

Abstract
The ability of Myc to promote cellular transformation is well established; however, a better understanding of the mechanisms through which Myc mediates tumorigenesis is essential for the development of therapeutic approaches to target this potent oncoprotein. Structure-function studies in rodent fibroblast cells have provided the basis for much of our current understanding of these mechanisms. To build on these approaches, we have characterized three novel human cell line models of Myc-dependent transformation: MCF10A, SH-EP Tet21/N-Myc, and LF1/TERT/LT/ST cells. We have also evaluated Myc family proteins (c-Myc and L-Myc), a naturally occurring isoform of Myc (MycS), and a set of N-terminal domain mutants (ΔMBII, W135E, T58A) for their ability to promote anchorage-independent growth in these models. Taken together these results provide the field with three new human cell-based models to study Myc activity, highlight the importance of cellular context, and challenge the paradigm that the ability of Myc to promote tumorigenesis is exclusively MBII-dependent.
Keywords: Myc, structure-function, cellular transformation, SV40, hTERT
Introduction
Deregulated expression of the Myc family of oncoproteins is well established and has a profound role in human malignancies. Despite the recognition that Myc is a transcriptional regulator associated with aggressive disease, the molecular mechanisms through which Myc is able to promote and maintain tumorigenesis are not yet fully defined. Our current understanding of Myc structure-function has been established largely using rodent fibroblast cell systems and in some cases murine bone marrow transplant models, with only limited work in human cells. These structure-function studies of Myc-dependent transformation are summarized in Supplementary Table 1. Owing to the established differences between murine and human cells (Drayton and Peters, 2002; Hahn et al., 1999; Prowse and Greider, 1995) and the prominent role of Myc in human cancers, it is valuable to expand and extend this analysis of Myc structure-function to more disease-relevant human models.
The three transforming members of the Myc protein family are c-Myc, N-Myc and L-Myc. Unless otherwise stated, we will use Myc to refer to human c-Myc, which is the only of these family members expressed in normal adult tissues. The human Myc proteins contain several functional regions (Supplementary Figure 1A). The C-terminal basic, helix-loop-helix, leucine zipper (B-HLH-LZ) domains of Myc are required for DNA binding and key protein-protein interactions, including the interaction between Myc and its obligate partner Max (Meyer and Penn, 2008). The N-terminus of Myc is comprised of several highly conserved regions, termed Myc homology boxes (MBI, MBII, MBIIIa, MBIIIb, MBIV) (Meyer and Penn, 2008). The best characterized of these regions are MBI and MBII. MBI contains critical phosphorylation sites and is a region frequently mutated in Burkitt’s Lymphomas (Bahram et al., 2000; Bhatia et al., 1993; Chang et al., 2000). The MBII region is essential for most biological functions, including transformation in all cells lines and mouse models reported (Cowling et al., 2007; Herbst et al., 2005; Oster et al., 2003; Stone et al., 1987). MBII is also a critical region for certain protein-protein interactions, including that with an important co-factor in transformation, TRRAP (McMahon et al., 1998). In addition to the full-length protein, Myc has a naturally occurring shorter isoform, MycS. MycS arises from a weak internal translation initiation AUG codon to produce a protein that lacks the first 100 amino acids (Spotts et al., 1997).
In this report, we established new human Myc-dependent transformation systems and then exploited these new models to advance the current understanding of Myc structure-function. Specifically we have evaluated Myc family proteins (c-Myc and L-Myc), a naturally occurring isoform of Myc (MycS), and a set of N-terminal domain mutants (ΔMBII, W135E, T58A) for their ability to promote anchorage-independent growth in three novel human cell line models, MCF10A, SH-EP Tet21/N-Myc, and LF1/TERT/LT/ST. Overall, our results highlight that Myc-dependent transformation is influenced by cellular context and provide evidence that the ability of Myc to promote oncogenesis is not exclusively mediated through Myc homology box II (MBII).
Results & Discussion
Myc induces transformation in human cell lines
Recognizing the importance of extending structure-function studies into more disease-relevant models, we employed specific criteria in selecting the human cell lines to characterize Myc-dependent transformation. First, the tissue of origin needed to represent a disease site commonly associated with deregulated Myc or Myc family members. Second, because Myc deregulation is evident both early and late in the malignant process, it was important to evaluate Myc at different stages of cellular transformation. We therefore evaluated Myc-dependent transformation in a non-tumorigenic breast epithelial cell line, MCF10A, as well as a tumor-derived system, SH-EP neuroblastoma cells. Finally, we chose to investigate a primary cell model immortalized and further transformed by the expression of viral oncoproteins, namely the LF1/TERT/LT/ST lung fibroblasts. Combined, these three models represent a spectrum of transformation states and provide renewable human systems to evaluate Myc activity in vitro.
The MCF10A cells were established through spontaneous immortalization of human mammary epithelium (Soule et al., 1990). The non-transformed nature of this cell line is supported by the absence of anchorage-independent growth and tumor formation in vivo (Soule et al., 1990). To evaluate the ability of deregulated Myc to drive transformation, we infected MCF10A cells with retrovirus containing human Myc cDNA or empty vector control. Myc protein expression is remarkably similar in cells transduced with empty vector control and ectoptic Myc (Figure 1A, compare lanes 1 and 2). To distinguish regulated and deregulated Myc expression, we evaluated Myc protein levels in response to one hour of serum and growth factor withdrawal. As expected, in MCF10A-GFP control cells, endogenous Myc protein levels dropped to a low but detectable level. The MCF10A-Myc cells, in contrast, were less responsive to this external cue and maintained a high level of deregulated Myc protein expression (Figure 1A, compare lanes 3 and 4). We next evaluated cellular transformation through anchorage-independent growth. Stable constitutive expression of Myc alone was sufficient to transform MCF10A cells, as measured by a 6-fold increase (p<0.001) in colony formation in soft agar(Figure 1B).
Figure 1. Human Myc promotes cellular transformation in MCF10A, SH-EP and LF1/3T human cell lines.

Control green fluorescent protein (GFP) or human Myc cDNA were introduced by infection with ecotropic, replication-incompetent retrovirus into human cell lines, MCF10A, SH-EP and LF1/3T, as described previously (Wu et al., 2004) and stable pools were isolated by fluorescence-activated cell sorting (FACS) for green fluorescent protein (GFP) expression. A) MCF10A cells (a kind gift from Senthil Muthuswamy, Ontatio Cancer Institute) were cultured as described previously (Debnath et al., 2003). Growth factor withdrawal was achieved by culturing cells in media containing 0.05% horse serum and supplemented with only 10 μL/mL insulin for one hour. Ectopic Myc expression was confirmed by immunoblotting of lysates isolated from asynchronously growing cells in full growth media and from cells subjected to 1 hour of serum and growth factor withdrawal. B) Transformation was evaluated by anchorage-independent colony growth in soft agar. Soft agar experiments were completed as previously described with the following modifications, 5 000 cells were seeded per 35 mm petri dish in triplicate, and colonies (greater than 6 cells) were counted at the end of a 2–3 week period (Oster et al., 2003). Transformation data is presented as a relative number of colonies compared to cells expressing wild-type Myc. Data represents the mean ± standard deviation from three independent experiments, **p<0.01, ***p<0.001, paired t-test. C) SH-EP Tet21/N-Myc cells (a kind gift from Manfred Schwab, German Cancer Research Center) were cultured in RPMI 1640 with 10% FBS (Breit and Schwab, 1989).
Ectopic Myc expression was confirmed by immunoblotting. This experiment was conducted both in the presence and absence of N-Myc expression. 1 μg/mL tetracycline (Sigma, St. Louis, MO) was added to growth media 48 hours prior to experiments to inactivate N-Myc expression. D) Soft agar transformation experiments were conducted as above, both in the absence (N-Myc ON) and presence (N-Myc OFF) of tetracycline. Data represents the mean ± standard deviation from two independent experiments. E) LF1/TERT/LT/ST cells (a kind gift from John Sedivy, Brown University) were grown in HAM F10 media and supplemented with 15% fetal bovine serum (FBS) (Wei et al., 2003). Ectopic Myc expression was confirmed by immunoblotting. F) Soft agar transformation experiments were conducted as above and data represents the mean ± standard deviation from three independent experiments, **p<0.01, paired t-test.
To contrast the non-transformed nature of MCF10A cells and evaluate a diversity of human cell types we evaluated Myc-dependent transformation in SH-EP neuroblastoma cells. Amplification of MYCN is a common event in neuroblastoma, and is associated with a particularly aggressive and prognostically poor subset of this paediatric cancer (Gustafson and Weiss, 2010; Schwab, 2004). Furthermore, tumors that lack MYCN amplification often have Myc deregulation (Breit and Schwab, 1989). The SH-EP cells used in this study have been modified to express a tetracycline (tet) regulatable N-Myc and are formally referred to as SH-EP Tet21/N-Myc cells (Lutz et al., 1996). The expression of N-Myc alone is able to promote anchorage-independent colony formation, which can be abolished by pre-treating cells with tetracycline to turn off expression of N-Myc (Kim et al., 2007). Myc and N-Myc protein expression in untreated (N-Myc ON) or tetracycline treated (N-Myc OFF) cells was confirmed by immunoblotting (Figure 1C). Endogenous Myc protein expression was not detectable in N-Myc ON cells, but increased upon loss of ectopic N-Myc expression in response to tetracycline treatment (Figure 1C compare lanes 1 and 3), consistent with the well established mechanism of negative regulation by Myc family members (Breit and Schwab, 1989; Cleveland et al., 1988; Penn et al., 1990). We then assayed transformation of these cells through colony formation in soft agar. Ectopic Myc robustly potentiated transformation relative to empty vector control in the absence of N-Myc expression (Figure 1D). This level of transformation was similar to that observed in the presence of N-Myc Figure 1D). Thus, SH-EP cells provide a second human model system to study Myc-dependent transformation.
To further complement non-transformed MCF10A and tumor-derived SH-EP cells, we characterized a primary cell that had been engineered to an immortal cell line, namely the LF1/TERT/LT/ST (herein referred to as LF1/3T) cells. Derived from primary human diploid lung embryonic fibroblasts, these cells have been incrementally immortalized and transformed by stable expression of human telomerase (TERT), simian virus 40 (SV40) Large T-antigen (LT) and SV40 Small T-antigen (ST) (Wei et al., 2003). Notably, LF1/3T cells express low levels of endogenous Myc and were previously demonstrated to exhibit enhanced anchorage-independent growth with ectopic expression of murine c-Myc (Wei et al., 2003). We confirmed that ectopic expression of human Myc could recapitulate this transformation phenotype. Ectopic expression of human Myc was confirmed by immunoblotting (Figure 1E) and significantly (p<0.01) promoted a four-fold increase in anchorage independent growth of LF1/3T cells (Figure 1F). Representative images of colony formation in Myc-transformed cells in provided in Supplementary Figure 2. Taken together, we have established that MCF10A, SH-EP and LF1/3T cells can all serve as model systems, representing a range of transformation states, to study human Myc-dependent transformation.
Transformation in MCF10A and SH-EP cells is MBII-dependent
To characterize Myc-dependent transformation in our novel cell line models we prioritized mutants encompassing the unique and highly conserved regions, MBI and MBII (Supplementary Figure 1). Specifically, within MBI, phosphorylation of threonine-58 (T58) by GSK3 targets Myc for proteosomal degradation (Vervoorts et al., 2006). Not surprisingly, T58 is one of the residues commonly mutated in Burkitt’s Lymphomas, and studies have confirmed that mutation to an alanine results in increased protein stability (Bahram et al., 2000; Chang et al., 2000; Gregory et al., 2003). Additionally, the T58A point mutant formed tumors at a significantly higher penetrance and shorter latency than wild-type Myc in a murine bone marrow transplant model (Hemann et al., 2005). Therefore, the T58A point mutant served as a strong positive control for this work, representing a mutant that is more transforming than wild-type Myc. As mentioned previously, MBII is essential in all reported models of Myc-dependent transformation. Tryptophan-135 (W135) is a key hydrophobic residue contained within MBII and mutation to glutamic acid (W135E) impaired binding to TRRAP and inhibited cellular transformation (Oster et al., 2003; Wood et al., 2000). We selected ΔMBII and W135E mutants to evaluate MBII dependence in these transformation models. In addition to the above-mentioned mutants, we extended our evaluation to include a naturally occurring isoform of Myc, MycS, as well as the Myc family member L-Myc. While these naturally occurring molecules have been evaluated previously, their contributions to cellular transformation remain poorly understood.
Briefly, we infected cells with retroviruses harbouring cDNAs for human wild-type Myc, L-Myc, MycS, Myc N-terminal domain deletion or point mutants, and vector control (Supplementary Figure 1). We conducted immunoblot analysis to ensure similar expression across our panel (Figure 2A and D). For MCF10A cells, we evaluated protein expression in both asynchronously growing cells, and cells subjected to one hour of serum and growth factor withdrawal in order to down-regulate endogenous Myc expression and then be able to compare and ensure similar expression between ectopic constructs. We next measured the proliferative potential of the cells by assaying their growth rate of over five days. The cells expressing the various Myc constructs had doubling times that were similar to the control and wild-type Myc-expressing cells (Figure 2B and E). Therefore, changes observed in anchorage-independent growth cannot be easily attributed to changes in the proliferative rate of the cells. Finally, we evaluated anchorage-independent growth in soft agar. Overall, results were consistent between the MCF10A and SH-EP cells (Figure 2C and F). We observed a statistically significant 1.6-fold increase (p<0.05) in colony formation over wild-type Myc with the T58A point mutant in MCF10A cells (Figure 2C). While not statistically significant, the T58A point mutant showed a trend with 1.5-fold increased transformation in SH-EP cells and formed colonies that were qualitatively larger than wild-type (Figure 2F and data not shown). In these two cell line models, transformation was dependent on MBII as both the ΔMBII and W135E mutants formed significantly fewer colonies (Figure 2C and F). MycS also formed significantly fewer colonies compared to full length Myc. As MycS is still not fully characterized, these two cell line models may provide useful tools to better understand its biology. Finally, L-Myc exhibited significantly reduced colony formation compared to Myc, and reinforced the outstanding question of what makes L-Myc distinct from the other two family members. We further evaluated the ability of N-Myc to promote transformation in MCF10A cells. Our results confirmed previous reports that N-Myc is functionally interchangeable with Myc as colony formation was indistinguishable from wild-type Myc (data not shown) (Landay et al., 2000; Malynn et al., 2000; Oster et al., 2003).
Figure 2. Transformation in MCF10A and SH-EP cells is MBII-dependent.

The panel of Myc cDNAs were introduced by infection with ecotropic, replication-incompetent retrovirus into MCF10A and SH-EP cells as described previously (Wu et al., 2004). For all SH-EP experiments, 1 μg/mL tetracycline (Sigma, St. Louis, MO) was added to the media 48 hours prior to experiments to inactivate N-Myc expression. A,D) Protein expression was evaluated by immunoblotting. MCF10A cells were harvested under both asynchronously growing and 1 hour serum and growth factor withdrawl conditions. * indicates non-specific bands. B,E) Cell proliferation was assessed by subconfluently seeding 4000 cells/well in a 24-well dish in triplicate. Cells were counted daily over a 5 day period using a Coulter Counter, or haemocytometer. Population doubling times were calculated using GraphPad Prism software (v2.0b) and are presented as mean ± standard deviation for 3–5 independent experiments. C,F) Soft agar experiments were completed as described in Figure 1. Transformation data is presented as a relative number of colonies compared to cells expressing wild-type Myc, with mean ± standard deviation for 3–6 independent experiments. *p<0.05, **p<0.01, ***p<0.001, paired t-test.
Previous reports have shown the T58A point mutant to induce apoptosis to a lesser extent than wild-type Myc, and thereby provides a possible explanation for the increase in soft agar colony formation (Chang et al., 2000; Conzen et al., 2000; Hemann et al., 2005). To evaluate Myc-dependent cell death we generated MCF10A cells expressing 4-hydroxytamoxifen (4-OHT) inducible Myc and Myc mutants (Supplementary Figure 3)(Callus et al., 2008). Cells were subjected to 24 hours of serum and growth factor withdrawal and then subsequently treated with 100 nM 4-OHT or ethanol control and 1 μg/mL tunicamycin for an additional 24 hours. Cell cycle analysis was performed and the pre-G1 population was measured to quantify cell death. Myc significantly induced cell death compared to empty vector (Supplementary Figure 3B), and this was shown to be dependent on MBII. Importantly, there was a modest, yet significant (p<0.05), decrease in cell death with the T58A point mutant, suggesting that the increase in transformation observed is, in part, a result of this mutant having a decreased ability to promote cell death. We also evaluated cell death in response to serum withdrawal in the SH-EP cells, and observed no remarkable differences between wild-type Myc and the T58A point mutant (data not shown). This observation helps explain why transformation by the T58A mutant was evident, but less robust in SH-EP cells compared to the MCF10A cells.
Combined, these data suggest that Myc-dependent transformation is evident and similar in both MCF10A and SH-EP cells (Table 1). Both models demonstrate enhanced transformation in the presence of the T58A point mutation and significantly reduced colony formation by the ΔMBII and W135E mutants. These two systems, therefore, provide the field with human models that can complement and extend studies in both the classic rat fibroblast models and bone marrow transplantation models of Myc-dependent transformation.
Table 1.
Summary of structure-function data for Myc-dependent transformation
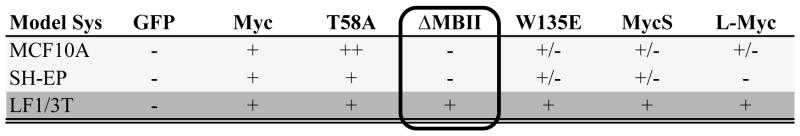
|
−, does not promote colony formation
+/−, promotes colony formation to a lesser extent than wild-type Myc
+, promotes colony formation to a similar extent as wild-type Myc
++, promotes colony formation to a greater extent than wild-type Myc
Transformation in LF1-3T cells is MBII-independent
We next conducted a similar structure-function analysis in LF1/3T cells. We demonstrated similar protein expression of stable cell pools by immunoblotting (Figure 3A). Additionally, there were no striking differences in proliferation between cells expressing the different Myc constructs (Figure 3B). We further investigated the ability of the Myc panel to potentiate transformation (Figure 3C). Surprisingly, the entire panel was able to promote anchorage-independent colony formation to a similar extent as wild-type Myc. Most notably, deletion of MBII and point mutation of W135E did not significantly impair Myc transformation of LF1/3T cells, in striking contrast to our MCF10A and SH-EP data.
Figure 3. Transformation in LF1/3T cells is MBII-independent.

The panel of Myc cDNAs were introduced by infection with ecotropic, replication-incompetent retrovirus into LF1/3T cells as described previously (Wu et al., 2004). A) Protein expression was confirmed by immunoblotting. * indicates non-specific bands. B) Doubling times were determined as described in Figure 2 and are presented as mean ± standard deviation from three independent experiments. C) Soft agar colony formation experiments were performed as described in Figure 1. Transformation data is presented as a relative number of colonies compared to cells expressing wild-type Myc, with mean ± standard deviation for three independent experiments. **p<0.01 paired t-test. D) Rat-1A cells were cultured in DMEM H21 with 10% FBS. Control GFP or Myc cDNAs were introduced by infection with ecotropic, replication-incompetent retrovirus. Protein expression was confirmed by immunoblotting. E,F) Soft agar colony formation experiments were completed as described in Figure 1 with the following modifications, 250 Rat-1A cells were seeded per 35 mm petri dish in triplicate. Data represents the mean ± standard deviation for two independent experiments.
To confirm this result we rigorously tested for any potentially confounding effects. As an additional control, Rat-1A cells were simultaneously infected, selected, and analyzed for protein expression and transformation. Immunoblot analysis confirmed successful transduction and similar expression of the ectopic constructs (Figure 3D). Confirming our initial observation, there were remarkable differences in transformation between the two cell lines with respect to the MBII deletion mutant (Figure 3E and F). Specifically, in the Rat-1A cells the induction of colony formation was MBII-dependent, whereas transformation in the LF1/3T cells was MBII-independent.
Taken together, these data suggest that the mechanisms of Myc-dependent transformation may be context and/or cell-type dependent. More specifically, they suggest for the first time that there may be MBII-independent mechanisms of transformation. One hypothesis is that MBII is essential for tumor initiation, but is dispensable for further tumor progression. This may have been previously missed because Myc’s role in initiation has been the focus of the majority of previous work (Adams et al., 1985; Hemann et al., 2005; Land et al., 1983; Langdon et al., 1986; Stewart et al., 1984). This hypothesis is further supported through the identification of MBII point mutations in Burkitt’s Lymphomas (Kuttler et al., 2001). When introduced into Rat-1A cells, these mutants in MBII were not able to promote anchorage-independent growth to the same extent as wild-type Myc; however, their contributions to tumor progression are not yet fully appreciated. Importantly, our results and the presence of MBII mutations in Burkitt’s Lymphomas further support the model that there may be MBII-independent modes through which Myc promotes tumorigenesis. A comprehensive evaluation of the ability of ectopic Myc to enhance transformation throughout the spectrum of cancer initiation and progression has not previously been reported, and our work suggests that it will be instructive to characterize MBII-dependence as a defining feature as these models are identified.
The LF1/3T cells were incrementally transformed with TERT, LT and ST. We do not believe that MBII-independence is mediated through the overexpression of TERT as HMECs immortalized with TERT have been recently shown to undergo transformation in an MBII-dependent manner (Cowling et al., 2007). Overexpression of the viral proteins LT and ST were employed to inactivate the tumor supressors p53, pRb and PP2A (Chen and Hahn, 2003). While these correspond to common mutations in human cancers, it is possible that expression of LT and ST confers MBII-independence. This possibility has broader implications as these viral oncogenes are widely applied throughout the field of cancer biology. The future evaluation of MBII-dependence in the different stages of transformation of LF1/3T and other cells, such as HMECs, and animal models of tumor progression will provide further insight into this novel observation.
In conclusion, we have characterized three new human models that clearly show that cellular context influences Myc-dependent transformation. In addition, we provide evidence of MBII-independent transformation, which challenges the current understanding of Myc structure-function and Myc-dependent transformation. Taken together, these data highlight the importance of defining the nature of cell-type and molecular make-up as we strive to delineate the mechanisms of Myc-dependent transformation and advance Myc as a therapeutic target in cancer.
Supplementary Material
Acknowledgments
The authors would like to acknowledge technical assistance from Ms. Caitlin Latimer. The authors thank the members of the Penn Lab for helpful discussions and critical review of this manuscript. This research was funded by a grant from the Canadian Cancer Society Research Institute (LZP), an Ontario Graduate Scholarship (AS), and a Canadian Breast Cancer Foundation Ontario Region Doctoral Fellowship (ARW). Additional support was provided by the Ontario Ministry of Health and Long Term Care. The views expressed do not necessarily reflect those of the OMOHLTC.
Footnotes
Conflict of Interest
The authors declare no conflict of interest
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–8. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Bahram F, von der Lehr N, Cetinkaya C, Larsson LG. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood. 2000;95:2104–10. [PubMed] [Google Scholar]
- Bhatia K, Huppi K, Spangler G, Siwarski D, Iyer R, Magrath I. Point mutations in the c-Myc transactivation domain are common in Burkitt’s lymphoma and mouse plasmacytomas. Nat Genet. 1993;5:56–61. doi: 10.1038/ng0993-56. [DOI] [PubMed] [Google Scholar]
- Breit S, Schwab M. Suppression of MYC by high expression of NMYC in human neuroblastoma cells. J Neurosci Res. 1989;24:21–8. doi: 10.1002/jnr.490240105. [DOI] [PubMed] [Google Scholar]
- Callus BA, PGE, JEH, AMJ, AK, JEV, et al. Cytoplasmic p53 is not required for PUMA-induced apoptosis. Cell Death and Differentiation. 2008;15:213–215. doi: 10.1038/sj.cdd.4402245. [DOI] [PubMed] [Google Scholar]
- Chang DW, Claassen GF, Hann SR, Cole MD. The c-Myc transactivation domain is a direct modulator of apoptotic versus proliferative signals. Mol Cell Biol. 2000;20:4309–19. doi: 10.1128/mcb.20.12.4309-4319.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Hahn WC. SV40 early region oncoproteins and human cell transformation. Histol Histopathol. 2003;18:541–50. doi: 10.14670/HH-18.541. [DOI] [PubMed] [Google Scholar]
- Cleveland JL, Huleihel M, Bressler P, Siebenlist U, Akiyama L, Eisenman RN, et al. Negative regulation of c-myc transcription involves myc family proteins. Oncogene Res. 1988;3:357–75. [PubMed] [Google Scholar]
- Conzen SD, Gottlob K, Kandel ES, Khanduri P, Wagner AJ, O’Leary M, et al. Induction of cell cycle progression and acceleration of apoptosis are two separable functions of c-Myc: transrepression correlates with acceleration of apoptosis. Mol Cell Biol. 2000;20:6008–18. doi: 10.1128/mcb.20.16.6008-6018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowling VH, D’Cruz CM, Chodosh LA, Cole MD. c-Myc transforms human mammary epithelial cells through repression of the Wnt inhibitors DKK1 and SFRP1. Mol Cell Biol. 2007;27:5135–46. doi: 10.1128/MCB.02282-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–68. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Drayton S, Peters G. Immortalisation and transformation revisited. Curr Opin Genet Dev. 2002;12:98–104. doi: 10.1016/s0959-437x(01)00271-4. [DOI] [PubMed] [Google Scholar]
- Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem. 2003;278:51606–12. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene. 2010;29:1249–59. doi: 10.1038/onc.2009.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–11. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst A, Hemann MT, Tworkowski KA, Salghetti SE, Lowe SW, Tansey WP. A conserved element in Myc that negatively regulates its proapoptotic activity. EMBO Rep. 2005;6:177–83. doi: 10.1038/sj.embor.7400333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SS, Shago M, Kaustov L, Boutros PC, Clendening JW, Sheng Y, et al. CUL7 is a novel antiapoptotic oncogene. Cancer Res. 2007;67:9616–22. doi: 10.1158/0008-5472.CAN-07-0644. [DOI] [PubMed] [Google Scholar]
- Kuttler F, Ame P, Clark H, Haughey C, Mougin C, Cahn JY, et al. c-myc box II mutations in Burkitt’s lymphoma-derived alleles reduce cell-transformation activity and lower response to broad apoptotic stimuli. Oncogene. 2001;20:6084–94. doi: 10.1038/sj.onc.1204827. [DOI] [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- Landay M, Oster SK, Khosravi F, Grove LE, Yin X, Sedivy J, et al. Promotion of growth and apoptosis in c-myc nullizygous fibroblasts by other members of the myc oncoprotein family. Cell Death Differ. 2000;7:697–705. doi: 10.1038/sj.cdd.4400701. [DOI] [PubMed] [Google Scholar]
- Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in E-mu-myc transgenic mice. Cell. 1986;47:11–8. doi: 10.1016/0092-8674(86)90361-2. [DOI] [PubMed] [Google Scholar]
- Lutz W, Stohr M, Schurmann J, Wenzel A, Lohr A, Schwab M. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene. 1996;13:803–12. [PubMed] [Google Scholar]
- Malynn BA, de Alboran IM, O’Hagan RC, Bronson R, Davidson L, DePinho RA, et al. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000;14:1390–9. [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998;94:363–74. doi: 10.1016/s0092-8674(00)81479-8. [DOI] [PubMed] [Google Scholar]
- Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976–90. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- Oster SK, Mao DY, Kennedy J, Penn LZ. Functional analysis of the N-terminal domain of the Myc oncoprotein. Oncogene. 2003;22:1998–2010. doi: 10.1038/sj.onc.1206228. [DOI] [PubMed] [Google Scholar]
- Penn LJ, Brooks MW, Laufer EM, Land H. Negative autoregulation of c-myc transcription. Embo J. 1990;9:1113–21. doi: 10.1002/j.1460-2075.1990.tb08217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prowse KR, Greider CW. Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc Natl Acad Sci U S A. 1995;92:4818–22. doi: 10.1073/pnas.92.11.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M. MYCN in neuronal tumours. Cancer Lett. 2004;204:179–87. doi: 10.1016/S0304-3835(03)00454-3. [DOI] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–86. [PubMed] [Google Scholar]
- Spotts GD, Patel SV, Xiao Q, Hann SR. Identification of downstream-initiated c-Myc proteins which are dominant-negative inhibitors of transactivation by full-length c-Myc proteins. Mol Cell Biol. 1997;17:1459–68. doi: 10.1128/mcb.17.3.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart TA, Pattengale PK, Leder P. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell. 1984;38:627–37. doi: 10.1016/0092-8674(84)90257-5. [DOI] [PubMed] [Google Scholar]
- Stone J, de Lange T, Ramsay G, Jakobovits E, Bishop JM, Varmus H, et al. Definition of regions in human c-myc that are involved in transformation and nuclear localization. Mol Cell Biol. 1987;7:1697–709. doi: 10.1128/mcb.7.5.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoorts J, Luscher-Firzlaff J, Luscher B. The ins and outs of MYC regulation by posttranslational mechanisms. J Biol Chem. 2006;281:34725–9. doi: 10.1074/jbc.R600017200. [DOI] [PubMed] [Google Scholar]
- Wei W, Jobling WA, Chen W, Hahn WC, Sedivy JM. Abolition of cyclin-dependent kinase inhibitor p16Ink4a and p21Cip1/Waf1 functions permits Ras-induced anchorage-independent growth in telomerase-immortalized human fibroblasts. Mol Cell Biol. 2003;23:2859–70. doi: 10.1128/MCB.23.8.2859-2870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, McMahon SB, Cole MD. An ATPase/helicase complex is an essential cofactor for oncogenic transformation by c-Myc. Mol Cell. 2000;5:321–30. doi: 10.1016/s1097-2765(00)80427-x. [DOI] [PubMed] [Google Scholar]
- Wu J, Wong WW, Khosravi F, Minden MD, Penn LZ. Blocking the Raf/MEK/ERK pathway sensitizes acute myelogenous leukemia cells to lovastatin-induced apoptosis. Cancer Res. 2004;64:6461–8. doi: 10.1158/0008-5472.CAN-04-0866. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.