 (d, J = 2.0 Hz, 1H), 11.36 (s, 1H), 12.32 (s, 1H). MS (ES) C15H12ClNO2 requires 273, found 272 [M-H]-; retention time on analytical chiral HPLC: 8.295 min.
(2R)-2-(6-chloro-9H-carbazol-2-yl)propanoic acid (-)-1
[α]D = - 58.25 (c = 0.1, MeOH), ee > 99.5 % (detector UV 240 nm); 1H NMR (400 MHz, DMSO-d6) δ 1.45 (d, J = 7.1 Hz, 3H), 3.84 (q, J = 7.0 Hz, 1H), 7.12 (dd, J = 8.1, 1.3 Hz, 1H), 7.37 (dd, J = 8.6, 2.1 Hz, 1H), 7.41 (s, 1H), 7.49 (d, J = 8.6 Hz, 1H), 8.09 (d, J = 8.1 Hz, 1H), 8.18 (d, J = 2.0 Hz, 1H), 11.36 (s, 1H), 12.29 (s, 1H). MS (ES) C15H12ClNO2 requires 273, found 272 [M-H]-; retention time on analytical chiral HPLC: 10.173 min.
(2S)-(6-chloro-9-(4-chlorobenzoyl)-9H-carbazol-2-yl)propanoic acid (+)-15c
[α]D = + 28.42 (c = 0.1, EtOAc), ee > 99.5 % (detector UV 240 nm); 1H NMR (400 MHz, DMSO-d6) δ 1.32 (d, J = 7.1 Hz, 3H), 3.76 (q, J = 7.0 Hz, 1H), 7.33–7.40 (m, 2H), 7.39–7.47 (m, 2H), 7.69 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.5 Hz, 2H), 8.22 (d, J = 8.0 Hz, 1H), 8.34 (d, J = 2.1 Hz, 1H), 12.35 (s, 1H). MS (ES) C22H15Cl2NO3 requires 411, found 366 [M-H-CO2]-; retention time on analytical chiral HPLC: 16.834 min.
(2R)-(6-chloro-9-(4-chlorobenzoyl)-9H-carbazol-2-yl)propanoic acid (-)-15c
[α]D = -28.72 (c = 0.1, EtOAc), ee > 99.5 % (detector UV 240 nm); 1H NMR (400 MHz, DMSO-d6) δ 1.32 (d, J = 7.1 Hz, 3H), 3.76 (q, J = 7.0 Hz, 1H), 7.33–7.40 (m, 2H), 7.39–7.47 (m, 2H), 7.69 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.5 Hz, 2H), 8.22 (d, J = 8.0 Hz, 1H), 8.34 (d, J = 2.1 Hz, 1H), 12.35 (s, 1H). MS (ES) C22H15Cl2NO3 requires 411, found 366 [M-H-CO2]-; retention time on analytical chiral HPLC: 23.442 min.
(2S)-(6-chloro-9-(oxazole-4-carbonyl)-9H-carbazol-2-yl)propanoic acid (+)-15i
[α]D = + 39.85 (c = 0.1, EtOAc), ee > 99.5 % (detector UV 240 nm) ; 1H NMR (400 MHz, DMSO-d6) δ 1.38 (d, J = 7.1 Hz, 3H), 3.82 (q, J = 7.0 Hz, 1H), 7.37 (dd, J = 8.1, 1.1 Hz, 1H), 7.44 (dd, J = 8.9, 2.2 Hz, 1H), 7.58–7.63 (m, 2H), 8.21 (d, J = 8.0 Hz, 1H), 8.33 (d, J = 2.1 Hz, 1H), 8.69 (s, 1H), 9.06 (s, 1H), 12.38 (s, 1H). MS (ES) C19H13ClN2O3S requires 384, found 323 [M-H-CO2]-; retention time on analytical chiral HPLC: 24.425 min.
(2R)-(6-chloro-9-(oxazole-4-carbonyl)-9H-carbazol-2-yl)propanoic acid (-)-15i
[α]D = - 39.38 (c = 0.1, EtOAc), ee > 99.5 % (detector UV 240 nm); 1H NMR (400 MHz, DMSO-d6) δ 1.38 (d, J = 7.1 Hz, 3H), 3.82 (q, J = 7.0 Hz, 1H), 7.37 (dd, J = 8.1, 1.1 Hz, 1H), 7.44 (dd, J = 8.9, 2.2 Hz, 1H), 7.58–7.63 (m, 2H), 8.21 (d, J = 8.0 Hz, 1H), 8.33 (d, J = 2.1 Hz, 1H), 8.69 (s, 1H), 9.06 (s, 1H), 12.38 (s, 1H). MS (ES) C19H13ClN2O3S requires 384, found 323 [M-H-CO2]-; retention time on analytical chiral HPLC: 37.257 min.
In vitro assays
FAAH activity was measured by incubating for 30 minutes at 37°C [3H] anandamide (1 uM cold AEA and 0.6 nM (1 mCi/mL) [3H]-AEA (Arachidonyl-[1-3H] ethanolamine, Specific activity 60 Ci/mmol; American radiolabel Chemicals, Inc. MO, USA) in the presence of 50 ug protein/sample of total rat brain homogenates in assay buffer (50 mM TRIS pH 7.4, 0.05 % fatty acid free BSA). The reaction was stopped with cold 1:1 CHCl3/MeOH. The aqueous phase was counted by liquid scintillation (Microbeta2 Lumijet, Perkin Elmer Inc., MA-USA: adapted from Kathuria et al, 2003). Inhibitors were pre-incubated with the enzyme preparation at the appropriate concentration for 10 minutes prior to substrate addition.
COX activity was measured using a commercial enzyme immunoassay kit (Cayman Chemical Co., Ann Arbor, MI, USA). The manufacturer protocol was followed except for the substrate concentration. Briefly, inhibitors were pre-incubated with either ovine COX-1 or human COX-2 for 10 min at 37 °C, and the reaction was carried out in the presence of 5 μM arachidonic acid for 2 minutes at 37 °C. The reaction was stopped with hydrochloric acid and COX-derived PGH2 was then converted to PGF2α with SnCl2. The PGF2α product is then quantified via enzyme immunoassay (EIA) using a PG-specific antibody and competing with a PG-acetylcholinesterase conjugate. Absorbance is measured at 412 nM with a Tecan Infinite M200 plate reader (Tecan Group Ltd., CH) and data processed according to manufacturer’s instructions.
Supplementary Material
Acknowledgments
We thank Silvia Venzano and Luca Goldoni, Paola Bisignano and Marino Convertino for their technical support.
Nonstandard abbreviations
- FAAH
Fatty acid amide hydrolase
- PGE2
prostaglandin E2
- DMAP
4-Dimethylaminopyridine
- POX
peroxidase activity
Footnotes
Supporting Information
Percentage distribution of the some molecular properties of the 382 COXs inhibitors; putative binding mode to FAAH of some known COXs inhibitors, including carprofen and some of its derivatives. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wilson KG, Eriksson MY, D’Eon JL, Mikail SF, Emery PC. Major depression and insomnia in chronic pain. Clin J Pain. 2002;18:77–83. doi: 10.1097/00002508-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Morin CM, Gibson D, Wade J. Self-reported sleep and mood disturbance in chronic pain patients. Clin J Pain. 1998;14:311–314. doi: 10.1097/00002508-199812000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Menefee LA, Frank ED, Doghramji K, Picarello K, Park JJ, Jalali S, Perez-Schwartz L. Self-reported sleep quality and quality of life for individuals with chronic pain conditions. Clin J Pain. 2000;16:290–297. doi: 10.1097/00002508-200012000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Stahl S, Briley M. Understanding pain in depression. Hum Psychopharmacol. 2004;19(Suppl 1):S9–S13. doi: 10.1002/hup.619. [DOI] [PubMed] [Google Scholar]
- 5.Linton SJ. A review of psychological risk factors in back and neck pain. Spine (Phila Pa 1976) 2000;25:1148–1156. doi: 10.1097/00007632-200005010-00017. [DOI] [PubMed] [Google Scholar]
- 6.Fishbain DA. The association of chronic pain and suicide. Semin Clin Neuropsychiatry. 1999;4:221–227. doi: 10.153/SCNP00400221. [DOI] [PubMed] [Google Scholar]
- 7.Tang NK, Crane C. Suicidality in chronic pain: a review of the prevalence, risk factors and psychological links. Psychol Med. 2006;36:575–586. doi: 10.1017/S0033291705006859. [DOI] [PubMed] [Google Scholar]
- 8.Kurumbail RG, Kiefer JR, Marnett LJ. Cyclooxygenase enzymes: catalysis and inhibition. Curr Opin Struct Biol. 2001;11:752–760. doi: 10.1016/s0959-440x(01)00277-9. [DOI] [PubMed] [Google Scholar]
- 9.Blobaum AL, Marnett LJ. Structural and functional basis of cyclooxygenase inhibition. J Med Chem. 2007;50:1425–1441. doi: 10.1021/jm0613166. [DOI] [PubMed] [Google Scholar]
- 10.Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res. 2009;50(Suppl):S29–S34. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marnett LJ. The COXIB experience: a look in the rearview mirror. Annu Rev Pharmacol Toxicol. 2009;49:265–290. doi: 10.1146/annurev.pharmtox.011008.145638. [DOI] [PubMed] [Google Scholar]
- 12.Naesdal J, Brown K. NSAID-associated adverse effects and acid control aids to prevent them: a review of current treatment options. Drug Saf. 2006;29:119–132. doi: 10.2165/00002018-200629020-00002. [DOI] [PubMed] [Google Scholar]
- 13.Topol EJ. Failing the public health--rofecoxib, Merck, and the FDA. N Engl J Med. 2004;351:1707–1709. doi: 10.1056/NEJMp048286. [DOI] [PubMed] [Google Scholar]
- 14.Prasit P, Wang Z, Brideau C, Chan CC, Charleson S, Cromlish W, Ethier D, Evans JF, Ford-Hutchinson AW, Gauthier JY, Gordon R, Guay J, Gresser M, Kargman S, Kennedy B, Leblanc Y, Leger S, Mancini J, O’Neill GP, Ouellet M, Percival MD, Perrier H, Riendeau D, Rodger I, Zamboni R, et al. The discovery of rofecoxib, [MK 966, Vioxx, 4-(4’-methylsulfonylphenyl)-3-phenyl-2(5H)-furanone], an orally active cyclooxygenase-2-inhibitor. Bioorg Med Chem Lett. 1999;9:1773–1778. doi: 10.1016/s0960-894x(99)00288-7. [DOI] [PubMed] [Google Scholar]
- 15.Piomelli D. The endocannabinoid system: a drug discovery perspective. Curr Opin Investig Drugs. 2005;6:672–679. [PubMed] [Google Scholar]
- 16.Gaetani S, Dipasquale P, Romano A, Righetti L, Cassano T, Piomelli D, Cuomo V. The endocannabinoid system as a target for novel anxiolytic and antidepressant drugs. Int Rev Neurobiol. 2009;85:57–72. doi: 10.1016/S0074-7742(09)85005-8. [DOI] [PubMed] [Google Scholar]
- 17.Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci U S A. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, Hohmann AG, Calignano A, Mor M, Tarzia G, Piomelli D. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 20.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 21.Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 22.Piomelli D. A thickening network of lipids. Pain. 2012;153:3–4. doi: 10.1016/j.pain.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 23.Fowler CJ. NSAIDs: eNdocannabinoid stimulating anti-inflammatory drugs? Trends Pharmacol Sci. 2012;33(9):468–473. doi: 10.1016/j.tips.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 24.Fowler CJ, Bjorklund E, Lichtman AH, Naidu PS, Congiu C, Onnis V. Inhibitory properties of ibuprofen and its amide analogues towards the hydrolysis and cyclooxygenation of the endocannabinoid anandamide. J Enzyme Inhib Med Chem. 2012 Jan 6; doi: 10.3109/14756366.2011.643304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seierstad M, Breitenbucher JG. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J Med Chem. 2008;51:7327–7343. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- 26.Otrubova K, Ezzili C, Boger DL. The discovery and development of inhibitors of fatty acid amide hydrolase (FAAH) Bioorg Med Chem Lett. 2011;21:4674–4685. doi: 10.1016/j.bmcl.2011.06.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palermo G, Branduardi D, Masetti M, Lodola A, Mor M, Piomelli D, Cavalli A, De Vivo M. Covalent inhibitors of fatty acid amide hydrolase: a rationale for the activity of piperidine and piperazine aryl ureas. J Med Chem. 2011;54:6612–6623. doi: 10.1021/jm2004283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Acevedo O, Guimaraes CR, Jorgensen WL, Cravatt BF. Discovery of a potent, selective, and efficacious class of reversible alpha-ketoheterocycle inhibitors of fatty acid amide hydrolase effective as analgesics. J Med Chem. 2005;48:1849–1856. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clapper JR, Vacondio F, King AR, Duranti A, Tontini A, Silva C, Sanchini S, Tarzia G, Mor M, Piomelli D. A second generation of carbamate-based fatty acid amide hydrolase inhibitors with improved activity in vivo. ChemMedChem. 2009;4:1505–1513. doi: 10.1002/cmdc.200900210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vacondio F, Silva C, Lodola A, Fioni A, Rivara S, Duranti A, Tontini A, Sanchini S, Clapper JR, Piomelli D, Mor M, Tarzia G. Structure-property relationships of a class of carbamate-based fatty acid amide hydrolase (FAAH) inhibitors: chemical and biological stability. ChemMedChem. 2009;4:1495–1504. doi: 10.1002/cmdc.200900120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, Beidler D, Liimatta MB, Smith SE, Dudley DT, Sadagopan N, Bhattachar SN, Kesten SJ, Nomanbhoy TK, Cravatt BF, Ahn K. Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor. ACS Med Chem Lett. 2011;2:91–96. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. Cyclohexylcarbamic acid 3’- or 4’-substituted biphenyl-3-yl esters as fatty acid amide hydrolase inhibitors: synthesis, quantitative structure-activity relationships, and molecular modeling studies. J Med Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 33.Naidu PS, Booker L, Cravatt BF, Lichtman AH. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther. 2009;329:48–56. doi: 10.1124/jpet.108.143487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sasso O, Bertorelli R, Bandiera T, Scarpelli R, Colombano G, Armirotti A, Moreno-Sanz G, Reggiani A, Piomelli D. Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol Res. 2012;65:553–563. doi: 10.1016/j.phrs.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M, Melchiorre C. Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem. 2008;51:347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 36.Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 37.Csermely P, Agoston V, Pongor S. The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci. 2005;26:178–182. doi: 10.1016/j.tips.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 38.Zimmermann GR, Lehar J, Keith CT. Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discov Today. 2007;12:34–42. doi: 10.1016/j.drudis.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 39.Holt S, Paylor B, Boldrup L, Alajakku K, Vandevoorde S, Sundstrom A, Cocco MT, Onnis V, Fowler CJ. Inhibition of fatty acid amide hydrolase, a key endocannabinoid metabolizing enzyme, by analogues of ibuprofen and indomethacin. Eur J Pharmacol. 2007;565:26–36. doi: 10.1016/j.ejphar.2007.02.051. [DOI] [PubMed] [Google Scholar]
- 40.Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, Djoumbou Y, Eisner R, Guo AC, Wishart DS. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011;39:D1035–1041. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berger L, Corraz AJ, Carbazoles 3869145 US.
- 43.Narlawar R, Perez Revuelta BI, Haass C, Steiner H, Schmidt B, Baumann K. Scaffold of the cyclooxygenase-2 (COX-2) inhibitor carprofen provides Alzheimer gamma-secretase modulators. J Med Chem. 2006;49:7588–7591. doi: 10.1021/jm0610200. [DOI] [PubMed] [Google Scholar]
- 44.Bishay P, Schmidt H, Marian C, Haussler A, Wijnvoord N, Ziebell S, Metzner J, Koch M, Myrczek T, Bechmann I, Kuner R, Costigan M, Dehghani F, Geisslinger G, Tegeder I. R-flurbiprofen reduces neuropathic pain in rodents by restoring endogenous cannabinoids. PLoS One. 2010;5:e10628. doi: 10.1371/journal.pone.0010628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fowler CJ, Borjesson M, Tiger G. Differences in the pharmacological properties of rat and chicken brain fatty acid amidohydrolase. Br J Pharmacol. 2000;131:498–504. doi: 10.1038/sj.bjp.0703569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duggan KC, Walters MJ, Musee J, Harp JM, Kiefer JR, Oates JA, Marnett LJ. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J Biol Chem. 2010;285:34950–34959. doi: 10.1074/jbc.M110.162982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fowler CJ, Tiger G, Stenstrom A. Ibuprofen inhibits rat brain deamidation of anandamide at pharmacologically relevant concentrations. Mode of inhibition and structure-activity relationship. J Pharmacol Exp Ther. 1997;283:729–734. [PubMed] [Google Scholar]
- 48.Fowler CJ, Janson U, Johnson RM, Wahlstrom G, Stenstrom A, Norstrom K, Tiger G. Inhibition of anandamide hydrolysis by the enantiomers of ibuprofen, ketorolac, and flurbiprofen. Arch Biochem Biophys. 1999;362:191–196. doi: 10.1006/abbi.1998.1025. [DOI] [PubMed] [Google Scholar]
- 49.Fowler CJ, Naidu PS, Lichtman A, Onnis V. The case for the development of novel analgesic agents targeting both fatty acid amide hydrolase and either cyclooxygenase or TRPV1. Br J Pharmacol. 2009;156:412–419. doi: 10.1111/j.1476-5381.2008.00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fowler CJ, Holt S, Tiger G. Acidic nonsteroidal anti-inflammatory drugs inhibit rat brain fatty acid amide hydrolase in a pH-dependent manner. J Enzyme Inhib Med Chem. 2003;18:55–58. doi: 10.1080/1475636021000049726. [DOI] [PubMed] [Google Scholar]
- 51.Hermanson DJ, Marnett LJ. Cannabinoids, endocannabinoids, and cancer. Cancer Metastasis Rev. 2011;30:599–612. doi: 10.1007/s10555-011-9318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marnett LJ. Inflammation and cancer: chemical approaches to mechanisms, imaging, and treatment. J Org Chem. 2012;77:5224–38. doi: 10.1021/jo300214d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nobeli I, Favia AD, Thornton JM. Protein promiscuity and its implications for biotechnology. Nat Biotechnol. 2009;27:157–167. doi: 10.1038/nbt1519. [DOI] [PubMed] [Google Scholar]
- 54.Congreve M, Carr R, Murray C, Jhoti H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov Today. 2003;8:876–877. doi: 10.1016/s1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- 55.Czarnobilski Z, Bem S, Czarnobilski K, Konturek SJ. Carprofen and the therapy of gastroduodenal ulcerations by ranitidine. Hepatogastroenterology. 1985;32:20–23. [PubMed] [Google Scholar]
- 56.Duggan KC, Hermanson DJ, Musee J, Prusakiewicz JJ, Scheib JL, Carter BD, Banerjee S, Oates JA, Marnett LJ. (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat Chem Biol. 2011;7:803–809. doi: 10.1038/nchembio.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 58.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 59.Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17:57–61. [PubMed] [Google Scholar]
- 63.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.