

Abstract
Recent investigations of renal cell carcinoma (RCC) have revealed several epigenetic modifications, as well as alterations in the gene and enzymes that regulate these changes. Preclinical models have revealed that histone gene modifiers and epigenetic alterations may play a critical role in RCC tumorigenesis. Specific changes in DNA methylation and mutations of histone modifiers have been identified and may be associated with an aggressive phenotype. In addition, the potential of reversing the effects of these enzymes, and hence reversing the cellular epigenetic landscape to a “normal phenotype” has led to an increasing interest in developing targeted chromatin remodeling agents. However, the translation of the understanding of these changes to the clinic for the treatment of RCC has posed significant challenges, also as consequence from tumor heterogeneity. This review describes the aberrant histone and DNA alterations recently reported in RCC and highlight the potential targeted chromatin remodeling therapies in the management of this disease.
Epigenetic modifications in cancer
DNA methylation, covalent histone alterations, non-covalent histone alterations, and non-coding RNAs (not discussed in this review) are the four primary types of epigenetic modifications [1]. DNA methylation leads to transcriptional silencing, whereas histone modifications can lead to either transcriptional activation or repression; moreover these changes can occur along with genetic aberrations that are found in cancer [1]. The potential of reversing these epigenetic variations has led to considerable interest in developing agents that target epigenetic variations [1]. In several tumors, global hypomethylation has been found to be a common occurrence with hypermethylation of CpG islands in the promoter region of certain genes [2–4]. Hypomethylation can occur in varying degrees in localized vs. invasive disease and is thought to lead to tumorigenesis by loss of imprinting, chromosomal instability, and activation of oncogenes [3]. Hypermethylation of CpG islands has been shown to result in silencing of tumor suppressors [3].
Histone modifications, on the other hand, are considered to be more dynamic changes with different alterations occurring depending on the cell context [2]. Histones are often acetylated (which neutralizes the lysine charge and leads to a more open chromatin and thus transcriptional activation), methylated (activator or repressor) or phosphorylated (altering the structure and function of chromatin)[2]. Modifications of the chromatin structure may lead to breaks in the chromosome which can result in tumorigenesis[5]. Histone alterations, for example at H3K27me3 (repressive) and H3K4me3 (active), have been shown to be important in the development process and genes with these alterations have been reported to be targeted for DNA methylation in tumorigenesis[2]. Studies have shown a loss of H4K16Ac and H4K20me3 in different malignancies. Interestingly, misinterpretation of the histone code can also lead to dysregulation of genes involved in normal cell functioning [5]. A review by You and Jones provides an extensive list of epigenetic changes occurring in cancer and agents that can target these changes [4]. This chapter discusses epigenetic modifications occurring in renal cell carcinomas and therapy targeted at these changes. The majority of enzymes involved in DNA methylation and histone modifications, their targets, and inhibitors that target these enzymes are listed in Table 1 and in Figure 1.
Table 1.
Epigenetic modifications, related enzymes and targeted agents.
| Modifications | Governing enzymes | Transcriptional effects | Targets | Targeted agents | References |
|---|---|---|---|---|---|
|
DNA methylation
| |||||
| De novo methylation | DNMT3a and DNMT3b | Silencing | Hemi-methylated DNA | 5-azacytidine (5-aza-CR), 5-aza-2′ deoxycytidine (5-aza-CdR), MG-98, RG108 | [1, 2, 4, 77–79] |
| Maintenance methylation | DNMT 1 | Silencing | Non-methylated DNA Methylated | ||
| Methyl binding proteins | MeCP2, MBD 1, 2 and 4 | Silencing | CpG islands | ||
|
| |||||
| Histone acetylation | |||||
|
| |||||
| p300/CBP GNAT family MYST family TIP 6 | Activation | Histones | L002 | [6, 80–82] | |
|
| |||||
| Histone deacetylation | |||||
|
| |||||
| Class I HDACs | HDAC 1, 2, 3 and 8 | Silencing | Histones | Depsipeptide, SK-7041, SK-7048, MS-275, Valproic acid, sodium butyrate, SAHA, TSA, LBH589 | [77–79] |
| Class IIb HDACs | HDAC 6 and 10 | Silencing/Activation | Histones and non-histone proteins | Tubacin, vorinostat, TSA and panobinostat | [77–79] |
| Class III (Sirtuins) | Sirt 1-7 | Primarily involved in metabolism | Nicotinamide, splitomicin | [77] | |
| Class IV | HDAC 11 | Not well understood | |||
|
| |||||
| Histone methyl transferases | |||||
|
| |||||
| SET domain HMTs | EZH2 | Silencing | H3K9me3, H4K20me3 | DZNep, Chaetocin, BIX-01294, Allantodapsone,Stilbamidine | [1, 80, 81, 83] |
| SUV39H1, SETDB1, G9a | Silencing | H3K9me3 | |||
| MLL1 and 4, SMYD3 | Activation | H3K4me3 | |||
| PRMT1 | Activation | Arg 3 of H4 | |||
| DOTL1L | Silencing | H3K79me3 | |||
|
| |||||
| Histone demethylases | |||||
|
| |||||
| UTX, JMJD3, JHDM2A | Activation | H3K9 | Monoamine oxidase A and B, N-oxalylglycine | [81, 83] | |
| LSD1/JARID1C | Activation/Silencing | H3K4 | |||
| JHDM1 | Silencing | H3K36 | |||
| JMJD2A | Activation/Silencing | H3K4, H3K9, H3K36, H4K20 | |||
Figure 1. Summary of histone modifications and enzymes involved.
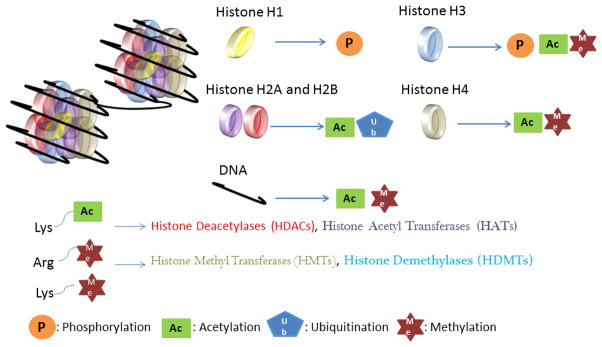
The nucleosome structure consists of the octamer core (H2A, H2B, H3, and H4) with the linker histone (H1). Histone tails can be modified through phosphorylation, acetylation, ubiquitination and methylation. DNA, however, can be only acetylated or methylated at different sites including promoter regions and CpG islands. Enzymes causing above mentioned modifications are Histone deacetylases (HDACs), Histone acetyl transferases (HATs), Histone methyl transferases (HMTs) and Histone demethylases (HDMTs). DNA can be methylated by a group of enzymes known as DNA methyl transferases and deacetylated by class II HDACs.
Histone deacetylases and HDAC inhibitors
Histone deacetylases are subdivided into different classes: class I HDACs (HDAC 1, 2, 3 and 8) that are related to the transcriptional regulator RPD3 in the yeasts; class II HDACs (HDAC 4, 5, 6, 7, 9 and 10) related to HDA1 in the yeasts, and class III HDACs Sirtuins (Sirt 1-7), that will not discussed in details in this chapter [6–8]. Class I HDACs are primarily located in the nucleus and depend on zinc for their activity; class II HDACs are able to translocate between the nucleus and the cytoplasm and also depend on zinc for their activity, whereas sirtuins are located in the nucleus, cytoplasm or the mitochondria and depend on NAD+ for their activity[6–8]. HDAC inhibitors primarily target the zinc domains in order to exert the biological effects through cell cycle arrest, differentiation and apoptosis in a variety of tumor models [6]. HDAC inhibitors are categorized based on their structure into hydroxamates(e.g. vorinostat), cyclic peptides (e.g. romidepsin), aliphatic acids (e.g. phenylbutyrate), and benzamides (entinostat)[8–10]. These small molecules are active at wide range of concentrations in vitro, ranging from millimolar, to micro and nanomolar values for butyrates, vorinostat/entinostat and TSA/tetrapeptides respectively [8].
HDACs and normal kidney cells
The use of HDAC inhibitors or sirtuin activation in some studies has been shown to have protective effects on renal tubular cells upon cisplatin treatment [11, 12] and prevention of TGF-β1 induced EMT [13]. Treatment with vorinostat during cisplatin treatment reduced apoptosis in renal proximal tubular cells through p53 suppression In addition, SIRT 1 activation during cisplatin treatment in vitro has been shown to prevent HK2 cell apoptosis through deacetylation of the p65 unit of NFκB[12]. Moreover, TSA treatment of RPTEC cells were found to prevent TGF-β1 induced EMT[13]. Using HEK293 cells, researchers have reported that induction of hypoxia leads to downregulation of RECK (gene involved in suppression of metastasis and angiogenesis[14]) both at the mRNA and protein levels. HDAC 1 and HIF-1α bind to the promoter region of RECK, thus promoting cell migration and invasion [15].
Histone modifications in RCC
Seglison et al., analyzed 192 localized renal tumors and 163 metastatic renal cell carcinomas for histone modifications by immunohistochemistry [16]. They found that patients with staining for H3K4me2 and H3K18Ac (associated with active transcription), as well as H3K9me2(associated with gene repression) correlated with poor prognosis and lower survival probability; these histone modifications were able to predict outcomes independent of the p53 and Ki67 status of the tumors [16]. However, only H3K9me2 was found to be a prognosticator of poorer outcome in metastatic patients [16]. Similarly, Moshavilli et al., showed that in 193 RCC patient tumors (inclusive of different histological subtype) reduction in total H3Ac and H4Ac was associated with worse clinical and pathological characteristics including metastasis [17]. Specifically, reduction in overall H3K18Ac strongly correlated with recurrence in those patients with localized RCC in this study [17]. In another large scale study that included 142 clear cell RCC samples, H3K4 methylation levels were inversely correlated with advanced stage, grade and metastatic parameter of the disease [18]. Lower H3K4 methylation levels were also related to shorter progression free survival with H3K4me2 significantly associated with cancer specific survival [18]. In another study by the same group using the same patient cohort, similar results were observed with H3K27 methylation status [19]. They showed lower H3K27me to be associated with worse clinical characteristics, including cancer specific and progression free survival [19]. In addition to changes in histone modifications, several changes occurring directly within the histone modifiers have been reported. Histone demethylases UTX, JMJD2, and EZH2 have been found to be upregulated at the protein and mRNA levels, with a corresponding decrease in H3K27me3 [20]. Similarly, the histone acetyltransferase MYST1 has been found to be downregulated in a small set of RCC samples with a corresponding decrease in H4K16Ac (its downstream histone target) [21]. In vitro models utilizing the RCC cell lines 786-O and C2 had confirmed lower acetylation levels at H4K16 with a decrease in MYST1 when compared to the human embryonic kidney cell lines (HEK293) [21]. In vitro analysis has also revealed mutations in the histone methyl transferase SETD2 with a corresponding decrease in H3K36me3 but not in H3K36me2 [22]. In a smaller set of ccRCC samples, truncating mutations of PBRM1 were found in 5 out of 10 tumors, which led to a loss of function in PBRM1 (located at the 3p21 region)[23]. BAP1 mutations involved in ubiquitination of histone H2A have been also found in the same set of samples[23]suggesting a role for BAP1 as a putative tumor suppressor gene [24]. Loss of PBRM1 expression [25] was observed in a greater percentage of higher stage tumors; PBRM1 also served as a prognostic marker with PBRM1 positive patients having better overall survival as compared to those patients with negative staining [26]. Knockdown analysis of PBRM1 in vitro increased cell proliferation, colony formation, and cell migration, leading to speculate that PBRM1 may also function as a tumor suppressor gene [25]. In a large scale study by Dagliesh et al, over 400 tumors were analyzed and results showed mutations in chromatin remodeling genes including SETD2, JARID1C, UTX, and MLL2, in 3–5% of tumors with some correlation with VHL mutations and tumors with a high hypoxic signature[27]. Renal tumors have been shown to have a higher percentage of global methylation and reduced histone acetylation as compared to non-tumor counterparts[28]. Interestingly, in a cluster study by Avissar-Whiting et al EZH2 was overexpressed in tumors that were less aggressive, and these patients had better prognosis [29]. In addition, class I HDACs specifically HDAC 1 and HDAC 2 have been shown to be overexpressed in clear cell renal cell carcinomas but no correlation with clinical parameters was observed[30].
HDAC inhibitors and renal cell carcinoma
HDAC inhibitors and protein ubiquitination
In vitro analyses of five renal tumor cell lines treated with either vorinostat or ritonavir alone and in combination showed consistent dephosphorylation of Rb along with a reduction in histone deacetylase 1 (HDAC 1) expression (with no change in HDAC 6 expression) and an increase in sub G1 fraction in combination treatment [31]. The combination also inhibited proliferation and colony formation in renal tumor cells, while having minimal effect on normal renal proximal tubular cells [31]. The same group has also showed that the combination of ritonavir and bortezomib induced the acetylation of histone H3 with a similar increase in the subG1 fraction [32]. However, the combinations in this model also led to a reduction in HDAC 6 expression, unlike the previous study, thereby with increasing protein ubquitination[32]. This group also combined vorinostat and bortezomib and observed similar inhibitory effects on cell proliferation, induction of ubiquitination and apoptosis, and tumor growth inhibition[33]. These studies suggest a potential therapeutic effect in combining HDAC and proteasome inhibitors in RCC.
HDAC and mTOR inhibition
Chronic single agent in vitro treatment of human ccRCC Caki-1 cells with either the mTOR inhibitor everolimus or the HDAC inhibitor valproic acid (VPA) led to drug resistance, but not with drug combination [34]. This loss of sensitivity to single agents was associated with an increase in epithelial–mesenchymal transition (EMT) markers, whereas combination treatment resulted in stabilization of the EMT markers [34]. In a study using in vitro and in vivo models of 786-0 and Caki-1 established tumors showed greater reduction in cell proliferation and tumor growth using a combination of temsirolimus and vorinostat[35], In addition, xenografts showed greater reduction in angiogenesis, an increase in apoptosis and a downregulation of survivin[35]. Chronic application of VPA has been shown to induce resistance through an increase in HDAC expression and a reduced histone acetylation[36]. Hypoxia inducible factor 1α and 2α are critical transcriptional factors in clear cell RCC as discussed in other chapters. HDAC inhibitors have been reported to inhibit HIFs by affecting its stability [37]. Qian et al have showed that HDAC 4 and HDAC 6 were present in the same complex as HIF-1α and knockdown of both HDAC 4 and 6 lowered HIF-1α expression and activity [37]. A rational combination strategy is to inhibit HIF stability with HDAC inhibitors and HIF protein synthesis by mTOR inhibitors. Using C2 cells and xenografts, the combination treatment showed greater inhibitory effects as well as increased anti angiogenic effects and downregulation of p-Akt[38]. In an ongoing phase I/II clinical trial (NCT01582009) the combination of panobinostat and everolimusis being evaluated in patients with metastatic renal cell carcinomas who have progressed following anti-VEGF therapies. A similar phase I study (NCT00169532) is assessing the combination of vorinostat and ridaforolimus in advanced renal cell carcinoma. Figure 2 summarizes the studies with HDAC inhibitors in combination with other agents.
Figure 2. Examples of HDAC inhibitor activity and clinical studies in RCC.
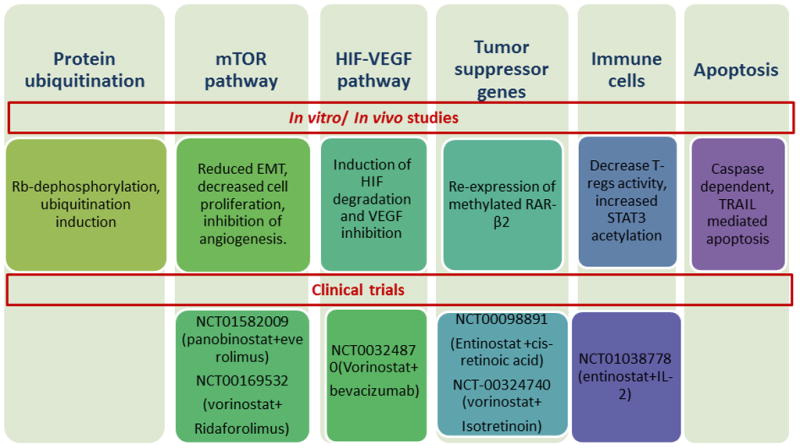
Summary of the different studies conducted in vitro, in vivo and clinical trials combining HDACs with other agents. Upper panel: genes and pathways affected by HDAC inhibitors. Lower panels: combination clinical trials with HDAC inhibitors.
Histone marks, HDAC inhibitors and the VHL-HIF axis in RCC
Sirt 1 has been shown to activate HIF-2α leading to activation of its downstream target Epo, a prosurvival factor that protecting stem and progenitor cells [39]. In addition to Sirt 1, other histone modifying genes such as the JMJC family of histone demethylases have been shown to be targets of the HIF pathway [40]. The enzymes activated under low oxygen concentrations though global changes in H3K9 were not measurable [40]. Similar results showed that HIF1α specifically regulates JMJD1A and JARID1B[41, 42]. The authors analyzed specific targets of hypoxia, including ADM, EDN1 and others to show that although global H3K9 changes were not observed upon hypoxic induction, the promoters at these specific genes showed a decrease in H3K9 methylation [42]. In vitro analyses have also revealed that RCC4 and RCC10 (VHL null) cell lines have higher H3K27me3, and K3K9me3 status and lower H3K4me3 status as compared to VHL wildtype counterparts [43]. In another study, 786-O cell line derived metastatic tumors had higher H3K4me3 levels and lower H3K27me3 levels in genes that were upregulated in the metastatic sites [44]. In particular, introduction of VHL in the metastatic cell lines reduced the activation mark H3K4me3 at the CXCR4 loci [44], a chemokine receptor involved in migration and metastasis [45]. In 5/10 ccRCCs clinical tumors, FHIT was found to be hypermethylated in ccRCC, however due to the limited number of samples the authors were not able to establish a correlation of FHIT methylation to any clinical parameters [46].
Based on the preclinical evidence that HDAC inhibitors induce HIF degradation, the combination of HDAC and VEGF inhibition has been recently tested. A clinical trial evaluated the combination of vorinostat (200mg twice daily for 2 weeks) and the VEGF blocker bevacizumab (15mg/kg IV ever 3 weeks) in patients with previously treated RCC. This combination was well tolerated and promising clinical benefit was observed. In 34 patients previously treated with a wide range of targeted therapies six objective responses were observed and nineteen patients had prolonged stable disease [47]. Future studies will evaluate the angiogenic and epigenetic signatures that may be associated with resistance to anti VEGF therapies and more suitable for treatments with HDAC inhibitors.
HDAC inhibitors and tyrosine kinase inhibitors
In vitro studies using renal tumor cell lines with different VHL status showed synergistic effects and an increase in caspase dependent apoptosis when cells were pretreated with the HDAC inhibitor belinostat followed by sorafenib[48]. The HDAC inhibitor VPA also acted in concert with another tyrosine kinase inhibitor, AEE788, to inhibit cell proliferation by Akt inhibition [49]. Combination of HDAC and VEGF receptor tyrosine kinase inhibition has been shown to be effective in inhibiting the HIF-VEGF axis suggesting a rational combination strategy to be tested in clinical trials [50].
HDAC inhibitors and differentiation agents
HDAC inhibitors have also been combined with differentiation agents. The retinoic acid receptor RAR-β2 is commonly methylated in epithelial tumors including RCC and retinoid resistance has been a major hurdle in differentiation therapy. RCC cell lines have been found to re-expressed following treatment with all-trans retinoic acid (ATRA) and TSA, with the combination having enhanced effects on inhibition of cell proliferation and tumor growth, in vitro and in vivo, respectively[51]. In another study, vorinostat and 13-cis retinoic acid (CRA) treatment of a different set of renal cell lines showed similar results with greater growth inhibition of cells in vitro and an induction of a different set of genes in the combination treated cells, including RAR-β2[52]. Re-expression of RAR-β2 in renal cells inhibited cell proliferation in vitro and tumor growth inhibition in vivo[53]. Entinostat treatment in vivo restored CRA sensitivity, in part through re-expression of RAR-β2, thus resulting in greater tumor growth inhibition with combination treatment[53]. In a Phase I clinical trial (NCT00098891) nineteen patients were treated with the combination of entinostat (4mg/m2 weekly) and CRA (1mg/kg daily). The treatment was well tolerated and though no objective responses were not observed, some patients including one with RCC benefitted from prolonged stables disease after failure of prior therapies [54]. A phase I/II trial (NCT-00324740)has tested the combination of vorinostat and CRA in patients with advanced renal cell carcinoma with evidence of clinical benefit in patients for refractory RCC[55]. These evidences support further testing of differentiation agents and HDAC inhibitors in RCC.
HDAC inhibitors and immunomodulation
Preclinical and clinical experience with HDAC inhibitors has suggested a possible immunomodulatory activity of these agents. The combination treatment of entinostat and high dose intereleukin-2 has shown a greater inhibitory effect in the RENCA model as compared to treatment with single agents with an observed decrease in regulatory T cells (Tregs) and increased cytotoxicity in the combination treated xenografts[56]. The combination also increased survival and reduced metastasis formation in this model [56]. Furthermore, Shen et al have reported that entinostat inhibited IL-2-induced Foxp3 induction and Tregs activity [57]. This study suggests that class I specific HDAC inhibition has greater inhibitory effect on Tregs than a pan HDAC inhibitor through the increased acetylation of STAT 3 and thereby its regulation of Foxp3[57]. Another study compared the in vitro and in vivo effects of single agents VPA and IFN-gamma to the combination treatments, and found that the combination induced enhanced tumor inhibition with greater acetylation of histone H3 and inhibition of HDAC activity[58]. An ongoing multicenter phase I/II clinical trial (NCT01038778) is studying the safety and tolerability of high dose IL-2 with class I HDAC inhibitor entinostat in patients with metastatic renal cell carcinomas. Preliminary data suggest acceptable toxicity and early signs of promising clinical benefit as compared to historical data[59]. Future preclinical and clinical studies will further exploit the immunomodulatory activity of HDAC inhibitors and will likely test novel combination strategies including immune checkpoint inhibitors and vaccines.
HDAC inhibitors and apoptosis inducing agents
Apoptosis was induced through induction of TRAIL and caspases on treatment of renal cell lines infected with Ad5-TRAIL and treated with class I specific HDAC inhibitors[60]. HDAC inhibitors, including romidepsin, vorinostat, and apcidicin decreased CXCL12 induced cell migration, with an upregulation of p21 and acetylated histone H3, classical markers of HDAC inhibitors[61]. In addition, class I specific HDAC inhibitors did not induce apoptosis in renal proximal tubular cells which led the authors to speculate that the HDAC inhibitors are selective for tumor cells and have less of an effect on normal cells[60]. Treatment of different cell lines with differential acetylated H3 status showed different sensitivity to romidepsin, with an induction of p21 either inducing cell cycle arrest or apoptosis [62]. Romidespin has been evaluated in a clinical trial as a monotherapy in refractory renal cell carcinoma. However, this treatment did not result in objective clinical response[63].
Methylation in RCC
Several reports have suggested the presence of numerous methylated genes in RCC[64–67]. In the analysis of 67 RCC specimens the lowest degree of methylation was associated with lower stage tumors [29]. sFRP2, an antagonist of the Wnt pathway has been found to be silenced in tumors as compared to non-tumor counterparts, as well as renal tumor cell lines when compared to normal human kidney cells [68]. sFRP2 is involved in anchorage independent growth, invasion and apoptosis. It was highly methylated at the promoter region and its expression was induced by treatment by the demethylating agent 5-Aza-dc, but with greater induction by TSA as it increased acetylation of histone H3 and H4 at the promoter regions[68, 69]. However, these effects of TSA and combination varied with different cell lines [69]. DLEC 1, a tumor suppressor gene, has been found to be silenced by methylation in gastric and colon cancer [70], and similar results was found in renal cell lines and tumors [71]. DLEC 1 is re-expressed in cell lines when treated with Aza and TSA and DLEC 1 re-expression reduced colony formation in renal cell lines [71]. The combination of vinblastine (a cytotoxic agent) and 5-Aza-dC induced expression of p21 with an accumulation of subG1 populations, reduction of anti-apoptotic gene Bcl2, as well as a reduction in tumor growth in xenografts[72]. Microarray analysis of cell lines treated with 5-Aza-Dc and TSA for 24 hours, revealed upregulation of several genes, including genes involved in migration, adhesion, and growth promoting pathways [73]. The authors also confirmed the methylation of these genes by methylation specific PCR of both the cell lines analyzed as well as 32 localized renal tumors [73]. In a study with a different set of renal cell lines, upon treatment with 5-Aza-Dc a large set of genes were upregulated and the some of these genes had tumor suppressor activity [74]. In a study of 51 patient clear cell RCC tumors, tumors were classified into two subgroups based on the methylation profiles of their adjacent non tumor tissue and one of the subgroups had tumors with more nodular, vascular and renal vein involvement and higher tumor stages and grades and higher tumor recurrence [75, 76]. Overall these data support the testing of demethylating agents and HDAC inhibitors in RCC. Further preclinical studies hopefully will identify the methylome signature most suitable for this therapeutic approach.
Conclusions
Alterations in histone status and DNA methylation occur in RCC and are rational targets for therapeutic interventions by using agents against enzymes regulating these changes. However, clinical translation of these agents has been challenging as the use of HDAC inhibitors as single agents has not yielded significant clinical benefit. However, the rational combination of different compounds and the identification of those patients with significant modifications in clinically relevant epigenetic enzymes will likely provide a beneficial effect. In addition, the utilization of histone and methylation changes as potential biomarkers of response to different therapies including antiangiogenics and immunotherapies is exciting. A greater understanding of the epigenetic alterations will provide new effective therapies for the treatment of RCC.
References
- 1.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dawson Mark a, Kouzarides T. Cancer Epigenetics: From Mechanism to Therapy. Cell. 2012;150(1):12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 3.Kristensen LS, Nielsen HM, Hansen LL. Epigenetics and cancer treatment. European Journal of Pharmacology. 2009;625(1–3):131–142. doi: 10.1016/j.ejphar.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 4.You Jueng s, Jones Peter a. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell. 2012;22(1):9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waldmann T, Schneider R. Targeting histone modifications—epigenetics in cancer. Current Opinion in Cell Biology. 2013;25(2):184–189. doi: 10.1016/j.ceb.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Acharya MR, Sparreboom A, Venitz J, Figg WD. Rational Development of Histone Deacetylase Inhibitors as Anticancer Agents: A Review. Molecular Pharmacology. 2005;68(4):917–932. doi: 10.1124/mol.105.014167. [DOI] [PubMed] [Google Scholar]
- 7.De Ruijter AJM, Van Gennip AH, Caron HN, Kemp S, Van Kuilenburg ABP. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370(3):737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1(3):194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Manero G, Issa J-P. Histone deacetylase inhibitors: a review of their clinical status as antineoplastic agents. Cancer Investigation. 2005;23(7):635–642. doi: 10.1080/07357900500283119. [DOI] [PubMed] [Google Scholar]
- 10.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6(1):38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 11.Dong G, Luo J, Kumar V, Dong Z. Inhibitors of histone deacetylases suppress cisplatin-induced p53 activation and apoptosis in renal tubular cells. American Journal of Physiology - Renal Physiology. 2010;298(2):F293–F300. doi: 10.1152/ajprenal.00410.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung YJ, Lee JE, Lee AS, et al. SIRT1 overexpression decreases cisplatin-induced acetylation of NF-κB p65 subunit and cytotoxicity in renal proximal tubule cells. Biochemical and Biophysical Research Communications. 2012;419(2):206–210. doi: 10.1016/j.bbrc.2012.01.148. [DOI] [PubMed] [Google Scholar]
- 13.Yoshikawa M, Hishikawa K, Marumo T, Fujita T. Inhibition of Histone Deacetylase Activity Suppresses Epithelial-to-Mesenchymal Transition Induced by TGF-β1 in Human Renal Epithelial Cells. Journal of the American Society of Nephrology. 2007;18(1):58–65. doi: 10.1681/ASN.2005111187. [DOI] [PubMed] [Google Scholar]
- 14.Sasahara RM, Brochado SM, Takahashi C, et al. Transcriptional control of the RECK metastasis/angiogenesis suppressor gene. Cancer Detection and Prevention. 2002;26(6):435–443. doi: 10.1016/s0361-090x(02)00123-x. [DOI] [PubMed] [Google Scholar]
- 15.Lee KJ, Lee KY, Lee YM. Downregulation of a tumor suppressor RECK by hypoxia through recruitment of HDAC1 and HIF-1α to reverse HRE site in the promoter. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2010;1803(5):608–616. doi: 10.1016/j.bbamcr.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Seligson DB, Horvath S, Mcbrian MA, et al. Global Levels of Histone Modifications Predict Prognosis in Different Cancers. The American Journal of Pathology. 2009;174(5):1619–1628. doi: 10.2353/ajpath.2009.080874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mosashvilli D, Kahl P, Mertens C, et al. Global histone acetylation levels: Prognostic relevance in patients with renal cell carcinoma. Cancer Science. 2010;101(12):2664–2669. doi: 10.1111/j.1349-7006.2010.01717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellinger J, Kahl P, Mertens C, et al. Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. International Journal of Cancer. 2010;127(10):2360–2366. doi: 10.1002/ijc.25250. [DOI] [PubMed] [Google Scholar]
- 19.Rogenhofer S, Kahl P, Mertens C, et al. Global histone H3 lysine 27 (H3K27) methylation levels and their prognostic relevance in renal cell carcinoma. BJU International. 2012;109(3):459–465. doi: 10.1111/j.1464-410X.2011.10278.x. [DOI] [PubMed] [Google Scholar]
- 20.Shen Y, Guo X, Wang Y, et al. Expression and significance of histone H3K27 demethylases in renal cell carcinoma. BMC Cancer. 2012;12(1):470. doi: 10.1186/1471-2407-12-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Zhang R, Wu D, et al. Epigenetic change in kidney tumor: downregulation of histone acetyltransferase MYST1 in human renal cell carcinoma. Journal of Experimental & Clinical Cancer Research. 2013;32(1):8. doi: 10.1186/1756-9966-32-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duns G, Van Den Berg E, Van Duivenbode I, et al. Histone Methyltransferase Gene SETD2 Is a Novel Tumor Suppressor Gene in Clear Cell Renal Cell Carcinoma. Cancer Research. 2010;70(11):4287–4291. doi: 10.1158/0008-5472.CAN-10-0120. [DOI] [PubMed] [Google Scholar]
- 23.Duns G, Hofstra RMW, Sietzema JG, et al. Targeted exome sequencing in clear cell renal cell carcinoma tumors suggests aberrant chromatin regulation as a crucial step in ccRCC development. Human Mutation. 2012;33(7):1059–1062. doi: 10.1002/humu.22090. [DOI] [PubMed] [Google Scholar]
- 24.Pena-Llopis S, Vega-Rubin-De-Celis S, Liao A, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012;44(7):751–759. doi: 10.1038/ng.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varela I, Tarpey P, Raine K, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469(7331):539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pawłowski R, Mühl SM, Sulser T, Krek W, Moch H, Schraml P. Loss of PBRM1 expression is associated with renal cell carcinoma progression. International Journal of Cancer. 2013;132(2):E11–E17. doi: 10.1002/ijc.27822. [DOI] [PubMed] [Google Scholar]
- 27.Dalgliesh GL, Furge K, Greenman C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minardi D, Lucarini G, Filosa A, et al. Prognostic role of global DNA-methylation and histone acetylation in pT1a clear cell renal carcinoma in partial nephrectomy specimens. Journal of Cellular and Molecular Medicine. 2009;13(8b):2115–2121. doi: 10.1111/j.1582-4934.2008.00482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Avissar-Whiting M, Koestler DC, Houseman EA, Christensen BC, Kelsey KT, Marsit CJ. Polycomb group genes are targets of aberrant DNA methylation in renal cell carcinoma. Epigenetics. 2011;6(6):703–709. doi: 10.4161/epi.6.6.16158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fritzsche F, Weichert W, Roske A, et al. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer. 2008;8(1):381. doi: 10.1186/1471-2407-8-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato A, Asano T, Horiguchi A, Ito K, Sumitomo M, Asano T. Combination of Suberoylanilide Hydroxamic Acid and Ritonavir is Effective Against Renal Cancer Cells. Urology. 2010;76(3):764.e767–764.e713. doi: 10.1016/j.urology.2010.04.042. [DOI] [PubMed] [Google Scholar]
- 32.Sato A, Asano T, Ito K, Asano T. Ritonavir Interacts With Bortezomib to Enhance Protein Ubiquitination and Histone Acetylation Synergistically in Renal Cancer Cells. Urology. 2012;79(4):966.e913–966.e921. doi: 10.1016/j.urology.2011.11.033. [DOI] [PubMed] [Google Scholar]
- 33.Sato A, Asano T, Ito K, Sumitomo M, Asano T. Suberoylanilide hydroxamic acid (SAHA) combined with bortezomib inhibits renal cancer growth by enhancing histone acetylation and protein ubiquitination synergistically. BJU International. 2012;109(8):1258–1268. doi: 10.1111/j.1464-410X.2011.10533.x. [DOI] [PubMed] [Google Scholar]
- 34.Juengel E, Dauselt A, Makarević J, et al. Acetylation of histone H3 prevents resistance development caused by chronic mTOR inhibition in renal cell carcinoma cells. Cancer Letters. 2012;324(1):83–90. doi: 10.1016/j.canlet.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 35.Mahalingam D, Medina EC, Esquivel JA, et al. Vorinostat Enhances the Activity of Temsirolimus in Renal Cell Carcinoma Through Suppression of Survivin Levels. Clinical Cancer Research. 2010;16(1):141–153. doi: 10.1158/1078-0432.CCR-09-1385. [DOI] [PubMed] [Google Scholar]
- 36.Juengel E, Makarević J, Tsaur I, et al. Resistance after Chronic Application of the HDAC-Inhibitor Valproic Acid Is Associated with Elevated Akt Activation in Renal Cell Carcinoma In Vivo. PLoS ONE. 2013;8(1):e53100. doi: 10.1371/journal.pone.0053100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qian DZ, Kachhap SK, Collis SJ, et al. Class II Histone Deacetylases Are Associated with VHL-Independent Regulation of Hypoxia-Inducible Factor 1α. Cancer Research. 2006;66(17):8814–8821. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 38.Verheul HMW, Salumbides B, Van Erp K, et al. Combination Strategy Targeting the Hypoxia Inducible Factor-1α with Mammalian Target of Rapamycin and Histone Deacetylase Inhibitors. Clinical Cancer Research. 2008;14(11):3589–3597. doi: 10.1158/1078-0432.CCR-07-4306. [DOI] [PubMed] [Google Scholar]
- 39.Dioum EM, Chen R, Alexander MS, et al. Regulation of Hypoxia-Inducible Factor 2α Signaling by the Stress-Responsive Deacetylase Sirtuin 1. Science. 2009;324(5932):1289–1293. doi: 10.1126/science.1169956. [DOI] [PubMed] [Google Scholar]
- 40.Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P. The Histone Demethylases JMJD1A and JMJD2B Are Transcriptional Targets of Hypoxia-inducible Factor HIF. Journal of Biological Chemistry. 2008;283(52):36542–36552. doi: 10.1074/jbc.M804578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wellmann S, Bettkober M, Zelmer A, et al. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochemical and Biophysical Research Communications. 2008;372(4):892–897. doi: 10.1016/j.bbrc.2008.05.150. [DOI] [PubMed] [Google Scholar]
- 42.Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ. Regulation of the Histone Demethylase JMJD1A by Hypoxia-Inducible Factor 1α Enhances Hypoxic Gene Expression and Tumor Growth. Molecular and Cellular Biology. 2010;30(1):344–353. doi: 10.1128/MCB.00444-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu J, Wang B, Xu Y, et al. Epigenetic regulation of HIF-1[alpha] in renal cancer cells involves HIF-1[alpha]/2[alpha] binding to a reverse hypoxia-response element. Oncogene. 2012;31(8):1065–1072. doi: 10.1038/onc.2011.305. [DOI] [PubMed] [Google Scholar]
- 44.Vanharanta S, Shu W, Brenet F, et al. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat Med. 2013;19(1):50–56. doi: 10.1038/nm.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 46.Kvasha S, Gordiyuk V, Kondratov A, et al. Hypermethylation of the 5′CpG island of the FHIT gene in clear cell renal carcinomas. Cancer Letters. 2008;265(2):250–257. doi: 10.1016/j.canlet.2008.02.036. [DOI] [PubMed] [Google Scholar]
- 47.Pili R, Lodge M, Verhuel H, et al. Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in pretreated patients with renal cell carcinoma: safety, efficacy, and pharmacodynamic results. ASCO, GU meeting 2010 genitourinary cancers symposium; 2010. [Google Scholar]
- 48.Kim MJ, Kim DE, Jeong IG, et al. HDAC Inhibitors Synergize Antiproliferative Effect of Sorafenib in Renal Cell Carcinoma Cells. Anticancer Research. 2012;32(8):3161–3168. [PubMed] [Google Scholar]
- 49.Juengel E, Engler J, Mickuckyte A, et al. Effects of combined valproic acid and the epidermal growth factor/vascular endothelial growth factor receptor tyrosine kinase inhibitor AEE788 on renal cell carcinoma cell lines in vitro. BJU International. 2010;105(4):549–557. doi: 10.1111/j.1464-410X.2009.08759.x. [DOI] [PubMed] [Google Scholar]
- 50.Qian DZ, Wang X, Kachhap SK, et al. The Histone Deacetylase Inhibitor NVP-LAQ824 Inhibits Angiogenesis and Has a Greater Antitumor Effect in Combination with the Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitor PTK787/ZK222584. Cancer Research. 2004;64(18):6626–6634. doi: 10.1158/0008-5472.CAN-04-0540. [DOI] [PubMed] [Google Scholar]
- 51.Touma SE, Goldberg JS, Moench P, et al. Retinoic Acid and the Histone Deacetylase Inhibitor Trichostatin A Inhibit the Proliferation of Human Renal Cell Carcinoma in a Xenograft Tumor Model. Clinical Cancer Research. 2005;11(9):3558–3566. doi: 10.1158/1078-0432.CCR-04-1155. [DOI] [PubMed] [Google Scholar]
- 52.Tavares TS, Nanus D, Yang XJ, Gudas LJ. Gene microarray analysis of human renal cell carcinoma: The effects of HDAC inhibition and retinoid treatment. Cancer Biology & Therapy. 2008;7(10):1607–1618. doi: 10.4161/cbt.7.10.6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X-F, Qian DZ, Ren M, et al. Epigenetic Modulation of Retinoic Acid Receptor β2 by the Histone Deacetylase Inhibitor MS-275 in Human Renal Cell Carcinoma. Clinical Cancer Research. 2005;11(9):3535–3542. doi: 10.1158/1078-0432.CCR-04-1092. [DOI] [PubMed] [Google Scholar]
- 54.Pili R, Salumbides B, Zhao M, et al. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br J Cancer. 2012;106(1):77–84. doi: 10.1038/bjc.2011.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nanus D, Tagawa S, Dutcher Jp, et al. A phase I trial of suberoylanilide hydroxamic acid (SAHA) and 13-cis trinoic acid in the treatment of patients with advcanced renal cell carcinoma (RCC) Journal of Clinical Oncology. 2011;29(7):abstract 349. [Google Scholar]
- 56.Kato Y, Yoshimura K, Shin T, et al. Synergistic In vivo Antitumor Effect of the Histone Deacetylase Inhibitor MS-275 in Combination with Interleukin 2 in a Murine Model of Renal Cell Carcinoma. Clinical Cancer Research. 2007;13(15):4538–4546. doi: 10.1158/1078-0432.CCR-07-0014. [DOI] [PubMed] [Google Scholar]
- 57.Shen L, Ciesielski M, Ramakrishnan S, et al. Class I Histone Deacetylase Inhibitor Entinostat Suppresses Regulatory T Cells and Enhances Immunotherapies in Renal and Prostate Cancer Models. PLoS ONE. 2012;7(1):e30815. doi: 10.1371/journal.pone.0030815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones J, Juengel E, Mickuckyte A, et al. Valproic acid blocks adhesion of renal cell carcinoma cells to endothelium and extracellular matrix. Journal of Cellular and Molecular Medicine. 2009;13(8b):2342–2352. doi: 10.1111/j.1582-4934.2008.00603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pili R, Shen L, George S, et al. Phase I study of high-dose interleukin 2, aldesleukin, in combination with the histone deacetylase inhibitor, entinostat, in patients with metastatic renal cell carcinoma: Safety and tolerability results. Journal of Clinical Oncology (ASCO) (supplement 6):Abstract 369. (2013 Genitourinary Cancers Symposium) [Google Scholar]
- 60.Vanoosten RL, Earel JK, Jr, Griffith TS. Enhancement of Ad5-TRAIL cytotoxicity against renal cell carcinoma with histone deacetylase inhibitors. Cancer Gene Ther. 2006;13(6):628–632. doi: 10.1038/sj.cgt.7700939. [DOI] [PubMed] [Google Scholar]
- 61.Ierano C, Basseville A, To KKW, et al. Histone deacetylase inhibitors induce CXCR4 mRNA but antagonize CXCR4 migration. Cancer Biology & Therapy. 2013;14(2):175–183. doi: 10.4161/cbt.22957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanao K, Mikami S, Mizuno R, Shinojima T, Murai M, Oya M. Decreased acetylation of histone H3 in renal cell carcinoma: a potential target of histone deacetylase inhibitors. The Journal of urology. 2008;180(3):1131–1136. doi: 10.1016/j.juro.2008.04.136. [DOI] [PubMed] [Google Scholar]
- 63.Stadler WM, Margolin K, Ferber S, Mcculloch W, Thompson JA. A Phase II Study of Depsipeptide in Refractory Metastatic Renal Cell Cancer. Clinical Genitourinary Cancer. 2006;5(1):57–60. doi: 10.3816/CGC.2006.n.018. [DOI] [PubMed] [Google Scholar]
- 64.Morris M, Maher E. Epigenetics of renal cell carcinoma: the path towards new diagnostics and therapeutics. Genome Med. 2010;2(9):59. doi: 10.1186/gm180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wozniak MB, Le Calvez-Kelm F, Abedi-Ardekani B, et al. Integrative Genome-Wide Gene Expression Profiling of Clear Cell Renal Cell Carcinoma in Czech Republic and in the United States. PLoS ONE. 2013;8(3):e57886. doi: 10.1371/journal.pone.0057886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ricketts CJ, Morris MR, Gentle D, et al. Genome-wide CpG island methylation analysis implicates novel genes in the pathogenesis of renal cell carcinoma. Epigenetics. 2012;7(3):278–290. doi: 10.4161/epi.7.3.19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arai E, Chiku S, Mori T, et al. Single-CpG-resolution methylome analysis identifies clinicopathologically aggressive CpG island methylator phenotype clear cell renal cell carcinomas. Carcinogenesis. 2012;33(8):1487–1493. doi: 10.1093/carcin/bgs177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kawakami K, Yamamura S, Hirata H, et al. Secreted frizzled-related protein-5 is epigenetically downregulated and functions as a tumor suppressor in kidney cancer. International Journal of Cancer. 2011;128(3):541–550. doi: 10.1002/ijc.25357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kawamoto K, Hirata H, Kikuno N, Tanaka Y, Nakagawa M, Dahiya R. DNA methylation and histone modifications cause silencing of Wnt antagonist gene in human renal cell carcinoma cell lines. International Journal of Cancer. 2008;123(3):535–542. doi: 10.1002/ijc.23514. [DOI] [PubMed] [Google Scholar]
- 70.Ying J, Poon FF, Yu J, et al. DLEC1 is a functional 3p22.3 tumour suppressor silenced by promoter CpG methylation in colon and gastric cancers. Br J Cancer. 2009;100(4):663–669. doi: 10.1038/sj.bjc.6604888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Q, Ying J, Li J, et al. Aberrant Promoter Methylation of DLEC1, a Critical 3p22 Tumor Suppressor for Renal Cell Carcinoma, is Associated With More Advanced Tumor Stage. The Journal of urology. 2010;184(2):731–737. doi: 10.1016/j.juro.2010.03.108. [DOI] [PubMed] [Google Scholar]
- 72.Iwata H, Sato H, Suzuki R, et al. A demethylating agent enhances chemosensitivity to vinblastine in a xenograft model of renal cell carcinoma. Int J Oncol. 2011;38(6):1653–1661. doi: 10.3892/ijo.2011.999. [DOI] [PubMed] [Google Scholar]
- 73.Ibanez De Caceres I, Dulaimi E, Hoffman AM, Al-Saleem T, Uzzo RG, Cairns P. Identification of Novel Target Genes by an Epigenetic Reactivation Screen of Renal Cancer. Cancer Research. 2006;66(10):5021–5028. doi: 10.1158/0008-5472.CAN-05-3365. [DOI] [PubMed] [Google Scholar]
- 74.Morris MR, Gentle D, Abdulrahman M, et al. Functional epigenomics approach to identify methylated candidate tumour suppressor genes in renal cell carcinoma. Br J Cancer. 2008;98(2):496–501. doi: 10.1038/sj.bjc.6604180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arai E, Kanai Y. Genetic and epigenetic alterations during renal carcinogenesis. Int J Clin Exp Pathol. 2010;4(1):58–73. [PMC free article] [PubMed] [Google Scholar]
- 76.Arai E, Ushijima S, Fujimoto H, et al. Genome-wide DNA methylation profiles in both precancerous conditions and clear cell renal cell carcinomas are correlated with malignant potential and patient outcome. Carcinogenesis. 2009;30(2):214–221. doi: 10.1093/carcin/bgn268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grønbaek K, Hother C, Jones PA. Epigenetic changes in cancer. APMIS: Acta Pathologica, Microbiologica, Et Immunologica Scandinavica. 2007;115(10):1039–1059. doi: 10.1111/j.1600-0463.2007.apm_636.xml.x. [DOI] [PubMed] [Google Scholar]
- 78.Tsai H-C, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Cell Res. 2011;21(3):502–517. doi: 10.1038/cr.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laird PW. Cancer epigenetics. Human Molecular Genetics. 2005;14(suppl 1):R65–R76. doi: 10.1093/hmg/ddi113. [DOI] [PubMed] [Google Scholar]
- 80.Lund AH, Van Lohuizen M. Epigenetics and cancer. Genes & Development. 2004;18(19):2315–2335. doi: 10.1101/gad.1232504. [DOI] [PubMed] [Google Scholar]
- 81.Suvà ML, Riggi N, Bernstein BE. Epigenetic Reprogramming in Cancer. Science. 2013;339(6127):1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang H, Pinello CE, Luo J, et al. Small-Molecule Inhibitors of Acetyltransferase p300 Identified by High-Throughput Screening Are Potent Anticancer Agents. Molecular Cancer Therapeutics. 2013;12(5):610–620. doi: 10.1158/1535-7163.MCT-12-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Spannhoff A, Hauser A-T, Heinke R, Sippl W, Jung M. The Emerging Therapeutic Potential of Histone Methyltransferase and Demethylase Inhibitors. Chem Med Chem. 2009;4(10):1568–1582. doi: 10.1002/cmdc.200900301. [DOI] [PubMed] [Google Scholar]