

Abstract
Biliary atresia (BA) is a rare disease of unknown etiology and unpredictable outcome, even when there has been timely diagnosis and exemplary surgery. It has been the commonest indication for liver transplantation during childhood for the past 20 years. Hence much clinical and basic research has been directed at elucidating the origin and pathology of BA. This review summarizes the current clinical variations of BA in humans, its occasional appearance in animals and its various manifestations in the laboratory as an experimental model.
Keywords: Biliary atresia, Neonatal cholestasis, Animal models, Etiology, Neonatal immunology
Background
There are few diseases where so much is known yet so little understood than the condition of biliary atresia (BA). Nonetheless, as our current strategy (Kasai portoenterostomy (KPE) and liver transplantation where necessary) for dealing with BA is relatively successful and about 80-90% of currently affected infants will survive to adolescence and adulthood-does it matter that the cause in most cases is still obscure? In most probably not, although there is a still considerable risk of significant morbidity related to cirrhosis in those who have had Kasai portoenterostomy alone. About half of even biochemically normal adolescent survivors will have histological cirrhosis [1] and have the potential for decompensation, portal hypertension and even malignancy [2]. Furthermore, survivors with successfully transplanted organs still need to remain pharmacologically immunosuppressed and therefore have an increased risk of infection and again malignancy in the form of post-transplant lymphoproliferative disorders.
The key question that this review aims to address is what is biliary atresia and what may cause this rare disease? Increasingly it is evident that BA is not a single entity with a single aetiology but rather appears to be a phenotype characterized by an obliterated (or absent) biliary tree presenting in the first few weeks after birth [3].
The scope and limits of review are to collate observable evidence both from the human condition and the animal laboratory and try to stitch these two disparate views into one distinct picture.
Biliary atresia in the clinic
Although most older textbooks usually talk in terms of only two variants, embryonic and perinatal, this is far too simplistic and it also assumes that we know a lot more about when the disease starts than we actually do [4]. We prefer to use more verifiably descriptive names with less assumption on cause. Thus, there are probably at least four clinical variants which can be defined (Figure 1).
Figure 1.
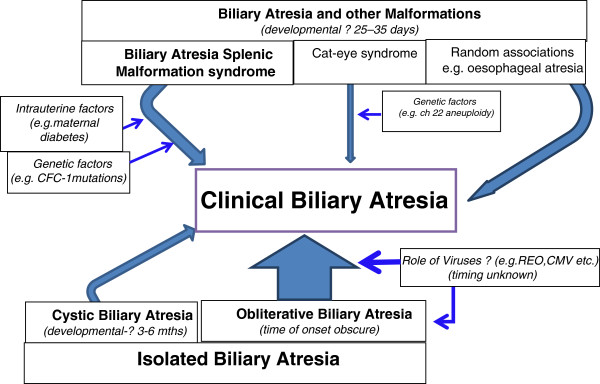
Patterns of Clinical Biliary Atresia.
Biliary atresia and other congenital malformations
Firstly there are those infants with BA who have other congenital anomalies. These can be further sub-divided into three groups. Firstly there are those who have elements of the Biliary Atresia Splenic Malformation (BASM) syndrome [5,6]. These might include splenic malformation (usually polysplenia, but also asplenia and double spleen), disorders of visceral symmetry (e.g. situs inversus and malrotation), malformations of the intra-abdominal veins (e.g. absent inferior vena cava, preduodenal portal vein) and cardiac anomalies. These infants are usually girls, and some seem to come from an abnormal intrauterine environment (e.g. maternal diabetes, maternal thyrotoxicosis). It is almost certain that their bile duct pathology occurs at the same time as their other developmental anomalies (i.e. during the embryonic phase of organ development, perhaps at 5-6 weeks of gestation) and therefore at the time that the hepatic diverticulum is pushing into the mesenchyme of the prehepatic septum transversum. This is well before any intrahepatic duct system has formed (7-10 weeks) and at operation concurs with the observation that the extrahepatic biliary tree is often atrophic with little inflammation and there is usually absence of the common bile duct. Surprisingly, given this timeline the liver parenchyma at the time of birth is actually normal [7] and there is no difference in their birth weight or gestational age compared to those with isolated BA [8]. There are also clear racial differences in that this type is infrequent in the high incidence areas of the world for BA (e.g. China, Japan, Korea). Variation in BASM incidence can also be shown in multiethnic populations such as that of England and Wales [8]. It is this group which seems likely to have a clear genetic basis for their spectrum of anomalies (pace maternal diabetes). So of the range of possible bile duct development genes (see later); CFC-1 mutations have been have identified in 50% of a French series of infants with BASM [9]. This gene, present at Ch 2q 21.1, encodes for CRYPTIC protein and mutations have been related to heterotaxy syndromes (i.e. randomised situs) and cardiac anomalies such as transposition of the great vessels/ double outlet right ventricle [10].
There is a second group of infants, also with BA, who have other features of other distinct syndromes. The example for this is the so-called cat-eye syndrome (coloboma, ano-rectal atresia etc.) and in these chromosomal aneuploidy (Ch 22) has been shown [11].
Finally, some infants with BA have non-syndromic congenital anomalies such as oesophageal atresia, jejunal atresia, ano-rectal atresia etc. but none of the peculiar anomalies listed above and for which we have no convincing genetic explanation.
Cystic biliary atresia
The extrahepatic component of BA is usually characterised by atrophy or absence (particularly in BASM) or by inflammatory obliteration of an intact tree. However, in about 10% of cases, cyst formation (bile or mucus) can occur, and may lead to diagnostic confusion with early obstructed cystic choledochal malformation.
Cholangiography at surgery invariably shows that the intrahepatic ducts are grossly abnormal with irregularity and pruning if there is preservation of a tree-like pattern or a cloud-like appearance caused by multiple interconnections of filamentous intrahepatic biliary ductules. Such infants belong within the BA spectrum family and should be termed cystic biliary atresia (CBA) [12]. These don’t seem to have any racial, genetic or epidemiological peculiarities but what is clear is that they are observable on antenatal ultrasound scanning–if looked for. In our experience about half are detectable–usually the large ones–and from at least 18-20 weeks gestation [13]. They also have a better outcome following surgery, probably because of better quality of luminal continuity with those intrahepatic ducts.
Isolated biliary atresia
This is the largest clinical grouping and epidemiologically there is wide geographical (and presumed racial) variation across the world from about 1 in 5000 births in Taiwan to about 1 in 18,000 in Northern Europe with no overt seasonal variation and an equal gender split [8,14-16].
So what do we know of their aetiology? The initial hypothesis was that these infants also had a developmental problem of their bile ducts. The timing of onset must be later than the syndromic groups as no other system appeared abnormal and perhaps at one of the key stages in biliary development–the juxtaposition of the intrahepatic and extrahepatic bile ducts. Both elements have different origins, develop along separate lines from different structures and biliary continuity is only established by 10-12 weeks gestation. Tan et al. compared the appearance of serial sections at the level of the porta hepatis in normal fetuses at this gestation with the histological appearance of the porta of infants with established isolated BA [17]. They were remarkably similar showing multiple biliary ductules around branches of the portal vein undergoing selection, deletion and then morphing into the larger fully-formed ducts by 13-14 weeks. So, the hypothesis suggests that even isolated BA might be a form of 1st trimester arrested development–for whatever reason.
The alternate hypothesis is that such infants once had a completely formed intact biliary tree but that obliteration occurs as a secondary, indeed perinatal, phenomenon [18,19]. The clinical evidence supporting a patent bile duct system and then obliteration is difficult to obtain. Anecdotal reports of an initial “neonatal hepatitis” picture on investigation but evolving into biliary atresia later have been suggested. Postnatal biliary obliteration can certainly happen and we previously described 3 infants who became jaundiced secondary to perinatal events (proximity surgery and probable healed bile duct perforations). Their extrahepatic bile ducts were completely obliterated although they retained a normal intrahepatic biliary tree which had dilated by the time of definitive surgery [20].
Amniotic fluid has been assayed for the hepatic-exclusive enzyme γ-glutamyl transpeptidase in one large screening study. The lowest values were found in 3 infants who ultimately proved to have BA which clearly implied a prenatal onset and absence of bile from the fetal gastrointestinal tract [21]. The level of bile acids in the Guthrie blood spots (taken in first few days of life) has also been measured [22] and showed that 47 (77%) of 61 infants who later proved to have BA had elevated total bile acids (>97th percentile, 33 μmol/L) with no difference if the blood had been taken at <7 days or >10 days. This suggests that cholestasis is already obvious (if looked for) during the first post-natal week in at least three-quarters of those who prove to have isolated BA. A similar North American study of split bilirubin levels in 31 infants obtained when they were < 48 hours old and who later turned out to have BA also showed levels distinctly higher than controls [23], again implying established biliary pathology at the time of birth.
Our last, and perhaps most controversial clinical variant has been that of viral-associated BA–which some might say is simply the latter group plus a culprit caught in the act. The role of viruses in the animal model (whether as instigators or genuine pathogens) will be outlined in detail later but clinically this entity has been the subject of much debate since the 1980s and 90s. Initially, work focused on serology of REOvirus type 3 [24], but a more sophisticated search evolved for actual viral footprints within the liver and specifically for group C rotavirus (positive in 50% of BA infants) [25] and cytomegalovirus [26,27]. Most recently our Hannover group [28] examined liver biopsies in 74 infants with BA against a panel of possible hepatotropic viruses and showed that viral RNA/DNA could be found in just less than half (REOvirus 33%; cytomegalovirus 11%; adenovirus 1% and enterovirus 1.5%). The debate revolves around whether these are simply (not so) innocent bystanders or integral to the cholangiodestructive mechanism. The faecal flora of infants with BA is different [29] and enteric absence of bile salts may allow selection of such enteroviruses. Does it make a difference? In the follow-up to the German study there was no difference in outcome between virus + ve and virus–ve infants[30]. However, in a separate study using only infants who tested IgM + ve for CMV (n = 20) from our London group compared to 111 infants with negative IgM serology we have shown reduced clearance of jaundice and a significantly increased mortality in the CMV IgM + ve group [31].
Cellular kinetics, inflammation and clinical biliary atresia
There is a potent inflammatory reaction in the livers of infants with isolated BA, focused on the portal tracts [32-34]. There is abnormal expression of MHC Class II antigens and the adhesion molecules ICAM and VCAM on both vascular and biliary endothelium, together with a sometimes dense population of activated and proliferating mononuclear cells [35], [36]. Among these are mast cells [37] and Kupffer cells [38] but they seem to be predominantly CD4+ T cells and NK cells [39,40] orchestrating a Th1 cytokine response [41,42] with increased expression of IL-2, IFNγ, TNFα and IL-12 and other proinflammatory factors, e.g. iNOS and NO [43,44]. The cell lines appear to be oligoclonal at least in the CD4+ and CD8+ subsets identified by Mack et al. [45]. Alternatively, Shinkai et al. observed a predominantly CD8+ infiltrate in their infants with BA [46,47] though their study group was much smaller; and previous work has suggested poor CD8+ cytotoxic function [48]. Muraji et al. has also described significantly more CD8+ T cells in the livers of BA infants, and interestingly, these appear to be of maternal origin [49]. This is the interesting concept of maternal microchimerism, whereby transplacental passage of immune active cells (seemingly CD8+ CD45+) occurs, localizing within the liver and initiating a form of graft-versus-host reaction. Such cells may also presumably provide an alternative target for an autoimmune response. Finally, a third subset of helper T cells, Th17+ cells also seem to play a significant role in mucosal host defence and have been implicated in some autoimmune cholangiopathies such as primary biliary cirrhosis in adults. Such Th17+ cells, can be shown to accumulate at the bile duct epithelial interface and infiltration of this subset in portal tracts and also appears to be a characteristic of BA [50,51].
There is also a pronounced soluble inflammatory component observable at the time of presentation. Initially this with increased levels of sICAM and sVCAM [52,53] and the cytokines IL-2 and Il-10 compared to controls which then actually increases following the KPE. In the case of IL2, IL-18 and TNFα this increase is very marked to about 9-12 months. If there has then been resolution of jaundice then it tends to abate but only sICAM appears to be closely correlated with plasma bilirubin levels [54].
Early-onset of liver fibrosis appears to be a defining characteristic of BA, perhaps not seen in other cholestatic conditions of infancy such as Alagille’s syndrome or α-1-antitrypsin deficiency. Harada et al. cultured human intrahepatic biliary epithelial cells (BECs) and showed that exposure to a viral RNA analogue triggered an immune reaction which persisted long after removal of the virus [55]. Excessive activation of Hedgehog (Hh) signaling pathways and increased expression of matrix metalloproteinases (MMP) [56] can be shown in BA infants promoting epithelial–to–mesenchymal transition of BECs and leading to enhanced hepatic fibrogenesis. Resident (Kupffer cells) or systemic / recruited macrophages /monocytes seem to play a dual role in this process. Initially they may act as antigen presenting cells (APCs) early on in the process but they also appear to be the key factor in this development of fibrosis. Tracy et al. in 1996 first highlighted the increase in the resident macrophage population in BA and increased levels of both CD68+ cells and its circulating markers (TNFα and IL-18) have been shown [57,58].
Biliary atresia in the animal world
The lamprey is an eel like creature which parasitizes and feeds on the blood of fish and is unique amongst vertebrates in that its liver does not have a gallbladder, biliary tree or even canaliculi. It does have them in the larval stage but then they disappear on metamorphosis to the adult stage. The only real effect on their hepatocytes appears to be increased numbers of cytoplasmic vacuoles presumably where some form of bile degradation occurs [59,60]. However, despite a series of papers, which looked at the lamprey as a potential animal model for the congenital form of BA, this direction is no longer pursued.
There have been sporadic reports in the veterinary literature of biliary atresia or at least BA-like lesions in lambs, foals, dogs, calves and even a Rhesus monkey (associated with REOvirus) [61,62]. An amazing epidemic of BA in over 200 lambs and 9 calves was attributed to maternal grazing and ingestion of a toxic weed known as the red crumbweed (Dysphania glomulifera subsp. glomulifera) growing on the newly exposed silt foreshores of Burrinjuck Dam, New South Wales, Australia [63].
Biliary atresia in the laboratory
Bile duct development
There is much ignorance about the steps of bile duct development during the embryonic period, and what is known has been largely derived from work in mice and zebrafish [64] and is somewhat outside the remit of this review. The “ductal plate” refers to differentiation of hepatoblasts into BECs and then condensation around in-growing portal venules starting at the porta hepatis and occurs at around 8-10 weeks with later remodeling and definitive tubularisation via selection and deletion. This appears to be induced in the fetal liver by a periportal gradient of activin/transforming growth factor-beta (TGFβ) signaling. The current consensus is that the entire intrahepatic system is derived from this mechanism as suggested by Desmet [65]. The establishment of biliary continuity with the lumen of an already established foregut diverticulular structure occurs although when and how is indistinct. The hepatocytes start to secrete bile from about the 12th week.
Cholangiocyte specification is promoted by the Wnt/β-catenin signaling pathway and initiation and overall control appears to be a function of the Notch signaling pathway, Notch 2 rather more so than Notch 1 [66,67]. Certainly, mutations in the latter’s ligand (Jagged1) appear responsible for bile duct hypoplasia of Alagille’s syndrome (and murine equivalents). Various transcription factors (e.g. Hes-1, Hnf-6, Foxa1/2, TGF-β) are also involved in remodeling of the ductal plate and prevention of excess cholangiocyte proliferation.
Inversin (Inv) is a gene localised to Ch 4 in humans. The inv mutant mouse, created by insertional mutagenesis, was first reported in 1993 by Yokoyama et al. [68], who described situs inversus and severe jaundice in homozygous mutants. The actual biliary pathology has been disputed however, with Mazziotti et al. [69] suggesting extrahepatic discontinuity but more recently Shimadera et al. [70] showing periportal ductular proliferation (perhaps akin to ductal plate malformation) and anomalous development of the intrahepatic bile ducts but a normal extrahepatic system. Neither group demonstrated inflammatory, ischemic or cystic features.
Inducing cholestasis and atresia in prenatal experiments
Ligation of the common bile duct in fetal lambs at about 80 days of gestation produces cholestasis and cystic change in the bile duct evident at birth and, perhaps, resembling those infants with CBA [71,72]. Similar findings can be shown in fetal rabbits when the hepatic artery has been ligated and in one study absence or hypoplasia of the intrahepatic bile ducts was also seen in 5 out of 14 five-week old pups [73].
The effect of drugs administered during pregnancy has also been studied. Hosada et al. administered intraperitoneal phalloidin, an actin-binding toxin derived from the Death Cap mushroom (Amanita phalloides) to Wistar rats on gestational day 15. After birth, the pups presented with cholestasis and an increased volume of actin filaments around bile ducts, but without typical BA features [74]. In another study, the anti-helminthic drug, 1,4-phenylenediisothiocyanate, was given orally to pregnant, newborn and adult Wistar rats. Peribiliary inflammation and dilatation was found in the post-natal drug groups but the most convincing BA-like findings with almost complete obliteration but no dilatation was found in those exposed to the drug during the fetal period and at one month after birth [75].
Inducing cholestasis and atresia in postnatal experiments
A number of manoeuvres, e.g. ligation of common bile duct, intrabiliary injection of sclerosants and superglue, in a variety of animal models, e.g. mice [76], rat [77], and minipigs [78], have been reported with the intention of reproducing early neonatal cholestasis. These studies seem to have more value in replicating the metabolic consequences of cholestasis in this age-group rather than providing a real insight into aetiology. One exception may be that of Schmeling et al. [79], who infused phorbol myristate acetate (PMA) into the gallbladders of golden hamsters. PMA is not in itself toxic but it does activate peribiliary neutrophils which release potent free-radical oxygen species thereby setting up a marked peribiliary inflammatory process and ultimately fibrosis. Actual obliteration and the effects on the extrahepatic biliary tree were not commented upon.
A possible immune-mediated mechanism was first investigated by Schreiber et al. in 1992 [80]. They transplanted fetal, neonatal and adult bile ducts taken from C57BL/6 into the adult B10 mice and observed a dynamic pattern of rejection and fibrosis. They found that prenatally harvested bile ducts were relatively “immunoprivileged” because of diminished expression of HLA class I and II antigens and that in adult mice, graft rejection could be stopped by cyclosporin A.
The viral model of biliary atresia
Initial mice models used intraperitonal inoculation of REOviruses with varying serotypes, e.g. type 3 (Abney) [81,82]. All seemed to induce liver and biliary inflammation with jaundice as a consequence, but without real features of atresia.
Oral administration of REOvirus to newborn ND4 Swiss Webster mice showed that hepatotropism was higher in those strains which have the capacity to bind sialic acid as a co-receptor. These mice presented with oily fur syndrome, portal lymphoid infiltrates in the liver and sporadic bile duct necrosis. However, mortality appeared to be due to encephalitic disease, and surviving pups recovered completely from their hepatobiliary pathology [83]. Similar observations were made when other hepatotropic Rotavirus and REOvirus strains were used [84,85].
A better model with peribiliary inflammation leading to features of actual irreversible extrahepatic atresia was developed by infecting newborn Balb/c-mice with Rhesus Rotavirus (RRV) [86]. In the initial study [87] about half of the orally RRV infected pups developed cholestasis, and most died within 3 weeks. Dissection showed obstruction of the common bile duct and BA-like changes in the liver, e.g. inflammatory infiltrates and bile duct proliferation. Our Hannover group modified this model using intraperitoneal RRV given at 24-48 hours with an increase in the proportion becoming cholestatic and the development of atresia-like biliary segments (Figure 2) [88-90]. Still, older 3 week old pups failed to show much in the way of liver fibrosis, and using picosirius red staining collagen deposition around portal triads was much less obvious than in the human condition [91,92].
Figure 2.
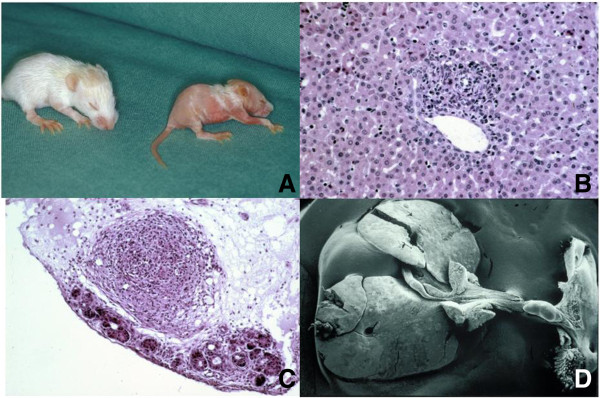
The Rhesus Rotavirus induced biliary atresia model in newborn Balb/c-mice. (A) Two 14 day old Balb/c mice, left, healthy control, right, jauniced with oily fur syndrome. (B) inflammatory infiltration in a portal field (9 day old, H x E 400×). (C) Section of the hepatoduodenal ligament at the level of the common bile duct atresia (16 day old, H x E 250×) (59). (D) Scanning electron microscopy of a whole specimen, showing a small gallbladder, a vanishing common bile duct along with the hepatoduodenal ligament a prestenotic cystic dilatation near to the duodenum (15 day old) (56).
To work, however, three preconditions must be fulfilled; but it offers a way of understanding a mechanism for the observable pathology. The preconditions are such that the strain of mouse, the timing and the dose of virus inoculation must be perfect to achieve BA-like pathology [89,93,94]. Maternal infection during late pregnancy does not induce jaundice, but does prevent the offspring from developing cholestasis following postnatal RRV application [95,96]. Post-natal inoculation at 12-24 hours after birth produces BA-like lesions in over 90% of animals compared to less than 20% if the inoculation is given at 48-72 hours. Table 1 illustrates this effect of mouse strain time of virus administration and viral load on outcome, and shows that too high a dose is lethal before the onset of biliary pathology. On the other hand, changing of the RRV-dosage or the mouse strains, respectively, does not necessarily prevent infected pups from cholestasis, but from developing destruction and atresia of extrahepatic bile ducts. Those mice mostly catch up weight and recover completely. The versatility of the model is also limited as sequential investigations cannot be performed in the same animal. Diseased pups are extremely unstable and too small for repetitive biopsies or blood sampling. However various stages of the assumed immune cascade can be studied by extraction of cell lines, e.g. cholangiocytes taken from RRV infected pups can be isolated [97-99]. One advantage of this model is that modification of each of the preconditions allows investigation of subsequent immunological or cellular changes.
Table 1.
The incidence of virally induced cholestasis and biliary atresia depends on three variable parameters, mouse strain, time (hours post partum) and dosage of Rhesus rotavirus application
| Variable parameter | Cholestasis | Biliary atresia |
|---|---|---|
|
Mouse strain | ||
| Balb/c 55, 71 |
67%-85% |
67%-91% |
| CD 59 |
33% |
46% |
| NMRI 59 |
19% |
50% |
| C57Bl6 67 |
13% |
100% |
| Balb/c-IFN-γ–/–65 |
90%-100% |
20% |
| Balb/c-TNF-α–/–68 |
No data |
86% |
| Balb/c-Mx + −A2G 71 |
65% |
65% |
| WT 129 70 |
30% |
50% |
| A 129 IFN-α/β receptor–/–70 |
79% |
96% |
| G 129 IFN-γ receptor–/–70 |
39% |
86% |
| AG 129 IFN-α/β/ γ receptor–/–70 |
70% |
96% |
|
Age at infection | ||
| 12-24 hours 65, 83 |
80%-86% |
90%-100% |
| 24-48 hours 55 |
61%-85% |
69%-91% |
| 48-72 hours 52, 53 |
13%-42% |
0-17% |
|
Infective dose | ||
| 107* pfu 62 |
100% |
100% |
| 106 pfu 62 |
86% |
100% |
| 105 pfu 62 |
38% |
100% |
| 104 pfu 62 | 0 | 0 |
pfu–plaque forming unit.
* >106 pfu is not recommended, because the early lethality is inacceptably high.
The dynamic pattern of RRV infection can be monitored on the basis of the expression of mRNA encoding RRV non-structural and structural proteins, e. g. VP6, VP4, and viral plaque assays. So, the peak of viral load is seen at day 7 after inoculation, and is mostly cleared from the liver by day 14 [97-99]. However, the cellular and humoral immune response persists even after complete viral clearance.
The steps of the immune reaction to RRV inoculation appear to be as follows (Figure 3). The viruses target cholangiocytes and macrophages [100], while the initial response depends upon innate NK cells and APCs inducing a T-cell mediated immune response. In this murine model, cholangiocytes and dendritic cells [86], appear to act as the APCs via expression of both MHC class I antigens [97,98,101] (to cytotoxic CD8+ cells) and MHC class II antigens (to CD4+ T-helper cells). Activated CD4+ cells, driven by IL-12, produce a bewildering range of cytokines, such as IL-2, IFN-γ, IFN-α, IFN-β, CXCL 9, CXCL 10, CCL2, CCL5, Stat 1, Granzyme A and B, Osteopontin, etc. [102,103]. Lack of IL-12 does not seem to abolish this particular response, as Th1 dependent cytokines, such as IFN-γ and TNF-α, are not reduced in IL-12 deficient mice, suggesting the presence of alternative pathways [103-105]. Recently, a Th2 response has been shown to induce epithelial injury and inflammatory obstruction of extrahepatic bile ducts in the same mouse model [106]. Cholestasis becomes evident one week after infection and the infiltration of CD4+ and CD8+ cells increased three-fold with a peak on day 14.
Figure 3.
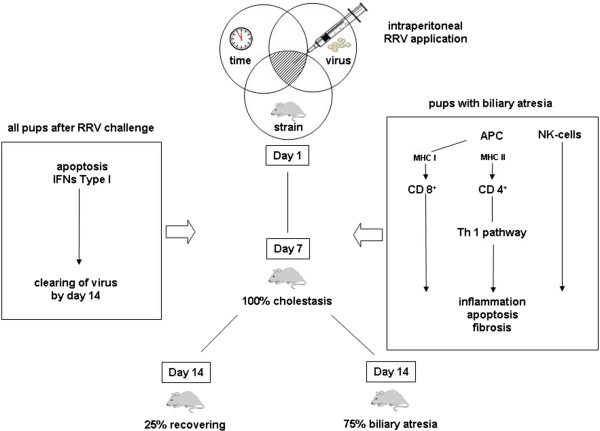
Schematic network of cellular dynamics and immunological cascade in mouse model of biliary atresia. For more details see Table 2.
Interferons seem to play a crucial role in this model, as Mx protein (an IFN type I specific indicator) persists in hepatocytes, bile ducts and intrahepatic endothelial cells of cholestatic mice beyond the second week after RRV infection [107,108]. In other studies, for instance with IFN-receptor knock-out mice, it can be shown that IFN type II attenuates the tropism of lymphocytes to bile ducts and is somehow imperative to the development of BA [99,109].
Depletion of CD8+ cells, as well as loss of IFN-γ, reduces the incidence of experimental BA. IFN-type II regulated chemokines such as Mig, IP-10 and I-Tac can be shown to peak at day 7 only in BA-developing Balb/c but not in IFN-γ–/–mice [110]. Thus in these knock-out mice, early lobular and portal inflammation with periductal infiltration by neutrophils is still seen at day 3 and is followed by bile duct proliferation. But in contrast, they did not go on to develop bile duct obstruction or atresia and indeed recovered in most cases. Administration of recombinant IFN-γ restores this detrimental consequence. We hypothesis that both IFN-γ and CD8–T cells constitute two complementary mechanisms in which IFN-γ and other Th-1 dependent cytokines serves as a molecular signal that promotes the infiltration of injured bile ducts by inflammatory cells, while CD8- T cells, together with NK-cells, trigger the transformation of an ongoing inflammatory process into fibrosis and occlusion of the bile ducts [111,112].
The role of CD19+ B-cells and activation of Th2 effector cells remains unclear and the expression of Th2 related cytokines such as IL-4 and IL-5 has not been uniform [99,113].
Regulatory T-cells (T-reg) seem to be involved in this auto-immune process. In mice, T-regs are absent in the newborn and become activated during the first week of life. Initially substitution of T-regs in the mouse model was supposed to reduce the incidence of BA and shift the focus onto NK-cells [114,115]. Adoptive transfer of T-cells from mice with RRV-induced BA to SCID recipients results in periductular inflammation, and a bile duct epithelia specific T-cell mediated autoimmunity [116]. The hypothesis that humoral autoimmunity plays some role in the pathology of BA [117] has been endorsed by detection of anti-α-enolase autoantibodies in both experimental and human BA and found in patients with typical autoimmune liver disease, such as autoimmune hepatitis and primary sclerosing cholangitis.
Apoptosis of the biliary epithelium (as shown by TUNEL assay and expression of caspase 1 and 4) and activation of the complement cascade appears to peak at day 7 and can be induced by proinflammatory cytokines such as TNF-α and IFN-γ [109]. Additionally, NF-κB, which is a first responder to harmful cellular stimuli as a rapid-acting primary transcription factor inducing apoptosis, shows an increasing level from day 5 with a peak at day 15 [84,118]. It still remains unclear if apoptosis is part of the clearing mechanism after viral infection or a hyper-responsiveness consistent with immature immunity [119].
There have also been attempts to modify pathology using this model. For instance, repeated administration of IFN-α at onset of jaundice, has been shown to ameliorate the development of BA-like lesions and increase survival. It remains unclear if this boosts viral clearance or modulates the immune response [120,121]. Immunisation of dams with RRV before mating and during pregnancy protects their offspring from cholestasis and development of BA [95,96]. Dams, which were preconceptionally and orally vaccinated with commercially available rotavirus vaccines, did not prevent their RRV infected newborns from becoming jaundiced, but did prevent the development of BA [122]. While these preliminary results are far from clinical application in humans, the potential for at least some form of prophylaxis has been shown.
Table 2 illustrates a chronological overview of the key immunological observations mentioned in the text in both human and experimental BA to date.
Table 2.
All papers, which have so far been published concerning BA and immunology in both, human and experimental BA
| Effector | Subset/Cytokine | Human (Reference) | Murine (Reference) | Key Message |
|---|---|---|---|---|
| T-regs |
|
[123] |
|
Circulating and local T-regs in lymph nodes of the porta hepatis are reduced in CMV positive patients with BA, compared to age matched controls with other cholestatic liver diseases |
| CD4 + −T-Cells |
Osteopontin |
[43] |
[124] |
Osteopontin mRNA expression was shown in liver biopsies of BA patients and also in experimental BA |
| Cholangiocytes |
TLRs |
[125] |
|
TLRs 2 and 8 mRNA expression was higher in early states of BA-patients and significantly elevated to age matched controls |
| CD4 + −T-Cells |
Th2/IL-13 |
[106] |
[106] |
STAT1–/–mice infected with RRV exhibit a Th2 dominated inflammation, evidence can be found in humans, too, also mediating experimental biliary atresia if not accompanied by IL-13 blockade |
| CD4 + −T-Cells |
|
[41] |
|
DNA-Hypomethylation throughout the genome of CD4 + −T-Cells is negatively correlated with IFN-ɣ mRNA-levels compared to healthy controls |
| Dendritic Cells |
|
[86] |
[86] |
RRV-primed DC, also present in human livers, boost lymphocyte expansion and mediate epithelial injury whereas depletion of DC or blocking IL-15 reduced NK-Cell activation |
| Macrophages |
Mip2/Cxcl2 |
|
[100] |
RRV-infected Macrophages influence neutrophil chemotaxis |
| |
iNOS |
|
[44] |
Liver samples show a strong correlation between NF-κB -activation and iNOS hyperexpression |
| T-regs |
|
|
[114,126] |
Activated T-regs reduce inflammatory cytokine production and suppress NK-cell activation in vitro and in vivo |
| Cholangiocytes |
|
|
[101] |
Release of CXC–and CC-Chemokines by cholangiocytes markedly increase upon onset of disease |
| Cholangiocytes |
|
|
[97] |
In the setting of RRV-infection and Th1 inflammation cholangiocytes produce inflammatory cytokines and chemokines but do not function as APC despite expressing all necessary surface markers |
| NK-Cells |
|
[127] |
[115] |
NK-Cells populate the liver of mice, and humans, and their depletion or blocking Nkg2d prevents cholangiocyte lysis in vitro and in vivo |
| |
IFN-ɣ /TNF-α |
|
[109] |
Inhibition of caspases reduces apoptosis induced by synergism of IFN-ɣ and TNF- α |
| CD8 + −T-Cells |
|
[49] |
|
Primarily CD8 + −T-Cells of the lymphocyte infiltrate suffer from maternal micochimerism |
| |
NF-κB gene products |
[55] |
|
Intrahepatic Biliary Epithelium reacts with NF-κB activation if treated with a viral-dsRNA-analogon |
| |
TNF-α |
|
[103] |
Although highly elevated in experimental biliary atresia blocking TNF- α exerts no effect |
| T-Cells |
|
[45] |
|
CD4+ and CD8 + −T-Cells show oligoclonal expansion of TCR Vβ with Vβ20 dominating in the latter |
| |
NF-κB gene products |
[117] |
|
Biliary Epithelium reacts similar to a viral infection if treated with a viral-dsRNA-analogon |
| CD8 + −T-Cells |
|
|
[110] |
Depletion of RRV-primed CD8 + −T-Cells reduces disease incidence and mediate the epithelial injury |
| |
IL-12 |
|
[105] |
Loss of IL-12 in mice does not prevent experimental biliary atresia but shifts the Th1-phenotype of inflammation towards neutrophils |
| CD4 + −T-Cells |
IFN-ɣ |
|
[116] |
Although not being able to elicit experimental biliary atresia in SCID mice RRV-primed CD4 + −T-Cells are the source of IFN-ɣ |
| |
IFN-ɣ |
|
[107] |
IFN-ɣ-RII is without influence on development of experimental biliary atresia |
| CD8 + −T-Cells |
|
[46] |
|
Elevated numbers of CD8 + −T-Cells populate the portal tract in biliary atresia |
| T-Cells |
|
[47] |
|
T-Cells within the livers of diseased express more CXCR3 than controls |
| |
IFN-ɣ |
|
[128] |
RRV-challenge of mice upregulates Interferoninducers followed by the IFN-ɣ network genes with onset of disease in the biliary transcriptome |
| CD4 + −T-Cells Macrophages |
Th1 TNF-α |
|
[113] |
With RRV present one week p.i. the portal tract infiltrate is predominated by CD4 + −T-Cells producing IFN-ɣ and Macrophages producing TNF-α, whereas both effectors produce TNF-α after virus clearance |
| Macrophages |
|
[129] |
|
Increased infiltration of Macrophages in the liver after Kasai is associated with a favourable outcome |
| |
IFN-ɣ |
|
[99] |
IFN-ɣ depletion prevents bile duct obstruction while administering IFN-ɣ to IFN-ɣ–/–induces experimental biliary atresia after RRV-injection |
| CD4 + −T-Cells |
Th1 |
[33] |
|
The portal tract inflammatory response is dominated by CD4 + −T-Cells with a Th1-cytokine response and an increased number of Kupffer cells |
| CD4 + −T-Cells |
|
[130] |
|
The reduced number of naïve-CD4 + −T-Cells in patients persists after transplantation |
| |
Costimulatory factors |
[35] |
|
Costimulatory factors of APC are highy expressed on bile duct epithelium |
| Lymphocytes |
|
[36] |
|
Leukocyte infiltration of livers often do not include immunocompetent lymphocytes |
| |
Th1 |
[43] |
|
Within the transcriptome of pooled mRNA genes pointing towards a Th1-profile are often upregulated |
| Lymphocytes |
|
[40] |
|
Activated and proliferating lymphocytes populate the liver |
| CD8 + −T-Cells |
|
[48] |
|
CD8+ T-Cell infiltrate of the proliferating bile ducts after Kasai operation lack the phenotype of activated cytoxic lymphocytes |
| CD4 + −T-Cells |
|
[42] |
|
A decreased number of naïve-CD4 + −T-Cells is accompanied by reduced receptor density |
| Mast cells |
|
[37] |
|
The number of intrahepatic mast cells negatively correlates with liver function |
| Macrophages |
IL-18 |
[38] |
|
Proliferation of Kupffer-Cells is found in the liver accompanied of elevated IL-18 levels in serum |
| |
IFN-α |
|
[121] |
IFN-α prevents experimental biliary atresia |
| Macrophages |
|
[58] |
|
Increased infiltration of Macrophages in the liver after Kasai is correlated with a bad prognosis |
| Macrophages |
|
[57] |
|
Macrophages coexpress CD68 and CD14 in biliary atresia |
| Lymphocytes |
|
[34] |
|
Lymphocyte infiltration into biliary epithelium is quite similar to GVHD |
| Mononuclear cells |
|
[131] |
|
Mononuclear infiltrate in BA is more similar to normal livers than to those suffering from chronic infection or autoimmunity |
| T-regs | [114,126,132] | The RRV-induced murine model of BA is associated with defects in the production and function of T-regs, probably suppressing DC-dependent activation of naive NK cells |
References are listed in reverse chronological order and the key message of each paper is summed up as well as the key effector cells and/or corresponding cytokines.
The key question, of course, is does this experimental model of BA in mice really mirror the human disease and can the putative mechanism really be applied outside of the mouse [16,41]? One problem is that cholestatic pups do not survive longer than 21 days. About 80% of the RRV infected newborn mice develop jaundice by the 7th day, stop gaining weight and die within three weeks. Although lethality is 100%, the actual cause of death is still unclear, in as much as these mice no longer have the virus. It is likely that dams no longer feed their diseased pups and one task would be to improve the survival of diseased pups, perhaps by means of artificial nourishment, in order to simulate better the natural history of BA. One final fundamental dissimilarity between murine and human BA is that mice do not develop severe liver fibrosis.
Optimizing the infective mouse model is a promising outlook for understanding of the pathological mechanism in mice but it needs to be balanced by effective translational research. We first see infants with BA when a good deal of the damage has already been done and do not know whether their immune response is primary and etiological or simply a secondary phenomenon.
Conclusion
Donald Rumsfeld served as the American secretary of state for defense from 2001-6 and highlighted rather a neat philosophical principle, although it was widely derided at the time. He said: “There are known knowns. These are things we know that we know. There are known unknowns. That is to say, there are things that we know we don’t know. But there are also unknown unknowns. There are things we don’t know we don’t know.” In terms of BA what do we know? We know it is an exceptionally rare disease and its onset is confined to the newborn period. We know that there are distinct clinical variants and therefore more than one mechanism for obliteration. Primary failure to develop luminal continuity seems obvious for “genetic” conditions such as BASM and similarly some local fetal insult or event perhaps causes CBA. What little clinical evidence there is suggests that most of the remainder are cholestatic in the first week of life. What are the known unknowns then? We struggle to relate what the best available experimental model tells us about the pathological mechanism evident in the human condition with any degree of consistency or accuracy [40,45,54]. That there is an integrated, multilayered, harmonised and structured inexorable inflammatory response to something (possibly viral) is clear, and that it occurs crucially within a very narrow window just after birth also appears obvious from the mouse model. The response is aberrant and peculiar to this period and may be auto-immune in its effect–although there is no suggestion that it continues much beyond the first few months (otherwise KPE and transplanted livers would invariably fail) or has any effect on other organs. Something makes the immature biliary tract susceptible in a way that the older child’s or adult’s liver is not. But viruses are ubiquitous, and it seems that many disparate viruses fit the offender profile laid out by the experimentalists; and if their role is crucial rather than incidental then why is this disease so rare? Improved synchronicity between those model-based workers with those able to furnish clinical material would help resolve some of these known unknowns. What then of those unknown unknowns, well we shall have to wait and see as they will surely declare themselves to a future audience.
Abbreviations
APC: Antigen presenting cells; BA: Biliary atresia; BASM: Biliary atresia splenic malformation; CBA: Cystic biliary atresia; CCL: Chemokine ligand; CCR-5: C-C chemokine receptor type 5; CD: Dendritic cell; CMV: Cytomegalovirus; CXCL: C-X-C chemokine ligand; dsRNA: Double-stranded RNA; Foxa 1: Forkhead box protein A1; Hh: Hedgehog; GVHD: Graft-versus-host disease; Hes-1: Hairy enhancer of split 1; HNF-6: Hepatocyte nuclear factor 6; HLA: Human leukocyte antigen; IF-10: Interferon inducible protein-10; IFN: Interferon; IL: Interleukin; iNOS: Cytokine-inducible mitric oxide synthase; MHC: Major histocompatibility complex; MIP2: Macrophage inflammatory protein; MMPs: Matrix metalloproteinases; mRN: Messenger ribonucleic acid; NF-κB: Nuclear factor kappa-light-chain-enhancer of activated B cells; NK-cells: Natural killer cells; PMA: Phorbol myristate acetate; RRV: Rhesus rotavirus; SCID: Severe combined immunodeficiency; SERPINA: Serpin peptidase inhibitor; STAT: Signal transducer and activator of transcription; TCR: T-cell receptor; TGF-β: Transforming growth factor beta; TNF: Tumor necrosis factor; T-reg: Regulatory T-cells; TUNEL: TdT-mediated dUTP-biotin nick end labeling; VP6: Viral protein 6.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CP and MD designed and wrote the paper, contributing equally to this work. Both authors approved the final version of the manuscript.
Contributor Information
Claus Petersen, Email: petersen.claus@mh-hannover.de.
Mark Davenport, Email: markdav2@ntlworld.com.
Disease name/synonyms
Biliary Atresia (BA), ORPHA30391, OMIM 210500.
Acknowledgement
The authors thank J. Kuebler, C. Klemann and A. Schröder for reviewing the manuscript concerning the immunological pathways in human and experimental BA.
Publication fees are supported by Deutsche Forschungsgemeinschaft.
References
- Hadzic N, Davenport M, Tizzard S, Singer J, Howard ER, Mieli-Vergani G. Long-term survival following Kasai portoenterostomy: is chronic liver disease inevitable? J Pediatr Gastroenterol Nutr. 2003;37(4):430–433. doi: 10.1097/00005176-200310000-00006. [DOI] [PubMed] [Google Scholar]
- Hadzic N, Quaglia A, Portmann B, Paramalingam S, Heaton ND, Rela M, Mieli-Vergani G, Davenport M. Hepatocellular carcinoma in biliary atresia: King’s College Hospital experience. J Pediatr. 2011;159(4):617–622. doi: 10.1016/j.jpeds.2011.03.004. e611. [DOI] [PubMed] [Google Scholar]
- Perlmutter DH, Shepherd RW. Extrahepatic biliary atresia: a disease or a phenotype? Hepatology. 2002;35(6):1297–1304. doi: 10.1053/jhep.2002.34170. [DOI] [PubMed] [Google Scholar]
- Davenport M. Biliary atresia: clinical aspects. Semin Pediatr Surg. 2012;21(3):175–184. doi: 10.1053/j.sempedsurg.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Davenport M, Savage M, Mowat AP, Howard ER. Biliary atresia splenic malformation syndrome: an etiologic and prognostic subgroup. Surgery. 1993;113(6):662–668. [PubMed] [Google Scholar]
- Davenport M, Tizzard SA, Underhill J, Mieli-Vergani G, Portmann B, Hadzic N. The biliary atresia splenic malformation syndrome: a 28-year single-center retrospective study. J Pediatr. 2006;149(3):393–400. doi: 10.1016/j.jpeds.2006.05.030. [DOI] [PubMed] [Google Scholar]
- Makin E, Quaglia A, Kvist N, Petersen BL, Portmann B, Davenport M. Congenital biliary atresia: liver injury begins at birth. J Pediatr Surg. 2009;44(3):630–633. doi: 10.1016/j.jpedsurg.2008.10.069. [DOI] [PubMed] [Google Scholar]
- Livesey E, Cortina Borja M, Sharif K, Alizai N, McClean P, Kelly D, Hadzic N, Davenport M. Epidemiology of biliary atresia in England and Wales (1999-2006) Arch Dis Child Fetal Neonatal Ed. 2009;94(6):F451–F455. doi: 10.1136/adc.2009.159780. [DOI] [PubMed] [Google Scholar]
- Davit-Spraul A, Baussan C, Hermeziu B, Bernard O, Jacquemin E. CFC1 gene involvement in biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr. 2008;46(1):111–112. doi: 10.1097/01.mpg.0000304465.60788.f4. [DOI] [PubMed] [Google Scholar]
- Bamford RN, Roessler E, Burdine RD, Saplakoglu U, Dela Cruz J, Splitt M, Goodship JA, Towbin J, Bowers P, Ferrero GB. et al. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nat Genet. 2000;26(3):365–369. doi: 10.1038/81695. [DOI] [PubMed] [Google Scholar]
- Allotey J, Lacaille F, Lees MM, Strautnieks S, Thompson RJ, Davenport M. Congenital bile duct anomalies (biliary atresia) and chromosome 22 aneuploidy. J Pediatr Surg. 2008;43(9):1736–1740. doi: 10.1016/j.jpedsurg.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Caponcelli E, Knisely AS, Davenport M. Cystic biliary atresia: an etiologic and prognostic subgroup. J Pediatr Surg. 2008;43(9):1619–1624. doi: 10.1016/j.jpedsurg.2007.12.058. [DOI] [PubMed] [Google Scholar]
- Hinds R, Davenport M, Mieli-Vergani G, Hadzic N. Antenatal presentation of biliary atresia. J Pediatr. 2004;144(1):43–46. doi: 10.1016/j.jpeds.2003.09.027. [DOI] [PubMed] [Google Scholar]
- Chardot C, Carton M, Spire-Bendelac N, Le Pommelet C, Golmard JL, Auvert B. Epidemiology of biliary atresia in France: a national study 1986-96. J Hepatol. 1999;31(6):1006–1013. doi: 10.1016/S0168-8278(99)80312-2. [DOI] [PubMed] [Google Scholar]
- Hsiao CH, Chang MH, Chen HL, Lee HC, Wu TC, Lin CC, Yang YJ, Chen AC, Tiao MM, Lau BH. et al. Universal screening for biliary atresia using an infant stool color card in Taiwan. Hepatology. 2008;47(4):1233–1240. doi: 10.1002/hep.22182. [DOI] [PubMed] [Google Scholar]
- Leonhardt J, Kuebler JF, Leute PJ, Turowski C, Becker T, Pfister ED, Ure B, Petersen C. Biliary atresia: lessons learned from the voluntary German registry. Eur J Pediatr Surg. 2011;21(2):82–87. doi: 10.1055/s-0030-1268476. [DOI] [PubMed] [Google Scholar]
- Tan CE, Driver M, Howard ER, Moscoso GJ. Extrahepatic biliary atresia: a first-trimester event? Clues from light microscopy and immunohistochemistry. J Pediatr Surg. 1994;29(6):808–814. doi: 10.1016/0022-3468(94)90377-8. [DOI] [PubMed] [Google Scholar]
- Sokol RJ, Mack C. Etiopathogenesis of biliary atresia. Semin Liver Dis. 2001;21(4):517–524. doi: 10.1055/s-2001-19032. [DOI] [PubMed] [Google Scholar]
- Sokol RJ, Mack C, Narkewicz MR, Karrer FM. Pathogenesis and outcome of biliary atresia: current concepts. J Pediatr Gastroenterol Nutr. 2003;37(1):4–21. doi: 10.1097/00005176-200307000-00003. [DOI] [PubMed] [Google Scholar]
- Davenport M, Saxena R, Howard E. Acquired biliary atresia. J Pediatr Surg. 1996;31(12):1721–1723. doi: 10.1016/S0022-3468(96)90062-7. [DOI] [PubMed] [Google Scholar]
- Muller F, Oury JF, Dumez Y, Boue J, Boue A. Microvillar enzyme assays in amniotic fluid and fetal tissues at different stages of development. Prenat Diagn. 1988;8(3):189–198. doi: 10.1002/pd.1970080305. [DOI] [PubMed] [Google Scholar]
- Mushtaq I, Logan S, Morris M, Johnson AW, Wade AM, Kelly D, Clayton PT. Screening of newborn infants for cholestatic hepatobiliary disease with tandem mass spectrometry. Bmj. 1999;319(7208):471–477. doi: 10.1136/bmj.319.7208.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpavat S, Finegold MJ, Karpen SJ. Patients with biliary atresia have elevated direct/conjugated bilirubin levels shortly after birth. Pediatrics. 2011;128(6):e1428–e1433. doi: 10.1542/peds.2011-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morecki R, Glaser JH, Cho S, Balistreri WF, Horwitz MS. Biliary atresia and reovirus type 3 infection. N Engl J Med. 1982;307(8):481–484. doi: 10.1056/NEJM198208193070806. [DOI] [PubMed] [Google Scholar]
- Riepenhoff-Talty M, Gouvea V, Evans MJ, Svensson L, Hoffenberg E, Sokol RJ, Uhnoo I, Greenberg SJ, Schakel K, Zhaori G. et al. Detection of group C rotavirus in infants with extrahepatic biliary atresia. J Infect Dis. 1996;174(1):8–15. doi: 10.1093/infdis/174.1.8. [DOI] [PubMed] [Google Scholar]
- De Tommaso AM, Andrade PD, Costa SC, Escanhoela CA, Hessel G. High frequency of human cytomegalovirus DNA in the liver of infants with extrahepatic neonatal cholestasis. BMC Infect Dis. 2005;5:108. doi: 10.1186/1471-2334-5-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischler B, Ehrnst A, Forsgren M, Orvell C, Nemeth A. The viral association of neonatal cholestasis in Sweden: a possible link between cytomegalovirus infection and extrahepatic biliary atresia. J Pediatr Gastroenterol Nutr. 1998;27(1):57–64. doi: 10.1097/00005176-199807000-00010. [DOI] [PubMed] [Google Scholar]
- Rauschenfels S, Krassmann M, Al-Masri AN, Verhagen W, Leonhardt J, Kuebler JF, Petersen C. Incidence of hepatotropic viruses in biliary atresia. Eur J Pediatr. 2009;168(4):469–476. doi: 10.1007/s00431-008-0774-2. [DOI] [PubMed] [Google Scholar]
- Kobayashi AKS, Benno Y. In: Biliary atresia. Ohi R, editor. Tokyo: Icom associates; 1991. Fecal flora of infants with biliary atresia: effects of the absence of bile on fecal flora. [DOI] [PubMed] [Google Scholar]
- Schukfeh N, Al-Gamrah A, Petersen C, Kuebler JF. Detection of hepatotropic viruses has no impact on the prognosis after Kasai procedure. J Pediatr Surg. 2012;47(10):1828–1832. doi: 10.1016/j.jpedsurg.2012.04.024. [DOI] [PubMed] [Google Scholar]
- Zani AQA, Hadzic N, Davenport M. 3rd World Congress of Pediatric Surgery (WOFAPS) New Delhi, India: ; 2010. Cytomegalovirus-associated biliary atresia is a distinct pathological entity. [Google Scholar]
- Feldman AG, Mack CL. Biliary atresia: cellular dynamics and immune dysregulation. Semin Pediatr Surg. 2012;21(3):192–200. doi: 10.1053/j.sempedsurg.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack CL, Tucker RM, Sokol RJ, Karrer FM, Kotzin BL, Whitington PF, Miller SD. Biliary atresia is associated with CD4+ Th1 cell-mediated portal tract inflammation. Pediatr Res. 2004;56(1):79–87. doi: 10.1203/01.PDR.0000130480.51066.FB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya T, Fujimoto T, Shimomura H, Miyano T. Degeneration of intrahepatic bile duct with lymphocyte infiltration into biliary epithelial cells in biliary atresia. J Pediatr Surg. 1995;30(4):515–518. doi: 10.1016/0022-3468(95)90120-5. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Li Z, Yamataka A, Lane GJ, Miyano T. Role of immunologic costimulatory factors in the pathogenesis of biliary atresia. J Pediatr Surg. 2003;38(6):892–896. doi: 10.1016/S0022-3468(03)00117-9. [DOI] [PubMed] [Google Scholar]
- Schweizer P, Petersen M, Jeszberger N, Ruck P, Dietz K. Immunohistochemical and molecular biological investigations regarding the pathogenesis of extrahepatic biliary atresia: (Part 1: immunohistochemical studies) Eur J Pediatr Surg. 2003;13(1):7–15. doi: 10.1055/s-2003-38287. [DOI] [PubMed] [Google Scholar]
- Uddin Ahmed AF, Ohtani H, Nio M, Funaki N, Sasaki H, Nagura H, Ohi R. Intrahepatic mast cell population correlates with clinical outcome in biliary atresia. J Pediatr Surg. 2000;35(12):1762–1765. doi: 10.1053/jpsu.2000.19245. [DOI] [PubMed] [Google Scholar]
- Urushihara N, Iwagaki H, Yagi T, Kohka H, Kobashi K, Morimoto Y, Yoshino T, Tanimoto T, Kurimoto M, Tanaka N. Elevation of serum interleukin-18 levels and activation of Kupffer cells in biliary atresia. J Pediatr Surg. 2000;35(3):446–449. doi: 10.1016/S0022-3468(00)90211-2. [DOI] [PubMed] [Google Scholar]
- Dillon PW, Belchis D, Minnick K, Tracy T. Differential expression of the major histocompatibility antigens and ICAM-1 on bile duct epithelial cells in biliary atresia. Tohoku J Exp Med. 1997;181(1):33–40. doi: 10.1620/tjem.181.33. [DOI] [PubMed] [Google Scholar]
- Davenport M, Gonde C, Redkar R, Koukoulis G, Tredger M, Mieli-Vergani G, Portmann B, Howard ER. Immunohistochemistry of the liver and biliary tree in extrahepatic biliary atresia. J Pediatr Surg. 2001;36(7):1017–1025. doi: 10.1053/jpsu.2001.24730. [DOI] [PubMed] [Google Scholar]
- Dong R, Zhao R, Zheng S. Changes in epigenetic regulation of CD4+ T lymphocytesin biliary atresia. Pediatr Res. 2011;70(6):555–559. doi: 10.1203/PDR.0b013e318232a949. [DOI] [PubMed] [Google Scholar]
- Socha P, Michalkiewicz J, Stachowski J, Pawlowska J, Jankowska I, Barth C, Socha J, Madalinski K. Deficiency of the expression of CD45RA isoform of CD45 common leukocyte antigen in CD4+ T lymphocytes in children with infantile cholestasis. Immunol Lett. 2001;75(3):179–184. doi: 10.1016/S0165-2478(00)00305-9. [DOI] [PubMed] [Google Scholar]
- Bezerra JA, Tiao G, Ryckman FC, Alonso M, Sabla GE, Shneider B, Sokol RJ, Aronow BJ. Genetic induction of proinflammatory immunity in children with biliary atresia. Lancet. 2002;360(9346):1653–1659. doi: 10.1016/S0140-6736(02)11603-5. [DOI] [PubMed] [Google Scholar]
- Huang L, Si XM, Feng JX. NF-kappaB related abnormal hyper-expression of iNOS and NO correlates with the inflammation procedure in biliary atresia livers. Pediatr Surg Int. 2010;26(9):899–905. doi: 10.1007/s00383-010-2683-5. [DOI] [PubMed] [Google Scholar]
- Mack CL, Falta MT, Sullivan AK, Karrer F, Sokol RJ, Freed BM, Fontenot AP. Oligoclonal expansions of CD4+ and CD8+ T-cells in the target organ of patients with biliary atresia. Gastroenterology. 2007;133(1):278–287. doi: 10.1053/j.gastro.2007.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai M, Shinkai T, Puri P, Stringer MD. Elevated expression of IL2 is associated with increased infiltration of CD8+ T cells in biliary atresia. J Pediatr Surg. 2006;41(2):300–305. doi: 10.1016/j.jpedsurg.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Shinkai M, Shinkai T, Puri P, Stringer MD. Increased CXCR3 expression associated with CD3-positive lymphocytes in the liver and biliary remnant in biliary atresia. J Pediatr Surg. 2006;41(5):950–954. doi: 10.1016/j.jpedsurg.2006.01.060. [DOI] [PubMed] [Google Scholar]
- Ahmed AF, Ohtani H, Nio M, Funaki N, Shimaoka S, Nagura H, Ohi R. CD8+ T cells infiltrating into bile ducts in biliary atresia do not appear to function as cytotoxic T cells: a clinicopathological analysis. J Pathol. 2001;193(3):383–389. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH793>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Muraji T, Hosaka N, Irie N, Yoshida M, Imai Y, Tanaka K, Takada Y, Sakamoto S, Haga H, Ikehara S. Maternal microchimerism in underlying pathogenesis of biliary atresia: quantification and phenotypes of maternal cells in the liver. Pediatrics. 2008;121(3):517–521. doi: 10.1542/peds.2007-0568. [DOI] [PubMed] [Google Scholar]
- Harada K, Nakanuma Y. Biliary innate immunity: function and modulation. Mediators Inflamm. p. 373878. Epub 2010 Jul 27. [DOI] [PMC free article] [PubMed]
- Harada K, Nakanuma Y. Biliary innate immunity in the pathogenesis of biliary diseases. Inflamm Allergy Drug Targets. 2010;9(2):83–90. doi: 10.2174/187152810791292809. [DOI] [PubMed] [Google Scholar]
- Minnick KE, Kreisberg R, Dillon PW. Soluble ICAM-1 (sICAM-1) in biliary atresia and its relationship to disease activity. J Surg Res. 1998;76(1):53–56. doi: 10.1006/jsre.1998.5285. [DOI] [PubMed] [Google Scholar]
- Davenport M, Gonde C, Narayanaswamy B, Mieli-Vergani G, Tredger JM. Soluble adhesion molecule profiling in preoperative infants with biliary atresia. J Pediatr Surg. 2005;40(9):1464–1469. doi: 10.1016/j.jpedsurg.2005.05.050. [DOI] [PubMed] [Google Scholar]
- Narayanaswamy B, Gonde C, Tredger JM, Hussain M, Vergani D, Davenport M. Serial circulating markers of inflammation in biliary atresia–evolution of the post-operative inflammatory process. Hepatology. 2007;46(1):180–187. doi: 10.1002/hep.21701. [DOI] [PubMed] [Google Scholar]
- Harada K, Sato Y, Isse K, Ikeda H, Nakanuma Y. Induction of innate immune response and absence of subsequent tolerance to dsRNA in biliary epithelial cells relate to the pathogenesis of biliary atresia. Liver Int. 2008;28(5):614–621. doi: 10.1111/j.1478-3231.2008.01740.x. [DOI] [PubMed] [Google Scholar]
- Omenetti A, Bass LM, Anders RA, Clemente MG, Francis H, Guy CD, McCall S, Choi SS, Alpini G, Schwarz KB. et al. Hedgehog activity, epithelial-mesenchymal transitions, and biliary dysmorphogenesis in biliary atresia. Hepatology. 2011;53(4):1246–1258. doi: 10.1002/hep.24156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy TF Jr, Dillon P, Fox ES, Minnick K, Vogler C. The inflammatory response in pediatric biliary disease: macrophage phenotype and distribution. J Pediatr Surg. 1996;31(1):121–125. doi: 10.1016/S0022-3468(96)90333-4. discussion 125-126. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Puri P, O’Briain DS, Surana R, Miyano T. Hepatic overexpression of MHC class II antigens and macrophage-associated antigens (CD68) in patients with biliary atresia of poor prognosis. J Pediatr Surg. 1997;32(4):590–593. doi: 10.1016/S0022-3468(97)90714-4. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sargent PA, Fisher MM, Youson JH. Periductal fibrosis and lipocytes (fat-storing cells or Ito cells) during biliary atresia in the lamprey. Hepatology. 1986;6(1):54–59. doi: 10.1002/hep.1840060111. [DOI] [PubMed] [Google Scholar]
- Youson JH, Sidon EW. Lamprey biliary atresia: first model system for the human condition? Experientia. 1978;34(8):1084–1086. doi: 10.1007/BF01915363. [DOI] [PubMed] [Google Scholar]
- Bastianello SS, Nesbit JW. The pathology of a case of biliary atresia in a foal. J S Afr Vet Assoc. 1986;57(2):117–120. [PubMed] [Google Scholar]
- Rosenberg DP, Morecki R, Lollini LO, Glaser J, Cornelius CE. Extrahepatic biliary atresia in a rhesus monkey (Macaca mulatta) Hepatology. 1983;3(4):577–580. doi: 10.1002/hep.1840030417. [DOI] [PubMed] [Google Scholar]
- Harper P, Plant JW, Unger DB. Congenital biliary atresia and jaundice in lambs and calves. Aust Vet J. 1990;67(1):18–22. doi: 10.1111/j.1751-0813.1990.tb07385.x. [DOI] [PubMed] [Google Scholar]
- Matthews RP, Eauclaire SF, Mugnier M, Lorent K, Cui S, Ross MM, Zhang Z, Russo P, Pack M. DNA hypomethylation causes bile duct defects in zebrafish and is a distinguishing feature of infantile biliary atresia. Hepatology. 2011;53(3):905–914. doi: 10.1002/hep.24106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskams T, Desmet V. Embryology of extra–and intrahepatic bile ducts, the ductal plate. Anat Rec. 2008;291:628–635. doi: 10.1002/ar.20710. [DOI] [PubMed] [Google Scholar]
- Lemaigre FP. Notch signaling in bile duct development: new insights raise new questions. Hepatology. 2008;48(2):358–360. doi: 10.1002/hep.22480. [DOI] [PubMed] [Google Scholar]
- Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, Lemaigre F, Stanger BZ. Notch signaling controls liver development by regulating biliary differentiation. Development. 2009;136(10):1727–1739. doi: 10.1242/dev.029140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama T, Copeland NG, Jenkins NA, Montgomery CA, Elder FF, Overbeek PA. Reversal of left-right asymmetry: a situs inversus mutation. Science. 1993;260(5108):679–682. doi: 10.1126/science.8480178. [DOI] [PubMed] [Google Scholar]
- Mazziotti MV, Willis LK, Heuckeroth RO, LaRegina MC, Swanson PE, Overbeek PA, Perlmutter DH. Anomalous development of the hepatobiliary system in the Inv mouse. Hepatology. 1999;30(2):372–378. doi: 10.1002/hep.510300223. [DOI] [PubMed] [Google Scholar]
- Shimadera S, Iwai N, Deguchi E, Kimura O, Fumino S, Yokoyama T. The inv mouse as an experimental model of biliary atresia. J Pediatr Surg. 2007;42(9):1555–1560. doi: 10.1016/j.jpedsurg.2007.04.018. [DOI] [PubMed] [Google Scholar]
- Holder TM, Ashcraft KW. Production of experimental biliary atresia by ligation of the common bile duct in the fetus. Surg Forum. 1966;17:356–357. [PubMed] [Google Scholar]
- Spitz L. Ligation of the common bile duct in the fetal lamb: an experimental model for the study of biliary atresia. Pediatr Res. 1980;14(5):740–748. doi: 10.1203/00006450-198005000-00007. [DOI] [PubMed] [Google Scholar]
- Morgan WW Jr, Rosenkrantz JC, Hill RB Jr. Hepatic arterial interruption in the fetus–an attempt to simulate biliary atresia. J Pediatr Surg. 1966;1(4):342–346. doi: 10.1016/0022-3468(66)90336-8. [DOI] [PubMed] [Google Scholar]
- Hosoda Y, Miyano T, Fujimoto T. Assay of gamma-glutamyl transpeptidase activity in amniotic fluid offers a possible prenatal diagnosis of biliary atresia in the rat model. Prenat Diagn. 1997;17(1):9–12. doi: 10.1002/(SICI)1097-0223(199701)17:1<9::AID-PD12>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Suruga K, Kojima Y, Kitahara T, Kuwabara N. Experimental study of the pathogenesis of infantile obstructive cholangiopathy and its clinical evaluation. J Pediatr Surg. 1983;18(2):131–135. doi: 10.1016/S0022-3468(83)80533-8. [DOI] [PubMed] [Google Scholar]
- Held MA, Cosme-Blanco W, Difedele LM, Bonkowski EL, Menon RK, Denson LA. Alterations in growth hormone receptor abundance regulate growth hormone signaling in murine obstructive cholestasis. Am J Physiol Gastrointest Liver Physiol. 2005;288(5):G986–G993. doi: 10.1152/ajpgi.00287.2004. [DOI] [PubMed] [Google Scholar]
- Sokal EM, Baudoux MC, Collette E, Hausleithner V, Lambotte L, Buts JP. Branched chain amino acids improve body composition and nitrogen balance in a rat model of extra hepatic biliary atresia. Pediatr Res. 1996;40(1):66–71. doi: 10.1203/00006450-199607000-00012. [DOI] [PubMed] [Google Scholar]
- Andrews HG, Shermeta DW, White JJ, Haller JA Jr. Hepatic artery interruption in fetal and neonatal miniature pigs: studies in the pathogenesis of biliary atresia. Surg Forum. 1970;21:384–385. [PubMed] [Google Scholar]
- Schmeling DJ, Oldham KT, Guice KS, Kunkel RG, Johnson KJ. Experimental obliterative cholangitis: a model for the study of biliary atresia. Ann Surg. 1991;213(4):350–355. doi: 10.1097/00000658-199104000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber RA, Kleinman RE, Barksdale EM Jr, Maganaro TF, Donahoe PK. Rejection of murine congenic bile ducts: a model for immune-mediated bile duct disease. Gastroenterology. 1992;102(3):924–930. doi: 10.1016/0016-5085(92)90178-2. [DOI] [PubMed] [Google Scholar]
- Papadimitriou JM. The biliary tract in acute murine reovirus 3 infection: light and electron microscopic study. Am J Pathol. 1968;52(3):595–611. [PMC free article] [PubMed] [Google Scholar]
- Phillips PA, Keast D, Papadimitriou JM, Walters MN, Stanley NF. Chronic obstructive jaundice induced by reovirus type 3 in weanling mice. Pathology. 1969;1(3):193–203. doi: 10.3109/00313026909071296. [DOI] [PubMed] [Google Scholar]
- Barton ES, Youree BE, Ebert DH, Forrest JC, Connolly JL, Valyi-Nagy T, Washington K, Wetzel JD, Dermody TS. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J Clin Invest. 2003;111(12):1823–1833. doi: 10.1172/JCI16303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Gu WZ, Si XM, Wei MF, Feng JX. Expression of NF-kappaB in rotavirus-induced damage to the liver and biliary tract in neonatal mice. Hepatobiliary Pancreat Dis Int. 2007;6(2):188–193. [PubMed] [Google Scholar]
- Szavay PO, Leonhardt J, Czech-Schmidt G, Petersen C. The role of reovirus type 3 infection in an established murine model for biliary atresia. Eur J Pediatr Surg. 2002;12(4):248–250. doi: 10.1055/s-2002-34477. [DOI] [PubMed] [Google Scholar]
- Saxena V, Shivakumar P, Sabla G, Mourya R, Chougnet C, Bezerra JA. Dendritic cells regulate natural killer cell activation and epithelial injury in experimental biliary atresia. Sci Transl Med. 2011;3(102):102ra194. doi: 10.1126/scitranslmed.3002069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riepenhoff-Talty M, Schaekel K, Clark HF, Mueller W, Uhnoo I, Rossi T, Fisher J, Ogra PL. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr Res. 1993;33(4 Pt 1):394–399. doi: 10.1203/00006450-199304000-00016. [DOI] [PubMed] [Google Scholar]
- Chan RY, Tan CE, Czech-Schmidt G, Petersen C. Computerized three-dimensional study of a rotavirus model of biliary atresia: comparison with human biliary atresia. Pediatr Surg Int. 2005;21(8):615–620. doi: 10.1007/s00383-005-1483-9. [DOI] [PubMed] [Google Scholar]
- Petersen C, Biermanns D, Kuske M, Schakel K, Meyer-Junghanel L, Mildenberger H. New aspects in a murine model for extrahepatic biliary atresia. J Pediatr Surg. 1997;32(8):1190–1195. doi: 10.1016/S0022-3468(97)90680-1. [DOI] [PubMed] [Google Scholar]
- Petersen C, Grasshoff S, Luciano L. Diverse morphology of biliary atresia in an animal model. J Hepatol. 1998;28(4):603–607. doi: 10.1016/S0168-8278(98)80283-3. [DOI] [PubMed] [Google Scholar]
- Nadler EP, Li X, Onyedika E, Greco MA. Differential expression of hepatic fibrosis mediators in sick and spontaneously recovered mice with experimental biliary atresia. J Surg Res. 2010;159(2):611–617. doi: 10.1016/j.jss.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler EP, Patterson D, Violette S, Weinreb P, Lewis M, Magid MS, Greco MA. Integrin alphavbeta6 and mediators of extracellular matrix deposition are up-regulated in experimental biliary atresia. J Surg Res. 2009;154(1):21–29. doi: 10.1016/j.jss.2008.05.023. [DOI] [PubMed] [Google Scholar]
- Petersen C, Kuske M, Bruns E, Biermanns D, Wussow PV, Mildenberger H. Progress in developing animal models for biliary atresia. Eur J Pediatr Surg. 1998;8(3):137–141. doi: 10.1055/s-2008-1071140. [DOI] [PubMed] [Google Scholar]
- Turowski C, Leonhardt J, Teichmann B, Heim A, Baumann U, Kuebler JF, Petersen C. Preconceptional oral vaccination prevents experimental biliary atresia in newborn mice. Eur J Pediatr Surg. 2010;20(3):158–163. doi: 10.1055/s-0030-1249700. [DOI] [PubMed] [Google Scholar]
- Bondoc AJ, Jafri MA, Donnelly B, Mohanty SK, McNeal MM, Ward RL, Tiao GM. Prevention of the murine model of biliary atresia after live rotavirus vaccination of dams. J Pediatr Surg. 2009;44(8):1479–1490. doi: 10.1016/j.jpedsurg.2009.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech-Schmidt G, Verhagen W, Szavay P, Leonhardt J, Petersen C. Immunological gap in the infectious animal model for biliary atresia. J Surg Res. 2001;101(1):62–67. doi: 10.1006/jsre.2001.6234. [DOI] [PubMed] [Google Scholar]
- Barnes BH, Tucker RM, Wehrmann F, Mack DG, Ueno Y, Mack CL. Cholangiocytes as immune modulators in rotavirus-induced murine biliary atresia. Liver Int. 2009;29(8):1253–1261. doi: 10.1111/j.1478-3231.2008.01921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafri M, Donnelly B, Allen S, Bondoc A, McNeal M, Rennert PD, Weinreb PH, Ward R, Tiao G. Cholangiocyte expression of alpha2beta1-integrin confers susceptibility to rotavirus-induced experimental biliary atresia. Am J Physiol Gastrointest Liver Physiol. 2008;295(1):G16–G26. doi: 10.1152/ajpgi.00442.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivakumar P, Campbell KM, Sabla GE, Miethke A, Tiao G, McNeal MM, Ward RL, Bezerra JA. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN-gamma in experimental biliary atresia. J Clin Invest. 2004;114(3):322–329. doi: 10.1172/JCI21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty SK, Ivantes CA, Mourya R, Pacheco C, Bezerra JA. Macrophages are targeted by rotavirus in experimental biliary atresia and induce neutrophil chemotaxis by Mip2/Cxcl2. Pediatr Res. 2010;67(4):345–351. doi: 10.1203/PDR.0b013e3181d22a73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafri M, Donnelly B, Bondoc A, Allen S, Tiao G. Cholangiocyte secretion of chemokines in experimental biliary atresia. J Pediatr Surg. 2009;44(3):500–507. doi: 10.1016/j.jpedsurg.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt J, Kuebler JF, Turowski C, Tschernig T, Geffers R, Petersen C. Susceptibility to experimental biliary atresia linked to different hepatic gene expression profiles in two mouse strains. Hepatol Res. 2010;40(2):196–203. doi: 10.1111/j.1872-034X.2009.00577.x. [DOI] [PubMed] [Google Scholar]
- Tucker RM, Hendrickson RJ, Mukaida N, Gill RG, Mack CL. Progressive biliary destruction is independent of a functional tumor necrosis factor-alpha pathway in a rhesus rotavirus-induced murine model of biliary atresia. Viral Immunol. 2007;20(1):34–43. doi: 10.1089/vim.2006.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt J, Stanulla M, von Wasielewski R, Skokowa J, Kübler J, Ure BM, Petersen C. Gene expression profile of the infective murine model for biliary atresia. Pediatr Surg Int. 2006;22(1):84–89. doi: 10.1007/s00383-005-1589-0. [DOI] [PubMed] [Google Scholar]
- Mohanty SK, Shivakumar P, Sabla G, Bezerra JA. Loss of interleukin-12 modifies the pro-inflammatory response but does not prevent duct obstruction in experimental biliary atresia. BMC Gastroenterol. 2006;6:14. doi: 10.1186/1471-230X-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Bessho K, Shivakumar P, Mourya R, Mohanty SK, Dos Santos JL, Miura IK, Porta G, Bezerra JA. Th2 signals induce epithelial injury in mice and are compatible with the biliary atresia phenotype. J Clin Invest. 2011;121(11):4244–4256. doi: 10.1172/JCI57728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuebler JF, Czech-Schmidt G, Leonhardt J, Ure BM, Petersen C. Type-I but not type-II interferon receptor knockout mice are susceptible to biliary atresia. Pediatr Res. 2006;59(6):790–794. doi: 10.1203/01.pdr.0000219860.96732.09. [DOI] [PubMed] [Google Scholar]
- Wehrmann F, Kuebler JF, Wienecke S, Al-Masri AN, Petersen C, Leonhardt J. Functional Mx protein does not prevent experimental biliary atresia in Balb/c mice. Eur J Pediatr Surg. 2008;18(5):318–321. doi: 10.1055/s-2008-1038720. [DOI] [PubMed] [Google Scholar]
- Erickson N, Mohanty SK, Shivakumar P, Sabla G, Chakraborty R, Bezerra JA. Temporal-spatial activation of apoptosis and epithelial injury in murine experimental biliary atresia. Hepatology. 2008;47(5):1567–1577. doi: 10.1002/hep.22229. [DOI] [PubMed] [Google Scholar]
- Shivakumar P, Sabla G, Mohanty S, McNeal M, Ward R, Stringer K, Caldwell C, Chougnet C, Bezerra JA. Effector role of neonatal hepatic CD8+ lymphocytes in epithelial injury and autoimmunity in experimental biliary atresia. Gastroenterology. 2007;133(1):268–277. doi: 10.1053/j.gastro.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty SK, Donnelly B, Bondoc A, Jafri M, Walther A, Coots A, McNeal M, Witte D, Tiao GM. Rotavirus replication in the cholangiocyte mediates the temporal dependence of murine biliary atresia. PLoS One. 2013;3;8(7):E69069. doi: 10.1371/journal.pone.0069069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack CL, Sokol RJ. Unraveling the pathogenesis and etiology of biliary atresia. Pediatr Res. 2005;57(5 Pt 2):87R–94R. doi: 10.1203/01.PDR.0000159569.57354.47. [DOI] [PubMed] [Google Scholar]
- Mack CL, Tucker RM, Sokol RJ, Kotzin BL. Armed CD4+ Th1 effector cells and activated macrophages participate in bile duct injury in murine biliary atresia. Clin Immunol. 2005;115(2):200–209. doi: 10.1016/j.clim.2005.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miethke AG, Saxena V, Shivakumar P, Sabla GE, Simmons J, Chougnet CA. Post-natal paucity of regulatory T cells and control of NK cell activation in experimental biliary atresia. J Hepatol. 2010;52(5):718–726. doi: 10.1016/j.jhep.2009.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivakumar P, Sabla GE, Whitington P, Chougnet CA, Bezerra JA. Neonatal NK cells target the mouse duct epithelium via Nkg2d and drive tissue-specific injury in experimental biliary atresia. J Clin Invest. 2009;119(8):2281–2290. doi: 10.1172/JCI38879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack CL, Tucker RM, Lu BR, Sokol RJ, Fontenot AP, Ueno Y, Gill RG. Cellular and humoral autoimmunity directed at bile duct epithelia in murine biliary atresia. Hepatology. 2006;44(5):1231–1239. doi: 10.1002/hep.21366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkow K, Loddenkemper C, Mintern J, Kruse N, Klugewitz K, Berg T, Wiedenmann B, Ploegh HL, Schott E. Differential priming of CD8 and CD4 T-cells in animal models of autoimmune hepatitis and cholangitis. Hepatology. 2007;46(4):1155–1165. doi: 10.1002/hep.21796. [DOI] [PubMed] [Google Scholar]
- Feng J, Li M, Cai T, Tang H, Gu W. Rotavirus-induced murine biliary atresia is mediated by nuclear factor-kappaB. J Pediatr Surg. 2005;40(4):630–636. doi: 10.1016/j.jpedsurg.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Lu BR, Brindley SM, Tucker RM, Lambert CL, Mack CL. Alpha-enolase autoantibodies cross-reactive to viral proteins in a mouse model of biliary atresia. Gastroenterology. 2010;139(5):1753–1761. doi: 10.1053/j.gastro.2010.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng N, Kim B, Fenaux M, Nguyen H, Vo P, Omary MB, Greenberg HB. Role of interferon in homologous and heterologous rotavirus infection in the intestines and extraintestinal organs of suckling mice. J Virol. 2008;82(15):7578–7590. doi: 10.1128/JVI.00391-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen C, Bruns E, Kuske M, Von Wussow P. Treatment of extrahepatic biliary atresia with interferon-alpha in a murine infectious model. Pediatr Res. 1997;42(5):623–628. doi: 10.1203/00006450-199711000-00013. [DOI] [PubMed] [Google Scholar]
- Turowski C, Leonhardt J, Teichmann B, Heim A, Baumann U, Kuebler JF, Petersen C. Preconceptional oral vaccination prevents experimental biliary atresia in newborn mice. Eur J Pediatr Surg. pp. 158–163. [DOI] [PubMed]
- Brindley SM, Lanham AM, Karrer FM, Tucker RM, Fontenot AP, Mack CL. Cytomegalovirus-specific T-cell reactivity in biliary atresia at the time of diagnosis is associated with deficits in regulatory T cells. Hepatology. 2012;55(4):1130–1138. doi: 10.1002/hep.24807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel PM, Crawford SE, Finegold MJ, Estes MK. Osteopontin upregulation in rotavirus-induced murine biliary atresia requires replicating virus but is not necessary for development of biliary atresia. Virology. 2011;417(2):281–292. doi: 10.1016/j.virol.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Hishiki T, Terui K, Mitsunaga T, Terui E, Nakata M, Yoshida H. Toll-like receptor mRNA expression in liver tissue from patients with biliary atresia. J Pediatr Gastroenterol Nutr. 2011;53(6):620–626. doi: 10.1097/MPG.0b013e3182307c9c. [DOI] [PubMed] [Google Scholar]
- Lages CS, Simmons J, Chougnet CA, Miethke AG. Regulatory T cells control the CD8 adaptive immune response at the time of ductal obstruction in experimental biliary atresia. Hepatology. 2012;56(1):219–227. doi: 10.1002/hep.25662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Zhu J, Pu CL, Deng YH, Zhang MM. Combinatory effects of hepatic CD8+ and NK lymphocytes in bile duct injury from biliary atresia. Pediatr Res. 2012;71(6):638–644. doi: 10.1038/pr.2012.17. [DOI] [PubMed] [Google Scholar]
- Carvalho E, Liu C, Shivakumar P, Sabla G, Aronow B, Bezerra JA. Analysis of the biliary transcriptome in experimental biliary atresia. Gastroenterology. 2005;129(2):713–717. doi: 10.1016/j.gastro.2005.05.052. [DOI] [PubMed] [Google Scholar]
- Kotb MA, El Henawy A, Talaat S, Aziz M, El Tagy GH, El Barbary MM, Mostafa W. Immune-mediated liver injury: prognostic value of CD4+, CD8+, and CD68+ in infants with extrahepatic biliary atresia. J Pediatr Surg. 2005;40(8):1252–1257. doi: 10.1016/j.jpedsurg.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Pawlowska J, Michalkiewicz J, Czubkowski P, Jankowska I, Teisseyre M, Socha P, Kalicinski P, Socha J. Reduction of naive CD4/CD45RA + T cells in children with biliary atresia before and after liver transplantation. Transplant Proc. 2003;35(8):3026–3028. doi: 10.1016/j.transproceed.2003.10.088. [DOI] [PubMed] [Google Scholar]
- Chen K, Gavaler JS, Van Thiel DH, Whiteside T. Phenotypic characterization of mononuclear infiltrate present in liver of biliary atresia. Dig Dis Sci. 1989;34(10):1564–1570. doi: 10.1007/BF01537111. [DOI] [PubMed] [Google Scholar]
- Tucker RM, Feldman AG, Fenner EK, Mack CL. Regulatory T Cells Inhibit Th1 cell-mediated bile duct injury in murine biliary atresia. J Hepatol. 2013. Epub ahead of print. [DOI] [PMC free article] [PubMed]