

Abstract
Purpose
Cancer chemoprevention is defined as the use of natural, synthetic, or biological agents to suppress, reverse or prevent the carcinogenic process from turning into aggressive cancer. Prostate apoptosis response-4 (Par-4) is a unique pro-apoptotic protein that selectively induces apoptosis in prostate cancer cells. However, its role in other malignancies has not been fully explored. This study tries to identify the functional significance of Par-4 in pancreatic cancer.
Methods
Multiple molecular techniques such as Western blot analysis, trypan blue assay for cell viability, MTT assay for cell growth inhibition and Histone/DNA ELISA for apoptosis were used.
Results
Western blot analysis revealed that 3,3′-diindolylmethane (DIM) a chemopreventive agent, specifically its more bioavailable formulation, B-DIM, at low doses (20 μmol/L) induces Par-4, in L3.6pl and Colo-357 pancreatic cancer cells. At similar doses, DIM reduced cell viability and caused cell growth inhibition and apoptosis. Moreover, DIM pre-treatment sensitized the cells to cytotoxic action of chemotherapeutic drug gemcitabine through up-regulation of Par-4.
Conclusion
The induction of Par-4 is indirectly related to increased sensitivity and cell death through apoptosis. To our knowledge the results reported here showed, for the first time, the induction of Par-4 by chemopreventive agents, in general, and DIM, in particular, in pancreatic cancer cells in vitro.
Keywords: apoptosis, B-DIM, chemoprevention, pancreatic cancer, Par-4
INTRODUCTION
The sustained magnitude of cancer problem and the failure of conventional therapy for treatment of advanced invasive disease indicate that new and alternative approaches to the control of cancer are critically needed. In this context, one has to re-evaluate some of the basic assumptions about the nature of cancer and the response that one might observe. Recent years have witnessed a tremendous increase in research on the use of chemopreventive agents in the modulation of cancer cell behavior toward the treatment of cancer. The definition of chemoprevention as given by National Cancer Institute is “the use of drugs, vitamins, or other agents to try to reduce the risk of, or delay the development or recurrence of, cancer”. Though the past decade has made quite substantial progress in introducing regimes of chemoprevention both, as preventive and adjuvant therapy yet it is still not fully explored for all cancers. Worldwide, the incidence of pancreatic carcinoma has increased steadily and it remains one of the major public health problems. Although surgery offers a low cure rate, it is also the only chance for cure. Unfortunately, the diagnosis of pancreatic adenocarcinoma often occurs very late and consequently less than 20% of patients are candidates for tumor resection (1,2). Contraindications for resective surgery are the presence of liver or distant metastases, peritoneal seeding, tumor infiltration into mesenteric vessel walls, and locoregional extension of the tumor into the mesentery. Conventional chemotherapy has shown only a minimal survival benefit when combined with surgical resection (3,4). Thus, only 1–3% of all patients diagnosed with pancreatic cancer can expect to survive 5 years (2,5). With such a poor progress made with conventional cancer therapies, chemoprevention against pancreatic cancer might be an alternative and very useful way of treating or increasing the survival changes of patients when combined with conventional therapeutics.
Pancreatic carcinoma arises through the accumulation of inherited and acquired genetic alterations (6,7). It is well established that k-ras mutations are one of the most common types of genetic abnormality in pancreatic adenocarcinomas. Point mutation in the k-ras gene occurs in 70–90% of cases and the vast majority occurs at codon 12 of the oncogene (8,9). Consequently, a considerable research effort has been made to define the function of Ras in normal and neoplastic cells and to target Ras for cancer treatment. Various strategies have been developed to target k-ras for the treatment of human cancers. As mentioned earlier that k-ras mutations occur very early (10) and are the most frequent mutations (9,11,12) in pancreatic cancer followed by mutation or silencing of p53 (13), p16 (14) and DPC4/smad4 (7). Addressing the contributions of these genes for the tumor cell survival after treatment may point to new ways for improving therapy for this disease. For pancreatic cancer, k-ras mutations have been reported as a negative prognostic factor after surgery and adjuvant chemo-radiation or surgery alone (15). Activated ras (mutant or oncogenic) are known to inactivate genes, which are directly involved in the regulation of growth inhibition and apoptosis. One such example is that oncogenic ras inhibits TGF-beta (TGF-β) signaling by down regulating TGF-β receptor II (RII) expression (16). A majority of pancreatic tumors show loss of expression of RII and are resistant to exogenous TGF-β mediated growth inhibition (17). In another example, oncogenic ras was also found to down regulate the pro-apoptotic gene, PAR-4 (18). Using rat prostate model, gene expression studies revealed that a gene designated as PAR-4 was exclusively induced by apoptotic stimuli but not by growth promoting, growth-inhibitory or necrotic stimuli (19). Gene function studies indicated that PAR-4 sensitizes cells to the action of apoptotic stimuli; however, PAR-4 by itself does not cause apoptosis. The gene product PAR-4 is widely expressed in normal tissues (20). Presence of this gene product in the normal tissue may act as important physiological mediator of cell apoptosis in either health or disease. Since PAR-4 is causally required for apoptosis, absence of this gene expression may render pancreatic tumors a selective resistance to apoptosis-inducing agents. PAR-4 is also known to selectively inhibit the oncogenic Ras-dependent NF-B function, the activation of which makes the tumors more resistant (21). In addition, PAR-4 binds the atypical protein kinase C isoforms (aPKCs), which serves to inhibit their enzymatic activity (22). Therefore, one of the mechanisms whereby PAR-4 induces apoptosis is through its ability to block the aPKC-IKK axis which, in turn, inhibits the anti apoptotic protein NF-κB. Based on the functional role of PAR-4 in cell death and because pancreatic tumors harbor high incidence of ras mutations, it has been hypothesized that the down regulation of PAR-4 due to mutations in k-ras will result in the impairment or dysregulation of apoptotic mechanism and thus render selective survival advantage to pancreatic tumors (23).
Studies from our laboratory and others have found that 3,3′-diindolylmethane (DIM) shows potent anti proliferative activities against a variety of cancers including pancreatic cancer (24,25). However, no studies have been reported, to date, elucidating the effect and molecular mechanisms of action of DIM on PAR-4 signaling in pancreatic cancer cells. For our studies, we used the formulation of DIM which has greater bioavailability, B-DIM obtained from BioResponse, Inc., and from this point forward we will use B-DIM to refer to 3,3′-diindolylmethane. In this paper we have tested our hypothesis whether B-DIM treatment prior to gemcitabine or cisplatin (cis-diaminedichloroplatinum-(II))-treatment in k-ras mutant and wild type pancreatic cancer cell lines could lead to enhanced cell killing in vitro. Here we report, for the first time, that B-DIM, in combination with standard chemotherapeutic drugs such as gemcitabine and cisplatin, is a potent combination treatment for causing increased cell growth inhibition and induction of apoptotic cell death which is partly due to activation of PAR-4 by B-DIM and via inactivation of Akt/Bcl-2 /Bcl-xL signaling pathways.
MATERIALS AND METHODS
Cell Lines
The human pancreatic carcinoma cell lines Colo-357 and L3.6pl were obtained from MD Anderson Cancer Center (Houston, TX). Rest of the cancer cell lines (Panc-1, BxPC-3 and HPAC) were obtained from American Type Culture Collection (Rockville, MD). The human pancreatic L3.6pl cells have been established from Colo-357 fast growing cells by injecting them into the pancreas of nude mice (26). Briefly, hepatic metastasis were harvested and re-injected into the pancreas. This process was repeated six times, resulting in the isolation of the L3.6pl cell line, which produces significantly higher incidence and number of lymph nodes and liver metastases than parental cells (26). The cell lines were maintained in continuous exponential growth by twice a week passage in Dulbecco modified Eagle's medium (DMEM, Life Technologies, Inc., Gaithersburg, MD) supplemented with 10% fetal bovine serum, 100 units/ml penicillin and 10 mg/ml streptomycin in a humidified incubator containing 5% CO2 in air at 37°C. Each cell line was split regularly before attaining 70–80% confluence.
Antibodies and Other Reagents
Antibodies were obtained from the following commercial sources: total Akt and Ser473-phosphorylated Akt was purchased from Cell Signaling (Beverly, MA); antibodies against Par-4, Bcl-2, and Bcl-xL antibody were procured from Santa Cruz Biotechnology (Santacruz, CA); anti-PARP antibody was from Biomol Research (Plymouth, PA). Anti-β actin antibody was from Sigma Chemical Co. (St. Louis, MO). A formulated 3,3′-diindolylmethane (B-DIM), which showed higher bioavailability, was kindly provided by Dr. Michael Zeligs (BioResponse, Boulder, CO) and was dissolved in DMSO to make 10 mmol/L stock solutions and stored at –20°C in multiple aliquots. Gemcitabine (Eli Lilly, Indianapolis, IN) was dissolved in sterile PBS to make 100 μmol/L stock solutions. Cisplatin (Sigma Chemical Co., St. Louis, MO) was dissolved in sterile PBS to make 100 μmol/L stock solutions.
Cell Growth Inhibition by Trypan Blue and 3-(4,5B-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT)
BxPC-3, Colo-357 and L3.6pl cells were seeded at a density of 3×103 cells per well in 96-well microtiter culture plates. After overnight incubation, medium was removed and replaced with fresh medium containing different concentrations of B-DIM (0–20 μmol/L) diluted from a 10 mmol/L stock. On completion of incubation, viability was assessed adding 50 μl of Trypan Blue solution (0.4% in PBS) in culture medium. After 1–2 min the number of dead cells, which retained the dye, was compared to the total number to calculate the viability percentage. MTT assay was performed by adding 20 μL of 3-(4,5B-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/mL in PBS) to each well and incubating further for 2 h. Upon termination, the supernatant was aspirated and the MTT formazan formed by metabolically viable cells was dissolved in 100 μL of isopropanol. The plates were gently rocked for 30 min on a gyratory shaker, and absorbance was measured at 595 nm using ULTRA Multifunctional Microplate Reader (TECAN, Durham, NC).
Cell Growth Inhibition by Cytotoxic Agents
Cells were plated as described above and allowed to attach overnight. The overnight media from individual wells was aspirated and replaced with fresh medium containing 20 μmol/L of B-DIM for 48 hours and then exposed to 100 nmol/L of the chemotherapeutic agent, gemcitabine or 1 μmol/L cisplatin, for an additional 48 h. Thus, for a single-agent treatment, cells were exposed to B-DIM for 96 h and gemcitabine or cisplatin for 48 h. The effect of B-DIM pretreatment on cell viability was examined by the MTT assay method as stated above.
Quantification of Apoptosis by ELISA
The Cell Apoptosis ELISA Detection Kit (Roche, Palo Alto, CA) was used to detect apoptosis in Colo-357 and L3.6pl cells with different treatments according to the manufacturer's protocol. Briefly, Colo-357 and L3.6pl cells were treated with 20 μmol/L B-DIM for 48 h, and then exposed to a chemotherapeutic agent, 100 nmol/L of gemcitabine or 1 μmol/L of cisplatin for an additional 48 h. For single-agent treatment, Colo-357 and L3.6pl cells were treated with B-DIM for 96 hours and gemcitabine/cisplatin for 48 h. After treatment, the cytoplasmic histone DNA fragments from Colo-357 and L3.6pl cells with different treatments were extracted and bound to immobilized anti-histone antibody. Subsequently, the peroxidase-conjugated anti-DNA antibody was used for the detection of immobilized histone DNA fragments. After addition of substrate for peroxidase, the spectrophotometric absorbance of the samples was determined using ULTRA Multifunctional Microplate Reader (TECAN, Durham, NC) at 405 nm.
Protein Extraction and Western Blot Analysis
The pancreatic cancer cells Colo-357 and L3.6pl were plated and allowed to attach for 36 h. B-DIM was directly added to cell cultures at 10 and 20 μmol/L concentrations and incubated for 48 h followed by the addition of gemcitabine or cisplatin. Control cells were incubated in the medium containing an equivalent amount of DMSO. After 48 h of incubation with gemcitabine, the cells were harvested in PBS and whole cell lysate was prepared by suspending the cells in 200 μL of lysis buffer [1 mol/L Tris–HCl (pH 7.5), 150 mmol/L NaCl, 1 mmol/L EGTA, 0.1% Triton X-100; 0.1 mmol/L sodium orthovanadate, 1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 2 μg/mL leupeptin, and 2 μg/mL aprotinin]. The cells were disrupted by sonication and the total proteins were extracted by centrifuging the tubes at 4°C for 30 min at maximal microfuge speed to remove debris. For immunoblotting, each extract was prepared as above and 50 μg total proteins were separated through SDS-PAGE, electrotransferred onto nitrocellulose membranes, and probed with specific antibodies. Detection of specific proteins was carried out with an enhanced chemiluminescence Western blotting kit according to manufacturer's instructions. The same membrane was re-probed with the anti-beta-actin (β-actin) antibody, which was used as an internal control for protein loading.
Statistical Analysis
Data are represented as mean±SD for the absolute values or percent of controls as indicated in the vertical axis of Figs. 4, 5, 6. The statistical significance of differential findings between experimental groups and control was determined by Student's t test as implemented by Excel 2000 (Microsoft Corp., Redmond, WA). P values<0.05 were considered statistically significant.
Fig. 4.

Evaluation of cell viability in B-DIM-treated Colo-357, L3.6pl and BxPC-3 cells by trypan blue staining. Cells were either untreated (DMSO) or treated with increasing concentration of B-DIM (10 and 20 μmol/L) for 96 h and then analyzed for viable cells by trypan blue staining assay as described under MATERIALS AND METHODS.
Fig. 5.

Sensitization of pancreatic cancer cells, (1) Colo-357, (2) L3.6pl and (3) BxPC-3 to B-DIM (20 μmol/L) and (A) Gemcitabine (Gz) or (B) cisplatin-induced cell growth inhibition as determined by MTT assay after 96 h treatment with B-DIM (20 μmol/L) alone or a combination of B-DIM (48 h) followed by Gemcitabine or cisplatin for 48 h. Increased growth inhibition was evident in combination treatment group relative to untreated control or individual treatment groups. Each point represents average of 4–5 independent experiments.
Fig. 6.

Sensitization of pancreatic cancer cells, (1) Colo-357, (2) L3.6pl and (3) BxPC-3 to B-DIM (20 μmol/L) and (A) Gemcitabine (Gz) or (B) cisplatin-induced apoptosis as determined by ELISA after 96 h pretreatment with B-DIM (20 μmol/L) alone or a combination of B-DIM (48 h) followed by gemcitabine/cisplatin for 48 h. Increased apoptotic response was evident in combination treatment group of the cells relative to untreated control or individual treatment groups. Each point represents average of 4–5 independent experiments.
RESULTS
Differential Expression of Par-4 in Pancreatic Cancer Cells
A recent study indicated that oncogenic ras down regulates endogenous PAR-4 protein (23), therefore, in order to confirm this observation we first analyzed six different pancreatic cancer cell lines for assessing the basal levels of PAR-4 protein. Four of the cell lines such as Panc1, L3.6pl, Colo-357 and MiaPaCa have mutations in k-ras while the other two, BxPC-3 and HS766T have wild type k-ras. As can be seen in Fig. 1, there is a differential expression of PAR-4 among the various cell lines. Although there was a slightly lower expression of PAR-4 in k-ras mutant cells (L-3.6pl, Colo-357and MiaPaCa), nevertheless our observations suggest that this is not a universal phenomenon suggesting that the level of PAR-4 expression does not directly correlate with mutational status of k-ras. The results of Fig. 1 clearly indicate that Panc-1 cells which have mutant k-ras still expresses PAR-4 equivalent to cell lines that have wild type k-ras gene.
Fig. 1.
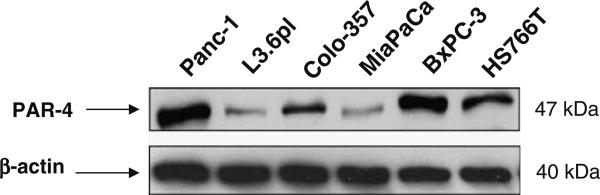
Western blot analysis of PAR-4 protein expression in pancreatic cancer cell lines. Panc-1 (Lane 1), L3.6pl (Lane 2), Colo-357 (Lane 3), MiaPaCa (Lane 4), BxPC-3 (Lane 5) and Hs766T (Lane 6). β-actin protein was used as protein loading control as shown for each blot. Cell extracts were prepared according to the procedure described under MATERIALS AND METHODS section.
B-DIM Induces Par-4 in Pancreatic Cancer Cells
Among all the natural compounds screened (such as genistein, resveratrol, EGCG, curcumin and B-DIM) only B-DIM was effective in inducing PAR-4 in different pancreatic cancer cell lines (Colo-357 and L3.6pl; results not shown). As can be seen in Fig. 2A and B, treatment with 10 μmol/L and 20 μmol/L of B-DIM resulted in significant increase in PAR-4 levels in the two cells lines tested. Further, we also tested whether gemcitabine or cisplatin could induce PAR-4 either alone or in combination with B-DIM. Results presented in Fig. 3A, B and C clearly demonstrate that therapeutic drugs such as gemcitabine and cisplatin, either alone or in combination with B-DIM, were able to induce PAR-4 in Colo-357, L3.6pl as well as BxPc-3 cell lines. In order to further test whether this induction in PAR-4 by B-DIM could lead to increased sensitivity to conventional chemotherapeutic agents in terms of growth arrest and apoptosis, we performed trypan blue staining, MTT and ELISA assays.
Fig. 2.

PAR-4 expression is up-regulated by B-DIM. (A) Western blot analysis of lysates of L3.6pl cells treated with: DMSO (untreated; Lane 1); 10 μmol/L B-DIM (Lane 2); 20 μmol/L (Lane 3), respectively. (B) Western blot analysis of lysates of Colo-357 cells treated with DMSO (untreated; Lane 1); 10 μmol/L B-DIM (Lane 2); 20 μmol/L (Lane 3), respectively. β-actin protein was used as loading control as shown for each blot.
Fig. 3.

PAR-4 expression is up-regulated by B-DIM in combination with gemcitabine. (A) Western blot analysis of lysate extracted from Colo-357 cells treated with; DMSO (untreated; Lane 1); B-DIM 20 μmol/L (Lane 2); Gemcitabine 100 nmol/L (Lane 3); B-DIM 20 μmol/L+Gemcitabine 100 nmol/L, respectively. (B) Western blot analysis of lysate extracted from L-3.6pl cells treated with; DMSO (untreated; Lane 1); B-DIM 20 μmol/L (Lane 2); Gemcitabine 100 nmol/L (Lane 3); B-DIM 20 μmol/L+Gemcitabine 100 nmol/L, respectively. Western blot analysis of lysate extracted from BxPC-3 cells treated with; DMSO (untreated; Lane 1); B-DIM 20 μmol/L (Lane 2); Gemcitabine 100 nmol/L (Lane 3); B-DIM 20 μmol/L+ Gemcitabine 100 nmol/L, respectively. Cell lysates were prepared according to the procedure described under MATERIALS AND METHODS section.
Effect of B-DIM on Gemcitabine or Cisplatin Induced Growth Inhibition
To test the effect of B-DIM on cell growth, two mutant (L3.6pl and Colo-357) and one k-ras wild type (BxPC-3) pancreatic cancer cell lines were treated with increasing concentrations of B-DIM (0–20 μmol/L) for 96 h. As shown in Fig. 4, cell growth was inhibited by B-DIM treatment in a dose dependant manner. In Colo-357 cells, treatment with 10 and 20 μmol/L of B-DIM for 96 hours resulted in 2% and 33% inhibition of cell growth inhibition relative to control, respectively. Similarly, treatment of L3.6pl cells resulted in 3% and 24% inhibition of cell growth, respectively, relative to control. In case of BxPC-3 cells, similar treatment resulted in 13% and 41% growth inhibition. These results indicate that B-DIM alone, at the dose tested, was not very effective in inhibiting the growth of pancreatic cancer cells. The effect of B-DIM on BxPC-3 cells was slightly more compared with Colo-357 cells and L3.6pl cells. We subsequently evaluated the effect of gemcitabine or cisplatin on cell growth in vitro and found that gemcitabine or cisplatin was effective as single agent in inhibiting cell growth with equal potency in all the cell lines tested above (results not shown). Next we examined whether the cells pretreated with B-DIM were more sensitive to the cytotoxic effect of gemcitabine/cisplatin and the results are presented below.
B-DIM Potentiates Growth Inhibition Induced by Gemcitabine or Cisplatin in Colo-357, L3.6pl and BxPC-3 Cells
We assessed the effect of pretreatment and co-treatment of a combination of B-DIM and gemcitabine and/or cisplatin on cell viability by MTT assay. For these studies, cells were either pretreated with B-DIM (20 μmol/L) alone or in combination with a single dose of gemcitabine (100 nmol/L) or cisplatin (1 μmol/L) and viable cells were evaluated at 96 h of treatment by MTT assay. The dose used here was chosen based upon a preliminary dose escalation study done by us prior to this experiment. We found that treatment with either B-DIM, gemcitabine or cisplatin alone for 96 h resulted in only 33%, 3% and 16% loss of viability, respectively, of Colo-357, and 24%, 8% and 13% loss of viability respectively of L3.6pl cells. While in case of BxPC-3 cells, treatment with B-DIM, gemcitabine or cisplatin alone for 96 hours resulted in 41%, 16% and 22% respectively. However, pretreatment with B-DIM for 48 h followed by treatment with gemcitabine or cisplatin resulted in the loss of 55% to 70% of viable cells in all three cell lines tested in our studies (Fig. 5A, B). Similarly, treatment with B-DIM plus gemcitabine simultaneously was also effective (data not shown), but pretreatment of cells with B-DIM was more effective in gemcitabine-induced killing. These results suggest that the pretreatment with low doses of B-DIM sensitizes the cells for better cell growth inhibition with conventional chemotherapeutic drugs such as gemcitabine and cisplatin. Although BxPC-3 cells appears to be more sensitive towards treatment with a single agent but the pretreatment observations do not point out to any significant differences in sensitivity based on the mutational status of k-ras. In order to assess whether inhibition of cell growth and viability as determined by MTT could also be due to the induction of apoptotic cell death, apoptosis studies were also performed and the results are presented below.
B-DIM Sensitizes Colo-357, L3.6pl and BxPc-3 Cells to Apoptosis Induced by Gemcitabine or Cisplatin
We observed induction of apoptosis in pancreatic cancer cells treated with B-DIM (20 μmol/L), gemcitabine (100 nmol/L) or cisplatin (1 μmol/L) alone. Relative to single agents, B-DIM pretreatment followed by either gemcitabine or cisplatin treatment induced much more apoptosis in both the cell lines as shown by histone DNA ELISA assay (Fig. 6A and B). These results are consistent with cell growth inhibition studies by MTT, suggesting that the loss of viable cells by B-DIM and gemcitabine or cisplatin is partly due to the induction of apoptotic cell death mechanism. Collectively, the above results clearly suggest that B-DIM sensitizes pancreatic cells towards more efficient killing induced by conventional chemotherapeutic agents such as gemcitabine and cisplatin.
Cellular Basis for B-DIM and Gemcitabine in Augmenting Apoptosis Processes
To identify the mechanism of enhanced apoptotic response by B-DIM and gemcitabine combination treatment, we assessed PARP cleavage by Western blotting and also determined the status of anti-apoptotic proteins using Colo-357 and L3.6pl cells. Preliminary experiments were done to determine the optimal treatment schedule and dose of individual agents. Cells were treated as described previously and the whole-cell extract were subjected to Western blotting analysis. Our data showed that combination treatment substantially inhibited the levels of all markers tested that favor cell survival (Fig. 7A, B). Because PARP is a substrate for caspase activity and a reliable marker of apoptosis, we assessed the level of cleaved PARP. Individually, these agents showed low levels of PARP cleavage; however, the combination of B-DIM and gemcitabine resulted in the appearance of a stronger cleaved PARP band (Fig. 7A, B). Previous studies from our laboratory have shown that B-DIM inactivates NF-κB and thus may down regulate its downstream target genes. We found a significant decrease in the levels of Bcl-2 and Bcl-xL, which is consistent with transcriptional inactivation of Bcl-2 and Bcl-xl which are downstream target of NF-κB as well as markers for cell survival. As expected, we also observed an increase in the expression of pro-apoptotic factor Bax (Fig. 7A, B), suggesting that the downstream down-regulation of Bcl-2 and Bcl-xl and up-regulation of Bax contributes to the induction of apoptosis. Several reports strongly suggest the critical role of Akt in anti-apoptotic phenomenon. Akt activation occurs due to phosphorylation of Akt protein at Thr 308 or Ser 473 by PI3K. Therefore, we tested whether B-DIM and gemcitabine-combination induced apoptosis could be associated with decreased Akt activity. As shown in Fig. 7, the expression level of phosphorylated Akt was found to be significantly attenuated in response to combination treatment while the total Akt was not affected.
Fig. 7.

(A) and (B) Western blot analysis of PARP, anti-apoptotic Bcl-xl, Bcl-2, p-Akt and the pro-apoptotic Bax in whole cell lysates of L3.6pl and Colo-357 cells after treatment with either only B-DIM (48 h), gemcitabine (48 h), or B-DIM pretreatment (48 h) followed by gemcitabine treatment (48 h). Down-regulation of anti-apoptotic markers is evident in both cell types pre-sensitized with B-DIM followed by gemcitabine treatment. β-actin protein was used as protein loading control as shown for each blot. Cell extracts were prepared according to the procedure described under MATERIALS AND METHODS section.
DISCUSSION
The need for newer agents as well as novel targets to prevent cancer is the urgent need of the hour. Although the proof of principle has been quite clearly validated still it has not been described in all cancers specially that of pancreas. Pancreatic tumors are the only solid tumors that show mutations of the k-ras gene at the highest incidence. The ras pathway in a cell is highly important for the transmission of growth promoting signals received from the cell surface receptors to nuclear compartment, leading to a signaling cascade that affects the production and the regulation of other key proteins. Most mutations in genes lead to inactivation, whereas, mutations in ras genes lead to active signaling (26). The pro-apoptotic prostate apoptosis response-4 gene product, PAR-4, sensitizes prostate cancer cells to the induction of programmed cell death. However not much is known about the role of PAR-4 in pancreatic cancer. Our first aim in this study was to examine the differential expression of PAR-4 in human pancreatic cell lines based on the status of k-ras mutations. The heterogeneous expression of PAR-4 was detected, which prompted us to investigate the biological relevance of PAR-4 in pancreatic cancer.
Our findings are in accordance with a recent study in which it was shown that oncogenic ras down regulates PAR-4 in different pancreatic cancer cell lines as well as in tumors (22). In recent years, the predictive antitumor therapeutic effect with combinations of different classes of chemotherapeutic agents has been attempted in the clinical setting, but owing to high toxicity and acquired chemo-resistance, the expected therapeutic benefit could not be accomplished. In our opinion both de novo and acquired resistance to therapy could be overcome by employing rational combination therapy, where toxic agents could be used in lower doses but the efficacy of treatment could be increased by novel non-toxic agent that may have different mechanism of action. Therefore we hypothesize that the activation of PAR-4 (an apoptosis inducing agent) by a novel non-toxic agent could lead to sensitization of pancreatic cancer cells to conventional chemotherapeutic agents such as gemcitabine or cisplatin. Based on this rationale, we sought to assess the efficacy of B-DIM, a compound which has been reported as a non-toxic agent, in combination with gemcitabine or cisplatin against three pancreatic cancer cell lines. The findings presented in this paper provide additional novel mechanistic insights underlying sensitization of pancreatic cancer cells to gemcitabine or cisplatin. In our study, we found that the treatment of different pancreatic cancer cell lines with low doses of B-DIM (10 and 20 μmol/L) resulted in the induction of PAR-4 (Fig. 2). Interestingly, the combination of B-DIM with conventional drugs resulted in enhanced cell killing. DNA/Histone ELISA revealed that the observed cytotoxicity of this combination treatment was apoptotic in nature. In order to determine the downstream targets involved in apoptosis, Western blot analysis was done which revealed that combination of B-DIM with gemcitabine or cisplatin resulted in the appearance of cleaved PARP. The Bcl-2 protein is a potent inhibitor of cell death (27). Our results clearly show a marked decrease in Bcl-2 upon treatment with B-DIM or drug alone as well as their combination. Similar decreased expression was observed for another anti-apoptotic factor Bcl-xl. On the other hand a significantly induced expression of proapoptotic factor Bax was observed which again confirms that the combination treatment results in apoptotic cell death. These observations strongly suggest that B-DIM sensitizes the pancreatic Colo-357, L3.6pl as well as BxPC-3 cells to gemcitabine as well as cisplatin-induced apoptosis, which is partly due to the activation of PAR-4. Several lines of evidence suggest that the Akt pathway is intricately linked to regulation of apoptosis as well as prognosis (28–31). The conventional notion is that Akt enhances survival of tumor cells through transduction of anti-apoptotic and proliferative signals (32). The mechanism of Akt activation in pancreatic cancer remains unknown, although a majority of pancreatic cancer cell lines that have been examined harbor constitutively activated Akt. Akt was found to be activated (i.e., phosphorylated at Ser 473) in both the Colo-357 as well as L3.6pl cell lines tested, which was abrogated by B-DIM treatment. It is therefore logical to speculate that the observed suppression of cell viability and augmentation of apoptotic cell death by B-DIM could be attributed to its dual effect on targeted disruption of Akt phosphorylation and its substrate, which could be another mechanism by which B-DIM sensitizes pancreatic cancer cells killing to conventional therapeutics. Although apoptosis by B-DIM, cisplatin/gemcitabine combination occurs with simultaneous induction of PAR-4, it is still quite premature to conclude whether PAR-4 plays any major or integral role in this event; although our results are encouraging and warrants further in-depth mechanistic studies in the future. In conclusion, our findings suggest that B-DIM at non-toxic doses induces PAR-4 in pancreatic cancer cells and when combined with a chemotherapeutic drug leads to cell death through apoptosis. This phenomenon can be exploited for future development of therapeutic strategies against pancreatic cancers.
REFERENCES
- 1.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Sankila R, Ferlay J, Parkin DM. Estimates of cancer incidence and mortality in Europe in 1995. Eur. J. Cancer. 2002;38:99–166. doi: 10.1016/s0959-8049(01)00350-1. [DOI] [PubMed] [Google Scholar]
- 3.Wray CJ, Ahmad SA, Matthews JB, Lowy AM. Surgery for pancreatic cancer: recent controversies and current practice. Gastroenterology. 2005;128:1626–1641. doi: 10.1053/j.gastro.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 4.Beger HG, Rau B, Gansauge F, Poch B, Link KH. Treatment of pancreatic cancer: challenge of the facts. World J. Surg. 2003;27:1075–1084. doi: 10.1007/s00268-003-7165-7. [DOI] [PubMed] [Google Scholar]
- 5.Berlin JD, Rothenberg M. Chemotherapy for resectable and advanced pancreatic cancer. Oncology (Williston Park) 2001;15:1241–1249. 1254. [PubMed] [Google Scholar]
- 6.Cowgill SM, Muscarella P. The genetics of pancreatic cancer. Am. J. Surg. 2003;186:279–286. doi: 10.1016/s0002-9610(03)00226-5. [DOI] [PubMed] [Google Scholar]
- 7.Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeo CJ, Kern SE. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57:1731–1734. [PubMed] [Google Scholar]
- 8.Sun C, Yamato T, Furukawa T, Ohnishi Y, Kijima H, Horii A. Characterization of the mutations of the K-ras, p53, p16, and SMAD4 genes in 15 human pancreatic cancer cell lines. Oncol. Rep. 2001;8:89–92. doi: 10.3892/or.8.1.89. [DOI] [PubMed] [Google Scholar]
- 9.Grunewald K, Lyons J, Frohlich A, Feichtinger H, Weger RA, Schwab G, Janssen JW, Bartram CR. High frequency of Ki-ras codon 12 mutations in pancreatic adenocarcinomas. Int. J. Cancer. 1989;43:1037–1041. doi: 10.1002/ijc.2910430614. [DOI] [PubMed] [Google Scholar]
- 10.Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57:2140–2143. [PubMed] [Google Scholar]
- 11.Shibata D, Capella G, Perucho M. Mutational activation of the c-K-ras gene in human pancreatic carcinoma. Baillieres Clin. Gastroenterol. 1990;4:151–169. doi: 10.1016/0950-3528(90)90044-h. [DOI] [PubMed] [Google Scholar]
- 12.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 13.Barton CM, Staddon SL, Hughes CM, Hall PA, O'Sullivan C, Kloppel G, Theis B, Russell RC, Neoptolemos J, Williamson RC. Abnormalities of the p53 tumour suppressor gene in human pancreatic cancer. Br. J. Cancer. 1991;64:1076–1082. doi: 10.1038/bjc.1991.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adeno-carcinoma. Nat. Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- 15.Bernhard EJ, McKenna WG, Hamilton AD, Sebti SM, Qian Y, Wu JM, Muschel RJ. Inhibiting Ras prenylation increases the radiosensitivity of human tumor cell lines with activating mutations of ras oncogenes. Cancer Res. 1998;58:1754–1761. [PubMed] [Google Scholar]
- 16.Zhao J, Buick RN. Regulation of transforming growth factor beta receptors in H-ras oncogene-transformed rat intestinal epithelial cells. Cancer Res. 1995;55:6181–6188. [PubMed] [Google Scholar]
- 17.Venkatasubbarao K, Ahmed MM, Mohiuddin M, Swiderski C, Lee E, Gower WR, Jr., Salhab KF, McGrath P, Strodel W, Freeman JW. Differential expression of transforming growth factor beta receptors in human pancreatic adenocarcinoma. Anticancer Res. 2000;20:43–51. [PubMed] [Google Scholar]
- 18.Barradas M, Monjas A, az-Meco MT, Serrano M, Moscat J. The downregulation of the pro-apoptotic protein Par-4 is critical for Ras-induced survival and tumor progression. EMBO J. 1999;18:6362–6369. doi: 10.1093/emboj/18.22.6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sells SF, Wood DP, Jr., Joshi-Barve SS, Muthukumar S, Jacob RJ, Crist SA, Humphreys S, Rangnekar VM. Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell Growth Differ. 1994;5:457–466. [PubMed] [Google Scholar]
- 20.Rangnekar VM. Apoptosis mediated by a novel leucine zipper protein Par-4. Apoptosis. 1998;3:61–66. doi: 10.1023/a:1009666705875. [DOI] [PubMed] [Google Scholar]
- 21.Nalca A, Qiu SG, El-Guendy N, Krishnan S, Rangnekar VM. Oncogenic Ras sensitizes cells to apoptosis by Par-4. J. Biol. Chem. 1999;274:29976–29983. doi: 10.1074/jbc.274.42.29976. [DOI] [PubMed] [Google Scholar]
- 22.az-Meco MT, Municio MM, Frutos S, Sanchez P, Lozano J, Sanz L, Moscat J. The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell. 1996;86:777–786. doi: 10.1016/s0092-8674(00)80152-x. [DOI] [PubMed] [Google Scholar]
- 23.Ahmed MM, Sheldon D, Fruitwala MA, Venkatasubbarao K, Lee EY, Gupta S, Wood C, Mohiuddin M, Strodel WE. Downregulation of PAR-4, a pro-apoptotic gene, in pancreatic tumors harboring K-ras mutation. Int. J. Cancer. 2008;122:63–70. doi: 10.1002/ijc.23019. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Wang Z, Kong D, Murthy S, Dou QP, Sheng S, Reddy GP, Sarkar FH. Regulation of FOXO3a/beta-catenin/GSK-3beta signaling by 3,3′-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in prostate cancer cells. J. Biol. Chem. 2007;282:21542–21550. doi: 10.1074/jbc.M701978200. [DOI] [PubMed] [Google Scholar]
- 25.Abdelrahim M, Newman K, Vanderlaag K, Samudio I, Safe S. 3,3′-diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis. 2006;27:717–728. doi: 10.1093/carcin/bgi270. [DOI] [PubMed] [Google Scholar]
- 26.Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reed JC. Bcl-2 and the regulation of programmed cell death. J. Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das A, Chendil D, Dey S, Mohiuddin M, Mohiuddin M, Milbrandt J, Rangnekar VM, Ahmed MM. Ionizing radiation down-regulates p53 protein in primary Egr-1–/–mouse embryonic fibroblast cells causing enhanced resistance to apoptosis. J. Biol. Chem. 2001;276:3279–3286. doi: 10.1074/jbc.M008454200. [DOI] [PubMed] [Google Scholar]
- 29.West KA, Castillo SS, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist. Updat. 2002;5:234–248. doi: 10.1016/s1368-7646(02)00120-6. [DOI] [PubMed] [Google Scholar]
- 30.Fahy BN, Schlieman MG, Virudachalam S, Bold RJ. Inhibition of AKT abrogates chemotherapy-induced NF-kappaB survival mechanisms: implications for therapy in pancreatic cancer. J. Am. Coll. Surg. 2004;198:591–599. doi: 10.1016/j.jamcollsurg.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 31.Bondar VM, Sweeney-Gotsch B, Andreeff M, Mills GB, McConkey DJ. Inhibition of the phosphatidylinositol 3′-kinase-AKT pathway induces apoptosis in pancreatic carcinoma cells in vitro and in vivo. Mol. Cancer Ther. 2002;1:989–997. [PubMed] [Google Scholar]
- 32.Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci USA. 2001;98:10983–10985. doi: 10.1073/pnas.211430998. [DOI] [PMC free article] [PubMed] [Google Scholar]