

Abstract
Background
RBFOX1 is an important splicing factor regulating developmental and tissue-specific alternative splicing in heart, muscle, and neuronal tissues. Constitutional genetic defects in RBFOX1 are implicated in multiple medical conditions.
Results
We identified 14 copy number variants (CNV) involving RBFOX1 from 2,124 consecutive pediatric patients referred for chromosomal microarray analysis (CMA), including 13 intragenic deletions and a single intragenic duplication. The clinical significances of the intragenic deletions of RBFOX1 were evaluated.
Conclusions
Our data strongly supports the associations of intragenic deletions of RBFOX1 with a diversity of neurodevelopmental and neuropsychiatric disorders, and possibly other clinical features.
Keywords: Microdeletion, Chromosome 16p13.3, RBFOX1, Chromosomal microarray analysis (CMA), Seizures, Developmental delay
Background
RBFOX1 [RNA binding protein, fox-1 homolog (C. elegans)], located at human chromosome 16p13.3, is one of the three member in Fox gene family encoding splicing factors [1,2]. It is specifically expressed in heart, muscle and neuronal tissues, regulating developmental and tissue-specific alternative splicing by binding with (U)GCAUG sequence in introns flanking alternative exons of its target genes [1,3-6]. RBFOX1 was initially identified as a binding partner of the brain-specific protein ATXN2, causing spinocerebellar ataxia 2 (SCA2) when the CAG repeats in this gene are expanded. Since then, constitutional genetic defects in RBFOX1 are implicated in multiple medical conditions including mental retardation and epilepsy [7], bipolar schizoaffective disorder [8], attention-deficit hyperactivity disorder (ADHD) [9], autism [10-13], hand osteoarthritis [14], congenital heart defects [15], obesity and diabetes [16,17], Intracranial arachnoid cysts [18], and primary biliary cirrhosis [19].
Results and discussion
In this report, we evaluated the clinical significances of the intragenic deletions/duplication of RBFOX1 in 12 out of 14 patients who carry the genomic imbalances. Fourteen copy number variants (CNV) involving RBFOX1, including 13 intragenic deletions and a single intragenic duplication, were identified in this cohort (Figure 1; Table 1), representing ~0.66% (14/2,124) of the patients analyzed in this study. There are no other clinically relevant CNVs in their genomes except in Patient 6 who carries an additional 178 kb deletion of 1p31.3 involving NF1A besides the intragenic deletion of RBFOX. Of the 13 deletions, 4 of them (in patients 1, 3, 5, and 6) involve exons while the remaining deletions involve only introns. The intragenic duplication in Patient 6 involves exons 4 and 5 of RBFOX1. The deletions/duplications were confirmed by qPCR methods (data not shown). The deletion in Patient 5 was maternally inherited while both deletions in Patient 6 occurred de novo. The inheritance patterns of the deletions/duplication could not be determined in remaining patients due to unavailability of DNA samples from either one parent in Patient 8 or both parents in other patients. By comparing breakpoints of the 14 intragenic deletions/duplication within the group and with the CNVs involving RBFOX1 documented in the DGV (http://projects.tcag.ca/variation/), multiple hot spots involving the generation of CNVs within RBFOX1 were observed, such as the proximal breakpoints of the CNVs in Patients 7, 8, and 11, and both proximal and distal breakpoints of the CNVs in Patients 9 and 10 (Figure 1; Table 1). In addition, G-banding analysis showed negative results for all 14 patients while a pathogenic mutation in ZEB2 and a pathogenic mutation in F8 were identified in Patient 1 and Patient 9 respectively.
Figure 1.
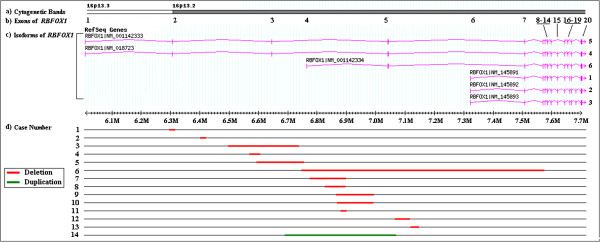
Genomic overview of the intragenic deletions (patients 1–13) and duplication of RBFOX1 (patient 14) in this study.
Table 1.
CNVs involving RBFOX1gene on chromosome 16
| Case | Start* (bp) | Stop*(bp) | Size (bp) | Del/Dup | Affected exon(s) | Inheritance and comments |
|---|---|---|---|---|---|---|
| 1 |
6,296,453 |
6,317,524 |
21,071 |
Del |
2 |
NT, a diagnosis of Mowat-Wilson syndrome |
| 2 |
6,398,588 |
6,417,167 |
18,579 |
Del |
- |
NT |
| 3 |
6,500,668 |
6,737,047 |
236,379 |
Del |
3 |
Inconclusive |
| 4 |
6,567,938 |
6,609,613 |
41,675 |
Del |
- |
NT |
| 5 |
6,597,626 |
6,759,803 |
162,177 |
Del |
3 |
Maternally inherited |
| 6 |
6,752,063 |
7,577,445 |
825,382 |
Del |
4 to 9 |
An additional 178 kb deletion of 1p31.3 (61,318,736-61,496,706); De novo for both deletions |
| 7 |
6,781,978 |
6,948,525 |
166,547 |
Del |
- |
NT |
| 8 |
6,829,209 |
6,904,158 |
74,949 |
Del |
- |
NT |
| 9 |
6,860,713 |
7,018,893 |
158,180 |
Del |
- |
NT, a diagnosis of mild hemophilia A |
| 10 |
6,866,819 |
7,011,349 |
144,530 |
Del |
- |
NT |
| 11 |
6,883,111 |
6,912,449 |
29,338 |
Del |
- |
NT |
| 12 |
7,079,544 |
7,133,209 |
53,665 |
Del |
- |
NT |
| 13 |
7,132,809 |
7,170,657 |
37,848 |
Del |
- |
NT |
| 14 | 6,689,120 | 7,062,757 | 373,637 | Dup | 4, 5 | NT |
Notes: *The genomic coordinates of these CNVs were based on the human genome annotation of hg18 build; Del deletion, Dup duplication, NT not tested.
Of the 14 patients containing intragenic deletions/duplication of RBFOX1 (Tables 1 and 2), two of them were excluded for further analysis of the genotype-phenotype correlations, including Patient 1 who was diagnosed with Mowat-Wilson syndrome both phenotypically and genotypically, and Patient 14, the single case with an intragenic duplication of RBFOX1 accompanied with multiple congenital anomalies. Although both Patients 6 and 9 contain additional genomic abnormalities in addition to the intragenic deletions of RBFOX1, these two cases were included for further analysis since the clinical significance of the 178 kb deletion of 1p31.3 (61,318,736-61,496,706) involving NFIA in Patient 6 has not been defined yet and the mild hemophilia A in Patient 9 is assumed not to interfere the phenotypic effects of the RBFOX1 deletion. Recurrent features in the 12 patients (Patients 2 to 13) with intragenic deletions of RBFOX1 include global developmental delay (GDD) (7/12), epilepsy (6/12), macrocephaly or microcephaly (6/12), renal problems (4/12), such as single kidney, renal insufficiency, hypospadias, voiding dysfunction and nocturnal enuresis, bone and muscle problems (4/12), such as hemivertebrae, kyphosis, equinovarus, contractures of knees and hips, hypotonia (2/12), and hearing impairment (2/12). Some isolated findings were also observed, including autism, paralyzed vocal cords, tracheomalacia, gastroesophageal reflux, complex congenital heart defect, short stature, type II diabetes, adenoid hypertrophy, hypothyroidism, lactic acidosis, and hypernatremia (Table 2).
Table 2.
Clinical findings in patients with intragenic deletions/duplication in this study
| Cases | Sex | Dysmorphism | Neurological problems | GDD/Autism/speech delay | Other organ abnormalities/disorders |
|---|---|---|---|---|---|
| 1 |
F |
None |
None |
GDD |
Hypotonia, feeding problems, eczema, bilateral renal reflux and dilatation, episodes of fussiness and gassiness, chronic constipation, a diagnosis of Mowat-Wilson syndrome |
| 2 |
M |
Macrocephaly |
None |
GDD, Autism |
Hypothyroid at birth, history of lactic acidosis |
| 3 |
M |
None |
Epilepsy |
None |
Gastroesophageal reflux, voiding dysfunction and nocturnal enuresis, and adenoid hypertrophy |
| 4 |
M |
Severe microcephaly |
Perinatal hypoxic ischemic encephalopathy with resultant severe cystic encephalomalacia, focal epilepsy, cerebral palsy, asymmetric spasticity |
GDD, especially speech delay |
None |
| 5 |
F |
None |
None |
None |
Bicuspid aortic valve with aortic dilation, anomalous superior left pulmonary venous return |
| 6 |
M |
Macrocephaly |
None |
Severe GDD |
Hypotonia, kyphosis |
| 7 |
M |
None |
Encephalopathy |
Alexia, dyslexia |
Short stature |
| 8 |
M |
None |
Epilepsy |
Mild developmental delay, speech delay |
Hypospadias |
| 9 |
M |
Macrocephaly |
Poorly developed corpus callosum |
Severe GDD |
Hearing impairment, hypernatremia and renal insufficiency, a diagnosis of mild hemophilia A |
| 10 |
F |
None |
None |
None |
Hypotonia, tracheal malasia, |
| 11 |
F |
Plagiocephaly and infantile torticolli |
Epilepsy, abnormal EEG with a slow background and temporal spikes, and staring spells |
GDD, especially speech delay |
Hearing impairment |
| 12 |
F |
None |
Epilepsy |
Intellectual disability, aggressive behavior |
None |
| 13 |
M |
Severe microcephaly and micrognathia |
Epilepsy, ventriculomegaly |
None |
Paralyzed vocal cords, bilateral equinovarus, contractures of knees and hips bilaterally |
| 14* | M | None | None | NA | Atrioventricular canal, transposition of the great arteries, pulmonary valve atresia, supraventricular tachycardia, and congenital absence of spleen |
Notes: *-newborn; M male, F female, GDD global developmental delay, EEG electroencephalogram, NA not applicable.
We believe the intragenic deletions of RBFOX1 identified in these patients are risk factors for a wide spectrum of neurodevelopmental disorders and other clinical features. 1. Comparing with healthy groups, the 0.61% (13/2,124) detection rate of intragenic deletions of RBFOX1 in our study is significantly higher than the rate observed in control populations, such as the 0.1% (2/1854) in Itsara’s report [20] and the 0.2% (4/2026) in Shaikh’s report [21]. 2. Genetic defects including point mutations or intragenic deletions of RBFOX1 were independently reported to be associated with a variety of clinical conditions [7,9-11,14-19]. Majority of the clinical findings in the 12 patients in our cohort, such as GDD, seizures, and other neurodevelopmental and neuropsychiatric disorders, are consistent with those reported cases who carried genetic defects in RBFOX1[7-11,22]. For example, a recent study identified exonic deletions of the RBFOX1gene in 5 of the 1,408 European patients with idiopathic generalized epilepsy (IGE), but in none of the 2,256 population controls [22]. Davis LK et al. presented a inherited RBFOX1 exon-2 deletion in a boy with autism and developmental hemiparesis [23]. Bone and muscle problems present in multiple cases in this study were also observed in a report [14]. 3. The intragenic deletions of RBFOX1 in patient 6 occurred de novo, providing additional support about its pathological relevance to severe GDD, macrocephaly, hypotonia, and kyphosis in this patient. 4. More importantly, functional studies on RBFOX1 strongly support the etiological roles of RBFOX1 for neurodevelopmental disorders [10,24,25]. For example, almost identical to the knockout mice (Rbfox1-/-) with central nervous system (CNS)-specific deletion of Rbfox1, mice with heterozygous deletion (Rbfox1+/-) showed a dramatically increased susceptibility to induced seizures [24]. The loss of Rbfox1 was found to cause an increase in excitability of the neuronal population of the dentate gyrus resulting from an altered synaptic function. Martin et al. reported a 160 kb cryptic deletion of RBFOX1 within the breakpoint involved in a translocation between the short arms of chromosomes 15 and 16 in a female with autism, epilepsy, and GDD [10]. Accompanied with this heterozygous deletion, reduced mRNA expression in the individual’s lymphocytes was showed, demonstrating the functional consequence of the RBFOX1 deletion in multiple tissues. In a mouse model of Facioscapulohumeral muscular dystrophy (FSHD), Pistoni et al. recently showed that reducing Rbfox1 expression inhibits muscle differentiation [25].
There are several weaknesses in this study. 1. Ascertainment bias exists in our cohort since many of the patients were referred for CMA analysis because of unexplained neurodevelopmental and neuropsychiatric presentations. 2. Intragenic deletions of RBFOX1 were observed in some individuals with apparently normal phenotypes although incomplete penetrance of RBFOX1 defects exist [20,21] (http://projects.tcag.ca/variation/). 3. Most of the intragenic deletions of RBFOX1 in our patients affected only introns or the noncoding exons although point mutations or intragenic deletions of RBFOX1 were considered pathogenic in majority of the previous studies [7,9-11,14-19]. 4. The inheritance pattern(s) of the intragenic deletions of RBFOX1 in this study could not be determined in majority of them due to unavailability of parental samples and Patient 5 who inherited the deletion from her mother, the only determined inheritance of the deletion in this study, showed a different phenotype compared with her mother [15] although variable expressivity might exist.
In conclusion, the current study strongly supports the associations of intragenic deletions of RBFOX1 with a diversity of neurodevelopmental and neuropsychiatric disorders, and possibly other clinical features. Our data expend the spectrums of both the genomic abnormalities of RBFOX1 and phenotypic presentations associated with mutated RBFOX.
Material and methods
The DNA samples used for the current study came from 2,124 consecutive pediatric patients referred for chromosomal microarray analysis (CMA) in our laboratory. Each child had one or more of the following clinical findings at the time of referral: intellectual disability, developmental delay, multiple congenital anomalies (MCA), autism spectrum disorders (ASD), dysmorphism, seizures, learning disabilities or speech delays. Informed consents of all patients analyzed in this study were obtained from the parents and the institutional review board of KingMed Genome Diagnostic Laboratory approved this study protocol. The CMA platform used in this study is the Agilent Human Genome Microarray Kit 244K which contains about 244,000 probes across human genome including 186 probes encompassing RBFOX1 (Agilent Technologies, Santa Clara, CA). All CMA tests were performed and analyzed following protocols described previously [26]. The genomic coordinates of these CNVs in this study were based on the human genome annotation of hg18 build. For confirmation of the abnormal CMA results, quantitative real time PCR (qPCR) methods were performed using three different sets of primers targeting to the deleted regions of RBFOX1 as described previously [27]. The qPCR method was also applied to test the 5 available parental samples (2 couples and 1 single parent) for determination of the inheritance pattern of the genomic abnormalities in their respective children (Table 1). In addition, all the 14 individuals were previously tested by standard chromosome analysis (G-banding) with negative results and some of them were evaluated by specific genetic tests.
Competing interests
The author declares that he has no competing interests.
References
- Auweter SD, Fasan R, Reymond L, Underwood JG, Black DL, Pitsch S, Allain FH. Molecular basis of RNA recognition by the human alternative splicing factor Fox-1. EMBO J. 2006;25(1):163–173. doi: 10.1038/sj.emboj.7600918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehl TR, Shibata H, Pulst SM. The ortholog of human ataxin-2 is essential for early embryonic patterning in C. elegans. J Mol Neurosci. 2000;15(3):231–241. doi: 10.1385/JMN:15:3:231. [DOI] [PubMed] [Google Scholar]
- Jin Y, Suzuki H, Maegawa S, Endo H, Sugano S, Hashimoto K, Yasuda K, Inoue K. A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 2003;22(4):905–912. doi: 10.1093/emboj/cdg089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood JG, Boutz PL, Dougherty JD, Stoilov P, Black DL. Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol Cell Biol. 2005;25(22):10005–10016. doi: 10.1128/MCB.25.22.10005-10016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata S, Kawamoto S. Tissue-dependent isoforms of mammalian Fox-1 homologs are associated with tissue-specific splicing activities. Nucleic Acids Res. 2005;33(7):2078–2089. doi: 10.1093/nar/gki338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehl TR, Shibata H, Vo T, Huynh DP, Pulst SM. Identification and expression of a mouse ortholog of A2BP1. Mamm Genome. 2001;12(8):595–601. doi: 10.1007/s00335-001-2056-4. [DOI] [PubMed] [Google Scholar]
- Bhalla K, Phillips HA, Crawford J, McKenzie OL, Mulley JC, Eyre H, Gardner AE, Kremmidiotis G, Callen DF. The de novo chromosome 16 translocations of two patients with abnormal phenotypes (mental retardation and epilepsy) disrupt the A2BP1 gene. J Hum Genet. 2004;49(6):308–311. doi: 10.1007/s10038-004-0145-4. [DOI] [PubMed] [Google Scholar]
- Hamshere ML, Green EK, Jones IR, Jones L, Moskvina V, Kirov G, Grozeva D, Nikolov I, Vukcevic D, Caesar S. et al. Genetic utility of broadly defined bipolar schizoaffective disorder as a diagnostic concept. Br J Psychiatry. 2009;195(1):23–29. doi: 10.1192/bjp.bp.108.061424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia J, Gai X, Xie HM, Perin JC, Geiger E, Glessner JT, D'Arcy M, de Berardinis R, Frackelton E, Kim C. et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry. 2009;15(6):637–646. doi: 10.1038/mp.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CL, Duvall JA, Ilkin Y, Simon JS, Arreaza MG, Wilkes K, Alvarez-Retuerto A, Whichello A, Powell CM, Rao K. et al. Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(7):869–876. doi: 10.1002/ajmg.b.30530. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnby G, Abbott A, Sykes N, Morris A, Weeks DE, Mott R, Lamb J, Bailey AJ, Monaco AP. Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am J Hum Genet. 2005;76(6):950–966. doi: 10.1086/430454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J. et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai G, van Meurs JB, Livshits G, Meulenbelt I, Valdes AM, Soranzo N, Hart D, Zhang F, Kato BS, Richards JB. et al. A genome-wide association study suggests that a locus within the ataxin 2 binding protein 1 gene is associated with hand osteoarthritis: the Treat-OA consortium. J Med Genet. 2009;46(9):614–616. doi: 10.1136/jmg.2009.067314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lale S, Yu S, Ahmed A. Complex Congenital Heart Defects in Association with Maternal Diabetes and Partial Deletion of the A2BP1 Gene. Fetal Pediatr Pathol. 2011;30(3):161–166. doi: 10.3109/15513815.2010.547555. [DOI] [PubMed] [Google Scholar]
- Ma L, Hanson RL, Traurig MT, Muller YL, Kaur BP, Perez JM, Meyre D, Fu M, Korner A, Franks PW. et al. Evaluation of A2BP1 as an obesity gene. Diabetes. 2010;59(11):2837–2845. doi: 10.2337/db09-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen AB, Cox AJ, Ziegler JT, Voruganti VS, Xu J, Freedman BI, Carr JJ, Comuzzie AG, Langefeld CD, Bowden DW. Genetic mapping of vascular calcified plaque loci on chromosome 16p in European Americans from the diabetes heart study. Ann Hum Genet. 2011;75(2):222–235. doi: 10.1111/j.1469-1809.2010.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarhus M, Helland CA, Lund-Johansen M, Wester K, Knappskog PM. Microarray-based gene expression profiling and DNA copy number variation analysis of temporal fossa arachnoid cysts. Cerebrospinal Fluid Res. 2010;7:6. doi: 10.1186/1743-8454-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshita S, Umemura T, Yoshizawa K, Katsuyama Y, Tanaka E, Ota M. A2BP1 as a novel susceptible gene for primary biliary cirrhosis in Japanese patients. Hum Immunol. 2010;71(5):520–524. doi: 10.1016/j.humimm.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI. et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84(2):148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K, O'Hara R, Casalunovo T, Conlin LK, D'Arcy M. et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19(9):1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal D, Trucks H, Moller RS, Hjalgrim H, Koeleman BP, de Kovel CG, Visscher F, Weber YG, Lerche H, Becker F. et al. Rare exonic deletions of the RBFOX1 gene increase risk of idiopathic generalized epilepsy. Epilepsia. 2013;54(2):265–271. doi: 10.1111/epi.12084. [DOI] [PubMed] [Google Scholar]
- Davis LK, Maltman N, Mosconi MW, Macmillan C, Schmitt L, Moore K, Francis SM, Jacob S, Sweeney JA, Cook EH. Rare inherited A2BP1 deletion in a proband with autism and developmental hemiparesis. Am J Med Genet A. 2012;158A(7):1654–1661. doi: 10.1002/ajmg.a.35396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehman LT, Stoilov P, Maguire J, Damianov A, Lin CH, Shiue L, Ares M Jr, Mody I, Black DL. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat Genet. 2011;43(7):706–711. doi: 10.1038/ng.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistoni M, Shiue L, Cline MS, Bortolanza S, Neguembor MV, Xynos A, Ares M Jr, Gabellini D. Rbfox1 downregulation and altered calpain 3 splicing by FRG1 in a mouse model of Facioscapulohumeral muscular dystrophy (FSHD) PLoS Genet. 2013;9(1):e1003186. doi: 10.1371/journal.pgen.1003186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Bittel DC, Kibiryeva N, Zwick DL, Cooley LD. Validation of the Agilent 244K oligonucleotide array-based comparative genomic hybridization platform for clinical cytogenetic diagnosis. Am J Clin Pathol. 2009;132(3):349–360. doi: 10.1309/AJCP1BOUTWF6ERYS. [DOI] [PubMed] [Google Scholar]
- Yu S, Kielt M, Stegner AL, Kibiryeva N, Bittel DC, Cooley LD. Quantitative real-time polymerase chain reaction for the verification of genomic imbalances detected by microarray-based comparative genomic hybridization. Genet Test Mol Biomarkers. 2009;13(6):751–760. doi: 10.1089/gtmb.2009.0056. [DOI] [PubMed] [Google Scholar]