

Abstract
The rabies virus (RABV) is highly neurotropic and it uses evasive strategies to successfully evade the host immune system. Because rabies is often fatal, understanding the basic processes of the virus-host interactions, particularly in the initial events of infection, is critical for the design of new therapeutic approaches to target RABV. Here, we examined the possible role of dendritic cells (DCs) in the transmission of RABV to neural cells at peripheral site of exposure. Viral replication only occurred at a low level in the DC cell line, JAWS II, after its infection with either pathogenic RABV (CVS strain) or low-pathogenic RABV (ERA strain), and no progeny viruses were produced in the culture supernatants. However, both viral genomic RNAs were retained in the long term after infection and maintained their infectivity. The biggest difference between CVS and ERA was in their ability to induce type I interferons. Although the ERA-infected JAWS II cells exhibited cytopathic effect and were apparently killed by normal spleen cells in vitro, the CVS-infected JAWS II cells showed milder cytopathic effect and less lysis when cocultured with spleen cells. Strongly increased expression of major histocompatibility complex classes I, costimulatory molecules (CD80 and CD86), type I interferons and Toll- like receptor 3, and was observed only in the ERA-inoculated JAWS II cells and not in those inoculated with CVS. During the silencing of the cellular immune response in the DCs, the pathogenic CVS strain cryptically maintained an infectious viral genome and was capable of transmitting infectious RABV to permissive neural cells. These findings demonstrate that DCs may play a role in the passive carriage of RABV during natural rabies infections.
Electronic supplementary material
The online version of this article (doi:10.1186/2193-1801-2-419) contains supplementary material, which is available to authorized users.
Keywords: Rabies virus, Dendritic cells, Immune invasion
Background
Innate immunity is the first line of defense against invading pathogens and encompasses several reactions that target the initial phases of viral infections. Dendritic cells (DCs) are the most efficient type of antigen-presenting cell and play a pivotal role in orchestrating both the innate and adaptive immune responses during viral infections (Steinman 1991). Upon the uptake of certain viral antigens, DCs undergo maturation, which involves the up-regulation of surface major histocompatibility complex (MHC) molecules and the production of type I interferon (IFN), including IFN-α and –β (Barchet et al. 2002; Cheng et al. 2004; Dhib-Jalbut and Cowan 1993). During viral infection, the activation of retinoic-acid-inducible gene I protein (RIG1), a cellular DExD/H box RNA helicase that induces the production of antiviral factors, following viral RNA detection, leads to increased type I IFN production (Hornung et al. 2006; Imaizumi et al. 2004; Rothenfusser et al. 2005; Sakaki et al. 2005). However, several viruses tend to elicit only weak immune responses or immune responses that are insufficient to eliminate the invading pathogen, and it has been suggested that certain RNA viruses specifically target RIG1 to allow immune evasion (Leung et al. 2012).
Rabies, although not preeminent among current infectious diseases, continues to afflict humans, with as many as 55,000 deaths annually. The fatality rate remains the highest among infectious diseases, and medical treatments have proven ineffective (Jackson 2013). Rabies virus (RABV) is a highly neurotropic negative-stranded RNA virus that spreads along the neural pathway and invades the central nervous system (CNS), where it causes an acute and often fatal infection (Finke and Conzelmann 2005). One of the characteristic features of RABV is its ability to reach the CNS in spite of inducing an immune response that can eliminate the virus (Feder et al. 2012). This ability is considered particularly important during the initial stages of peripheral infection following an animal bite, the most common route of transmission, as the virus moves from the muscle or subcutaneous tissue to the peripheral neuronal cells. Although reaching the CNS represents a critical stage in the establishment of the infection, the pathogenesis and detailed mechanism of host evasion and the preservation of RABV during these early stages are not well understood.
Several types of cells directly interact with RABV in the early stages of infection, at the site of initial exposure. A recent study has shown that vesicular stomatitis virus, which belongs to the same genus Rhabdoviridae, gains access to the CNS through the peripheral nerves in macrophage-depleted lymph nodes (Iannacone et al. 2010). RABV particles that are injected into the skin or muscles can slowly replicate in these tissues before they reach the CNS (Charlton et al. 1997; Murphy et al. 1973). It is unclear whether RABV directly invades neural cells or is sequestered by nonpermissive cells that subsequently migrate to neural cells. Recently, Li et al. demonstrated that infection with a pathogenic strain of RABV led to weak DC maturation, whereas a low-pathogenic strain caused the up-regulation of the NF- κB signaling pathway, resulting in high levels of IFN-α mRNA in immature DCs (Li et al. 2008). RABVs are generally classified into two categories: field-isolated virus (street viruses) and laboratory-adapted viruses (fixed strains). The former are known to be more pathogenic after peripheral infections and highly neuropathogenic. In contrast, the latter generally lose their infectivity after peripheral inoculation, despite the preservation of their neuropathogenicity (Lepine 1938). Among the fixed RABVs, representative low-pathogenic strains, including Evelyn-Rokitnicki-Abelseth (ERA), Street Alabama Dufferin and Vnukovo-32, also trigger apoptosis in both neural and nonneural cells, which acts as a potent activator of the immune response, leading to the effective elimination of RABV from the CNS (Préhaud et al. 2003; Thoulouze et al. 1997). In contrast, pathogenic RABV strains, such as the challenge virus standard (CVS), are postulated to evade the host immune surveillance system and to reach neural cells of the CNS by two different mechanisms (Lafon 2011; Rieder and Conzelmann 2011). Here, we hypothesize that immature DCs harboring infectious viruses are present for a long time at the site of initial exposure to RABV. In this study, we sought to clarify the possible role of RABV-infected DCs at the initial infection site in the early phase of RABV infection.
Results
Susceptibility of JAWS II cells to RABV
Previous studies have demonstrated that several pathogenic and non-pathogenic RABV strains can infect immune-related cells, including T cells and monocytes (Thoulouze et al. 1997; Nakamichi et al. 2004). Therefore, we examined whether the DC line, JAWS II, is susceptible to RABV infection using two types of RABV strains; pathogenic CVS and low -pathogenic ERA. We first examined whether RABV can replicate in JAWS II cells and produce progeny viruses in the culture supernatant. The growth kinetics of CVS and ERA in NA cells was determined and both RABV strains replicated significantly at an MOI of 0.1; at 48 h after infection, their titers were in the range of 106 ~107 ffu/well (Figure 1A). However, both CVS and ERA viral titers declined rapidly and neither had effectively replicated in the JAWS II cells at 48 h after infection, even at a high MOI of 10 or 30 (Figure 1A). Interestingly, infectious RABV at an MOI of 10 or 30 gradually decreased under cell-free conditions (CVS or ERA + medium, in Figure 1A) as a natural result because the virus could not survive without infection into the cells, although a more rapid decline in infectivity was observed in the presence of JAWS II cells (CVS or ERA + JAWS II, in Figure 1A), indicating that RABV was promptly ingested by the cells and that no infectious virus remained in the culture medium.
Figure 1.
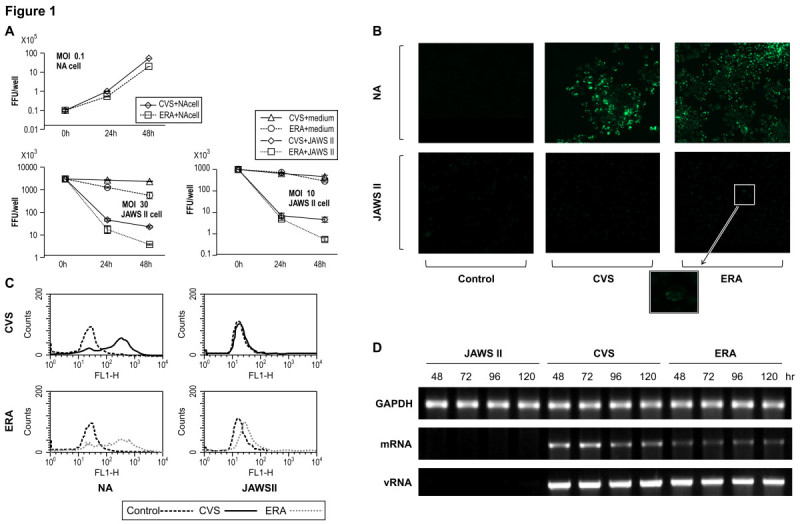
Virus growth in JAWS II and NA cells inoculated with rabies virus. (A) Virus growth curves in NA and JAWS II cells. CVS or ERA was infected at an MOI of 0.1 (NA) or 10,30 (JAWS II), and samples were collected 24 and 48 h after infection. n = 3. Error bars represent mean ± SEM. The black line and dotted line represent the viral titers of CVS and ERA, respectively. Decline in the infected viral titer was determined by using JAWS II cells (+JAWS II) and cell-free conditions (+medium). (B and C) The expression of the viral N protein was analyzed with both a laser scanning microscope and a flow cytometer. After the infection of JAWS II cells with CVS or ERA, the cells were permeabilized with IntraPrep Permeabilization Reagent, and then incubated with FITC-conjugated anti-RABV N MAb. (D) Detection of mRNA and genomic RNA in CVS- or ERA-infected JAWS II cells. The upper panel shows GAPDH, the middle panel shows the expression of mRNA from the RABV N gene detected by RT–PCR, using a poly (T)15 primer for the synthesis of the first-strand cDNA and the lower panel shows viral RNA. The experiments were performed three times and representative data are shown. CVS or ERA at an MOI of 10 was used for the experiments shown in (B to D).
Because RABV N protein is the most abundantly produced viral protein in infected cells, we examined the expression of RABV N protein in the JAWS II cells with laser scanning microscopy and FACSCalibur flow cytometry. Both CVS and ERA effectively propagated in the cells and strong expression of N protein was observed in the NA cells with fluorescence-activated cell sorting (FACS) and immunofluorescence analyses (Figure 1B and C). In contrast, no fluorescence activity was observed in the CVS-infected JAWS II cells (Figure 1B and C), and although a mild increase in fluorescence intensity was observed in the ERA-infected JAWS II cells (Figure 1B and C). Based on these observations, the JAWS II cells were slightly permissive to ERA infection, but were not susceptible to CVS infection.
Following the experiments described above, in which the cells were infected with CVS or ERA, we attempted to detect mRNA and viral genomic RNA in the cells using RT-PCR at 48, 72, 96, and 120 h after infection. Low levels of mRNA signals were detected in both the CVS- and ERA-infected JAWS II cells within 48 h (Figure 1D). Interestingly, viral genomic RNA was sustainably detected in the JAWS II cells infected with either CVS or ERA, even after 120 h (Figure 1D).
Cell death of ERA-infected JAWS II cells induced by viral infection and coculture with spleen cells
Previous studies have demonstrated that low -pathogenic RABV induces apoptosis in neural and nonneural cells (Préhaud et al. 2003; Thoulouze et al. 1997). ERA infected JAWS II cells at 120 h after re-plated to a new plate had decreased significantly (by nearly 80%), whereas that the number of CVS-infected JAWS II cells was slightly increased (Figure 2A). Light microscopic observation showed that the ERA-infected JAWS II cells had a relatively high cytopathic effect compared with the CVS-infected JAWS II cells (Figure 2A and B). Using a coculture assay, we examined whether RABV-infected JAWS II cells were recognized by and lysed in the presence of spleen cells. Normal spleen cells cocultured with uninfected JAWS II cells showed spontaneous cytotoxicity of 36 ± 4.03%. After infection with ERA, JAWS II cells showed ballooning, fragmentation, or condensation of their cytoplasm (Figure 2C and D). The ERA-infected JAWS II cells showed significantly higher cytotoxicity (92.5 ± 6.02%) than the CVS-infected JAWS II cells.
Figure 2.
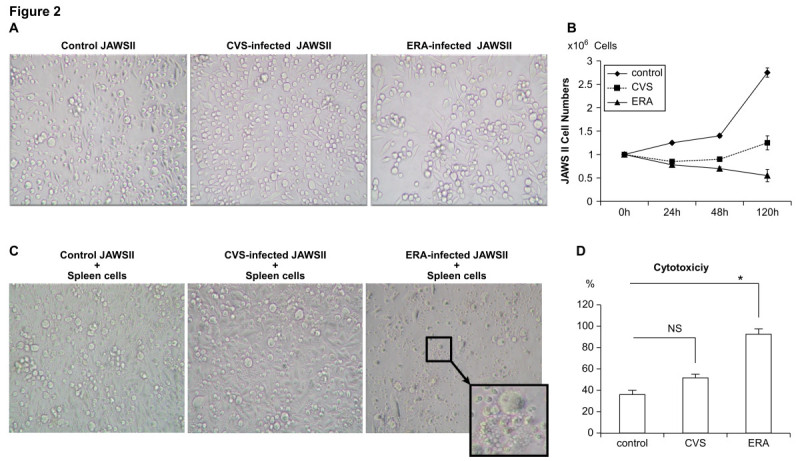
Morphological changes in JAWS II cells after RABV infection and coculture with mouse spleen cells. (A) Light-microscopic images of uninfected (left), CVS-infected (middle), and ERA-infected (right) JAWS II cells; (B) the numbers of viable cells were determined by Trypan blue staining 24, 48, 120 h after re-plated. (C and D) uninfected (left), CVS-infected (middle), and ERA-infected (right) JAWS II cells were cocultured with mouse spleen cells for a further 120 h and the cytotoxicity was calculated with a cell death kit. All the experiments were performed three times at an MOI of 10 and representative data are shown (A and C). Error bars represent mean ± SEM (B). *p < 0.01 (D).
Levels of MHC class I and II molecules costimulatory molecules, innate-immunity-related molecules, and the production of type I IFNs elevated in ERA-infected JAWS II cells
To determine whether CVS-infected JAWS II cells can avoid eliciting host immune responses, we examined the immune-related surface molecules on JAWS II cells at 48 h after their infection with CVS or ERA at an MOI of 10. Because MHC classes I and II are antigen-presenting molecules important in the activation of the immune responses, their expression on the surfaces of JAWS II cells was examined by flow cytometry. The analysis revealed that the expression of MHC class I increased more strongly on the surfaces of the cells infected with ERA (Figure 3). Similarly, the expression of the costimulatory molecules, CD80 and CD86 was significantly more strongly upregulated in the ERA-infected JAWS II cells than in the CVS-infected JAWS II cells.
Figure 3.

Expression of cell-surface molecules by flowcytometric analysis at 48 h after infection of CVS or ERA (MOI of 10). Profiles of the surface molecules on uninfected (control; white), CVS-infected (gray), and ERA-infected (black) JAWS II cells reacted with FITC conjugated anti-mouse MHC class I, MHC class II and CD80, PE conjugated anti-mouse CD86 and TLR3, and rat anti-mouse RIG1 antibodies, followed by a FITC-conjugated goat anti-rat IgG antibody. The relative fluorescence intensity of the surface molecules was determined with FACS. All cells including uninfected cells (control) were stained by the same procedure. The experiments were performed three times and error bars represent SEM. *p < 0.001, **p < 0.05, ***p < 0.01.
The phosphoprotein encoded by RABV is known to possess an antagonizing activity against the induction of IFN by RIG1 in host cells (Brzózka et al. 2006). Toll-like receptors (TLRs)-3, -7, -8, and −9 act as sensors of viral nucleic acids and, with the exception of the TLR-3 signaling pathway, also induce inflammatory cytokines and IFNs. Therefore, we also examined how innate immune sensor molecules are expressed in RABV-inoculated DCs, using flow cytometry. The expression of TLR-3 was more strongly elevated in the ERA-infected JAWS II cells than in the CVS-infected JAWS II cells, when examined flow cytometry (Figure 3). It is well known that the type I IFN response controls tissue tropism and pathogenicity during viral infection processes (Chopy et al. 2011). As well as analyzing the expression of MHC and costimulatory molecules after RABV infection, we examined the production of the type I IFNs, IFN-α and -β, in RABV-infected DCs using an ELISA. We determined that the production of IFN-α and -β was markedly higher in ERA-infected JAWS II cells than in cells infected with CVS (Figure 4).
Figure 4.
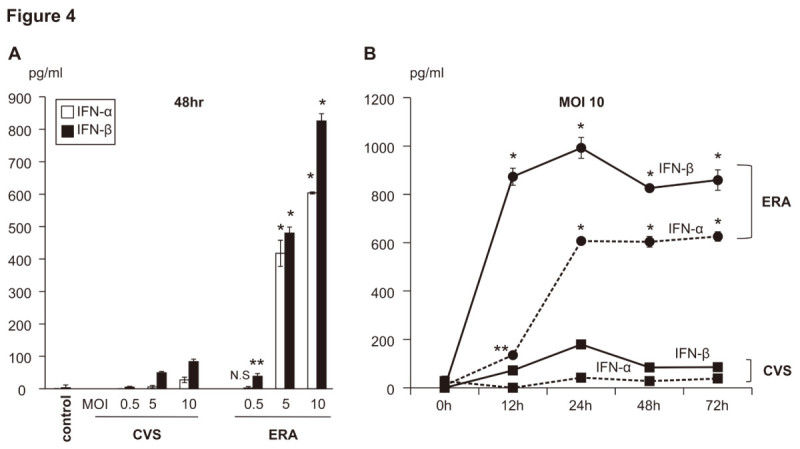
Production of IFN-α and IFN-β in CVS- and ERA-inoculated JAWS II cells. (A) The levels of IFN-α (white) and IFN-β (black) in the culture supernatant 48 h after infection in CVS- or ERA-infected JAWS II cells at various MOIs (0.5, 5, and 10). (B) Serial changes in IFN-α (dotted line) and IFN-β (black line) production in the CVS-infected (squares) and ERA-infected JAWS II cells (circles) at an MOI of 10. The levels of type I IFN were quantified by ELISA 72 h after infection. The experiments were performed three times and error bars represent SEM. *p < 0.001, **p < 0.01.
Possible cell-to-cell infection by RABV transmitted from JAWS II cells to neural cells
To confirm the hypothesis that DCs carry RABV and can cryptically transmit RABV to neural cells, we performed in vitro coculture assays to determine whether JAWS II cells harboring the CVS genome facilitate viral transmission via cell-to-cell infection. Neither productive viral replication nor viral antigen expression was demonstrated in the CVS-infected JAWS II cells in the analyses described above (Figure 1). Although CVS-infected JAWS II cells did not exhibit progeny viral production when assayed at the protein or genomic level, they did transmit “infectious viral genomes” to uninfected “naïve” NA cells, indicating the occurrence of cell-to-cell transmission (Figure 5). RABV easily replicated and produced progeny virus in the NA cells, so the CVS-infected NA cells were capable of transmitting cell-free virus or cell-associated virus (Figure 5). These results also indicate that the CVS genome was maintained in the JAWS II cells at detectable levels but avoided the host immune system, because it did not induce type I IFNs or upregulate the expression of MHC class I molecule. However, it retained the ability to infect neural cells in vitro through the process of cell-to-cell or “trans” infection.
Figure 5.
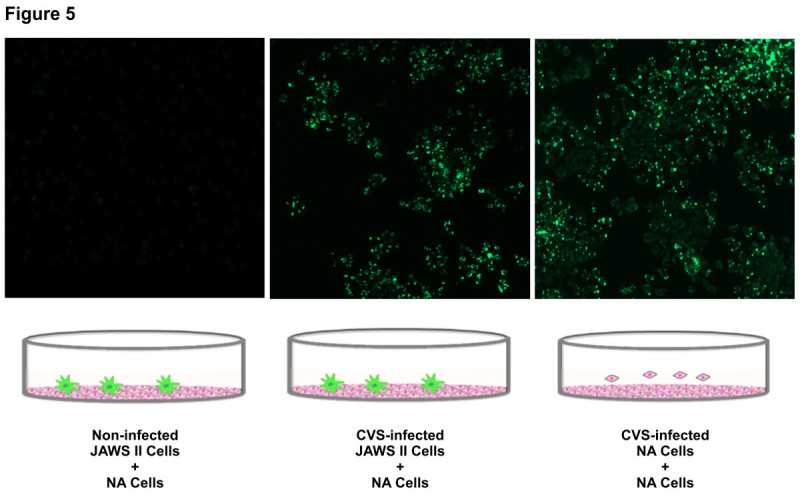
Cell-to-cell infection of RABV from JAWS II cells to neural cells. A coculture assay of NA cells (indicator cells) with CVS-infected JAWS II cells or NA cells (donor cells) was performed as described in the Methods. The expression of the viral N protein was analyzed with a laser scanning microscope. After the infection of JAWS II cells with CVS at an MOI of 10, the cells were permeabilized with IntraPrep Permeabilization Reagent, and then reacted with FITC-conjugated anti-RABV N MAb. The experiments were performed three times and representative data are shown.
Transmission of RABV to mouse brain via JAWS II cells harboring RABV
To examine whether cell-to-cell transmission occurs from DCs to neural cells in vivo, we directly examined the effects of the passive transfer of RABV-infected DCs to mice. Five-week-old C57BL/6 mice were injected intracerebrally with 106, 105, or 104 CVS- or ERA-infected JAWS II cells (n = 10 each group), and their survival was monitored for 14 days after transfer. No deaths were observed in the control group injected with RABV-uninfected DCs. Mice injected with either CVS- or ERA-infected JAWS II cells showed clinical symptoms and finally died in a dose-dependent manner (Figure 6A). However, the survival of the mice injected with CVS- or ERA-infected JAWS II cells differed. All mice injected with 106 CVS-infected JAWS II cells died within 10 days, whereas those injected with 106 ERA-infected JAWS II cells survived two days longer. Mice injected with 104 CVS-infected JAWS II cells had a survival rate of only 20%, whereas 70% survival was observed in mice injected with same number of ERA-infected JAWS II cells (Figure 6A). The mice injected with ERA-infected JAWS II cells showed less lethality and longer survival than those injected with CVS-infected JAWS II cells, indicating that ERA-infected JAWS II cells successfully induced the optimal immune responses required for viral elimination in vivo (Figure 6A). The surviving mice showed no apparent neurological manifestations or sequelae during the observation period. To confirm viral propagation in the mouse brain, the presence of mRNA and N protein was examined in the hippocampal tissues from mice injected with CVS-infected JAWS II cells. Viral mRNA was clearly detected by RT–PCR and viral N protein was confirmed with laser scanning microscopy and an immunochromatographic test (Figure 6B).
Figure 6.
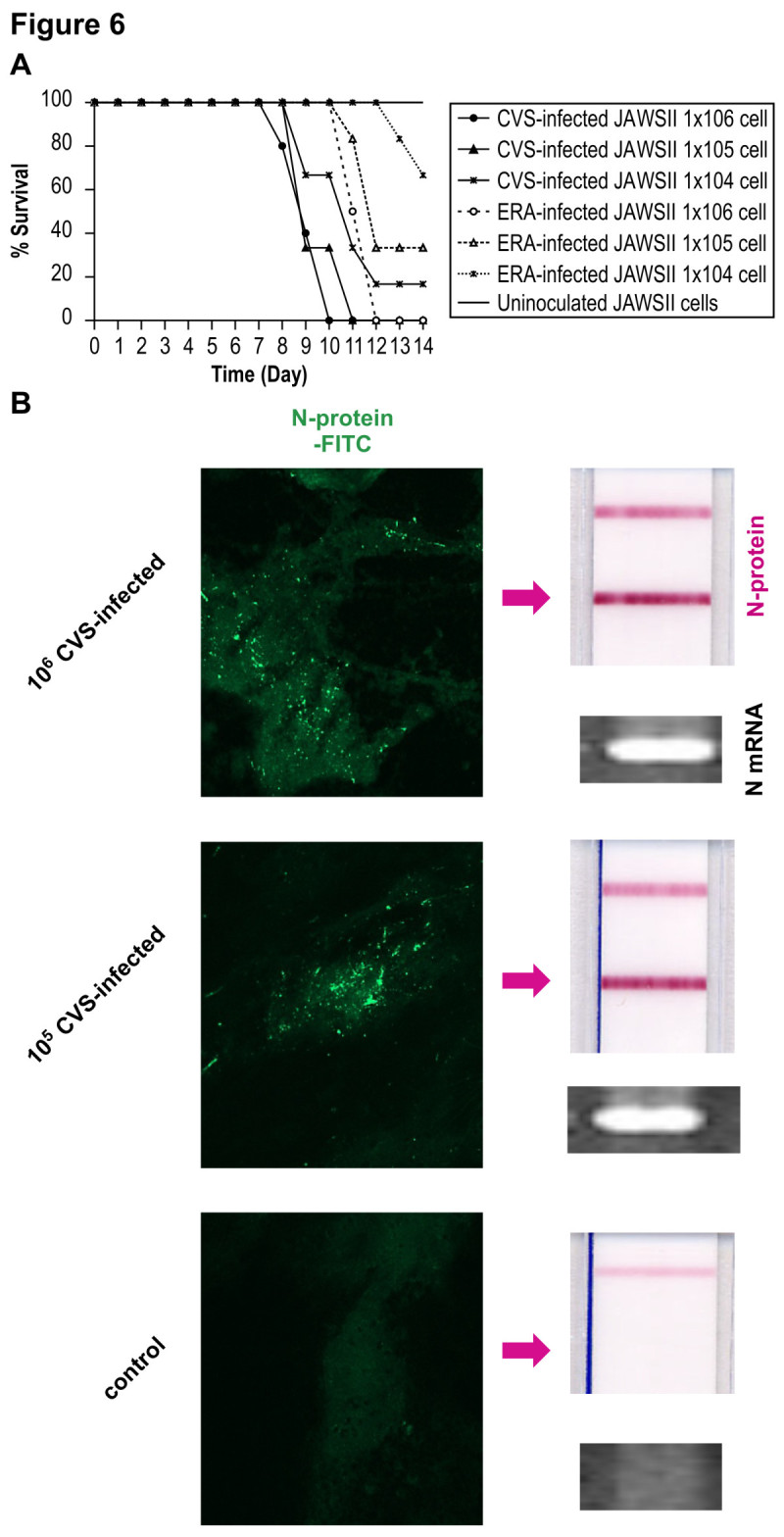
Transmission of RABV to mouse brainviaJAWS II cells harboring RABV. (A) Survival rates of five-week-old C57BL/6 mice injected intracerebrally with 106, 105, or 104 CVS- or ERA-infected JAWS II cells (n = 10 each group) at MOI of 30. Their survival was observed for 14 days after the transfer of the cells. The black and dotted lines represent the survival of the mice injected with CVS- and ERA-infected JAWS II cells, respectively. The circles indicate the survival of mice injected with 106 CVS- or ERA-infected JAWS II cells. The triangles indicate the survival of mice injected with 105 CVS- or ERA-infected JAWS II cells. The crosses indicate the survival of mice injected with 104 CVS- or ERA-inoculated JAWS II cells. The experiments were performed three times. The Kaplan-Meier method was used to analyze mouse survival. Statistical analyses were performed by log-rank test. (p < 0.01; 106 CVS: 106 ERA, 105 CVS: 105 ERA. p <0.05; 104 CVS: 104 ERA) (B) Half the mice from each group were sacrificed after seven days, and the presence of viral N mRNA and RABV N protein in the hippocampal tissues was determined by RT–PCR (mRNA) and a RABV N detection kit with FITC staining, respectively. The arrow indicates the positive band of N protein on the immunochromatographic test. The upper band corresponds to nonspecific control. The experiments were performed three times as shown in Figure 1B and representative pictures are shown.
Discussion
To examine the potential role of DCs in the transmission of RABV, we focused on the probable events that occur at the site of injury caused by rabid animals, by looking into the migration of RABV toward the CNS, which is the organ most permissive to RABV. Neither pathogenic CVS nor low-pathogenic ERA actively produced progeny viruses in JAWS II cells, which were used in this study. However, viral genomic RNA lodged persistently inside the virus-infected cells and was still infectious. Based on our observations, we propose two possible explanations of how RABV evades the host’s immune surveillance system and finally establishes a CNS infection, after which it reaches the brain cryptically. One possibility is that pathogenic RABV does not induce effective host responses for viral elimination in infected DCs; and the other is that the RABV hidden in these DCs plays a role in its cell-to-cell or trans infection, thus using DCs as a vehicle in the infection pathway.
In the early phase of the mediation between the innate and acquired immune responses, DCs predominantly reside in the peripheral tissues and play a role as sentinel cells in antigen capture. Immature DCs undergo maturation, characterized by the upregulation of surface MHC molecules and costimulatory molecules, and the subsequent release of numerous humoral factors, including cytokines and IFNs. Subsequently, the mature DCs migrate to the peripheral secondary lymphoid tissues, resulting in the presentation of optimally processed antigen to T lymphocytes via these immune synapses. Our flowcytometric and ELISA analyses revealed that the upregulated expression of MHC I molecule on the surface of JAWS II cells and the secretion of type I IFNs were much greater after they were infected with the low-pathogenic RABV strain, ERA, than when they were infected with the pathogenic CVS strain. Analyses of the surface immune molecules revealed that the JAWS II cells matured from the immature state after infection with ERA, but not after infection with CVS.
The mechanism through which JAWS II cells, which are nonpermissive to RABV, can induce this immunological maturation is explained as follows. The small amount of N protein produced in the ERA-infected JAWS II cells (Figure 1B and C) might not be utilized for viral production or morphogenesis, but may be degraded in cellular proteasome and finally assembled with MHC class I molecules, or the minimal N protein produced ERA may be presented directly on MHC class I molecules via a cross-presentation process. Another possibility is that in response to certain inhibitory molecules (eg. microRNAs) that are only produced during CVS infection, type I IFNs are not induced in the CVS-infected JAWS II cells (Zhao et al. 2012).
The inadequate immune response stimulated by CVS is also supported by the observation that the death of CVS-infected JAWS II cells was not induced in the presence of naïve spleen cells, whereas the ERA-infected cells were successfully lysed. Although we could not confirm that this was the result of apoptosis, a previous study has demonstrated that low-pathogenic strains of RABV can induce apoptotic responses in macrophages (Jackson and Rossiter 1997). Therefore, it appears that the pathogenic RABV remains hidden within DCs, where it effectively evades the host’s immune surveillance system and may prevent the migration of the infected DCs to the lymphoid tissues. Since we were not able to detect infectious viruses in supernatants of infected JAWS II cells, it is also possible that the rate of virus degradation and re-adsorption is higher than the rate of virus release.
The possibility that RABV exists within DCs below the detection limit of the immune surveillance system has important implications for the spread of RABV from the initial site of infection to the peripheral secondary lymphoid tissues and neural cells, and for the fate of RABV during the long-term incubation period that is often observed after the initial infection (Smith et al. 1991). Although the JAWS II cells were nonpermissive for RABV replication and could not produce progeny virus, in vitro coculture studies revealed that NA cells allowed viral transmission after direct cell-to-cell contact with CVS–infected JAWS II cells. This phenomenon is supported by the fact that the exposure of CVS- infected JAWS II cells to monoclonal antibodies directed against a RABV surface glycoprotein (G) 100% blocked the transmission of RABV to NA (KS and AN, unpublished observation). The relevance of viral transmission by a cell-to-cell contact process was demonstrated by the injection of CVS-infected JAWS II cells into the brains of mice, which led to the rapid spread of the virus and 100% mortality. In the establishment of immunodeficiency virus 1 (HIV-1) infection, the uptake of HIV-1 by immature DCs at the mucosal site occurs when it binds to either DC-SIGN or syndecan-3 (de Witte et al. 2007; Geijtenbeek et al. 2000). Following the migration of HIV-1 from the initial mucosal invasion site, DCs may carry the virus to the draining lymph nodes. It has been demonstrated that during the antigen presentation process, DCs cluster with T cells in the secondary lymphoid tissues (de Witte et al. 2007; Geijtenbeek et al. 2000; Lore and Larsson 2003). It is well known that abundant nerve-end fibers innervate the peripheral lymphoid tissue. Other studies have suggested that prions initially accumulate on follicular dendritic cells in the lymphoid tissues and subsequently spread via the peripheral nervous system to the brain (Raymond and Mabbott 2007; Sethi et al. 2007). Recently, it has been shown that micro RNAs secreted by Epstein-Barr virus-infected cells are transferred to uninfected cells via exosome structures (Pegtel et al. 2010). Our results also suggest that a mechanism similar to that discussed above is involved in the transmission of RABV from DCs to CNS.
DCs are heterogeneous cells and comprise several subsets characterized by unique morphological shapes and functions. Conventional or myeloid-type DCs (mDCs) have a dendritic shape, exhibit typical DC functions, such as antigen uptake, processing, and presentation, and are characterized as CD11c+, CD11b+, and B220-. In contrast, plasmacytoid DCs, which are nondendritic round cells, are defined as CD11low, CD11b-, and B220+ cells, and lack the ability to produce costimulatory molecules (Siegemund et al. 2009). Whereas mDC subsets migrate to the lymph nodes from peripheral tissue, plasmacytoid DCs directly enter the lymph nodes from the blood by crossing high endothelial venules (HEVs) (Banchereau and Steinman 1998; Shortman and Liu 2002) and are able to transform into mDCs in response to the surrounding conditions (Fukao et al. 2000; Fukao et al. 2001; Zuniga et al. 2004). When the cell-surface markers on JAWS II cells were analyzed by flow cytometry, CD11c and CD11b were detected, whereas CD8α was not, and the expression of costimulatory molecules was upregulated by stimulation with viral antigens. These results indicate that JAWS II cells are mDCs, which is consistent with a previous report (Otsu et al. 2006). It has also been speculated that the administration of RABV vaccine via an intradermal route in humans, as well as via the routine intramuscular route, induce adequate levels of viral neutralizing antibody (Shiota et al. 2008), indicating that mDC subset are recruited to the vaccination site and migrate to the lymph nodes for effective antigen presentation to naïve T cells. Therefore, we speculate that mDCs may play a critical role in RABV infection and pathogenesis.
Several limitations of our study warrant mention. Our attempts to determine whether RABV particles could be visualized in CVS-infected JAWS II cells by immunogold electron microscopy were unsuccessful. We were also unable to confirm the occurrence of apoptosis in the presence of spleen lymphocytes. However, high level of IFN was detected, indicating that cellular immunity was fully functional after the infection of JAWS II cells with ERA.
We have provided several lines of evidence that DCs allow RABV to escape the host immune surveillance system and facilitate the transmission of the virus to the neural cells. Following the uptake of pathogenic CVS, immature DCs do not properly process the viral antigens, as indicated by their lack of MHC class I and II expression. Because the RABV genome persists undetected in DCs and does not replicate or produce progeny virus, we propose that RABV uses these nonpermissive cells as a vehicle to reach the peripheral neural cells, where the virus can then replicate and spread to the CNS. This is the first report to demonstrate that DCs can transmit RABV to neural cells by cell-to-cell or trans infection, and sheds new light on the immune evasion strategies used by this deadly virus.
Methods
Cells
The DC cell line, JAWS II, and neuroblastoma (NA) cells were purchased from the American Type Culture Collection (Manassas, VA). JAWS II cells (CD11b+, CD11c+, CD8α-, TLR7+, and RIG-1+) were originally isolated from bone-marrow cultures from p53-deficient C57BL/6 mice. The cells were grown in a complete culture medium consisting of RPMI 1640 medium with GlutaMAX™ (Gibco BRL, Grand Island, NY) and supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, 50 μm 2-mercaptoethanol, and 5 ng/mL recombinant mouse granulocyte-macrophage colony-stimulating factor (BD Biosciences, San Jose, CA). Murine NA cells were grown in Eagle’s minimal essential medium (Gibco BRL, Grand Island, NY) containing 10% FBS and 1% penicillin-streptomycin.
Viruses
A pathogenic strain of RABV (CVS-11) and a low- pathogenic strain (ERA) were propagated in NA cells. The viruses in the culture supernatants of the RABV-inoculated NA cells were collected and centrifuged at 1200 × g for 10 min at 4°C (TOMY EX-125, TS-38, TOMY, Tokyo, Japan), and then purified by ultrafiltration through a 0.45-μm filter. The infection of NA or JAWS II cells with RABV was performed at 37°C at various multiplicities of infection (MOIs).
Antibodies
Fluorescein isothiocyanate (FITC)-conjugated goat anti-rat IgG antibody was obtained from Organon Teknika Corp. (Durham, NC). FITC-conjugated anti-mouse MHC class I, MHC class II, and CD80 antibodies, and phycoerythrin (PE)-conjugated anti-mouse CD86 antibody were purchased from eBioscience Inc. (San Diego, CA). PE-conjugated anti-mouse CD283 (TLR3) antibody and purified rat anti RIG1 antibody were obtained from BioLegend Inc. (San Diego, CA). FITC-conjugated anti-RABV N monoclonal antibody (MAb) was obtained from Fujirebio Diagnostics, Inc. (Malvern, PA).
Measurement of viral replication
NA and JAWS II cells were seeded in 48-well culture plates (105 cells/well) and infected with CVS or ERA at the appropriate MOI. The culture supernatants obtained from the RABV-infected NA or JAWS II cells were then separated by centrifugation at 1200 × g for 10 min 0, 24, or 48 h after infection. The virions in the supernatant were purified by ultrafiltration through a 0.45 μm filter and then titrated. The viruses in the culture supernatants were titrated with a focus assay on confluent monolayers of NA cells in 24-well plates, according to previously reported procedures (Yamada et al. 2012).
Flow cytometry and confocal analysis
The expression of cell-surface molecules was measured by immunofluorescence flow-cytometric analysis using a FACSCalibur flow cytometer (BD Immunocytometry Systems, San Jose, CA) and CellQuest software. For the analysis, NA and JAWS II cells were first seeded in six-well culture plates (106 cells/well) and infected with CVS or ERA at an MOI of 10. After 48 h at 37°C, the cells were collected and washed in phosphate-buffered saline (PBS), and then incubated on ice for 30 min with a FITC-conjugated anti-mouse MHC class I (1:50), MHC class II (1:50), or CD80 (1:50) antibody, or PE-conjugated anti-mouse CD86 antibody (1:50). The expression of the intracellular proteins, TLR-3 and RIG1 was also measured by immunofluorescence flow-cytometric analysis using a FACSCalibur flow cytometer and CellQuest software. After the infection of JAWS II cells with CVS or ERA (as described above), the cells were permeabilized with Intra Prep Permeabilization Reagent (Immunotech, Marseille Cedex, France), according to the manufacturer’s protocol, and then reacted with PE-conjugated anti-mouse TLR3 (1:50) or rat anti-mouse RIG-1 (1:50) antibody. An FITC-conjugated goat anti-rat IgG (1:200) was used as the secondary antibody to detected RIG1.
The expression of the viral N protein was analyzed with both a laser scanning microscope (Carl Zeiss MicroImaging, LSM 510; Carl Zeiss, Jena, Germany) and a FACSCalibur flow cytometer. After the infection of JAWS II cells with CVS or ERA (as described above), the cells were permeabilized with IntraPrep Permeabilization Reagent, and then reacted with FITC-conjugated anti-RV N MAb (1:50).
Enzyme-linked immunosorbent assays (ELISAs)
To measure the quantities of IFN-α and IFN-β secreted by the RABV-infected JAWS II cells, JAWS II cells seeded in six-well plates (106 cells/well) were inoculated with CVS or ERA at an MOI of 10 and the culture supernatants were then harvested at 0, 12, 24, 48, or 72 h after infection. The concentrations of IFN-α and IFN-β in the culture supernatants were assessed with sandwich ELISA kits (PBL Biomedical Laboratories, Piscataway, NJ), according to the manufacturer’s instructions.
Viral RNA extraction and reverse transcription (RT)-PCR
JAWS II cells in 12-well culture plates (5 × 105 cells/well) were infected with CVS or ERA at an MOI of 10. Following incubation for 48, 72, 96, or 120 h, the cells were collected and washed three times with PBS. The total RNA was then extracted from the cells using the acid-guanidinium thiocyanate -phenol -chloroform method (TRIzol®, Gibco BRL, Gaithersburg, MD). To detect the viral genomic RNA (negative polarity) or viral mRNA amplification, a partial sequence of the N gene was amplified using the sense primer NF2850 (nucleotides 28–50, 5′-ACAGACAGCGTCAATGGCAGAGC-3′) and antisense primer N660 (R) (nucleotides 660–676, 5′-GTTTGGTATAGTACTCC-3′) (Nishizono et al. 2002). The primer positions are given according to the N gene of CVS (GenBank accession number DQ286762). Total RNA (1 μg) was reverse transcribed to synthesize the first-strand cDNA of the viral genome RNA using the primer N660 (R) or the viral mRNA using the primer NF2850 and Moloney murine leukemia virus reverse transcriptase (Gibco BRL) at 37°C for 2 h. The first PCR reaction was performed with the following reaction mixture: 1 μg of sample cDNA, 50 mM KCl, 10 mM Tris–HCl (pH 8.4), 1.5 mM MgCl2, 20 mM each of primers NF2850 and N660 (R), 200 μM dNTPs, and 2 U of Taq DNA polymerase (Promega Corp., Madison, WI). The reaction mixture was subjected to 35 cycles of denaturation at 95°C for 30 s, annealing at 50°C for 30 s, and extension at 72°C for 90 s. The resulting amplicons were resolved electrophoresed on 1% agarose gels and stained with ethidium bromide. As the internal control mRNA, glyceraldehyde phosphate dehydrogenase (GAPDH) transcripts were amplified under the same conditions, with the sense primer 5′-TTCACCACCATGGAGAAGGC-3′ and the antisense primer 5′-GGCATGGACTGTGGTCATGA-3′.
Effect of naïve spleen cells on RABV-infected JAWS II cells
JAWS II cells were seeded in six-well culture plates (106 cells/well) and infected with CVS or ERA at an MOI of 10.The cells were collected after 48h incubation, washed three times in PBS, and then plated in a new six-well culture plate (105 cells/well). Spleen cells were obtained from the spleens of normal C57BL/6 mice (Otsu et al. 2006) (Charles River Japan Inc., Yokohama, Japan) and then cultured with or without the RABV-infected JAWS II cells (106 cells/well; final E/T ratio = 10:1). After 120 h of coculture, the cells were observed microscopically (Olympus CK30, Olympus, Tokyo, Japan) and a cytotoxicity assay was performed with the Cyto Tox 96® Non-Radioactive Cytotoxicity Assay (Promega Corp.). Cytotoxicity was calculated according to the formula: % cytotoxicity = (Experimental LDH release - Effector Spontaneous LDH release - Target Spontaneous LDH release / Target Maximum LDH release - Target Spontaneous LDH release × 100).
Cell-to-cell transmission of RABV from JAWS II cells to neural cells in vitro
JAWS II cells or NA cells in six-well culture plates (106 cells/well) were infected with CVS at an MOI of 10. After incubation for 48 h at 37°C, the cells were collected and washed thoroughly three times with PBS. A virus titration assay was performed to confirm that there were no remaining viruses in the supernatant, as described above. The cells were then added to a 24-well plate containing monolayers of cultured NA cells. After 48 h of coculture, the NA cells were stained with FITC-conjugated anti-RABV N protein MAb and observed under a laser scanning microscope.
Injection of CVS- or ERA-inoculated JAWS II cells into mouse brains
JAWS II cells in six-well culture plates (106 cells/well) were infected with CVS or ERA at an MOI of 30. Following incubation for 48 h, the cells were collected and washed three times with PBS. A virus titration assay was performed to confirm that no viruses remained in the supernatant, as described above. Specific pathogen-free five-week-old female C57BL/6 mice (Seac Yoshitomi, Fukuoka, Japan) were injected intracerebrally with 106, 105, or 104 CVS- or ERA-infected JAWS II cells in 0.03 mL with a 23-gauge needle. Half the mice in each injected group (n = 10) were observed for 2 weeks and their survival was recorded daily. The remaining mice were sacrificed on day 7 and the tissue samples taken from the hippocampus were analyzed for viral N protein using a laser scanning microscope, for viral mRNA and an immunochromatographic test that had been developed previously by us (Nishizono et al. 2008). All animal procedures conformed to animal care guidelines approved by Ethics Committee in Oita University.
Authors’ information
Kazuyo Senba: DDS, PhD
Kagoshima University Graduate School of Medical and Dental Sciences
Area of specialization: Virology, Immunology, Bacteriology
Takashi Matsumoto: PhD
Oita University Graduate School of Medical Sciences
Area of specialization: Virology, Immunology, Bacteriology
Kentaro Yamada: DVM, PhD
United Graduate School of Veterinary Sciences
Area of specialization: Virology
Seiji Shiota: MD, PhD
Oita University Graduate School of Medical Sciences
Area of specialization: Immunology,
Hidekatsu Iha: PhD
The Graduate University for Advanced Studies
Area of specialization: Cell biology
Yukari Date: MD, PhD
Miyazaki University Graduate School of Medical Sciences
Area of specialization: Biochemistry
Motoaki Ohtsubo: PhD
Graduate School of Medical Sciences, Kyushu University
Area of specialization: Cell biology
Akira Nishizono: MD, PhD
Oita University Graduate School of Medical Sciences
Area of specialization: Virology, Immunology, Bacteriology
Acknowledgements
We thank Kunimitsu Inoue and Mika Okamoto for technical assistance and helpful discussion. This work was supported by the Grant-in-Aid for Scientific Research from the Health and Labour Science Research Grants of Japan (Research on International and Cooperation in Medical Science, H23-, H24- KOKUI-SHITEI-003). This work was in part supported by The Naito foundation.
Abbreviations
- DC
Dendritic cells
- MHC
Major histocompatibility complex
- IFN
Interferon
- RIG 1
Retinoic-acid-inducible gene I
- RABV
Rabies virus
- CNS
Central nervous system
- ERA
Evelyn-Rokitnicki-Abelseth
- FACS
Fluorescence activated cell sorting
- TLR
Toll-like recepetor
- HIV
Human immunodeficiency virus
- MDC
Myeloid-type DC
- HEV
High endothelial venules
- FITC
Fluorescein isothiocyanate
- PE
Phycoerythrin
- MAb
Monoclonal antibody
- GADPH
Glyceraldehyde phosphate dehydrogenase.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Footnotes
Competing interests
The authors declare they have no competing interests in relation to this article.
Authors’ contributions
KS and AN conceive of the study and participated in its design and coordination and draft the manuscript. KS, TM and MO carried out the PCR and ELISA experiments. KS, HI and YD carried out the confocal microscopy and FACS analysis. KS, KY and SS performed animal studies. All authors participated in the preparation of the manuscript, and read and approved the final manuscript.
Contributor Information
Kazuyo Senba, Email: kazutan@nm.beppu-u.ac.jp.
Takashi Matsumoto, Email: tmatsumoto9@oita-u.ac.jp.
Kentaro Yamada, Email: kentaro-y@oita-u.ac.jp.
Seiji Shiota, Email: sshiota@oita-u.ac.jp.
Hidekatsu Iha, Email: hiha@oita-u.ac.jp.
Yukari Date, Email: dateyuka@med.miyazaki-u.ac.jp.
Motoaki Ohtsubo, Email: mohtsubo@nm.beppu-u.ac.jp.
Akira Nishizono, Email: a24zono@med.oita-u.ac.jp.
References
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Barchet W, Cella M, Odermatt B, Asselin-Paturel C, Colonna M, Kalinke U. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J Exp Med. 2002;195:507–516. doi: 10.1084/jem.20011666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzózka K, Finke S, Conzelmann KK. Inhibition of interferon signaling by rabies virus phosphoprotein P: activation-dependent binding of STAT1 and STAT2. J Virol 80:2675–2683 extracellular signal-regulated kinases 1 and 2. J Virol. 2006;78:9376–9388. doi: 10.1128/JVI.80.6.2675-2683.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton KM, Nadin-Davis S, Casey GA, Wandeler AI. The long incubation period in rabies: delayed progression of infection in muscle at the site of exposure. Acta Neuropathol. 1997;94:73–77. doi: 10.1007/s004010050674. [DOI] [PubMed] [Google Scholar]
- Cheng Y, King NJ, Kesson AM. Major histocompatibility complex class I (MHC-I) induction by West Nile virus: involvement of 2 signaling pathways in MHC-I up-regulation. J Infect Dis. 2004;189:658–668. doi: 10.1086/381501. [DOI] [PubMed] [Google Scholar]
- Chopy D, Pothlichet J, Lafage M, Mégret F, Fiette L, Si-Tahar M, Lafon M. Ambivalent role of the innate immune response in rabies virus pathogenesis. J Virol. 2011;85:6657–6668. doi: 10.1128/JVI.00302-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Witte L, Bobardt M, Chatterji U, Degeest G, David G, Geijtenbeek TB, Gallay P. Syndecan-3 is a dendritic cell-specific attachment receptor for HIV-1. Proc Natl Acad Sci USA. 2007;104:19464–19469. doi: 10.1073/pnas.0703747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhib-Jalbut SS, Cowan EP. Direct evidence that interferon-beta mediates enhanced HLA-class I expression in measles virus-infected cells. J Immunol. 1993;151:6248–6258. [PubMed] [Google Scholar]
- Feder HM, Jr, Petersen BW, Robertson KL, Rupprecht CE. Rabies: still a uniformly fatal disease? Historical occurrence, epidemiological trends, and paradigm shifts. Curr Infect Dis Rep. 2012;4:408–422. doi: 10.1007/s11908-012-0268-2. [DOI] [PubMed] [Google Scholar]
- Finke S, Conzelmann KK. Replication strategies of rabies virus. Virus Res. 2005;111(2):120–131. doi: 10.1016/j.virusres.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Fukao T, Matsuda S, Koyasu S. Synergistic effects of IL-4 and IL-18 on IL-12-dependent IFN-gamma production by dendritic cells. J Immunol. 2000;164:64–71. doi: 10.4049/jimmunol.164.1.64. [DOI] [PubMed] [Google Scholar]
- Fukao T, Frucht DM, Yap G, Gadina M, O’Shea JJ, Koyasu S. Inducible expression of Stat4 in dendritic cells and macrophages and its critical role in innate and adaptive immune responses. J Immunol. 2001;166:4446–4455. doi: 10.4049/jimmunol.166.7.4446. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GCF, Middel J, Cornelissen ILMHA, Nottet HSLM, KewalRamani VN, Littman DR, Figdor CG, van Kooyk Y. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-Infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/S0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann K, Schlee M, Endres S, Hartmann G. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, Whelan SP, Guidotti LG, von Andrian UH. Sub capsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature. 2010;465:1079–1083. doi: 10.1038/nature09118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi T, Hatakeyama M, Yamashita K, Yoshida H, Ishikawa A, Taima K, Satoh K, Mori F, Wakabayashi K. Interferon-gamma induces retinoic acid-inducible gene-I in endothelial cells. Endothelium. 2004;11:169–173. doi: 10.1080/10623320490512156. [DOI] [PubMed] [Google Scholar]
- Jackson AC. Current and future approaches to the therapy of human rabies. Antiviral Res. 2013;99(1):61–67. doi: 10.1016/j.antiviral.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Jackson AC, Rossiter JP. Apoptosis plays an important role in experimental rabies virus infection. J Virol. 1997;71:5603–5607. doi: 10.1128/jvi.71.7.5603-5607.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon M. Evasive strategies in rabies virus infection. Adv Virus Res. 2011;79:33–53. doi: 10.1016/B978-0-12-387040-7.00003-2. [DOI] [PubMed] [Google Scholar]
- Lepine P. On the evolution of fixed strains of rabies virus. J Hyg (Lond) 1938;38:180–184. doi: 10.1017/S0022172400011013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung DW, Basler CF, Amarasinghe GK. Molecular mechanisms of viral inhibitors of RIG-I like receptors. Trends Microbiol. 2012;20(3):139–146. doi: 10.1016/j.tim.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McGettigan JP, Faber M, Schnell MJ, Dietzschold B. Infection of monocytes or immature dendritic cells (DCs) with an attenuated rabies virus results in DC maturation and a strong activation of the NFκB signaling pathway. Vaccine. 2008;17:419–426. doi: 10.1016/j.vaccine.2007.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lore K, Larsson M. The role of dendritic cells in the pathogenesis of HIV-infection. APMIS. 2003;111:776–788. doi: 10.1034/j.1600-0463.2003.11107809.x. [DOI] [PubMed] [Google Scholar]
- Murphy FA, Baur SP, Harrison AK, Winn WC. Comparative pathogenesis of rabies and rabies-like viruses. Lab Invest. 1973;28:361–376. [PubMed] [Google Scholar]
- Nakamichi K, Inoue S, Takasaki T, Morimoto K, Kurane I. Rabies virus stimulates nitric oxide production and CXC chemokine ligand 10 expression in macrophages through activation of extracellular signal-regulated kinases 1 and 2. J Virol. 2004;78:9376–9388. doi: 10.1128/JVI.78.17.9376-9388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizono A, Mannen K, Elio-Villa LP, Tanaka S, Li KS, Mifune K, Arca BF, Cabanban A, Martinez B, Rodriguez A, Atienza VC, Camba R, Resontoc N. Genetic analysis of rabies virus isolates in the Philippines. Microbiol Immunol. 2002;46:413–417. doi: 10.1111/j.1348-0421.2002.tb02714.x. [DOI] [PubMed] [Google Scholar]
- Nishizono A, Khawplod P, Ahmed K, Goto K, Shiota S, Mifune K, Yasui T, Takayama K, Kobayashi Y, Mannen K, Tepsumethanon V, Mitmoonpitak C, Inoue S, Morimoto K. A simple and rapid immunochromatographic test kit for rabies diagnosis. Microbiol Immunol. 2008;52:243–249. doi: 10.1111/j.1348-0421.2008.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu S, Gotoh K, Yamashiro T, Yamagata J, Shin K, Fujioka T, Nishizono A. Transfer of antigen-pulsed dendritic cells induces specific T-cell proliferation and a therapeutic effect against long-term Helicobacter pylori infection in mice. Infect Immun. 2006;74:984–993. doi: 10.1128/IAI.74.2.984-993.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegtel DM, Cosmopoulos K, Thorly-Lawson DA, van Eijndhoven MA, Hospmans ES, Lindenberg JL, de Gruijl TD, Wurdinger T, Middeldorp JM. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci USA. 2010;107:6328–6333. doi: 10.1073/pnas.0914843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Préhaud C, Lay S, Dietzschold B, Lafon M. Glycoprotein of nonpathogenic rabies viruses is a key determinant of human cell apoptosis. J Virol. 2003;77:10537–10547. doi: 10.1128/JVI.77.19.10537-10547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Mabbott NA. Assessing the involvement of migratory dendritic cells in the transfer of the scrapie agent from the immune to peripheral nervous systems. J Neuroimmunol. 2007;187:114–125. doi: 10.1016/j.jneuroim.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Rieder M, Conzelmann KK. Interferon in rabies virus infection. Adv Virus Res. 2011;79:91–114. doi: 10.1016/B978-0-12-387040-7.00006-8. [DOI] [PubMed] [Google Scholar]
- Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175:5260–5268. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- Sakaki H, Imaizumi T, Matsumiya T, Kusumi A, Nakagawa H, Kubota K, Nishi N, Nakamura T, Hirashima M, Satoh K, Kimura H. Retinoic acid-inducible gene-I is induced by interleukin-1β in cultured human gingival fibroblasts. Oral Microbiol Immunol. 2005;20:47–50. doi: 10.1111/j.1399-302X.2005.00181.x. [DOI] [PubMed] [Google Scholar]
- Sethi S, Kerksiek KM, Brocker T, Kretzschmar H. Role of the CD8+ dendritic cell subset in transmission of prions. J Virol. 2007;81:4877–4880. doi: 10.1128/JVI.02345-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota S, Khawplod P, Ahmed K, Mifune K, Nishizono A. A pilot study on intradermal vaccination of Japanese rabies vaccine for pre-exposure immunization. Vaccine. 2008;26:6441–6444. doi: 10.1016/j.vaccine.2008.08.068. [DOI] [PubMed] [Google Scholar]
- Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- Siegemund S, Hartl A, von Buttlar H, Dautel F, Raue R, Freudenberg MA, Fejer G, Būttner M, Köhler G, Kirschning CJ, Sparwasser T, Alber G. Conventional bone marrow-derived dendritic cells contribute to toll-like receptor-independent production of alpha/beta interferon in response to inactivated parapoxvirus ovis. J Virol. 2009;83:9411–9422. doi: 10.1128/JVI.02362-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Fishbein DB, Rupprecht CE, Clark K. Unexplained rabies in three immigrants in the United States. A virologic investigation. N Engl J Med. 1991;324:205–211. doi: 10.1056/NEJM199101243240401. [DOI] [PubMed] [Google Scholar]
- Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- Thoulouze MI, Lafage M, Montano-Hirose JA, Lafon M. Rabies virus infects mouse and human lymphocytes and induces apoptosis. J Virol. 1997;71:7372–7380. doi: 10.1128/jvi.71.10.7372-7380.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Park CH, Noguchi K, Kojima D, Kubo T, Komiya N, Matsumoto T, Mitui MT, Ahmed K, Morimoto K, Inoue S, Nishizono A. Serial passage of a street rabies virus in mouse Neuroblastoma cells resulted in attenuation: potential role of the additional N-glycosylation of a viral glycoprotein in the reduced pathogenicity of street rabies virus. Virus Res. 2012;165:34–45. doi: 10.1016/j.virusres.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Zhao P, Ahao L, Zhang K, Feng H, Wang H, Wang T, Xu T, Feng N, Wang C, Gao Y, Huang G, Qin C, Yang S, Xia X. Infection with street strain rabies virus induces modulation of the microRNA profile of the mouse brain. Virol J. 2012;9:159–171. doi: 10.1186/1743-422X-9-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga EI, McGavern DB, Pruneda-Paz JL, Teng C, Oldstone MB. Bone marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat Immunol. 2004;5:1227–1234. doi: 10.1038/ni1136. [DOI] [PMC free article] [PubMed] [Google Scholar]