

Abstract
A combined (triplex) immunoassay for the simultaneous detection of three mycotoxins in grains was developed with superparamagnetic colour-encoded microbeads, in combination with two bead-dedicated flow cytometers. Monoclonal antibodies were coupled to the beads, and the amounts of bound mycotoxins were inversely related to the amounts of bound fluorescent labelled mycotoxins (inhibition immunoassay format). The selected monoclonal antibodies were tested for their target mycotoxins and for cross-reactivity with relevant metabolites and masked mycotoxins. In the triplex format, low levels of cross-interactions between the assays occurred at irrelevant high levels only. All three assays were influenced by the sample matrix of cereal extracts to some extent, and matrix-matched calibrations are recommended for quantitative screening purposes. In a preliminary in-house validation, the triplex assay was found to be reproducible, sensitive and sufficiently accurate for the quantitative screening at ML level. The triplex assay was critically compared to liquid chromatography–tandem mass spectrometry using reference materials and fortified blank material. Results for the quantification of ochratoxin A and zearalenone were in good agreement. However, the fumonisin assay was, due to overestimation, only suitable for qualitative judgements. Both flow cytometer platforms (Luminex 100 and FLEXMAP 3D) performed similar with respect to sensitivity with the advantages of a higher sample throughput and response range of the FLEXMAP 3D and lower cost of the Luminex 100.
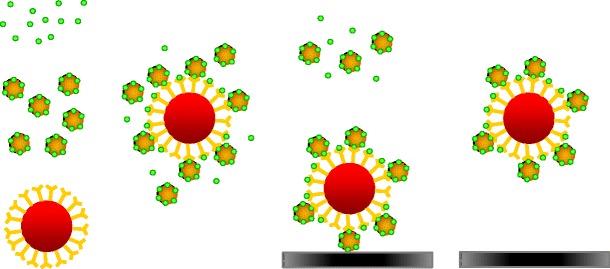
The priciple of the direct inhibition microbead immunoassay using fluorescent mycotoxin-reporter conjugates
Electronic supplementary material
The online version of this article (doi:10.1007/s00216-013-7095-7) contains supplementary material, which is available to authorized users.
Keywords: Mycotoxins, Masked, Multiplex, Immunoassay, Flow cytometer, Microspheres
Introduction
Mycotoxins are secondary metabolites produced by fungi as a part of their plant-invasive actions. Their toxic effects can range from nausea to cancer. In 2010, BIOMIN monitored the mycotoxin prevalence in raw materials and animal feed worldwide as part of their annual mycotoxin survey program. From more than 3,300 samples analyzed, 55, 26 and 42 % tested positive for the mycotoxins fumonisins, ochratoxin A and zearalenone, respectively [1]. Co-occurrence of mycotoxins in cereals can lead to increased toxicity by additive and synergistic effects [2]. Therefore, the risk for humans and animals increases, and this calls for multiplex mycotoxin testing.
Fumonisins (FBs) most likely to contaminate commodities are, in decreasing order of occurrence, fumonisin B1 (FB1), fumonisin B2 (FB2) and fumonisin B3 (FB3) [3]. Maize and sorghum are the main commodities where FBs naturally occur [4]. The maximum level (ML) set by the EU for FB1 + FB2 in unprocessed maize is 2,000 μg/kg [5]. Ochratoxin A (OTA) is the most common and most toxic ochratoxin [6] and primarily occurs in grain products, although its presence in coffee, cacao, grapes and indirect contamination in pork have been noted [7, 8]. The ML set by the EU for OTA is 5 μg/kg for all cereals [5]. Zearalenone (ZEN) is a non-steroidal oestrogenic compound [9] The main ZEN derivatives include α-zearalenol (α-ZEL), β-zearalenol (β-ZEL), zearalanone, α-zearalanol and β-zearalanol [10, 11]. ZEN is a frequent contaminant of maize, oats, wheat, barley, sorghum, millet and rice [9]. The ML set by the EU for ZEN in unprocessed maize is 200 μg/kg and for other cereals 100 μg/kg [5]. Conjugated mycotoxins, in which the toxin is usually bound to a more polar substance like glucose, are referred to as masked mycotoxins. Because of their conjugation, they escape routine analysis, provided that there is no reference substance available. They can emerge by metabolisation of the original molecule by living plants or food processing. Known plant conjugates for ZEN are zearalenone-14-β-d-glucopyranoside (Z14G), α-zearalenol-14-β-d-glucopyranoside (α-ZELG) and β-zearalenol-14-β-d-glucopyranoside (β-ZELG) and zearalenone 14-sulphate (Z14S) [12, 13] and can appear in a variety of food and feed products [14].
Methods for the detection of mycotoxins are mainly based on chromatography and immunochemistry. A number of liquid chromatography–tandem mass spectrometry (LC-MS/MS) methods detect a large range of mycotoxins and their metabolites in a variety of food [15–17] and feed commodities [18]. In 2007, Sulyok et al. [19] reported an LC-MS/MS method capable of detecting 87 analytes with just a single extraction step after which the diluted crude extract was measured directly. When applied, this method was able to detect 37 different metabolites in mouldy food samples. This method was then further extended with 99 fungal and bacterial metabolites [20]. Although the multiplex capacity and precision is high, and the need for clean-up is not always necessary [21], it cannot be considered as a rapid technology for a selection of mycotoxins.
The main immunochemical method used for the detection of mycotoxins is the enzyme-linked immunosorbent assay (ELISA) [22]. The ELISA format is robust, fast and has a high sample throughput. Sample purification is often minimal, even when screening raw materials. ELISAs for FB1, OTA and ZEN were previously developed [23–25], and ELISA kits for common mycotoxins are available from a variety of suppliers [26]. Nowadays, lateral flow tests and dipstick tests are other popular immunochemical techniques for the detection of mycotoxins. They are rapid, can be carried out in just a few minutes, the sample preparation is often very short and they do not need equipment. However, they are less sensitive when compared to instrumental methods, and the results are mostly qualitative, providing an answer on the presence or absence of a certain mycotoxin only [22]. Recently, a multiplex dipstick immunoassay for the indirect detection of ZEN, deoxynivalenol (DON), T-2/HT-2 toxin and FB was developed, but omitting the most toxic mycotoxins. A photometric strip reader was used to get semi-quantitative results [27]. Using conventional surface plasmon resonance (SPR), several applications are known for single [28] and for multiple (4) mycotoxins [29]. A new approach is the multiplex detection of mycotoxins using the label-free imaging SPR (iSPR) technique. Using an IBIS iSPR, capable of reading out a spotted microarray sensor chip, Dorokhin et al. [30] developed a method for the simultaneous detection of ZEN and DON. The used iSPR technology facilitates higher multiplexing capacity as was shown for allergens [31].
The demand for faster multiplex testing is high and new techniques are emerging. The proprietary xMAP® (Multi-Analyte Profiling) technology is a suspension array platform based upon colour-encoded microspheres, more often referred to as beads [32]. The beads have a carboxylated surface to facilitate the covalent coupling of biomolecules such as peptides, proteins, antibodies, polysaccharides, lipids and nucleotides. These beads can then be analysed in a bead-dedicated flow cytometer [33]. Previously, two bead-based indirect immunoassays for mycotoxins were described [7, 33] in which the mycotoxins were immobilised on the beads. In contrast, direct immunoassays having immobilised antibodies on the beads are rare. Very recently, Czeh et al. [34] described a similar approach as in the present work, but using a different analyser with non-paramagnetic beads. Unfortunately, experimental data as well as detailed procedures are lacking in [34], thus hampering a direct comparison with our results. Moreover, no LC-MS/MS or other confirmatory method was carried out using certified reference samples to verify or pre-validate that assay.
In our direct triplex assay for OTA, FB1 and ZEN described here, we created three unique mAb-coupled paramagnetic bead sets and mycotoxin-R-Phycoerythrin (RPE) conjugates as reporter molecules. Samples were all-in-one incubated with antibody-coupled beads and reporter molecules in a single well. The free mycotoxins in the samples compete with the mycotoxin–RPE conjugates for interaction with the antibodies on the beads (Fig. 1). This triplex format was tested for the application in wheat, maize and cereal-based feed. Two different bead-dedicated flow cytometers were explored. Moreover, results were critically compared with multi-mycotoxin LC-MS/MS.
Fig. 1.
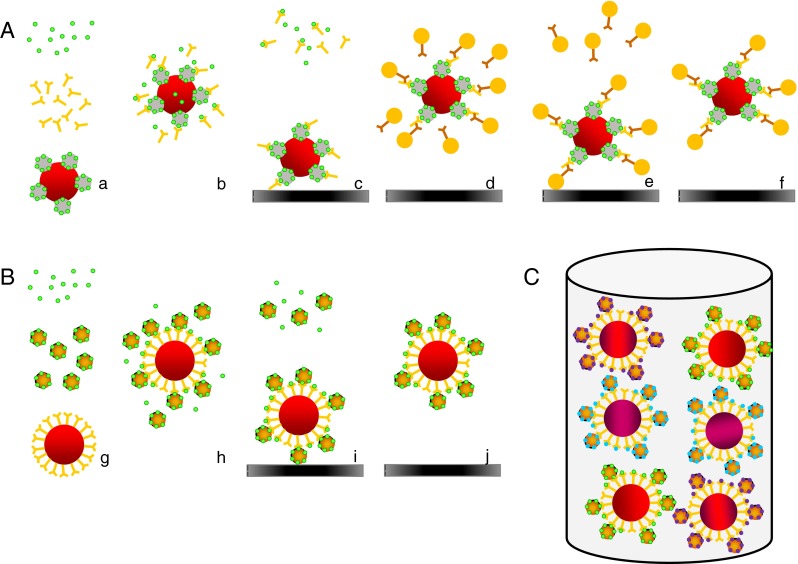
Comparison of the direct assay (this work) and indirect microsphere immunoassay [33]. In an indirect assay (A), sample, antibodies and mycotoxin-BSA conjugated beads (a) are incubated so that there is competition between the conjugated mycotoxins on the bead and the free mycotoxins in the sample (b). After incubation, the beads are trapped by a magnet and the non-bound reagents washed away (c). The beads are released and an anti-mouse-RPE is added (d). After incubation, the beads are trapped again and non-bound anti-mouse-RPE is washed away (e). After release, the beads are measured (f). In the much simpler direct assay presented in this work (B), sample, mycotoxin-RPE conjugate labels and antibody-coupled beads are incubated (g). Labelled and free mycotoxins compete for antibodies on the beads (h). Then beads are trapped by a magnet and the non-bound reagents washed away (i). Beads are released and measured (j). This is done all-in one for three different mycotoxins in one well (C)
Materials and methods
Instrumentation
For the measurement of the xMAP immunoassays, two different flow cytometers from Luminex (Austin, TX, USA) were used. The Luminex-100 (consisting of a LX-100TM analyser, a sheath fluid delivery system and the XY platform) and the new FLEXMAP 3D which integrates all of these components in one machine. The LX-100 operates on XPONENT software version 4.0 and the FM3D on version 4.1. A Bio-Plex II Wash Station with magnetic plate support (Bio-Rad Laboratories, Veenendaal, The Netherlands) was used for all washing steps. For the retention of the MagPlex beads during the antibody-microsphere coupling process, a DynaMag-2 magnetic separator stand (Invitrogen Dynal, Oslo, Norway) was used. A Bühler TiMix 2 (Salm en Kipp, Breukelen, The Netherlands) was used for all microtiter plate incubation steps. A REAX 2 overhead shaker (Heidolph, Schwabach, Germany) was utilised for the agitation of samples during mycotoxin extraction. Centrifugation of 50-ml Greiner tubes was done in an Eppendorf 5810R centrifuge using a A-4-62 rotor (VWR International, Amsterdam, The Netherlands) and high speed centrifugation of Eppendorf tubes with a Bio-Rad Model 16K Microcentrifuge (Bio-Rad Laboratories, Veenendaal, The Netherlands). A Vortex Genie 2 (Scientific Industries, New York, USA) was used to mix samples. Bead counting was done using a Bio-Rad TC10 automated cell counter (Bio-Rad Laboratories). For LC-MS/MS analysis, a Shimadzu Prominence high performance liquid chromatography system (Kyoto, Japan) was coupled with an AB SCIEX (Framingham, MA, USA) QTRAP 5500 triple quadrupole mass spectrometer, run in multiple reaction monitoring mode. The probe temperature was set at 400 °C. Additional MS/MS acquisition details are provided in Table S1 (see Electronic Supplementary Material). A Restek (Bellefonte, PA, USA) Ultra Aqueous C18 (100 × 2.1 mm) LC column was used. The chromatograms were integrated automatically with the Signal Finder integration algorithm of MultiQuant V2.0 software.
Chemicals and reagents
The MagPlex bead sets MC10026, MC10036, MC10038 and sheath fluid were obtained from Luminex. Cellstar 96-wells culture microtiter plates (Greiner, Alphen a/d Rijn, The Netherlands) were used for all assays. Centrifugal filter units (50 kDa), used for buffer exchange, and 30 kDa Amicon Ultra 4 centrifugal filter devices were purchased from Millipore (Bedford, MA, USA). Monoclonal antibodies (mAbs) against FB1 and OTA were purchased from Soft Flow Biotechnology Ltd. (Gödöllő, Hungary). The anti-ZEN mAb as well as the FB1 and OTA mycotoxins were purchased from Aokin AG (Berlin, Germany). Mycotoxin solutions of FB1, FB2, FB3, OTA, OTB, ZEN, α-ZEL and β-ZEL were purchased from Coring System Diagnostix (Gernsheim am Rhein, Germany). Z14G, α-ZELG, β-ZELG and Z14S were a kind gift of Dr. Franz Berthiller. The R-Phycoerythrin (RPE)-FB1 and RPE-OTA conjugates were produced in-house using RPE from Moss (Pasadena, MD, USA). For RPE conjugate purification, Amicon Ultra 4 centrifugal filter devices were used. The RPE-ZEN conjugate was custom made by Aokin AG using the same Moss RPE. MES (2-(N-morpholino) ethanesulfonic acid), sulfo-NHS (N-hydroxysulfosuccinimide) and EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide and caffeine-(trimethyl-13C3) were purchased from Sigma-Aldrich (Zwijndrecht, The Netherlands). Ethanol and all other solid chemicals were purchased from VWR International (Amsterdam, The Netherlands). Syringeless filter devices for sample clean-up were purchased from GE Healthcare (Rotterdam, The Netherlands). Acetonitrile (ACN) and methanol (MeOH) were purchased from Biosolve (Valkenswaard, The Netherlands), formic acid (FA) from Merck (Whitehouse Station, NJ, USA) and ammonium formate (AMF) from Fluka Analytical (Steinheim, Germany).
Matrix and reference samples
The blank maize and wheat materials were previously analysed at RIKILT using an accredited confirmatory LC-MS/MS method for feed samples (FB1, FB2 and FB3 < 0.1 mg/kg, OTA < 0.025 mg/kg, ZEN, α-ZEL and β-ZEL < 0.05 mg/kg). Naturally contaminated maize and wheat materials were either laboratory stock or purchased from Coring System Diagnostix (Gernsheim am Rhein, Germany) and R-Biopharm/Trilogy (Darmstadt, Germany) as reference materials. Additionally, naturally contaminated cereal-based feed samples were obtained from the Institute for Reference Materials and Measurements (Geel, Belgium).
Methods
mAb coupling to the beads
The mAbs against the three mycotoxins were separately coupled to three different sets of the paramagnetic MagPlexTM beads according to a modified protocol originally provided by Luminex. Initially, the antibody stock solutions (1 mg/ml) were diluted to a concentration of 100 μg/ml using 50 mM MES buffer (pH 5.0), transferred to a 50-kDa filtering unit and centrifuged at 10,000×g for 10 min at RT. The concentrated antibody solution was then reconstituted in 500 μl using the same MES buffer. The original bead stock was vortexed for 1 min to resuspend the beads. From the bead suspension, 1 ml (approximately 1.25 × 107 beads) was pipetted into an Eppendorf tube. The Eppendorf tube was then placed into the magnetic separator, and after a 1-min magnetic separation period, the supernatant was carefully removed without disturbing the beads. The Eppendorf tube was removed from the magnetic separator, and the beads were resuspended in 100 μl double distilled water. The Eppendorf tube was placed back into the magnetic separator, and after another 1 min separation, the supernatant was removed. This wash step was repeated twice, and the beads were resuspended in 100 μl of 100 mM monobasic sodium phosphate buffer (pH 6.2) and placed in the magnetic separator. The supernatant was discarded and the beads resuspended in 80 μl of the phosphate buffer. To this microsphere suspension, 10 μl of sulfo-NHS and 10 μl of EDC were added (both previously prepared at a concentration of 50 mg/ml with double distilled water). This microsphere suspension was vigorously mixed by vortex and incubated in the dark at RT for a total of 20 min with a vortex pulse at 5 min intervals. Following the incubation period, the tube was shortly centrifuged to make sure that all beads were at the bottom of the tube. After a 1-min incubation in the magnetic stand, the supernatant was removed. Beads were resuspended in 250 μl 50 mM MES (pH 5.0) and mixed by vortex. After magnetic separation, this step was repeated and the previously prepared mAb solution (500 μl) was added to the microsphere pellet and mixed by vortex. The activated beads and antibodies were then incubated in the dark at RT on a rotational shaker for 2 h. Following the incubation, the tube was centrifuged for 30 s, placed into the magnetic separator stand and, after a 45-s separation period, the supernatant was removed. The tube was then removed from the magnetic stand, and the beads were resuspended in 1 ml of phosphate buffered saline-TBN (PBS, 0.1 % bovine serum albumin (BSA), 0.02 % Tween-20 and 0.05 % sodium azide, pH 7.4). The bead suspension was placed into the magnetic stand, and after a 45-s separation period, the supernatant removed. This wash step was repeated twice. Finally, the covalently modified beads were suspended in 1 ml PBS-TBN and stored in the dark at 4 °C.
Bead counting
To make sure that the right number of beads of each set was added to the assay, coupled beads were counted with the Bio-Rad TC10™ automated cell counter. The bead suspensions were diluted 10-fold with PBST (PBS and 0.02 % Tween-20, pH 7.4), and 10 μl of each of the diluted suspension was applied to one of the counting chambers of a counter-dedicated slide. The slide was then inserted into the cell counter for charge-coupled device imaging. The number of beads in the readout was then multiplied by a factor 10.
Coupling of OTA and FB1 to RPE
The OTA-RPE conjugation procedure was identical to the one described previously by Aqai et al. [35]. The conjugation of FB1 to RPE was based on the method of Szurdoki et al. [31] with slight modifications: A glutaraldehyde buffer was prepared just before use by adding 400 μl of a 25 % glutaraldehyde solution, 0.4 g of NaCl and 5 ml of a 0.1 M sodium phosphate solution (pH 7.5) to a 50-ml tube, and the volume was adjusted to 50 ml using fresh double distilled water. From this buffer, 3 ml was added to a dark glass vial. From a fresh 10 mg/ml RPE solution, 50 μl was added to the same glass vial. The reaction mixture was incubated for 24 h with constant slow stirring and exchanged to PBS using 30-kDa centrifugal filter devices with a total of four centrifuge steps at 3,000×g and a total volume of 12 ml PBS. The volume of the final concentrate was adjusted to 3 ml with PBS. To the modified RPE solution, 200 μl of FB1 (10 mg/ml in methanol) was added dropwise. This reaction mixture was incubated overnight at room temperature. The next day, 16 μl of 0.2 M of l-lysine was added to the reaction, vortexed and placed in the fridge again for overnight incubation. The conjugate was purified by transferring the reaction mixture to a 30-kDa centrifugal filter device. The volume was adjusted to 4 ml with PBS and concentrated to 50 μl. This step was repeated three times, and the remaining conjugate solution volume was adjusted to 500 μl with PBS.
Sample fortification
Blank maize and wheat materials were fortified at the MLs for each mycotoxin for unprocessed cereals. For the fumonisins, we chose to fortify with FB1 at 2,000 μg/kg, which is the combined ML for FB1 + FB2 in unprocessed maize. Fortification with OTA was done at 5 μg/kg, which is the ML for all unprocessed cereals. For ZEN, we chose to fortify at 100 μg/kg, which is valid for almost all unprocessed cereals. For the sample fortification, 2.5 g of sample was weighed in a 50-ml tube. The mycotoxin solutions (in 10 % MeOH) were pipetted to the side of a near-horizontally positioned 50 ml tube (50 μl of 100 μg/ml for FB1, 125 μl of 0.1 μg/ml for OTA and 25 μl of 10 μg/ml for ZEN) and the lid carefully placed on. The tubes were then shaken to allow the mycotoxins to mix with the sample material. The tubes were then un-capped and allowed to air dry for 60 min.
Sample extraction for xMAP assays
In a 50-ml tube, 10 ml of extraction solvent (80 % MeOH) was added to 2.5 g of sample material. The tubes were then vortexed vigorously for 10 s and placed in the overhead shaker at a moderate speed setting for 30 min. The tubes were centrifuged at room temperature (RT) for 10 min at 4,000×g. The supernatant was transferred to another tube without disturbing the pellet. Before analysing, the supernatant was diluted 8-fold with double distilled water in order to reduce the influence of matrix effects and to ensure that quantitation (at MLs) occurred within the linear part of the curve. After dilution, the extracts were centrifuged once more at 12,000×g for 10 min at RT to remove non-soluble components.
Calibration standards and matrix-matched standards
Initially, calibration standard solutions containing individual mycotoxins were prepared and tested in monoplex assay format. For multiplex analysis, a multi-standard was prepared including the three mycotoxins. The calibration curve ranges were determined according to the sensitivities of the assays. Serial dilutions were made from 1 mg/ml stock solutions. For FB1, the highest calibration point was 10 μg/ml and for OTA and ZEN 1 μg/ml. From these initial calibration points, the other calibration points were prepared by 10-fold serial dilutions with 10 % MeOH in six steps. The eighth—and final—calibration point was a negative control consisting of 10 % MeOH. For the matrix-matched standards, the blank sample extract was diluted 4-fold with double distilled water and combined (1:1) with the mycotoxin calibration standard solutions, thus resulting in an 8-fold dilution of the matrix material in total and a 2-fold dilution of the standards. Finally, the diluted samples with and without standard solutions were centrifuged at 12,000×g for 10 min at RT to remove non soluble components. Curve fitting and calculation of the fitting parameters was done using GraphPad Prism 5 (La Jolla, CA, USA).
Determination of specificity
To determine the specificity of the selected antibodies for a wide range of known metabolites and masked forms of the three target mycotoxins, calibration curves were made with the following compounds: ochratoxin B (OTB), FB2, FB3, α-ZEL, β-ZEL Z14G, α-ZELG, β-ZELG and Z14S. Stock solutions were diluted in 10 % MeOH to get calibration curves under similar conditions as the original mycotoxins. These calibration curves of individual compounds were analysed in buffer and maize extracts using all beads and all mycotoxin-RPE conjugates. The cross-reactivity of these metabolites was calculated by dividing the concentration at 50 % inhibition (IC50 value) of the target compound by the IC50 of the metabolite/masked form.
Triplex immunoassay
The final triplex immunoassay procedure started with the addition of 40 μl of a matrix-matched calibration standard solution (containing OTA, FB1 and ZEN) or sample extract to a well of a low-protein binding 96-well microtiter plate. Then, 10 μl of bead suspension containing 1,000 mAb-coupled beads for each of the three assays (3,000 beads in total per sample) was added to each well. After a 2-min pre-incubation on a plate shaker, 10 μl of the mixture of three mycotoxin-RPE conjugates (diluted 100–200 times) was added to each well using PBST pH 7.4 as the assay buffer. The microtiter plate was then incubated at RT on a shaker for 15 min and subjected to a single pre-programmed automated wash with PBST (previously optimised to retain the beads). To the residual volume left by the washer (30 μl), 70 μl PBST was added, and the plate was mixed on a shaker for 1 min at RT. Finally, the plate was measured in the Luminex platform counting 100 beads per bead set. The overall measurement time for the assay (incubations, washing steps and the actual measurement) was 50 min for a 96-well plate on the FM3D platform.
LC-MS/MS analysis
The concentrations of FB1, OTA, ZEN and their metabolites were determined by an in-house ISO 17025 accredited LC-MS/MS confirmatory method using multiple reaction monitoring. Briefly, the LC-MS/MS method was as follows. Electrospray ionisation was used as the soft ionisation mode in negative (ZEN and associated metabolites) and positive (the remaining) modes. Reference samples were analysed in parallel with fortified blank sample material in order to assess the validity of the results via monitoring of extraction efficiencies. Retention time and ion ratio were used as quality control parameters according to the SANCO/12495/2011 document for method validation and quality control procedures for pesticide residues analysis in food and feed [36]. LC-MS/MS sample preparation was as follows. An internal standard of 13C3-caffeine (10 μg/ml) was added to the sample prior to extraction in order to monitor extraction and injection of each sample. From each sample, 2.5 g of material was weighed and extracted with 10 ml ACN/H2O/FA [84:15:1 (v/v/v)] for 2 h on a horizontal shaker at room temperature. Following extraction, the tubes were centrifuged at 3,000×g for 10 min at RT. The supernatant was diluted (1:1) with Milli-Q water, vortexed and refrigerated at 4 °C for 30 min to allow for fat content to settle. Following refrigeration, the samples were filtered in polypropylene filter devices. The LC-MS/MS system was run with 5-μl sample injections. Mycotoxins were eluted from the LC column with an aqueous/organic gradient consisting of mobile phase (A) water and (B) MeOH/H2O (95/5) at a column temperature of 35 °C. To each mobile phase, 1 mM AMF and 0.53 mM formic acid (FA) was added. The flow rate was set at 0.4 ml/min with a total runtime of 15 min.
Results and discussion
Building the triplex assay
The principle of the direct triplex assay is shown in Fig. 1B. It shows clearly that the direct format is much simpler than the indirect formats (Fig. 1A) previously developed [7, 33]. After extensive testing of a variety of mAbs from different sources coupled at a fixed concentration to the beads, three of them were selected based on the following criteria: good responses at low concentrations of the mycotoxin-RPE conjugates, low cross-interactions between the assays, desired cross-reactivity with other metabolites and dynamic range of the assays, which should match with the MLs established by the EU (Commission regulation No 1881/2006 foodstuffs) [37]. PBST was chosen as the optimal buffer for the triplex assay. The optimal dilutions of the mycotoxin-RPE conjugates were determined in monoplex assay format and in all cases the highest dilution (approximately 1 ng reporter each, per well), while still retaining substantial absolute signal, gave the best dynamic range (data not shown). At this stage, the monoplex assays were combined to create the triplex format. This means that three beads, three RPE reporter conjugates and three mycotoxin calibrants were present in a single well (Fig. 1C). This had an impact on the sensitivities of the assays. It resulted in a 3-fold decrease in sensitivity for the ZEN assay and a 2-fold decrease for the FB1 and OTA assay based on the IC50s of the dose–response curves (see Electronic Supplementary Material Fig. S1) most likely caused by combining the calibration curves and reporter conjugates all in one well. The incubation time was set at 15 min (the shortest incubation time tested) to comply with rapid testing. Longer incubation times (30 and 60 min) did not result in improved sensitivities. To check for cross-interactions between the assays, this triplex assay was also tested using the three antibody-coupled beads and the individual mycotoxin-RPE conjugates. The three bead sets were found to respond to their corresponding conjugates only (data not shown).
Cross-reactivity with metabolites and masked forms
Although the antibodies used in the assay were raised against specific mycotoxins (FB1, ZEN and OTA), depending on the chemistry of the conjugates used in immunisation, cross-reactivity with metabolites and masked forms can occur because of strong homologies. To check for this, the triplex assay was used to record calibration curves of the designated mycotoxins and other relevant metabolites in buffer, as well as in maize extract. Percentages of cross-reactivityies were calculated at 50 % inhibition.
The FB1 antibody shows a desirable high cross-reaction with FB2 (61 %) since legislation is for the sum of FB1 and FB2. The cross-reaction with FB3 might result in an overestimation, but fortunately, that metabolite occurs at much lower concentrations than FB1 (see section on naturally contaminated reference materials). The cross-reactivity of FB3 is lower in maize matrix (44 %) than in buffer (63 %). Only OTA at a very high concentration (1 μg/ml, corresponding with 32 mg/kg in a cereal product) showed an impact on the FB1 assay causing 40 % inhibition. The OTA antibody showed high cross-reactivity for OTB (43 % in maize), which is less desired since OTB is not as hazardous as OTA [35]. However, OTB occurs in much lower concentrations than OTA and therefore will not cause a significant problem [38]. FB1 at a high concentration of 10 μg/ml and β-ZEL at 1 μg/ml showed inhibition in the OTA assay (20 and 30 %).The ZEN antibody reacts in the ZEN assay with α-ZEL (66 %) and to a much lesser extent with β-ZEL (13 %). There is no legislation for α-ZEL and β-ZEL in cereals, so their cross-reaction might be considered undesirable. However, their occurrence is in much lower concentrations than ZEN itself [39]. All the other metabolites with a glucose or sulphate group located at the 14 position of the molecule have no cross-reactivity at all. Apparently, the epitopes of the conjugated ZEN, α-ZEL and β-ZEL molecules used in our research are blocked for antibody recognition. These results are comparable to Dorokhin et al. [30] who used the same antibody in a label-free iSPR approach, except the result for β-ZEL, which differs substantially (10-fold). This might be due to the entirely different technique and the indirect assay approach used in that work. In the ZEN assay, inhibition of the signal is observed (30 %) when FB1 is present, but at a very high concentration (10 μg/ml) only.
Comparing the LX-100 and FM3D flow cytometers
In general, the new FM3D flow cytometer shows higher absolute responses (6–10 times, in high PMT voltage mode) when compared to the LX-100. As can be seen in Fig. S2 (see Electronic Supplementary Material), the dose–response curves for all three mycotoxins are almost identical when the relative responses are plotted against the concentrations of the mycotoxins. Because of these results, we decided to conduct further research on the newer FM3D system because of its faster throughput, knowing that the developed assay would also perform well on the LX-100 when necessary. Previously, Bienenmann et al. [40] showed that both machines had good correlation when absolute responses were compared for a five-plex immunoassay for coccidiostats. Also other assays developed in our lab showed equal sensitivities on both machines. The FB1 antibody initially used in Fig. S2 was later replaced by a more sensitive one.
Effects of wheat and maize matrix on calibration curves
Matrix-matched calibration curves are frequently used to compensate for any matrix effects and to avoid inaccurate quantitation. To check for the possible effects of maize and wheat extracts in our newly developed direct triplex assay method, we compared calibration curves in buffer with those in sample extracts. From Fig. 2, it becomes clear that only maize has a strong suppressive effect on the relative responses of the FB1 and ZEN assays. The effect of wheat on the FB1 curve is much less pronounced. OTA is not influenced by the maize extract, but the wheat matrix yielded some sensitivity enhancement. These results show that matrix-matched calibration curves are to be preferred for quantitative data in this multiplex screening assay. According to the IC50 and limit of detection (LOD) data, the triplex assay developed shows LODs for maize and wheat that are adequate for routine monitoring at ML. For OTA, the sensitivities were 0.7 and 3.4 μg/kg, respectively (ML = 5 μg/kg); for ZEN, 5.8 and 32 μg/kg, respectively (ML = 100 μg/kg); and for FB1 + FB2, 170 and 1270 μg/kg, respectively (ML = 2000 μg/kg). Maize, the material with the highest matrix effect on the dose–response curves, was not tested (or presented) in the work of Czeh et al. [34]; therefore, we compared our wheat sensitivities with their results. Our OTA assay was two times more sensitive, while their ZEN and FB1 assays were respectively four and five times more sensitive. Most importantly, our method met with the EU required MLs in grains [5].
Fig. 2.

Dose–relative response curves for FB1 (a), OTA (b) and ZEN (c) in the triplex microsphere immunoassay in buffer and maize and wheat extracts (n = 3)
Preliminary in-house validation using wheat and maize samples
Grinded blank maize and wheat samples were fortified in triplicate by the addition of individual mycotoxins and their mixture. FB1 was added at 2,000 ng/g, OTA at 5 ng/g, and ZEN at 100 ng/g, corresponding with the MLs targeted by EU legislation [37]. The same samples were also left unfortified and extracted, being the blank controls. After extraction, all samples were quantified in triplicate using the triplex immunoassay. The obtained results showed satisfying inhibition levels for all added mycotoxins and hardly any difference between the single and mixture fortification in maize and wheat (Table 1). The reproducibility of the triplicate analyses was ranging from 1 to 10 % relative standard deviation (RSD) with an exception for the OTA mixture fortification in wheat (23 % RSD). In general, variations were slightly higher for the wheat extracts. The accuracy of the method was determined by quantitating samples using the aforementioned matrix-matched calibration curves. The experimental mycotoxin levels were compared with the initial fortification levels. The screening accuracies for OTA in wheat and maize were 50 and 70 %, respectively. For ZEN, there was a 1.5-fold overestimation in maize, while the accuracy in wheat was 84 and 74 %, respectively. The accuracies for FB1 were higher than expected, leading to a 4-fold overestimation in maize and 3-fold in wheat. The overestimation is reproducible and remarkable since the matrix-matched calibration curves have the same level of matrix components as the fortified samples analysed. To test for sample variance, six independent blank wheat samples (tested for feed MLs) were fortified at food ML levels (FB1 at 2,000 ng/g, OTA at 5 ng/g and ZEN at 100 ng/g). One sample a day was fortified in triplicate with each single mycotoxin and extracted. As a blank control, no mycotoxin was added. In addition, the controls were extracted in triplicate. The extracts were measured in triplicate in the triplex immunoassay. The results (Fig. 3) show that there is satisfactory inhibition in each assay and only slight variation in the relative responses between samples. Variation is probably due to the different blank wheat samples used for fortification. They were considered blanks according to an in-house feed LC-MS/MS method, which does not exclude the presence of mycotoxins below the limits of detection. Another reason for the response variation could be that each independent sample was fortified, extracted and measured on a different day so inter-day extraction variation may occur.
Table 1.
Reproducibility and accuracy of the triplex immunoassay (n = 9) using single and mixed (all three mycotoxins) fortifications to wheat and maize with FB1, ZEN and OTA at MLs
| Mycotoxin fortification | Maize | Wheat | ||||||
|---|---|---|---|---|---|---|---|---|
| Fortification level (μg/kg) | Experimental level (μg/kg) | RSD (%) | Accuracy (%) | Fortification level (μg/kg) | Experimental level (μg/kg) | RSD (%) | Accuracy (%) | |
| FB1 single | 2,000 | 8,100 | 2 | 405 | 2,000 | 5,900 | 10 | 295 |
| FB1 mixed | 2,000 | 8,020 | 2 | 401 | 2,000 | 5,200 | 4 | 260 |
| OTA single | 5 | 3.46 | 4 | 69 | 5 | 2.5 | 4 | 50 |
| OTA mixed | 5 | 3.30 | 10 | 66 | 5 | 2.1 | 23 | 42 |
| ZEN single | 100 | 155 | 1 | 155 | 100 | 84 | 4 | 84 |
| ZEN mixed | 100 | 147 | 1 | 147 | 100 | 74 | 7 | 74 |
Fig. 3.

Relative responses (n = 9) obtained in the triplex microsphere immunoassay by multi-fortification of six different blank wheat samples with FB1 (2,000 μg/kg, blue bars), OTA (5 μg/kg, red bars) and ZEN (100 μg/kg, green bars)
Application to the analysis of incurred reference materials
An ISO 17025 accredited LC-MS/MS multi-mycotoxin method was used to analyse naturally contaminated maize (n = 11) and wheat (n = 6) reference samples and cereal-based feed samples (n = 3) using matrix-matched calibration curves. This instrumental multi-mycotoxin method determines 33 mycotoxins and metabolites. A summary of the results, and critical comparison with the triplex screening assay, is presented in Table 2.
Table 2.
Comparison of the results obtained with the triplex microsphere immunoassay (n = 3) and with LC-MS/MS (n = 2) using extracts of naturally contaminated wheat, maize and feed samples
| Sample ID | Material | Assigned mycotoxin (ng/g) | Mycotoxin concentrations (ng/g) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LC-MS/MS | Triplex immunoassay | |||||||||||
| FB1 | FB2 | FB3 | OTA | ZEN | α-ZEL | β-ZEL | FBa | OTA | ZENb | |||
| BRM 003022/M10482A | Wheat | DON/877 ± 23 | 155 | 38 | 16 | 0 | 122.7 | 5.7 | 0 | 1,763 | 1.0 | 142.6 |
| TR-O100/O-W-813 | Wheat | OTA/23.3 ± 3.2 | 141 | 41 | 22 | 27.1 | 0 | 0 | 8.8 | 2,304 | 14.0 | 1.0 |
| TR-O100/O-W-816 | Wheat | OTA/101.8 ± 12.2 | 7 | 2 | 1 | 120.2 | 4.1 | 0 | 0 | 320 | 77.0 | 5.0 |
| TR-O100/O-W-805 | Wheat | OTA/3.2 ± 0.6 | 15 | 4 | 2 | 4.3 | 0 | 0 | 0 | 480 | 2.2 | 3.0 |
| TR-D100/D-W-153 | Wheat | DON/2,100 ± 200 | 1 | 1 | 0 | 0 | 3.4 | 0 | 18.8 | 203 | 0 | 21.4 |
| TR-D100/D-W-163 | Wheat | DON/500 ± 100 | 2 | 1 | 0 | 0 | 0.9 | 0.2 | 8.0 | 164 | 0 | 28.0 |
| Mixed cereal 1 | Feed | OTA/NAc | 100 | 23 | 16 | 76.1 | 6.6 | 0 | 0 | 1,104 | 30.0 | 33.0 |
| Mixed cereal 2 | Feed | OTA/NAc | 79 | 21 | 13 | 266.0 | 7.9 | 0 | 19.0 | 1,412 | 171.0 | 54.0 |
| Mixed cereal 3 | Feed | OTA/NAc | 142 | 36 | 23 | 706.6 | 9.9 | 0 | 0 | 1,859 | 403.0 | 43.0 |
| BRM 003012/M10195A | Maize | DON/2010 ± 290 | 703 | 197 | 82 | 0 | 133.5 | 3.5 | 0. | 3,104 | 0 | 358 |
| BRM 003018/M10203C | Maize | FB1/270 ± 110 | 248 | 61 | 25 | 0 | 45.2 | 3.3 | 0 | 5,181 | 0 | 79.0 |
| FB2/<80 | ||||||||||||
| FB3/<80 | ||||||||||||
| TR-D100/D-C-606 | Maize | DON/1,100 ± 100 | 106 | 27 | 12 | 0.2 | 74.0 | 0 | 0 | 544 | 0 | 101 |
| TR-Z100/Z-C-310 | Maize | ZEN/59.4 ± 10.5 | 7,988 | 2,196 | 824 | 0 | 66.3 | 0 | 0 | 17,728 | 0 | 68.0 |
| BRM 003003/M09452Z | Maize | FB1/2406 ±630 | 2,123 | 644 | 271 | 0.2 | 21.6 | 0 | 0 | 15,040 | 0 | 156 |
| FB2/630 ± 116 | ||||||||||||
| TR-MT100/T-C-973 | Maize | T-2/1,153 ± 160 | 7,894 | 2,192 | 943 | 0 | 16.7 | 0 | 0 | 12,384 | 0 | 49.3 |
| H-T2/1381 ± 177 | ||||||||||||
| TR-A100/A-C-274 | Maize | AFB1/7.3 ± 0.9 | 1,552 | 412 | 161 | 0 | 60.4 | 1.3 | 0 | 5,376 | 0 | 77.4 |
| BRM 003017/M10203B | Maize | FB1/2,630 ± 740 | 2,841 | 811 | 310 | 1.1 | 206.6 | 12.7 | 6.3 | 15,424 | 1.3 | 184 |
| FB2/690 ± 340 | ||||||||||||
| FB3/310 ± 210 | ||||||||||||
| BRM 003019/M10205A | Maize | ZEN/177.3 ± 64.8 | 1,842 | 560 | 215 | 0 | 161.5 | 28.4 | 27.6 | 15,542 | 0 | 377 |
| TR-A100/A-C-268 | Maize | AFB1/4.3 ± 0.5 | 1,548 | 309 | 170 | 0 | 29.4 | 0 | 0 | 5,504 | 0 | 57.9 |
| AFB2/0.3 ± 0.1 | ||||||||||||
| TR-A100/A-C-276 | Maize | AFB1/1.7 ± 0.3 | 1,165 | 300 | 125 | 0 | 18.9 | 0 | 0 | 4,521 | 0 | 31.7 |
aSum of FB1, FB2 and FB3 according to antibody specificity
bSum of ZEN, α-ZEL and β-ZEL according to antibody specificity
cNA means not assigned for target mycotoxins
The immunoassay results for the OTA-containing reference and mixed cereal samples showed similar accuracy’s (∼50 %) as reported in the fortification experiments. In sample BRM 003022/M10482A, the immunoassay detects OTA at a concentration of 1 ng/g, while it is not present in the LC-MS/MS measurement. This is probably not due to the sensitivity of the LC-MS/MS method, since in two samples, concentrations below 1 ng/g are detected. It may be caused by OTB, which was not included in the LC-MS/MS method but will cross-react in the immunoassay. For ZEN, concentrations found in samples BRM 003022/M10482A and TR-O100/O-W-816 for the immunoassay correlate very well with the data found in the LC-MS/MS method. Also for sample TR-O100/O-W-813, the correlation is good since the immunoassay value (1 ng/g) is based on the 13–17 % cross-reaction of the mAb with the β-ZEL metabolite detected by the LC-MS/MS (8.8 ng/g). In contrast, much higher values for the immunoassay were found in samples TR-D100/D-W-153, TR-D100/D-W-163 and the mixed cereal samples (4–7-fold). For those samples, the overestimation cannot be explained by the presence of the α-ZEL and/or β-ZEL metabolites. Furthermore, it cannot be explained by the presence of the masked metabolites Z14G, α-ZELG, β-ZELG or Z14S since the ZEN mAb does not have any cross-reaction with those. For sample TR-O100/O-W-805, a concentration of 3 ng/g is determined with the immunoassay, while the LC-MS/MS does not detect any ZEN metabolite. These overestimations might be caused by other possible masked forms with which the ZEN mAb might cross-react. In addition, Thongrussamee et al. [25] found overestimations when two ELISA kits and high-performance liquid chromatography (HPLC) were compared. In the six wheat reference samples, FBs were found by LC-MS/MS (ranging from 0 to 155 ng/g). As expected, these concentrations were largely overestimated by the triplex immunoassay. The highest overestimation (100-fold) occurred at the lowest (according to LC-MS/MS) concentration (2 ng/g for FB1 + FB2). At the highest concentration found by LC-MS/MS, 193 ng/g (for FB1 + FB2), the immunoassay overestimation was approximately 10-fold. These overestimations are much higher when compared to the fortification experiments done for FB1. Note that the FB1 mAb is unable to distinguish between the metabolites, and therefore, the FB screening assay is indicative for the total level of fumonisins present. To study this overestimation issue further, a different antibody was tested under the same circumstances, but again, this resulted in high overestimations. After testing a range of buffers, PBST with a pH of 7.4 was chosen as the most optimal buffer. A more basic buffer (NaHCO3, pH 9.6) had dramatic consequences for the ZEA assay although it was beneficial for the sensitivity for the OTA assay. Using MES buffer pH 5, as a more acidic approach, we found a 50-fold decrease for the OTA sensitivity and a 10-fold decrease for the ZEA sensitivity. Furthermore, pHs outside the 5–10 regio will destabilise the RPE making it lose its reporter fluorescence. Using this standard PBST buffer, a range of additives [fish gelatin (1 %), Ficoll (1 %), polyethylene glycol (1 %), polyvinyl alcohol (1 %), polyvinylpyrrolidone (3 %) and skimmed milk powder (1 %)] were tested but without improved results. Omitting Tween-20 from the buffer is not an option since this is necessary to keep the beads from clustering. Furthemore, the same extraction method but with acidified solvent had no effect. Extraction with ACN/water generally showed decreased overestimation for maize samples, but this improvement (reduced FB1 overestimation) was not seen for the wheat samples (results not shown). Moreover, the ACN/water extraction had a negative impact on the sensitivities of the OTA and ZEN assay. Using MeOH/water/FA (80:20:0.1 v/v/v) showed the same overestimation as the MeOH/water extraction. Overestimation of FB1 in immunoassays has been widely documented. Tejada-Simon et al. [41] found consistently higher concentrations of FB1 when compared to HPLC. Kulisek et al. [42] described that extensively diluted samples yielded higher interpolated values for ELISA. Another issue causing inaccuracies might be the presence of bound fumonisins. Dall’Asta et al. [43] detected bound FBs in gluten-free food products in even higher concentrations than the free forms. Furthermore, hidden FBs were also found in unprocessed food but in a non-covalent bound form [43]. These non-covalent bound hidden FBs are referred to as extractable hidden FBs. To date, physical characterisation of the non-covalent interaction of FBs with matrix components was not carried out yet [44]. However, if we would apply a decision level (DL) for FB1 of 4,000 μg/kg for the triplex screening assay, then it correlates quiet well with the LC-MS/MS data for maize. Looking at the maize samples that have concentrations of FB1 and FB2 around or above the ML of 2,000 ng/g, we would then see no false-negative results and just one false positive (BRM 003018/M10203C) (Table 2).
Although the triplex assay principle, described in this work, seems similar to the work of Czeh et al. [34], there are some crucial differences. Besides sensitivity (discussed above), we used certified reference materials designated to single mycotoxins (and in case of FB1 also the FB2 and FB3 metabolites) in combination with a confirmatory LC-MS/MS method to get full information about all relevant metabolites present for a comprehensive comparison of the triplex assay results. The cross-reactivity of the coupled antibodies with known metabolites of the targeted mycotoxins were not researched nor mentioned in [34], while they can lead to undesired under- or overestimation of the original target mycotoxins.
To conclude, the developed direct inhibition multiplex immunoassay approach is faster and requires less procedural steps than the previously developed indirect assay format [33]. Moreover, because of the colour-encoded microsphere concept, more mycotoxins can be easily added at a later stage in order to extend the application range of this rapid assay.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 184 KB)
Acknowledgements
The authors would like to thank Frank Mallwitz from Aokin AG for fruitful discussions considering the RPE conjugate production. This research was carried out in the MultiTox project (PID091008/2.6A), financed by the Dutch Ministry of Economic Affairs, SenterNovem and the Province of Gelderland.
Footnotes
Published in the topical collection Rapid Detection in Food and Feed with guest editors Rudolf Krska and Michel Nielen.
References
- 1.Rodrigues I, Naehrer K. Biomin Survey 2010: mycotoxins inseparable from animal commodities and feed. AllAboutFeed. 2011;2(2):17–20. [Google Scholar]
- 2.Pedrosa K, Borutova R. Synergistic effects of mycotoxins discussed. Feedstuffs. 2011;83(19):1–3. [Google Scholar]
- 3.Jackson LS, Hlywka JJ, Senthil KR, Bullerman LB, Musser SM. Effects of time, temperature, and pH on the stability of fumonisin B1 in an aqueous model system. J Agric Food Chem. 1996;44(3):906–912. doi: 10.1021/jf950364o. [DOI] [Google Scholar]
- 4.Shetty PH, Bhat RV. Natural occurrence of fumonisin B1 and its co-occurrence with aflatoxin B1 in Indian sorghum, maize, and poultry feeds. J Agric Food Chem. 1997;45(6):2170–2173. doi: 10.1021/jf960607s. [DOI] [Google Scholar]
- 5.Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs (2006) J Eur Union L364:5–24
- 6.Belli N, Marin S, Sanchis V, Ramos AJ. Review: ochratoxin A (OTA) in wines, musts and grape juices: occurrence, regulations and methods of analysis. Food Sci Technol Int. 2002;8(6):325–335. [Google Scholar]
- 7.Anderson GP, Kowtha VA, Taitt CR. Detection of fumonisin B1 and ochratoxin A in grain products using microsphere-based fluid array immunoassays. Toxins. 2010;2(2):297–309. doi: 10.3390/toxins2020297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Brien E, Dietrich DR. Ochratoxin A: the continuing enigma. Crit Rev Toxicol. 2005;35(1):33–60. doi: 10.1080/10408440590905948. [DOI] [PubMed] [Google Scholar]
- 9.Zinedine A, Soriano JM, Molto JC, Manes J. Review on the toxicity, occurrence, metabolism, detoxification, regulations and intake of zearalenone: an oestrogenic mycotoxin. Food Chem Toxicol. 2007;45(1):1–18. doi: 10.1016/j.fct.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 10.Rai M, Varma A, Golinski P, Waskiewicz A, Gromadzka K (2010) Zearalenone and its derivatives: known toxins in new aspects. In: Rai M, Varma A (eds) Mycotoxins in food, feed and bioweapons. Springer, Berlin, Heidelberg, pp 113–129
- 11.Metzler M. Proposal for a uniform designation of zearalenone and its metabolites (Mycotoxin Research) Mycotoxin Res. 2011;27(1):1–3. doi: 10.1007/s12550-010-0075-2. [DOI] [PubMed] [Google Scholar]
- 12.Berthiller F, Schuhmacher R, Adam G, Krska R. Formation, determination and significance of masked and other conjugated mycotoxins. Anal Bioanal Chem. 2009;395(5):1243–1252. doi: 10.1007/s00216-009-2874-x. [DOI] [PubMed] [Google Scholar]
- 13.Berthiller F, Crews C, Dall'Asta C, De Saeger S, Haesaert G, Karlovsky P, Oswald IP, Seefelder W, Speijers G, Stroka J. Masked mycotoxins: a review. Mol Nutr Food Res. 2013;57(1):165–186. doi: 10.1002/mnfr.201100764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Boevre M, Di Mavungu JD, Landschoot S, Audenaert K, Eeckhout M, Maene P, Haesaert G, De Saeger S. Natural occurrence of mycotoxins and their masked forms in food and feed products. World Mycotoxin J. 2012;5(3):207–219. doi: 10.3920/WMJ2012.1410. [DOI] [Google Scholar]
- 15.Diana Di Mavungu J, Monbaliu S, Scippo ML, Maghuin-Rogister G, Schneider YJ, Larondelle Y, Callebaut A, Robbens J, Van Peteghem C, De Saeger S. LC-MS/MS multi-analyte method for mycotoxin determination in food supplements. Food Addit Contam Part A. 2009;26(6):885–895. doi: 10.1080/02652030902774649. [DOI] [PubMed] [Google Scholar]
- 16.Sulyok M, Berthiller F, Krska R, Schuhmacher R. Development and validation of a liquid chromatography/tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. Rapid Commun Mass Spectrom. 2006;20(18):2649–2659. doi: 10.1002/rcm.2640. [DOI] [PubMed] [Google Scholar]
- 17.Spanjer MC, Rensen PM, Scholten JM. LC-MS/MS multi-method for mycotoxins after single extraction, with validation data for peanut, pistachio, wheat, maize, cornflakes, raisins and figs. Food Addit Contam Part A. 2008;25(4):472–489. doi: 10.1080/02652030701552964. [DOI] [PubMed] [Google Scholar]
- 18.Monbaliu S, Van Poucke C, Detavernier C, Dumoulin F, Van De Velde M, Schoeters E, Van Dyck S, Averkieva O, Van Peteghem C, De Saeger S. Occurrence of mycotoxins in feed as analyzed by a multi-mycotoxin LC-MS/MS method. J Agric Food Chem. 2010;58(1):66–71. doi: 10.1021/jf903859z. [DOI] [PubMed] [Google Scholar]
- 19.Sulyok M, Krska R, Schuhmacher R. A liquid chromatography/tandem mass spectrometric multi-mycotoxin method for the quantification of 87 analytes and its application to semi-quantitative screening of moldy food samples. Anal Bioanal Chem. 2007;389(5):1505–1523. doi: 10.1007/s00216-007-1542-2. [DOI] [PubMed] [Google Scholar]
- 20.Vishwanath V, Sulyok M, Labuda R, Bicker W, Krska R. Simultaneous determination of 186 fungal and bacterial metabolites in indoor matrices by liquid chromatography/tandem mass spectrometry. Anal Bioanal Chem. 2009;395(5):1355–1372. doi: 10.1007/s00216-009-2995-2. [DOI] [PubMed] [Google Scholar]
- 21.Sulyok M, Krska R, Schuhmacher R. Application of an LC-MS/MS based multi-mycotoxin method for the semi-quantitative determination of mycotoxins occurring in different types of food infected by moulds. Food Chem. 2010;119(1):408–416. doi: 10.1016/j.foodchem.2009.07.042. [DOI] [Google Scholar]
- 22.Koppen R, Koch M, Siegel D, Merkel S, Maul R, Nehls I. Determination of mycotoxins in foods: current state of analytical methods and limitations. Appl Microbiol Biotechnol. 2010;86(6):1595–1612. doi: 10.1007/s00253-010-2535-1. [DOI] [PubMed] [Google Scholar]
- 23.Usleber E, Straka M, Terplan G. Enzyme immunoassay for fumonisin B1 applied to corn-based food. J Agric Food Chem. 1994;42(6):1392–1396. doi: 10.1021/jf00042a027. [DOI] [Google Scholar]
- 24.Yu FY, Chi TF, Liu BH, Su CC. Development of a sensitive enzyme-linked immunosorbent assay for the determination of ochratoxin A. J Agric Food Chem. 2005;53(17):6947–6953. doi: 10.1021/jf0513922. [DOI] [PubMed] [Google Scholar]
- 25.Thongrussamee T, Kuzmina NS, Shim WB, Jiratpong T, Eremin SA, Intrasook J, Chung DH. Monoclonal-based enzyme-linked immunosorbent assay for the detection of zearalenone in cereals. Food Addit Contam Part A. 2008;25(8):997–1006. doi: 10.1080/02652030701823159. [DOI] [PubMed] [Google Scholar]
- 26.Krska R, Schubert-Ullrich P, Molinelli A, Sulyok M, MacDonald S, Crews C. Mycotoxin analysis: an update. Food Addit Contam Part A. 2008;25(2):152–163. doi: 10.1080/02652030701765723. [DOI] [PubMed] [Google Scholar]
- 27.Lattanzio VMT, Nivarlet N, Lippolis V, Gatta SD, Huet A-C, Delahaut P, Granier B, Visconti A. Multiplex dipstick immunoassay for semi-quantitative determination of Fusarium mycotoxins in cereals. Anal Chim Acta. 2012;718:99–108. doi: 10.1016/j.aca.2011.12.060. [DOI] [PubMed] [Google Scholar]
- 28.Hodnik V, Anderluh G. Toxin detection by surface plasmon resonance. Sensors. 2009;9(3):1339–1354. doi: 10.3390/s9031339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Gaag B, Spath S, Dietrich H, Stigter E, Boonzaaijer G, van Osenbruggen T, Koopal K. Biosensors and multiple mycotoxin analysis (World Mycotoxin Forum, May 2001) Food Control. 2003;14(4):251–254. doi: 10.1016/S0956-7135(03)00008-2. [DOI] [Google Scholar]
- 30.Dorokhin D, Haasnoot W, Franssen MC, Zuilhof H, Nielen MW. Imaging surface plasmon resonance for multiplex microassay sensing of mycotoxins. Anal Bioanal Chem. 2011;400(9):3005–3011. doi: 10.1007/s00216-011-4973-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rebe Raz S, Haasnoot W. Multiplex bioanalytical methods for food and environmental monitoring. Trends Anal Chem. 2011;30(9):1526–1537. doi: 10.1016/j.trac.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu HY, Joos TO, Koga H. Multiplex microsphere-based flow cytometric platforms for protein analysis and their application in clinical proteomics—from assays to results. Electrophoresis. 2009;30(23):4008–4019. doi: 10.1002/elps.200900211. [DOI] [PubMed] [Google Scholar]
- 33.Peters J, Ploum ME, Rijk TC, Haasnoot W. Development of a multiplex flow cytometric microsphere immunoassay for mycotoxins and evaluation of its application in feed. Mycotoxin Res. 2011;27(1):63–72. doi: 10.1007/s12550-010-0077-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Czeh A, Mandy F, Feher-Toth S, Torok L, Mike Z, Koszegi B, Lustyik G. A flow cytometry based competitive fluorescent microsphere immunoassay (CFIA) system for detecting up to six mycotoxins. J Immunol Methods. 2012;384(1–2):71–80. doi: 10.1016/j.jim.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 35.Aqai P, Peters J, Gerssen A, Haasnoot W, Nielen MF. Immunomagnetic microbeads for screening with flow cytometry and identification with nano-liquid chromatography mass spectrometry of ochratoxins in wheat and cereal. Anal Bioanal Chem. 2011;400(9):3085–3096. doi: 10.1007/s00216-011-4974-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.SANCO/12495/2011 (2011) Method validation and quality control procedures for pesticides residues analysis in food and feed. http://www.crl-pesticides.eu/library/docs/fv/SANCO12495-2011.pdf. Accessed 15 Mar 2013
- 37.Commission Regulation (EC) No 1881/2006 maximum levels for certain contaminants in foodstuffs (2006)
- 38.Nielsen K, Mogensen J, Johansen M, Larsen T, Frisvad J. Review of secondary metabolites and mycotoxins from the Aspergillus niger group. Anal Bioanal Chem. 2009;395(5):1225–1242. doi: 10.1007/s00216-009-3081-5. [DOI] [PubMed] [Google Scholar]
- 39.Bottalico A, Visconti A, Logrieco A, Solfrizzo M, Mirocha CJ. Occurrence of zearalenols (diastereomeric mixture) in corn stalk rot and their production by associated Fusarium species. Appl Environ Microbiol. 1985;49(3):547–551. doi: 10.1128/aem.49.3.547-551.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bienenmann-Ploum ME, Huet AC, Campbell K, Fodey TL, Vincent U, Haasnoot W, Delahaut P, Elliott CT, Nielen MW. Development of a five-plex flow cytometric immunoassay for the simultaneous detection of six coccidiostats in feed and eggs. Anal Bioanal Chem. 2012;404(5):1361–1373. doi: 10.1007/s00216-012-6214-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tejada-Simon MV, Marovatsanga LT, Pestka JJ. Comparative detection of fumonisin by HPLC, ELISA, and immunocytochemical localization in Fusarium cultures. J Food Prot. 1995;58(6):666–672. doi: 10.4315/0362-028X-58.6.666. [DOI] [PubMed] [Google Scholar]
- 42.Kulisek ES, Hazebroek JP. Comparison of extraction buffers for the detection of fumonisin B1 in corn by immunoassay and high-performance liquid chromatography. J Agric Food Chem. 1999;48(1):65–69. doi: 10.1021/jf990696j. [DOI] [PubMed] [Google Scholar]
- 43.Dall'Asta C, Galaverna G, Mangia M, Sforza S, Dossena A, Marchelli R. Free and bound fumonisins in gluten-free food products. Mol Nutr Food Res. 2009;53(4):492–499. doi: 10.1002/mnfr.200800088. [DOI] [PubMed] [Google Scholar]
- 44.Scott PM. Recent research on fumonisins: a review. Food Addit Contam Part A. 2012;29(2):242–248. doi: 10.1080/19440049.2010.546000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 184 KB)