

Abstract
The trimer of a Bradykinin derivative displayed more than five-fold increase in binding affinity for phosphatidylserine-enriched nanovesicles as compared to its monomeric precursor. The nanovesicle selection is directly correlated to multivalency, which amplified the electrostatic attraction. This strategy may lead to novel molecular probes for detecting highly curved membrane bilayers.
Membrane curvature generation plays a crucial role in cell signalling, endo- and exocytosis, membrane fusion, and cell growth, division and movement.1 One critical consequence of membrane curvature generation is the secretion of extracellular nanovesicles with diameters of 30 – 100 nm whose outer leaflet contains high amount of anionic phospholipids. These vesicles are known to function as carriers of chemical signals that mediate intercellular communication. Several reports have demonstrated that the dynamic modulation of membrane curvature is brought about by changes in lipid composition, scaffolding proteins, and insertion of protein regions into the membranes.2-5 Proteins that sense highly curved plasma membranes include the Bin-Amphiphysin-Rvs (BAR) domain and ArfGAP1 Lipid Packing Sensor (ALPS) motif-bearing proteins.4-7 Other examples of curvature sensing proteins/peptides include Synaptotagmin-I,8 the origin of a new macrocyclic curvature-sensing peptide C2BL3C9 and the effector domain of the myristoylated alanine-rich C-kinase substrate protein (MARCKS), MARCKS-ED.10
Our goal is to discover short peptides that recognize and select for lipid vesicles based on size and lipid composition.9,10 These peptides have advantages over protein-based curvature sensors including stability, availability, and modifiability. Herein, we report a trimeric peptide BKKG-tri (3) prepared from a Bradykinin derivative BKKG (1, Scheme 1) that recognizes membrane surfaces. We demonstrate that this peptide preferentially binds to highly curved surfaces of both synthetic lipid vesicles and natural extracellular vesicles from rat blood plasma. We also show that multimerization resulted in a significant increase in preference for lipid vesicles with diameters < 100 nm and with high palmitoyl oleoyl phosphatidylserine (POPS) content. The RtoA mutant peptides (2 and 4) lost lipid vesicle affinity, demonstrating a primarily electrostatics-mediated mechanism of 1 and 3 in membrane association. Other interactions that stabilize the lipid packing defects found in highly curved bilayers, such as the insertion of the aromatic side chains of the Phe residues, could also contribute to the preferential binding.
Scheme 1.
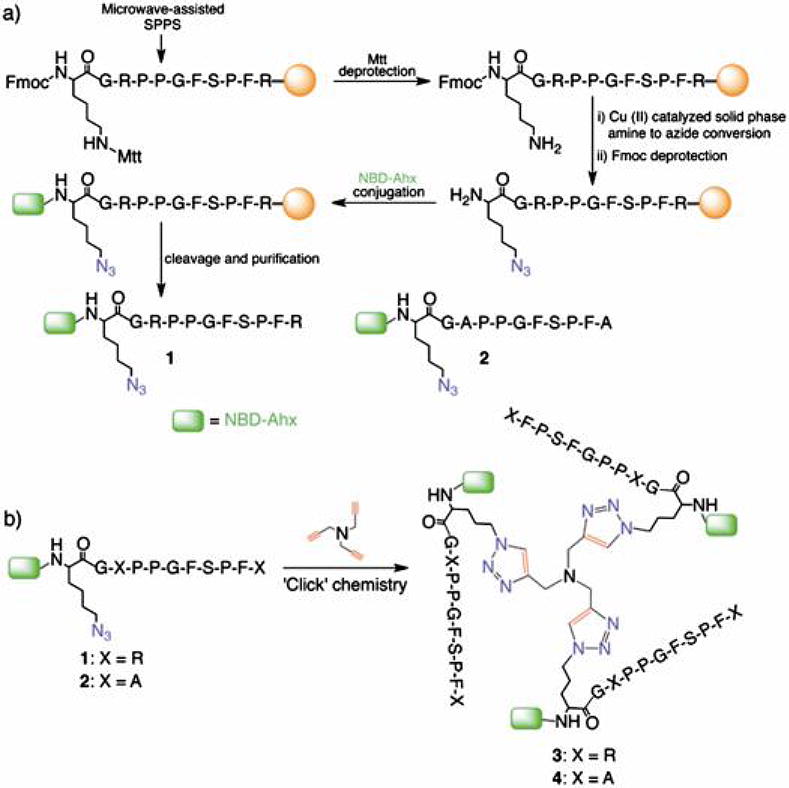
General reaction scheme: a) Preparation of monomers on solid support and b) solution phase trimerization by ‘Click’ chemistry.
Bradykinin (BK) is a cationic peptide ligand for B1 and B2 G-protein coupled receptors. Its amino acid sequence is RPPGFSPFR. BK was selected as the core molecule in our peptide design for the following reasons. First, it is believed that the conformation adopted by peptide ligands like BK is facilitated by interactions with membrane phospholipids prior to receptor binding and activation,11-14 suggesting that BK interacts with lipid bilayers. The general consensus suggests that BK exists as a random coil in aqueous solution but undergoes conformational modifications upon interaction with membrane lipids, which allows the accumulation of the active peptide conformation in the extracellular environment.15 Second, the BK proline residues induce a β-turn conformation that orients the arginine residues to a preferred claw-like “C” and “Z” active conformations, as demonstrated in the structures determined using solid-state NMR spectroscopy and molecular dynamics simulations shown in Fig. 1.12-14 Although the side chain orientation of the terminal arginine residues vary in both models, the peptide backbone was not perturbed from its preferred conformation. This conformation is believed to be critical for lipid recognition and docking on the membrane-bound receptors. Third, previous reports have shown that BK penetrates membrane bilayers and manifests differential interactions with lipid vesicles and micelles,12,16 with a strong preference for mixtures with higher anionic components. These findings were interpreted as that an increase in the local peptide concentration at the vesicle surface is strongly dictated by the cationic arginine residues at positions R1 and R9 (Fig. 1),17 suggesting electrostatic-driven attraction.
Fig. 1.
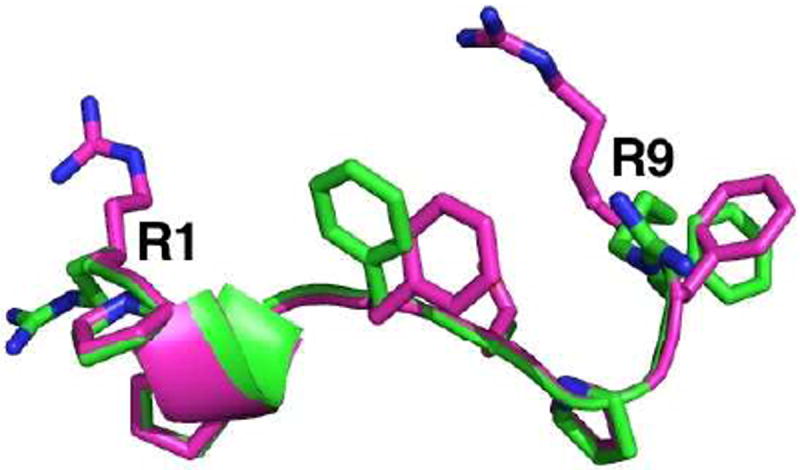
Overlay of “C” (magenta) and “Z” (green) active conformations of BK as determined by a combination of solid-state NMR spectroscopy and molecular dynamics simulations, showing the claw-like shape of the molecule and the crucial orientation of terminal arginine residues on the same face of the peptide. (Coordinates courtesy of Profs. H. Schwalbe, C. Glaubitz, and A. Gieldon. See references 13 and 14).
BKKG (1) was designed based on reports that N-terminal alteration on BK does not affect its receptor binding activity,18 suggesting a conserved conformation. Peptide 1 was prepared by adding the following residues at the N-terminus: (a) glycine as a spacer, (b) ε-azidolysine (Lys(ε-N3)) as the residue that bears an orthogonal group for ‘Click’ chemistry, and (c) 6-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)aminohexanoic acid (NBD-Ahx) as a reporter molecule in fluorescence assays. We prepared the 11-mer precursor of 1 using standard Fmoc chemistry and microwave-assisted solid phase peptide synthesis (Scheme 1a). The ε-amino group of the lysine residue was converted to an azide by solid phase Cu(II)-catalyzed amine-to-azide conversion19 to produce the Lys(ε-N3) residue. To accomplish this conversion, the methyltrityl (Mtt) protection was selectively removed using 94% CH2Cl2, 1% TFA, and 5% triisopropylsilane (TIPS) (10 mL × 3 min × 10) at r.t. and followed by thorough washing with CH2Cl2 and MeOH (5 mL × 3). After deprotection, the resin was swelled in a mixture of CH2Cl2/MeOH/H2O/Et3N, drained, and treated with CuSO4 and freshly prepared TfN3.19 The mixture was stirred for 24 h at r.t. and followed by washing with MeCN, DMF, H2O, 0.1 M EDTA, H2O, MeOH, CH2Cl2, and MeOH. A Kaiser test, FT-IR, and mass analyses confirmed the conversion of NH2 to N3. The peptide N-terminus was deprotected using 20% piperidine in 1-Methyl-2-pyrrolidinone (NMP) and conjugated with NBD-Ahx. The peptide was cleaved from the resin using 85/5/5/5 TFA/H2O/phenol/TIPS, purified by reversed phase HPLC, lyophilized, and characterized by mass spectrometry. Peptide 2 (BKKG-mut, R3A R11A mutant) was prepared in an analogous manner to investigate the effects of replacing arginine with a neutral residue.
To confirm that N-terminal modification of BK does not lead to loss of lipid vesicle affinity, a binding assay by fluorescence enhancement (FE) was performed. In FE, the fluorescence intensity of a fluorophore-labeled peptide increases due to the change from an aqueous polar solvent to a hydrophobic phospholipid environment that surrounds the fluorophore attached to the BK peptides via flexible linkers upon binding, concurring with a blue shift in the maximum emission wavelength.5 Synthetic lipid vesicle models of various sizes were prepared to mimic different membrane curvatures. Three kinds of vesicles were designed to closely resemble natural biological membranes, including lipid components such as cholesterol, POPC and POPE,20 that were named lipid model (LM) 1, 2, and 3 and contain 0, 10, and 20% POPS, respectively. These vesicles were prepared by a pressure-controlled extrusion method21 through polycarbonate membranes with pore sizes of 30, 100, and 400 nm to cover a wide range of curvatures (Supp. S8).5 Since curvature is defined as the inverse of radius, the membrane with the smallest pore size produced vesicles with the highest curvature. The extrusion process yielded vesicles with average diameters of 454 ± 25 nm (V454), 116 ± 8 nm (V116), and 58 ± 5 nm (V58), as analyzed by dynamic light scattering (DLS). These vesicles were further characterized by transmission electron microscopy (TEM), (Supp. Fig. S8), which visually confirmed the vesicle sizes.
Peptides 1 and 2 (125 μL, 1 μM) were treated with the vesicles (125 μL, 1 mM), incubated for 1 h at 4 °C, and their fluorescence emission spectra recorded (λex = 480 nm). The protein C2AB from Synaptotagmin-1 (Syt1, residues 96-421), an established lipid vesicle curvature sensor,8 was used as the positive control following our previous work.9,10 Peptide 1 treated with V58 showed fluorescence intensity higher than the peptide treated with V116, or V464 across all three lipid models. This increase is about two-folds (1.85 ± 0.03/2.14 ± 0.04 RFU) relative to the untreated peptide, with highest intensity observed with LM3 (Fig. 2a). Furthermore, the fluorescence intensities with V58 and V116 treatment are significantly higher than the positive control C2AB treated with LM3 (Fig. 2b). Peptide 2 showed minimal fluorescence increase (1.24 ± 0.04/1.46 ± 0.05 RFU) upon vesicle treatment but did not show curvature differentiation across all lipid vesicle models tested.
Fig. 2.
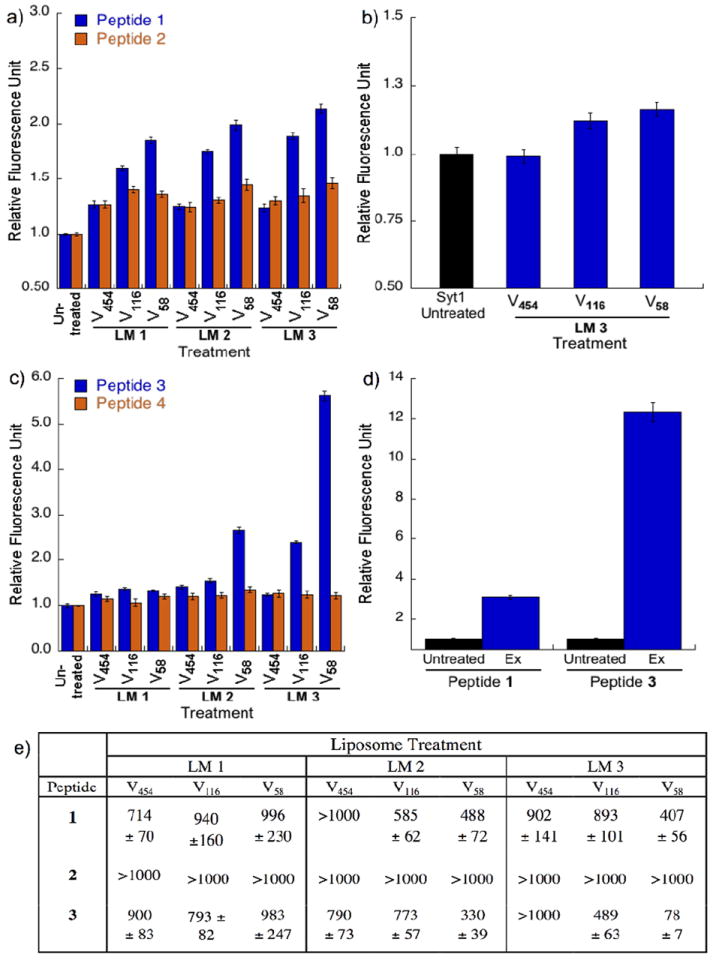
Fluorescence experiments probing the interaction of peptides with synthetic and natural lipid vesicles. a) plots for fluorescence enhancement (FE) assay of monomeric peptides 1 and 2 with lipid vesicles V58, V116, and V454 prepared from LM1, LM2, and LM3. b) plots for FE assay of Synaptotagmin-I with lipid vesicles prepared from LM3; c) plots for FE assay of trimeric peptides 3 and 4 using V58, V116, and V454 prepared from LM1, LM2, and LM3; d) plots for FE assay of peptides 1 and 3 with isolated exosomes (Ex) from rat blood plasma. Error bars represent SEM (n = 3); e) fluorescence anisotropy titration table showing the dissociation constants (Kd) of peptides 1-3, in μM, with LM1, LM2, and LM3. LM = liposome model. [Peptide] = 1 μM; [Lipid titrant] = 2 mM. Lower values indicate stronger binding.
Following on the premise that the conformation of lipid-binding peptides like 1 is stabilized upon binding to vesicles, we recorded the circular dichroism (CD) spectra of untreated and vesicle-treated 1 following reported methods for BK.16,23 This study investigated if the presence of a membrane mimetic environment that contains anionic polar head groups would perturb the peptide conformation in solution, as was previously seen for BK in the presence of sodium dodecyl sulfate (SDS) and ganglioside monosialylated type 1 (GM1) micelles.16,24 The CD spectra were measured in the region of 190-260 nm in 10 mM phosphate buffer (pH 7.4) with and without lipid vesicles. The CD spectrum of 1 in Fig. 3 shows a trough at 230 nm, a peak at 223 nm, and a deep trough at 202 nm. Overall, this profile is comparable to the CD profile of BK recorded in water that was described as a random coil.16,25,26 Upon treatment of 1 with LM3, we found a decrease in the CD bands at 230 and 223 nm and a ~10% increase in the trough depth at 202 nm. It is conceivable that the presence of liposomes perturbed the conformation of peptide 1 and that the increased depth in the trough at 202 nm is a consequence of the stabilization of the β-turn conformation, consistent with previous observations on BK that was treated with the anionic GM1 micelles.16 We did not observe a similar change in the CD spectrum of 2 after treatment with LM3, suggesting that 2 did not interact with LM3. This confirms that mutating arginine into alanine residues resulted in the loss of lipid affinity, consistent with our observations in FE assays that the guanidinium group in arginine residues is crucial for lipid recognition. This also confirms that an electrostatic-induced conformational change of 1 occurs in the presence of LM3. The above findings are in agreement with the general consensus that BK interaction with membrane lipids allows for the accumulation of the active peptide conformation in the extracellular environment.15
Fig. 3.
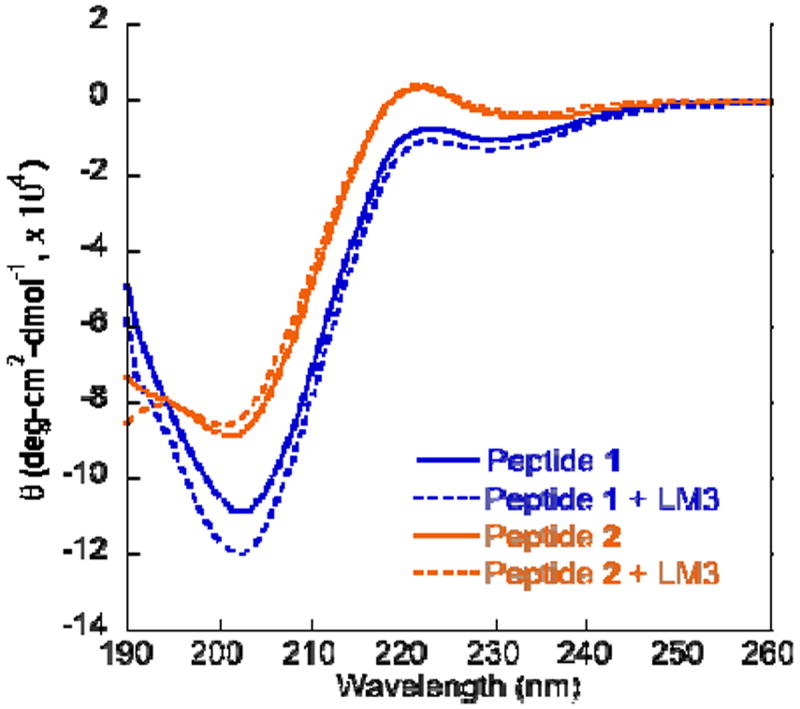
CD spectra showing an increase in trough depth at 202 nm for 1 in the presence of LM3 indicating stabilization of the β-turn conformation. Peptide 2 remains in polyproline conformation upon treatment with LM3.
Encouraged by these promising results and by previous reports that multimerization could amplify the affinity of peptides to various protein targets,27,28 we proceeded with the preparation of trimeric peptide 3 (Scheme. 1b) using the well-known, bioorthogonal strategy of copper-catalyzed azide-alkyne 1,3-dipolar cycloaddition (‘Click’ chemistry). We prepared peptide 3 using a modified method29 by reacting 1 with tripropargylamine as the alkyne core and using an excess amount of Cu (I) as [(CH3CN)4Cu]PF6 that does not require sodium ascorbate. In the same manner, the trimeric peptide 4 was prepared from 2 as a negative control. Based on the same FE assay as described above, peptide 3 treated with LM3 V58 showed almost six-fold fluorescence intensity when compared to the untreated peptide (5.6 ± 0.11 RFU), which is equivalent to 2.6 times from that observed with 1 (Fig. 2c). Treatment with LM3 V116 and LM2 V58 treatment did not show a large increase in fluorescence intensity at 2.38 ± 0.03 and 2.64 ± 0.07 RFU, respectively. The high fluorescence observed for vesicle-treated 3 is in good agreement with our hypothesis that peptide multimerization increases lipid vesicle binding and differentiation.
A FE assay is useful for screening by showing qualitative binding between peptide and lipid vesicles. However, the enhanced fluorescence may be due to either peptide-vesicle association or different fluorophore insertion depth, which requires further experimental elucidation. To dissect its respective contribution, we adopted a previously reported fluorescence anisotropy (FA) assay to specifically measure binding affinity of the BK peptides.9,10 Membrane-binding peptides partition between the hydrophobic lipid bilayer and the aqueous solvent. We could then calculate the dissociation constant (Kd) as the reciprocal of the molar partition coefficient.30 FA assay was done by titrating a constant concentration of fluorescently labelled peptide with increasing concentration of lipid vesicles. The FA increase is due to changes in tumbling rates of the fluorophore attached to either the bound peptide-vesicle complex or to the free peptide in aqueous solution. Table 1 shows that peptide 1 has modest binding to LM2 and LM3 V58 (Kd = 488 ± 72 and 407 ± 56 μM, respectively) and modest to negligible binding for V116 and V454 across all lipid models. Peptide 2 showed negligible binding across all lipid models and sizes. Meanwhile, peptide 3 showed a Kd of 78 ± 7 μM for LM3 V58, the strongest binding strength observed among the tested peptides, which corresponds to 6.3 folds higher than LM 3 V116. This affinity is 5.2 folds higher than 1 for LM3 V58. It showed modest to weak binding for other lipid models and sizes. For comparison, peptide 3 possesses a higher affinity than C2AB for 105 nm vesicles (Kd = 151 ± 6 μM).8 These data support our hypothesis that BK derivatives could sense membrane curvatures, and that electrostatic attraction plays a dominant role in their binding to anionic phospholipid-enriched vesicles. Taking into consideration that trimeric peptide 3 has three of peptide 1, it appears that there is still a significant synergistic effect that is not simply additive due to the increase in the concentration of peptide 1, as the binding affinity of peptide 3 was shown to be >5 times stronger than the monomeric peptide 1.
To examine if our findings on synthetic vesicles would translate to natural lipid vesicles, we tested exosomes (Ex) from blood plasma of rats that underwent inescapable tailshock stress as a model of exosome release.32 Ex have diameters of 30 - 100 nm (Supp. Fig. S11) and therefore provides a good biological model for testing our curvature-sensing peptides.9,33 The total number of Ex in the sample was counted using NanoSight nanoparticle tracking analysis9,34 to ensure that their concentration is comparable to the synthetic models (Supp. Fig. S12). As shown in Fig. 2d, Ex-treated peptides 1 and 3 have significantly higher fluorescence intensity at 3.09 ± 0.08 and 12.34 ± 0.47 RFU, respectively, than the untreated peptides, demonstrating that their synthetic lipid vesicle sensing property is translatable to the detection of Ex. Since exosomes contain the anionic lipids phosphatidylserine and phosphatidylinositol35 at ~16 – 19% that corresponds to the POPS concentration in LM3, these FE data directly correlate to our findings with synthetic lipid vesicles.
Previously, McMahon hypothesized that the curvature sensing and induction by the BAR domain is associated with its ability to form pseudo-multivalent clusters that is facilitated by their helices.36 This BAR domain clustering is enabled by an N-terminal extension domain called helix-0 that undergoes random coil to helix transition upon interaction with membrane mimetic environments.37 Our work provides a novel demonstration that a synthetic, covalently-linked cluster of a curvature sensing peptide possesses higher selection and stronger affinity for lipid nanovesicles enriched in anionic phospholipids. Based on our findings, we report solid evidence of observing curvature sensing ability of bradykinin derivatives. Binding interactions are primarily due to electrostatic attraction with the lipid vesicles, aided by a claw-like peptide conformation that favorably orients the arginine side chains towards the phospholipid head groups. Perhaps, this curvature sensing behavior may be due to influences made by the negatively charged PS lipid component, which aids in binding affinity as well as lipid packing effects in smaller vesicles or even hydrophobic Phe interactions with the membrane. The synthetic lipid vesicle binding ability of these peptides was also translated to the detection of exosomes, which launched an exciting new direction in the study of cell secreted lipid vesicles that carry membrane-protected information for intercellular communications.
Supplementary Material
Acknowledgments
We thank the HHMI (Collaborative Innovation Award for H.Y.), NIH (GM103843 for H. Y. and MH061876 for E.R.C.), NSF (IOS 1022451 for M.F.), and WSU (Startup fund for J.P.S.) for financial support. E.R.C. is an Investigator of the HHMI. L.A.M. is supported by a Ruth L. Kirschtein National Research Service Award (F31 CA165349) and a NIH training grant (T32 GM008759. S.K.C. is supported by an AACR Thomas J. Bardos scholarship.
Footnotes
Electronic Supplementary Information (ESI) available: [experimental details, MS, NMR, DLS, FE, FA, TEM, NTA data]. See DOI: 10.1039/b000000x/
Contributor Information
Jonel P. Saludes, Email: jonel.saludes@wsu.edu.
Hang Yin, Email: hubert.yin@colorado.edu.
Notes and references
- 1.Zimmerberg J, Kozlov MM. Nat Rev Mol Cell Biol. 2006;7:9. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
- 2.Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, Rapoport TA. Cell. 2010;143:774. doi: 10.1016/j.cell.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMahon HT, Gallop JL. Nature. 2005;438:590. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 4.Zimmerberg J, McLaughlin S. Curr Biol. 2004;14:R250. doi: 10.1016/j.cub.2004.02.060. [DOI] [PubMed] [Google Scholar]
- 5.Drin G, Casella JF, Gautier R, Boehmer T, Schwartz TU, Antonny B. Nat Struct Mol Biol. 2007;14:138. doi: 10.1038/nsmb1194. [DOI] [PubMed] [Google Scholar]
- 6.Bhatia VK, Madsen KL, Bolinger PY, Kunding A, Hedegard P, Gether U, Stamou D. EMBO J. 2009;28:3303. doi: 10.1038/emboj.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bigay J, Antonny B. Biochemistry. 2007;46:1779. doi: 10.1021/bi062288w. [DOI] [PubMed] [Google Scholar]
- 8.Hui EF, Johnson CP, Yao J, Dunning FM, Chapman ER. Cell. 2009;138:709. doi: 10.1016/j.cell.2009.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saludes JP, Morton LA, Ghosh N, Beninson L, Chapman ER, Fleshner M, Yin H. ACS Chem Biol. 2012;7:1629. doi: 10.1021/cb3002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morton LA, Yang H, Saludes JP, Fiorini Z, Beninson L, Chapman ER, Fleshner M, Xue D, Yin H. ACS Chem Biol. 2013;8:218. doi: 10.1021/cb300429e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langelaan DN, Rainey JK. Biochem Cell Biol. 2010;88:203. doi: 10.1139/O09-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonechi C, Ristori S, Martini G, Rossi C. Biochim Biophys Acta. 2009;1788:708. doi: 10.1016/j.bbamem.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 13.Lopez JJ, Shukla AK, Reinhart C, Schwalbe H, Michel H, Glaubitz C. Angew Chem Int Ed. 2008;47:1668. doi: 10.1002/anie.200704282. [DOI] [PubMed] [Google Scholar]
- 14.Gieldon A, Lopez JJ, Glaubitz C, Schwalbe H. ChemBioChem. 2008;9:2487. doi: 10.1002/cbic.200800324. [DOI] [PubMed] [Google Scholar]
- 15.Schwyzer R. Biopolymers. 1991;31:785. doi: 10.1002/bip.360310624. [DOI] [PubMed] [Google Scholar]
- 16.Chatterjee C, Mukhopadhyay C. Biochem Biophys Res Commun. 2004;315:866. doi: 10.1016/j.bbrc.2004.01.134. [DOI] [PubMed] [Google Scholar]
- 17.Turchiello RF, Lamy-Freund M, Hirata IY, Juliano L, Ito AS. Biopolymers. 2002;65:336. doi: 10.1002/bip.10238. [DOI] [PubMed] [Google Scholar]
- 18.Srivastava S, Phadke RS, Kamath SA, Coutinho EC. Eur J Med Chem. 1997;32:669. [Google Scholar]
- 19.Oyelere AK, Chen PC, Yao LP, Boguslavsky N. J Org Chem. 2006;71:9791. doi: 10.1021/jo0618122. [DOI] [PubMed] [Google Scholar]
- 20.Rothman JE, Lenard J. Science. 1977;195:743. doi: 10.1126/science.402030. [DOI] [PubMed] [Google Scholar]
- 21.Morton LA, Saludes JP, Yin H. J Vis Exp. 2012;64:e4151. doi: 10.3791/4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holthuis JCM, Burger KNJ. Develop Cell. 2003;5:821. doi: 10.1016/s1534-5807(03)00366-6. [DOI] [PubMed] [Google Scholar]
- 23.Hashimoto C, Nozaki S, Muramatsu I. Bull Chem Soc Jpn. 1991;64:3571. [Google Scholar]
- 24.Cann JR, Vatter A, Vavrek RJ, Stewart JM. Peptides. 1986;7:1121. doi: 10.1016/0196-9781(86)90142-7. [DOI] [PubMed] [Google Scholar]
- 25.Cann JR, Stewart JM, Matsueda GR. Biochemistry. 1973;12:3780. doi: 10.1021/bi00743a029. [DOI] [PubMed] [Google Scholar]
- 26.Mondal S, Ghosh S, Ghosh D, Saha A. P Phys Chem C. 2012;116:9774. [Google Scholar]
- 27.Tweedle MF. Acc Chem Res. 2009;42:958. doi: 10.1021/ar800215p. [DOI] [PubMed] [Google Scholar]
- 28.Ruzza P, Marchiani A, Antolini N, Calderan A. Anti-Cancer Agents Med Chem. 2012;12:416. doi: 10.2174/187152012800617849. [DOI] [PubMed] [Google Scholar]
- 29.Alleti R, Rao V, Xu L, Gillies RJ, Mash EA. J Org Chem. 2010;75:5895. doi: 10.1021/jo101043m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rusu L, Gambhir A, McLaughlin S, Radler J. Biophys J. 2004;87:1044. doi: 10.1529/biophysj.104.039958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3. Springer; 2006. [Google Scholar]
- 32.Johnson JD, Fleshner M. J Leukoc Biol. 2006;79:425. doi: 10.1189/jlb.0905523. [DOI] [PubMed] [Google Scholar]
- 33.Bobrie A, Colombo M, Raposo G, Thery C. Traffic. 2011;12:1659. doi: 10.1111/j.1600-0854.2011.01225.x. [DOI] [PubMed] [Google Scholar]
- 34.Dragovic RA, Gardiner C, Brooks AS, Tannetta DS, Ferguson DJP, Hole P, Carr B, Redman CWG, Harris AL, Dobson PJ, Harrison P, Sargent IL. Nanomedicine: NBM. 2011;7:780. doi: 10.1016/j.nano.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subra C, Laulagnier K, Perret B, Record M. Biochimie. 2007;89:205. doi: 10.1016/j.biochi.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 36.Gallop JL, McMahon HT. In: Lipids, rafts and traffic: Biochemical Society Symposium No 72. McIlhinney RAJ, Hooper NM, editors. Vol. 72. Portland Press; London: 2005. p. 223. [DOI] [PubMed] [Google Scholar]
- 37.Low C, Weininger U, Lee H, Schweimer K, Neundorf I, Beck-Sickinger AG, Pastor RW, Balbach J. Biophys J. 2008;95:4315. doi: 10.1529/biophysj.108.134155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.