

Abstract
Background
Velo-cardio-facial syndrome/DiGeorge syndrome (VCFS/DGS) is caused by a 1.5-3 Mb microdeletion of chromosome 22q11.2, frequently referred to as 22q11.2 deletion syndrome (22q11DS). This region includes TBX1, a T-box transcription factor gene that contributes to the etiology of 22q11DS. The requirement for TBX1 in mammalian development is dosage-sensitive, such that loss-of-function (LOF) and gain-of-function (GOF) of TBX1 in both mice and humans results in disease relevant congenital malformations.
Results
To further gain insight into the role of Tbx1 in development, we have targeted the Rosa26 locus to generate a new GOF mouse model in which a Tbx1-GFP fusion protein is expressed conditionally using the Cre/LoxP system. Tbx1-GFP expression is driven by the endogenous Rosa26 promoter resulting in ectopic and persistent expression. Tbx1 is pivotal for proper ear and heart development; ectopic activation of Tbx1-GFP in the otic vesicle by Pax2-Cre and Foxg1-Cre represses neurogenesis and produces morphological defects of the inner ear. Overexpression of a single copy of Tbx1-GFP using Tbx1Cre/+ was viable, while overexpression of both copies resulted in neonatal lethality with cardiac outflow tract defects. We have partially rescued inner ear and heart anomalies in Tbx1Cre/- null embryos by expression of Tbx1-GFP.
Conclusions
We have generated a new mouse model to conditionally overexpress a GFP-tagged Tbx1 protein in vivo. This provides a useful tool to investigate in vivo direct downstream targets and protein binding partners of Tbx1.
Keywords: VCFS/DGS, Tbx1, Rosa26, Mouse model, Gain-of-function
Background
Velo-cardio-facial syndrome/DiGeorge syndrome (VCFS/DGS), also known as 22q11.2 deletion syndrome (22q11DS), is the most common microdeletion syndrome occurring de novo in approximately 1/4,000 live births [1]. TBX1, encoding a T-box transcription factor, is located within the 1.5 Mb critically deleted region and haploinsufficiency of this gene is responsible for the congenital defects associated with 22q11DS [2]. Tbx1 null mutant mice have malformations of the heart, thymus/parathyroid, craniofacial region, and ear that are similar but more severe, to what is typically found in 22q11DS patients [2-4]. Previously generated BAC316.23 transgenic mice expressing 8–10 copies of human TBX1 also have developmental defects of the same tissues affected in Tbx1−/− null mice, resembling clinical features of a recently identified 22q11 duplication syndrome [5-9]. Ablation studies using Tbx1flox conditional null mutants have proven to be indispensible for understanding the tissue and cell-type specific roles of the Tbx1 gene [10,11]. COET, a transgenic mouse line with conditional overexpression of Tbx1, has previously been reported [12]. However, the COET mouse line does not contain a protein affinity tag on the Tbx1 protein. We have generated a conditional allele targeted at the Rosa26 locus for the purpose of expressing additional copies of Tbx1 fused to a GFP reporter in a tissue-specific manner. We chose a GFP tag in part to detect live GFP as a readout for activation of Tbx1-GFP in vivo and to use it for future biochemical or chromatin immunoprecipitation experiments. Tbx1 mutant rescue experiments demonstrate that Tbx1-GFP fusion protein functions in vivo in a manner similar to that of endogenous Tbx1. The Tbx1-GFP mouse allele provides a new gain-of-function model that is targeted to the constitutively active Rosa26 locus; targeting Tbx1-GFP to the endogenous Tbx1 locus would simultaneously disrupt the normal function of the targeted allele (as is the case for Tbx1Cre/+ mice, Huynh et al., 2007). As such, our model works to activate 1–2 ectopic copies of Tbx1 while simultaneously tracing cells with GFP, and should therefore be of high value in future experimental studies.
Results and discussion
To generate the Tbx1-GFP allele, we first generated a bacterial plasmid that expresses a Tbx1-GFP fusion protein of approximately 75 kilodaltons (kD) (Figure 1A), corresponding to the combined mass of Tbx1 (~50 kD) and GFP (~27 kD) (Figure 1B). The Tbx1-GFP fragment was inserted downstream of a triple polyadenylation (tpA) transcriptional stop sequence that is flanked by LoxP sites in the pBigT vector [13]. The loxP-tpA-loxP-Tbx1-GFP element was cloned into the pROSA26PA vector for targeting to the Rosa26 locus [13,14], and electroporated into WW6 (129/SvJ) mouse embryonic stem cells (ESC). We identified 5/56 correctly targeted ESC clones by Southern blot analysis (Figure 1C). Clone 45 was chosen for blastocyst injection in C57BL6 females to generate chimeras for germline transmission.
Figure 1.
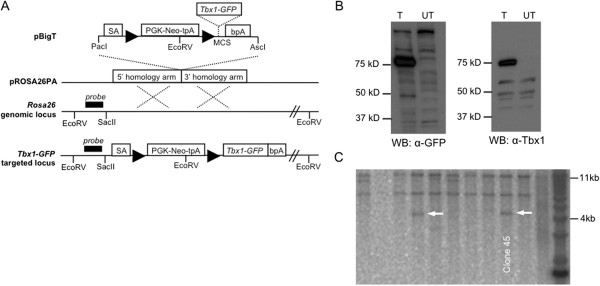
Generation of the Tbx1-GFP mouse. (A) Tbx1-GFP was inserted into the multiple cloning site (MCS) of pBigT downstream of a triple polyadenylation sequence (tpA) that is flanked by LoxP sites (triangles). Additional subcloning into the pROSA26PA plasmid resulted in the final targeting construct. 5’ and 3’ homology arms mediate recombination with the endogenous Rosa26 locus. (B) Whole cell lysates from COS7 cells transfected (T) with Tbx1-GFP plasmid or untransfected (UT). The fusion protein is ≈75 kD and is detected by anti-GFP and anti-Tbx1 on Western blot. (C) Southern blot was used to screen ESC clones. Genomic DNA was linearized with EcoRV and hybridized with a radiolabeled probe that binds upstream of the 5’ homology arm. Two of five positive clones are shown. The correctly targeted allele is 4,071 bp and the wildtype allele is 11,517 kb.
To test the Tbx1-GFP allele, we crossed Tbx1-GFPflox/flox mice to Foxg1-Cre and Pax2-Cre mice [15,16] and analyzed their respective phenotypes. Both Cre drivers have previously been used to inactivate Tbx1 by tissue-specific recombination of loxP sites [11,17]. Foxg1-Cre expression overlaps with endogenous Tbx1 expression in tissues such as the pharyngeal apparatus and otic vesicle (OV) [11,15,17]. In the study presented here, we show that Foxg1-Cre can ectopically activate Tbx1-GFP in the olfactory placode, forebrain, and optic vesicle resulting in morphological defects of these tissues during development (Figure 2A). In addition, by E15.5, Foxg1-Cre;Tbx1-GFPflox/+ mutants display thymic aplasia likely due to overexpression of Tbx1-GFP in the 3rd pharyngeal pouch, and abnormal formation of the nasal prominence. Pax2-Cre;Tbx1-GFPflox/+ mutants are perinatal lethal (Figure 2A).
Figure 2.
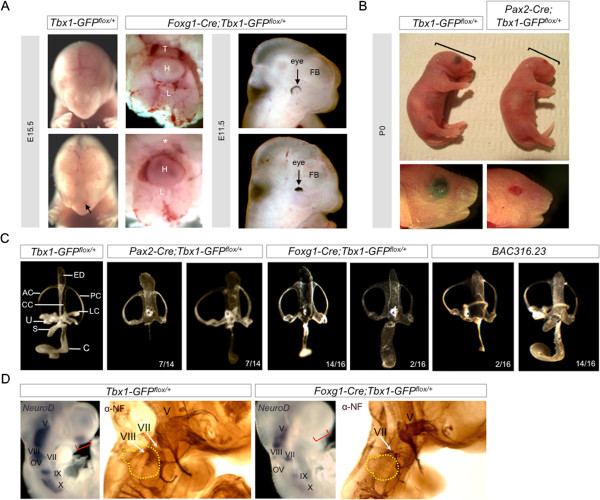
Activation of Tbx1-GFP by Foxg1-Cre and Pax2-Cre. (A)Foxg1-Cre;Tbx1-GFPflox/+ mutants have thymic aplasia and nasal malformations at E15.5, and eye and pharyngeal defects at E11.5. (B)Pax2-Cre;Tbx1-GFPflox/+ mutants die shortly after birth. They have microcephaly and ocular defects. Thymus (T), heart (H), lungs (L), forebrain (FB). (C) Paintfilling of inner ears in Pax2-Cre;Tbx1-GFPflox/+ and Foxg1-Cre;Tbx1-GFPflox/+ mutants. The endolymphatic duct (ED) and common crus (CC) are enlarged. The lateral semicircular canal (LC), utricle (U), and saccule (S) are missing. The cochlea (C) is shortened to varying degrees. BAC316.23 mice show similarities in inner ear morphological defects. Anterior canal (AC), posterior canal (PC). (D) Wholemount RNA in situ hybridization at E9.5 for NeuroD, expressed in neuroblasts of the cranial ganglia. Wholemount immunohistochemistry stains cranial sensory ganglia with anti-Neurofilament (NF) at E10.5. Red brackets indicate olfactory placode. Otic vesicle is circled in yellow.
Since inner ear and cardiac defects are two of the most prominent features in the Tbx1 null mutant, we decided to examine those structures in more detail in the Pax2-Cre;Tbx1-GFPflox/+ mutants and Foxg1-Cre;Tbx1-GFPflox/+ mutants, respectively. Foxg1-Cre and Pax2-Cre are both strongly expressed throughout the OV, from which the inner ear forms. Both loss or gain of Tbx1 function in the OV disrupts inner ear morphogenesis [5,18,19]. Paintfilling of the inner ear in Foxg1-Cre;Tbx1-GFPflox/+ and Pax2-Cre;Tbx1-GFPflox/+ mutants at E14.5 shows an enlarged endolymphatic duct (ED) and common crus (CC) that joins the anterior and posterior semicircular canals (SCC) (Figure 2B). The utricle, saccule, and lateral SCC are missing. The cochlea is hypoplastic with varying degree, but enlarged in two instances (Figure 2B). This phenotype is very similar to that of BAC316.23 transgenic mice [5]; with the exception that BAC316.23 mice still possess the lateral SCC, and in most cases the saccule and utricle are present, albeit malformed (Figure 2B).
Tbx1 is also known to repress neurogenesis of the VIIIth cranial ganglion that innervates the inner ear [19]. Here we show that in Foxg1-Cre;Tbx1-GFPflox/+ mutants, expression of Neurogenic differentiation factor 1 (NeuroD) is nearly abolished in the VIIIth cranial ganglion, and highly attenuated in cranial ganglia V, VII, IX, and X (Figure 2C). Immunohistochemistry with anti-Neurofilament confirmed loss of the VIIIth cranial ganglion and abnormal size and misguided projections from other cranial ganglia in Foxg1-Cre;Tbx1-GFPflox/+ mutants (Figure 2C). In addition, we found that NeuroD expression is completely missing from the olfactory placode of Foxg1-Cre;Tbx1-GFPflox/+ mutants; a region where Foxg1-Cre is active and there is no Tbx1 expression in a wild type context suggesting that gain of function of Tbx1 in the olfactory placode leads to ectopic repression of NeuroD. These findings demonstrate that Tbx1 could act directly or indirectly on neurogenic factors that are common to both otic and olfactory placodes.
Examination of SCC defects in the Pax2-Cre;Tbx1-GFPflox/+ mutants using paintfilling revealed a delay in SCC formation at earlier developmental stages (Figure 3A). In the wild type, clearing of the fusion plates is visible by E12.25 and complete by E12.5 [20]. In Pax2-Cre;Tbx1-GFPflox/+ mutants, the fusion plates have not yet joined at E12.25 although they appear to partially recover by E14.5 (Figure 3A). Histological sections at E12.25 confirm that the fusion plates in Pax2-Cre;Tbx1-GFPflox/+ mutants remain attached to the periotic mesenchyme compared to control littermates (Figure 3A). We examined expression of laminin and netrin-1, a secreted protein that is related to laminins and promotes basal lamina breakdown [21,22]. We observed decreased laminin protein levels along the otic epithelium, especially along the site of lateral SCC formation along with a corresponding decrease in netrin-1 mRNA expression along the fusion plates in Pax2-Cre;Tbx1-GFPflox/+ mutants (Figure 3B). It is possible that Tbx1 may act to repress netrin-1, thereby modulating cell adhesion properties. Complementation of these results occurred in Tbx1−/− null mice in which expression of netrin-1 is expanded in the mesenchyme surrounding the OV (Figure 3C). We did not observe expanded netrin-1 expression in the OV of Tbx1−/− null mice, possibly due to the severity of the OV phenotype and subsequent absence of vestibular structures.
Figure 3.
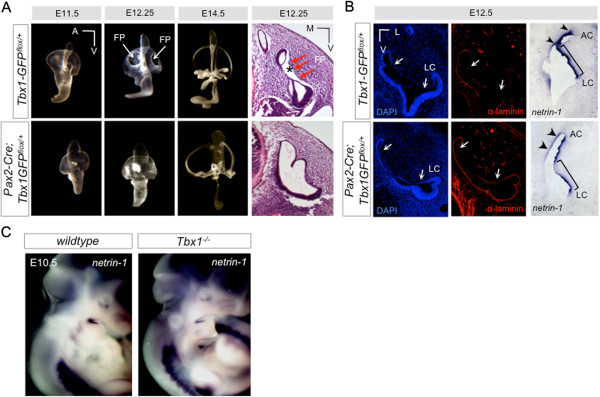
Altered cell adhesion properties in Tbx1 mutants. (A) Paintfilling of inner ears from Pax2-Cre;Tbx1-GFPflox/+ mutants at progressively earlier stages of development reveals a delay in SCC formation. At E12.25, fusion plates (arrows, FP) are visible in the controls, but do not appear to have formed in the mutants. Histological analysis confirms defects in the fusion plates (FP, red arrows). In controls, the vestibular epithelia can be seen to have separated from the mesenchyme (asterisks). In Tbx1-GFP mutants, there is no separation of the epithelium from surrounding tissue. (B) Immunofluorescence with anti-laminin (red). Arrows indicate the span of the fusion plate epithelia. There is more intact laminin in the Tbx1-GFP mutants. RNA in situ hybridization to netrin-1 on sections. Netrin-1 expression is reduced in the fusion plates of Tbx1-GFP mutants. Anterior canal (AC, lateral canal (LC). (C) Expression of netrin-1 mRNA on wildtype and Tbx1−/− null mice. Netrin-1 expression is increased in the mesenchyme surrounding the inner ear in the absence of endogenous Tbx1.
To test whether Tbx1-GFP could rescue inner ear defects that occur in Tbx1Cre/- null embryos, we crossed Tbx1-GFPflox/flox mice to Tbx1Cre/+ mice [23]. Tbx1Cre/- mice have been shown to recapitulate the Tbx1−/− null phenotype [23]. When Cre is expressed, cells from the Tbx1Cre/+ expressing domain are positive for GFP in a manner that represents the Tbx1 lineage in the OV in embryos (Figure 4A). Expression of Tbx1-GFP under the Rosa26 promoter alone is not sufficient for direct visualization of natural GFP, therefore Tbx1-GFP protein was detected using an antibody to GFP. Since expression of Tbx1-GFP is persistently driven by the endogenous Rosa26 promoter, we expect the domain of activated Tbx1-GFP to be more extensive than native Tbx1 expression (Figure 4A). Immunofluorescence with anti-GFP antiserum demonstrated that there is no Tbx1-GFP protein in the absence of Cre expression in the OV (Figure 4A). Tbx1Cre/+;Tbx1-GFPflox/+ mutants did not exhibit gross morphological defects of the inner ear based on paintfilling (Figure 4B), perhaps because one allele of Tbx1 is removed by knock-in of Cre. Paintfilling confirmed that the inner ears of Tbx1−/− null mice lack all discernible vestibular and auditory structures with the exception of what appears to be an enlarged ED (Figure 4C). When a single allele of Tbx1-GFP is activated in a Tbx1Cre/- background, there is rescue of the anterior and posterior SCCs in all embryos, and partial rescue of the saccule and cochlea (n=6) (Figure 4C). Complete rescue of the null phenotype is not expected with constitutive activation of Tbx1-GFP because temporal regulation of Tbx1 is required for normal inner ear development.
Figure 4.
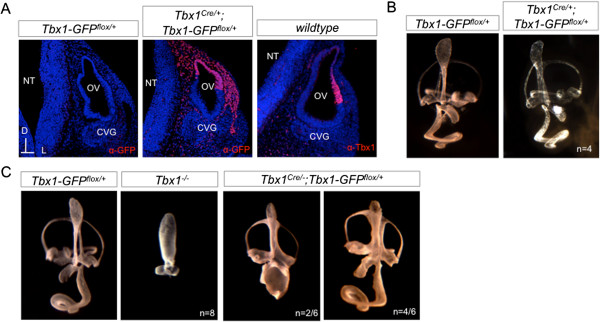
Activation of Tbx1-GFP in the inner ear using Tbx1Cre/+. (A) Immunofluorescence with GFP (red) and Tbx1 (red) antibodies on E10.5 transverse sections through the otic vesicle (OV). The VIIIth cranial ganglion, also known as the cochleovestibular ganglion (CVG) is adjacent to the OV. Neural tube (NT). (B) Activation of Tbx1-GFP by Tbx1Cre/+ does not produce major morphological defects of the inner ear, as visualized by paintfilling at E14.5. (C) Paintfilling shows failure of inner ear morphogenesis in Tbx1Cre/- null mice at E14.5 compared to the Tbx1-GFPflox/+ control. Activation of a single allele of Tbx1-GFP on a Tbx1Cre/- null background is capable of partially rescuing morphogenesis.
The Tbx1Cre/+ allele was also used to activate Tbx1-GFP in other endogenous sites of Tbx1 expression such as the pharyngeal mesoderm including the secondary heart field (Figure 5A). Histological analysis of Tbx1Cre/+;Tbx1GFPflox/+ embryos at E14.5 showed normal heart development (Figure 5B). Genotyping of mice at P10 confirmed presence of live Tbx1Cre/+;Tbx1GFPflox/+ mice in normal Mendelian ratios (Figure 5B). In contrast, mice that have both Tbx1-GFP alleles activated by Tbx1Cre/+ are not viable (Figure 5B), prompting us to assess the heart phenotype in Tbx1Cre/+;Tbx1GFPflox/flox embryos at E14.5. Tbx1Cre/+;Tbx1GFPflox/flox embryos exhibited defects such as persistent truncus arteriosus (PTA, 2/5 embryos) and double outlet right ventricle (DORV, 3/5 embryos), all with ventricular septal defects (VSD, 5/5 embryos) (Figure 5C). Overexpression of TBX1 has been previously associated with VSD in ~10% of BAC316.23 transgenic mice [2]. Heart malformations can account for the perinatal lethality of Tbx1Cre/+;Tbx1GFPflox/flox mice. We wanted to compare the protein expression levels of the Tbx1-GFP fusion protein activated in the Tbx1 domain compared to endogenous Tbx1 protein. To do this, we performed western blot on tissue samples (Figure 5D) and observed that the level of Tbx1-GFP protein expressed from a single allele was higher than that observed for endogenous wildtype Tbx1. However, this may reflect the larger domain of expression of Tbx1-GFP within the cell lineage that is labeled by Tbx1Cre/+.
Figure 5.
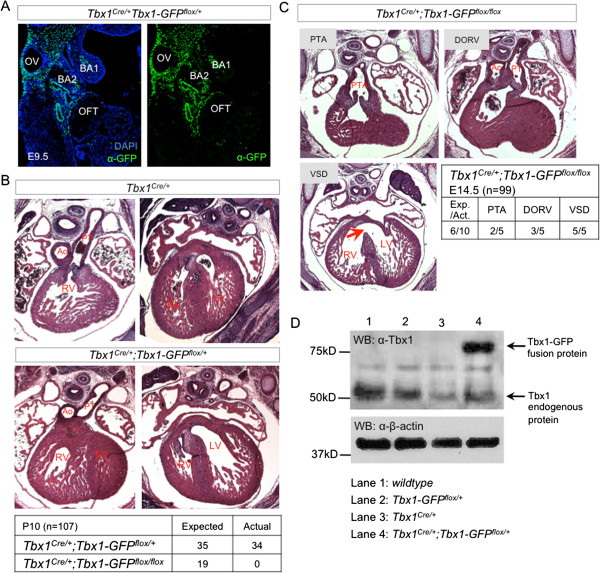
Dosage effects of Tbx1-GFP activation on heart development. (A) Immunofluorescence with a GFP antibody shows activation of Tbx1-GFP by Tbx1Cre/+ at E9.5. GFP is detected in the posterior otic vesicle (OV), 1st and 2nd branchial arches (BA1, BA2), and mesoderm including the secondary heart field proximal to the outflow tract (OFT). (B) Histological transverse sections at E14.5 shows that Tbx1Cre/+;Tbx1-GFPflox/+ mutants do not have outflow tract or ventricular defects. They survive in normal mendelian ratios, unlike Tbx1Cre/+;Tbx1-GFPflox/flox mutants which are not viable. (C) Activation of both alleles of Tbx1-GFP by Tbx1Cre/+ causes outflow tract septation (persistant truncus arteriosis, PTA) and alignment (double outlet right ventricle , DORV) defects with ventricular septal defect (VSD). (D) Western blot of proteins isolated from E9.5 pharyngeal regions to compare protein expression levels of Tbx1-GFP and endogenous Tbx1. For each genotype, we pooled 4 embryos (23–24 somite stage) and loaded equal amounts of protein per lane. The same membrane was probed with antibodies to Tbx1 and β-actin. The expected size of endogenous Tbx1 is ≈50 kD while the Tbx1-GFP fusion protein is ≈75kD. β-actin is detected at ≈40 kD.
Next, we wanted to test whether Tbx1-GFP could also rescue the cardiac defects that occur in Tbx1Cre/- embryos (Figure 6A), all of which have PTA and VSD (n=3) [12]. Analysis of Tbx1Cre/-;Tbx1GFPflox/+ embryos at E14.5 showed a complete rescue of outflow tract septation defects (n=8), although 50% still had a misalignment of the outflow tract resulting in a DORV (Figure 6B). VSD was observed in all Tbx1Cre/-;Tbx1GFPflox/+ embryos. It is not clear if these defects are due to failure to rescue the Tbx1 null phenotype, or if they are due to overexpression of Tbx1-GFP protein (Figure 5D), even though protein expression levels as detected by western blot do not provide information about the functional activity levels of Tbx1-GFP protein. It is also possible that failure of complete rescue is due to the fact that Tbx1-GFP is expressed constitutively under the control of the Rosa26 promoter and is therefore not subject to temporal regulation. Activation of both Tbx1-GFP alleles in Tbx1Cre/-; Tbx1GFPflox/flox embryos resulted in more severe heart phenotypes including PTA in 5/6 embryos in addition to VSD (Figure 6C) due to further Tbx1 gene dosage imbalance.
Figure 6.
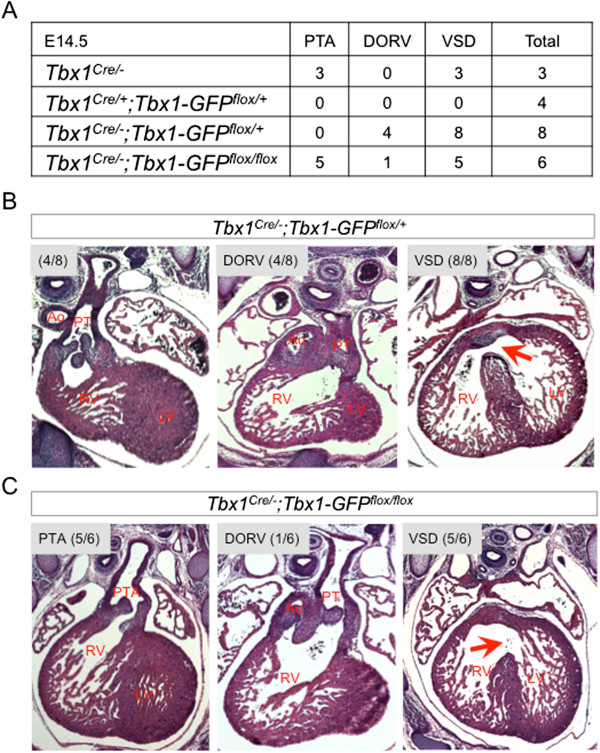
Tbx1-GFP can partially rescue endogenous Tbx1Cre/-loss-of-function. Histological transverse sections through the heart at E14.5. (A)Tbx1Cre/- mice are functionally null and have PTA and VSD. Tbx1Cre/+;Tbx1-GFPflox/+ mutants are viable with normal heart histology. (B)Tbx1Cre/-;Tbx1-GFPflox/+ mutants exhibit complete rescue of outflow tract septation although half of the embryos have double outlet right ventricle (DORV). (C)Tbx1Cre/-;Tbx1-GFPflox/flox mutants all have ventricular septal defect (VSD), primarily with persistant truncus arteriosus (PTA) and one example of DORV.
Conclusions
In summary, we have generated a new mouse model that conditionally expresses a Tbx1-GFP fusion protein. We show that Tbx1-GFP does not act in a dominant-negative manner and can functionally substitute for endogenous Tbx1 during heart and inner ear development by partial rescue of heart and inner ear morphological defects in a Tbx1Cre/- null background. We cannot completely rule out deleterious effects of the GFP tag protein on the ability of Tbx1 to act as a transcriptional regulator either via direct binding of DNA or via protein: protein interactions as we have not directly tested this. However, ectopic activation of Tbx1-GFP by Foxg1-Cre and Pax2-Cre causes morphological defects of the inner ear that resemble those occurring in BAC316.23 transgenic mice. Taken together with the partial rescue of heart and inner ear defects on a Tbx1−/− null background, it seems likely that the Tbx1-GFP fusion protein retains functional characteristics of the endogenous Tbx1 protein. As such, the Tbx1-GFP mouse line provides a genetic tool that is amenable to probing direct downstream targets of Tbx1 and identifying protein-protein interacting partners of Tbx1 in vivo.
Methods
Targeting vector
A pTbx1-EGFP-N1 plasmid was generated by cloning Tbx1 cDNA from a Tbx1-TOPO plasmid into pEGFP-N1 (Clontech 6085–1). The following oligonucleotide adaptor sequences were used for an in-frame ligation of the C-terminus of Tbx1 to the N-terminus of EGFP, simultaneously changing the Tbx1 stop codon from TAG to TTG and introducing NotI and BamHI restriction enzyme sites: 5’-GGCCGCGCCGCCCGGTGCCTACGACTACTGCCCCAGATTG-3’; 5’GATCCAATCTGGGGCAGTAGTCGTAGGCACCGGGCGGCGC-3’. Adaptors were annealed by combining 10 nmol of each oligonucleotide in 200 μL of water and heating to 90°C followed by cooling at room temperature to 40°C. For 20 μL ligation reactions (Roche 1243292, Roche 10799009001), 5 μL of 1:500 diluted annealed adaptors were used. The sequence encoding the Tbx1-GFP fusion protein was inserted into the multiple cloning site of pBigT [13]. The loxP-tpA-loxP-Tbx1-GFP fragment was excised by restriction enzyme digestion with PacI and AscI then cloned into the pROSA26PA plasmid [13] to generate the final targeting vector (Figure 1). The final targeting vector was linearized with BbvCI and purified by phenol chloroform extraction, then electroporated into WW6 ESC and selection performed with G418. Positively selected ESC clones were plated in duplicate for DNA isolation using the DNeasy Blood and Tissue Kit (Qiagen 69506). DNA was digested overnight at 37°C with EcoRV and Southern blot performed with a previously described 5’ probe [13].
Western blot
The Tbx1-GFP fusion protein was expressed in COS7 cells by transient transfection of the pTbx1-EGFP-N1 plasmid with Polyfect (Qiagen 301105). Protein from whole cell lysate and embryonic tissue was collected in RIPA Lysis Buffer (50 mM Tris HCl pH 7.5, 200 mM NaCl, 1% Triton X-100, 1 mM EDTA, 0.25% deoxycholic acid, protease inhibitor). For tissue samples, the pharyngeal region was isolated from E9.5 embryos and the head, first pharyngeal arch, heart, and caudal part of the embryos (below the fourth pharyngeal pouch) were removed. Tissue was homogenized and lysed in RIPA buffer for 2 hours at 4°C. Samples were run on 7.5 or 10% polyacrylimide gels. Tbx1-GFP fusion protein (≈75 kD) was detected using polyclonal anti-Tbx1 (Zymed, 1:500), anti-GFP (Invitrogen A6455, 1:600), mouse anti-βactin (Abcam ab6276, 1:5,000), donkey anti-Rabbit IgG-HRP (Amersham NA934V, 1:20,000) and sheep anti-Mouse IgG-HRP (Amersham NA931V, 1:5,000) antibodies. Detection was performed using the ECL Western Blotting Detection Kit (Amersham RPN2106) and exposed on Kodak film.
Mouse models
Tbx1-GFP mice were genotyped with the FastStart High Fidelity PCR System (Roche 03553361001) and the following primers: TB3F (5’-CTGCACCACCATCCCTACAA-3’) and GFPR (5’- TGAACTTCAGGGTCAGCTTG-3’) for a 421 bp product from the targeted allele, and RO1F (5’-GCAATACCTTTCTGGGAGTT-3’) and GFP-wtR (5’- CAATGCTCTGTCTAGGGGTT-3’) for a 605 bp product from the wildtype allele. Foxg1-Cre, Pax2-Cre, and Tbx1Cre/+ mice were kindly provided by Drs. Jean Hebert, Andrew K. Groves, and Antonio Baldini, respectively.. Embryos were dissected according to date of vaginal plug (E0.5). Embryonic stages <E11.5 were confirmed by counting pairs of somites. Animals were maintained in a 12 hr dark/12 hr light cycle in compliance with the Albert Einstein College of Medicine of Yeshiva University Institutional Animal Care and Use Committee (IACUC). Tbx1-GFP mice will become available to the research community upon acceptance of the manuscript.
RNA in situ hybridization
Embryos were fixed in 4% paraformaldehyde (PFA) at 4°C overnight then dehydrated in a methanol series. RNA in situ hybridization (ISH) was performed following rehydration to 0.1% PBS/0.1% Tween-20 (PBT) [24]. Anti-sense digoxigenin-labeled RNA probe for NeuroD[25] was generated from a plasmid by standard protocol. The RNA probe template for netrin-1 was generated from amplified E9.5 mouse cDNA using the following primers: Netrin1-Fwd (5’-GGGGAATTAACCCTCACTAAAGGGTGATCCTTGCTCGGATGAGA-3’), Netrin1-Rev (5’-GGGGTAATACGACTCACTATAGGGTTCTTCTCCCGTTGCTGGAA-3’).
Primer sequences introduced T7 RNA polymerase binding sites for generating antisense probes and T3 RNA polymerase binding sites for generating sense probes. For wholemount ISH, we rehydrated embryos to PBT and digested them with Proteinase K. This was followed by washes in Glycine solution and PBT. Embryos were then fixed in 4% PFA/0.2% gluteraldehyde for 15 min on ice followed by PBT washes. Embryos were incubated with RNA probes in hybridization buffer overnight at 70°C then washed in a series of SSC, maleic acid buffer, and PBT washes. Then embryos were incubated overnight at 4°C in antibody buffer with 1:10,000 dilution of anti-Digoxigenin-AP antibody (Roche 11093274910) followed by washes in 0.1% BSA/PBT and AP1 buffer. Staining was performed using BM Purple (Roche 11442074001) followed by fixation in 4% PFA. RNA in situ hybridization to netrin-1 was performed on tissue cryosections according to the David Anderson laboratory protocol. Briefly, tissue sections were fixed in 4% PFA for 20 min at RT and digested with Proteinase K followed by post-fixation in 4% PFA for 15 min. Acetylation was performed in TEA buffer with acetic anhydride followed by washes in PBS and air-drying. Hybridization with RNA probes was done overnight at 68°C in a hyb chamber humidified with 50% formamide/4X SSC. Tissue sections were washed with SSC and PBT then incubated overnight at 4°C in blocking buffer with 1:2,000 dilution of anti-Digoxigenin-AP antibody followed by washes in PBT and AP1 buffer and staining with BM Purple.
Immunofluorescence and whole mount immunohistochemistry
Embryos were fixed overnight in 4% paraformaldehyde, placed in 30% sucrose/PBS, embedded in O.C.T., then cryosectioned at 12 μm thickness. For immunofluorescence, tissue sections were washed in PBS, permeabilized for 5 min in 0.5% Triton X-100, then incubated in blocking solution (5% serum in PBS/0.1% Triton X-100 i.e. PBT) for 1 hour. Primary antibodies (rabbit anti-mouse pan-laminin 1:200, Chemicon AB2034; rabbit anti-Tbx1 1:500, goat anti-GFP 1:500, Abcam ab6673) were diluted in block and incubated on tissue for 1 hr at room temperature. Sections were washed in PBT then incubated with secondary antibody (Alexa Fluor 568 goat anti-rabbit IgG 1:500; Alexa Fluor 568 donkey anti-goat IgG 1:500) for 30 min. DAPI (1:500) was added to secondary antibody. Sections were washed in PBT mounted in Vectashield hard-set mounting medium (Vector Labs H-1400). For whole mount immunohistochemistry, embryos were fixed overnight in 4:1 methanol/DMSO at 4°C. Endogenous peroxidase activity was blocked in 4:1:1 methanol/DMSO/30% H2O2 for 6 hrs at room temperature. Embryos were then rehydrated in a methanol series and blocked in 2% milk/0.5% Triton X-100/PBS (PBSMT) for 1 hour. Primary antibody (anti-neurofilament 1:200, DSHB mAb2H3) was diluted in PBSMT and incubated overnight at 4°C. Embryos were washed five times for an hour each in PBSMT at 4°C and then incubated with [HRP]-sheep anti-mouse IgG (Amersham NA931V, 1:500) diluted in PBSMT overnight at 4°C. Embryos were washed again in PBSMT for 5 hrs at 4°C and then 30 min in 0.2% BSA (Sigma A4503)/0.5% Triton X-100/PBS. Staining was achieved with DAB substrate with chromagen (DAKO K3466) for 15 min at room temperature. Embryos were post-fixed in 4% paraformaldehyde overnight at 4°C, dehydrated in a methanol series, then cleared in 1:2 Benzyl alcohol: Benzyl Benzoate (BABB) for imaging and long-term storage.
Inner ear paintfilling and histology
Embryos were sliced below the forelimbs then fixed in 5% glacial acetic acid, 2% formaldehyde, and 75% ethanol overnight followed by an overnight dehydration in 100% ethanol. Methyl salicylate was then used for clearing of the tissue and long-term storage. Prior to paintfilling, embryos were bisected dorsally and the brain was removed to reveal the inner ear capsule. A micropipette was used to microinject 0.2% correction fluid diluted in methyl salicylate into the inner ear labyrinth. For histological analysis, embryos were fixed in 4% paraformaldehyde overnight at 4°C. They were then dehydrated to 70% ethanol and embedded in paraffin. Tissue sectioning was performed at 10-12 μm thickness. Tissues were cleared in xylene, stained with hematoxylin and eosin (H&E) and then mounted in Permount.
Competing interests
The authors declare no financial or non-financial competing interests in relation to this manuscript.
Authors’ contributions
LF performed experiments, analyzed data, and wrote the manuscript. SN generated the Tbx1-GFP fusion plasmid, designed the targeting vector, and commented on the manuscript. MP performed the Southern Blots to screen for targeted embryonic stem cells. AB provided the Tbx1Cre/+ mouse line used for rescue experiments. BEM formulated the project, supervised and funded the experiments, and revised the manuscript. All authors read and approved the manuscript.
Contributor Information
Laina Freyer, Email: laina.freyer@einstein.yu.edu.
Sonja Nowotschin, Email: nowotscs@mskcc.org.
Melinda K Pirity, Email: pirity.melinda@brc.mta.hu.
Antonio Baldini, Email: antonio.baldini@igb.cnr.it.
Bernice E Morrow, Email: bernice.morrow@einstein.yu.edu.
Acknowledgements
We thank Dr. Frank Constantini for providing the pBigT and pROSA26PA plasmids. We are grateful to the AECOM Transgenic and Gene Targeting Facility. We thank Raquel Castellanos for helpful advice. The work was supported by NIH R01 HL088698 and DC05186 (BEM).
References
- McDermid HE, Morrow BE. Genomic disorders on 22q11. Am J Hum Genet. 2002;70:1077–1088. doi: 10.1086/340363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/S0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet. 2002;11:915–922. doi: 10.1093/hmg/11.8.915. [DOI] [PubMed] [Google Scholar]
- Funke B, Epstein JA, Kochilas LK, Lu MM, Pandita RK, Liao J, Bauerndistel R, Schuler T, Schorle H, Brown MC, Adams J, Morrow BE. Mice overexpressing genes from the 22q11 region deleted in velo-cardio-facial syndrome/DiGeorge syndrome have middle and inner ear defects. Hum Mol Genet. 2001;10:2549–2556. doi: 10.1093/hmg/10.22.2549. [DOI] [PubMed] [Google Scholar]
- Liao J, Kochilas L, Nowotschin S, Arnold JS, Aggarwal VS, Epstein JA, Brown MC, Adams J, Morrow BE. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum Mol Genet. 2004;13:1577–1585. doi: 10.1093/hmg/ddh176. [DOI] [PubMed] [Google Scholar]
- Hassed SJ, Hopcus-Niccum D, Zhang L, Li S, Mulvihill JJ. A new genomic duplication syndrome complementary to the velocardiofacial (22q11 deletion) syndrome. Clin Genet. 2004;65:400–404. doi: 10.1111/j.0009-9163.2004.0212.x. [DOI] [PubMed] [Google Scholar]
- Brunet A, Gabau E, Perich RM, Valdesoiro L, Brun C, Caballin MR, Guitart M. Microdeletion and microduplication 22q11.2 screening in 295 patients with clinical features of DiGeorge/Velocardiofacial syndrome. Am J Med Genet A. 2006;140:2426–2432. doi: 10.1002/ajmg.a.31499. [DOI] [PubMed] [Google Scholar]
- Portnoi MF. Microduplication 22q11.2: a new chromosomal syndrome. Eur J Med Genet. 2009;52:88–93. doi: 10.1016/j.ejmg.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, Baldini A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. 2004;131:3217–3227. doi: 10.1242/dev.01174. [DOI] [PubMed] [Google Scholar]
- Arnold JS, Werling U, Braunstein EM, Liao J, Nowotschin S, Edelmann W, Hebert JM, Morrow BE. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development. 2006;133:977–987. doi: 10.1242/dev.02264. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Huynh T, Baldini A. Gain of function of Tbx1 affects pharyngeal and heart development in the mouse. Genesis. 2009;47:188–195. doi: 10.1002/dvg.20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2009;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- Ohyama T, Groves AK. Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis. 2004;38:195–199. doi: 10.1002/gene.20017. [DOI] [PubMed] [Google Scholar]
- Arnold JS, Braunstein EM, Ohyama T, Groves AK, Adams JC, Brown MC, Morrow BE. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Hum Mol Genet. 2006;15:1629–1639. doi: 10.1093/hmg/ddl084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitelli F, Viola A, Morishima M, Pramparo T, Baldini A, Lindsay E. TBX1 is required for inner ear morphogenesis. Hum Mol Genet. 2003;12:2041–2048. doi: 10.1093/hmg/ddg216. [DOI] [PubMed] [Google Scholar]
- Raft S, Nowotschin S, Liao J, Morrow BE. Suppression of neural fate and control of inner ear morphogenesis by Tbx1. Development. 2004;131:1801–1812. doi: 10.1242/dev.01067. [DOI] [PubMed] [Google Scholar]
- Morsli H, Choo D, Ryan A, Johnson R, Wu DK. Development of the mouse inner ear and origin of its sensory organs. J Neurosci. 1998;18:3327–3335. doi: 10.1523/JNEUROSCI.18-09-03327.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen M, Meyer BI, Bober E, Gruss P. Netrin 1 is required for semicircular canal formation in the mouse inner ear. Development. 2000;127:13–22. doi: 10.1242/dev.127.1.13. [DOI] [PubMed] [Google Scholar]
- Abraira VE, Del Rio T, Tucker AF, Slonimsky J, Keirnes HL, Goodrich LV. Cross repressive interactions between Lrig3 and netrin 1 shape the architecture of the inner ear. Development. 2008;135:4091–4099. doi: 10.1242/dev.029330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh T, Chen L, Terrell P, Baldini A. A fate map of Tbx1 expressing cells reveals heterogeneity in the second cardiac field. Genesis. 2007;45:470–475. doi: 10.1002/dvg.20317. [DOI] [PubMed] [Google Scholar]
- Franco D, de Boer PA, de Gier-de VC, Lamers WH, Moorman AF. Methods on in situ hybridization, immunohistochemistry and beta-galactosidase reporter gene detection. Eur J Morphol. 2001;39:3–25. doi: 10.1076/ejom.39.1.3.7982. [DOI] [PubMed] [Google Scholar]
- Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]